

Abstract
Background and Purpose
Sphingosine 1‐phosphate (S1P), an important inflammatory mediator, has been shown to regulate COX‐2 production and promote various cellular responses such as cell migration. Mevastatin, an inhibitor of 3‐hydroxy‐3‐methylglutaryl‐CoA reductase (HMG‐CoA), effectively inhibits inflammatory responses. However, the mechanisms underlying S1P‐evoked COX‐2‐dependent cell migration, which is modulated by mevastatin in human tracheal smooth muscle cells (HTSMCs) remain unclear.
Experimental Approach
The expression of COX‐2 was determined by Western blotting, real time‐PCR and promoter analyses. The signalling molecules were investigated by pretreatment with respective pharmacological inhibitors or transfection with siRNAs. The interaction between COX‐2 promoter and transcription factors was determined by chromatin immunoprecipitation assay. Finally, the effect of mevastatin on HTSMC migration and leukocyte counts in BAL fluid and COX‐2 expression induced by S1P was determined by a cell migration assay, cell counting and Western blot.
Key Results
S1P stimulated mTOR activation through the Nox2/ROS and PI3K/Akt pathways, which can further stimulate FoxO1 phosphorylation and translocation to the cytosol. We also found that S1P induced CREB activation and translocation via an mTOR‐independent signalling pathway. Finally, we showed that pretreatment with mevastatin markedly reduced S1P‐induced cell migration and COX‐2/PGE2 production via a PPARγ‐dependent signalling pathway.
Conclusions and Implications
Mevastatin attenuates the S1P‐induced increased expression of COX‐2 and cell migration via the regulation of FoxO1 and CREB phosphorylation and translocation by PPARγ in HTSMCs. Mevastatin could be beneficial for prevention of airway inflammation in the future.
Abbreviations
- BAL
bronchoalveolar lavage
- BCECF/AM
2′,7′‐bis‐(2‐carboxyethyl)‐5‐(and‐6)‐carboxyfluorescein, acetoxymethyl ester
- ChIP
chromatin immunoprecipitation
- DCFH‐DA
2′,7′‐dichlorodihydrofluorescein diacetate
- DHE
dihydroethidium
- DPI
diphenyleneiodonium chloride
- HTSMCs
human tracheal smooth muscle cells
- Nox
NADPH oxidase
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| S1P1 receptor | Akt (PKB) |
| S1P3 receptor | COX‐2 |
| Nuclear hormone receptors b | HO‐1 |
| ERα | mTOR |
| PPARγ | PI3K |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,2013b,2013c).
Introduction
Airway smooth muscle is considered as an end‐response effector regulating regional differences in ventilation by contracting in response to various pro‐inflammatory mediators and exogenous substances released under homeostatic or pathological conditions, such as asthma (Lee et al., 2008). Sphingosine1‐phosphate (S1P), a bioactive sphingolipid metabolite, regulates numerous cellular responses, including motility and cytoskeletal re‐arrangements, formation of adheres junctions, proliferation, survival, angiogenesis and the trafficking of immune cells (Ryan and Spiegel, 2008), and it plays an important role in asthma (Ammit et al., 2001). S1P was found to be increased in the bronchoalveolar lavage (BAL) fluid of asthmatics following allergen challenge (Ammit et al., 2001) and induces allergic inflammation and bronchial hyperresponsiveness in murine ‘models’ of the disease (Kleinjan et al., 2013; Price et al., 2013). Previously, we demonstrated that S1P‐induced IL‐6 secretion, not only a parameter for exacerbated inflammation, but also indicative of potential lung damage, was mediated through the COX‐2/PGE2 pathway in HTSMCs (Hsu et al., 2015). Therefore, we suggested that S1P‐induced COX‐2/PGE2 generation may enhance inflammatory responses and exacerbate airway damage. On the other hand, COX is the rate‐limiting enzyme for the production of eicosanoids PGs and thromboxanes from free arachidonic acid (AA), which is generated from membrane phospholipids by PLA2 (Lee et al., 2010). The transient expression of COX‐2 is transcriptionally regulated by various stimuli, including S1P (Nodai et al., 2007; Volzke et al., 2014). An increased expression of COX‐2 and PG production are involved in the inflammatory processes. Therefore, elucidating the signalling mechanisms underlying COX‐2 gene regulation and PGE2 secretion will be beneficial for the development of therapeutic, anti‐inflammatory strategies.
Inflammatory airway and lung diseases are characterized by chronic inflammation and an oxidant/antioxidant imbalance. ROS are widely recognized as important mediators of cell growth, adhesion, differentiation, senescence and apoptosis (Lee and Yang, 2012). Excessive production of ROS (termed ‘oxidative stress’) by NADPH oxidase (Nox) is commonly thought to be responsible for tissue injuries associated with respiratory inflammatory diseases (Lee and Yang, 2012). The induction of COX‐2 is mediated through Nox activation and ROS production induced by various stimuli (Lin et al., 2010; Lee et al., 2013). Recent studies have suggested that the PI3K/Akt pathway plays a crucial role in the expression of inflammatory mediators, inflammatory cell recruitment, immune cell function, airway remodelling and corticosteroid insensitivity in chronic inflammatory respiratory diseases (Lee and Yang, 2012). Our previous study indicated that ATPγS up‐regulates COX‐2 expression and PGE2 release via PI3K/Akt activation (Lin et al., 2012). Mammalian target of rapamycin (mTOR) protein complex functions as a hub for the integration of various intracellular signalling pathways involving proliferation, cell survival and angiogenesis (Oya, 2010). PGE2 also mediates prostate cancer cell migration and invasion through activation of the PI3K/Akt/mTOR pathway (Vo et al., 2013).
The forkhead box O (FoxO) family, downstream of Akt, in particular FoxO1 has been shown to be involved in various cellular functions, such as gluconeogenesis, cell cycle and autophagy (Tikhanovich et al., 2013). Recently, FoxO1 has been shown to participate in IL‐1β‐induced cytokine production and increased COX‐2 expression (Lappas, 2013). cAMP response element‐binding protein (CREB) has been shown to play a critical role in the transcriptional activation of the COX‐2 gene in the early phase of adipogenesis (Fujimori et al., 2014). Whether these signalling components also participate in the increased expression of COX‐2 and PGE2 release in HTSMCs challenged with S1P remains unknown.
Statins are a class of drugs used to lower cholesterol levels by inhibiting the enzyme HMG‐CoA reductase, which plays a central role in the production of cholesterol in the liver. Statins also have pleiotropic effects, including an improvement in bone metabolism, immunomodulatory, anti‐oxidant and anti‐inflammatory effects (Osmak, 2012; Shao et al., 2012). Statins have been shown to ameliorate the proliferation and migration of cells, the expression of inflammatory proteins and development of pulmonary hypertension via the up‐regulation of haem oxygenase‐1 (HO‐1), p21 or PPARs (Esposito et al., 2012; Li et al., 2012). Previous studies indicated that statin use was associated with improved asthma control in patients with severe asthma (Maneechotesuwan et al., 2010; Braganza et al., 2011). However, the mechanisms of statin‐attenuated S1P‐induced inflammatory responses are still unclear in HTSMCs.
In this study we found that the increased expression of COX‐2 and PGE2 release in S1P‐challenged HTSMCs are mediated through Nox2/ROS‐ and PI3K/Akt‐dependent mTOR/FoxO1 and CREB phosphorylation. Moreover, mevastatin was shown to inhibit S1P‐induced COX‐2/PGE2‐dependent cell migration via PPARγ in HTSMCs. These results provide new insights into the mechanisms by which mevastatin affects cell migration and thus alleviates the inflammatory response in HTSMCs.
Methods
Cell culture
HTSMCs were purchased from ScienCell Research Lab (San Diego, CA) and grown in DMEM/F‐12 containing 10% (v v‐1) FBS and antibiotics (100 U·mL−1 penicillin G, 100 µg·mL−1 streptomycin and 250 ng·mL−1 fungizone) at 37°C in a humidified 5% CO2. Experiments were performed with cells from passages 4 to 7.
Transient transfection with siRNAs
Human siRNAs of scrambled, p47phox (SASI_Hs02_00302212), Nox2 (SASI_Hs01_00086110), p110 (SASI_Hs01_00219338), Akt (SASI_Hs01_00205545), mTOR (SASI_Hs01_00203144), CREB (SASI_Hs01_00116985), FoxO1 (SASI_Hs01_00076732) and PPARγ (SASI_Hs01_00106498) were from Sigma (St. Louis, MO). Transient transfection of siRNAs was carried out using Lipofectamine 2000 transfection reagent from Invitrogen (Carlsbad, CA). siRNA (100 nM) was formulated with Lipofectamine 2000 transfection reagent according to the manufacturer's instruction.
Western blot
Growth‐arrested cells were incubated with S1P at 37°C for the indicated time intervals. The cells were washed, scraped, collected and centrifuged at 45 000× g at 4°C for 1 h to yield the whole cell extract, as previously described (Hsu et al., 2014). Samples were denatured, subjected to SDS‐PAGE using a 10% running gel, and transferred to nitrocellulose membrane. Membranes were incubated with an anti‐COX‐2 antibody for 24 h, and then membranes were incubated with an anti‐mouse HRP antibody for 1 h. The immunoreactive bands were detected by ECL reagents. The values of Western blot are quantified by UN‐SCAN‐IT gel version 6.1 (Utah, USA) and normalized to β‐actin for five individual experiments. The blot is labelled with fold change values which are compared to the basal.
Real‐time PCR
Total RNA was extracted using TRIzol reagent. To avoid the genomic DNA contamination, mRNA was reverse‐transcribed into cDNA with oligo‐dT primer and analysed by real‐time PCR using SYBR Green PCR reagents (Applied Biosystems, Branchburg, NJ) with primers specific for COX‐2 and GAPDH. The primers used were as follows: 5′‐CAAACTGAAATTTGACCCAGAACTAC‐3′ (sense), and 5′‐ACTGTTGATAGTTGTATTTCTGGTCATGA‐3′ (anti‐sense) for COX‐2; 5′‐GCCAGCCGAGCCACAT‐3′ (sense) and 5′‐CTTTACCAGAGTTAAAAGCAGCCC‐3′ (anti‐sense) for GAPDH. The level of COX‐2 expression was determined by normalizing it to that of GAPDH expression. The CT values of COX‐2 and GAPDH were used to measure the ΔCT.
Measurement of COX‐2 promoter activity
For construction of the COX‐2‐luc plasmid, human COX‐2 promoter, a region spanning −483 to +37 bp was cloned into pGL3‐basic vector (Promega, Madison, WI). The 5′‐flanking region of the human COX‐2 gene was cloned using the Genome Walker kit (Clontech, Heidelberg, Germany) with internal (upstream) and external (downstream) primers from the human COX‐2 cDNA as followed: COX‐2 internal primer: 5′‐GGTACCGACGTACAGACCAGACACGG‐3′ (with a KpnI site), COX‐2 external primer: 5′‐CTCGAGGTGCTCCTGACGCTCACTGC‐3′ (with an XHoI site). The point mutation in the CREB binding site of COX‐2 promoter was generated by site‐directed mutagenesis that splices by overlap extension. To generate CREB mut‐luc, two PCR fragments (A and B) were amplified from COX‐2‐luc. Fragment A‐CRE was generated from a forward primer with a KpnI site (5′‐GGTACCGACGTACAGACCAGACACGG‐3′) and reverse primer (5′‐CAGTCATTTAATCACATGGG‐3′). Fragment B‐CRE was generated from a forward primer (5′‐CCCATGTGATTAAATGACTG‐3′) and reverse primer with an XhoI site (5′‐CTCGAGGTGCTCCTGACGCTCACTGC‐3′). Fragment A was combined with B, and this combination was amplified by PCR. The combination of fragments was inserted into pCRII‐TOPO (Invitrogen, CA, USA) and then was digested with KpnI and XhoI. The digested product was inserted into KpnI/XhoI‐digested pGL3‐basic vector. The resulting plasmid introduced the CREB mutation (the mutated bases are underscored: TTCGTCA → TTAATCA). COX‐2‐luc activity was determined as previously described using a luciferase assay system (Promega, Madison, WI) (Hsu et al., 2015). Firefly luciferase activities were standardized for β‐gal activity.
Measurement of intracellular ROS accumulation
The intracellular H2O2 levels were determined by measuring fluorescence of 2′, 7′‐dichlorodihydrofluorescin diacetate (DCFH‐DA), and the O2 •− levels were determined by measuring the level of dihydroethidium (DHE). The fluorescence for DCF and DHE staining was detected at 495/529 and 518/605 nm, respectively, using a fluorescence microscope (Zeiss, Axiovert 200M). For the purpose of these experiments, HTSMCs were washed with warm HBSS and incubated in HBSS or cell medium containing 10 μM DCFH‐DA or DHE at 37°C for 45 min. Subsequently, HBSS or medium containing DCFH‐DA or DHE was removed and replaced with fresh medium. HTSMCs were then incubated with S1P (30 μM). Cells were washed twice with PBS and detached with trypsin/EDTA, and the fluorescence intensity of the cells was analysed using a FACScan flow cytometer (BD Biosciences, San Jose, CA) at 518 nm excitation and 605 nm emission for DHE and at 495 nm excitation and 529 nm emission for DCF.
Determination of NADPH oxidase activity by chemiluminescence assay
After being incubated, cells were gently scraped and centrifuged at 400× g for 10 min at 4°C. The cell pellet was resuspended with 35 μL per well of ice‐cold RPMI‐1640 medium, and the cell suspension was kept on ice. Then, 5 μL of cell suspension (0.2 × 105 cells) were added to a final 200 μL volume of pre‐warmed (37°C) RPMI‐1640 medium containing either NADPH (1 μM) or lucigenin (20 μM), to initiate the reaction, followed by immediate measurement of chemiluminescence in an Appliskan luminometer (Thermo®) in out‐of‐coincidence mode. Appropriate blanks and controls were established, and chemiluminescence was recorded. Neither NADPH nor NADH enhanced the background chemiluminescence of lucigenin alone (30–40 counts per min). Chemiluminescence was continuously measured for 12 min, and the activity of NADPH oxidase was expressed as counts 10‐6cells.
Isolation of cell fractions
Cells were harvested, sonicated for 5 s at output 1.5 with a sonicator (Misonix, Farmingdale, NY) and centrifuged at 6800× g for 15 min at 4°C. The pellet was collected as the nuclear fraction. The supernatant was centrifuged at 20,000× g at 4°C for 60 min to yield the pellet (membrane fraction) and the supernatant (cytosolic fraction). FoxO1 translocation from nucleus to cytosol and CREB translocation from cytosol to nucleus were determined by Western blot.
Chromatin immunoprecipitation assay
To detect the in vivo association of transcription factors with human COX‐2 promoter, chromatin immunoprecipitation (ChIP) analysis was performed as previously described (Yang et al., 2012). Briefly, HTSMCs were cross‐linked with 1% formaldehyde at 37°C for 10 min and washed thrice with ice‐cold PBS containing 1 mM PMSF and 1% aprotinin. Soluble chromatin was prepared using a ChIP assay kit (Upstate) according to the manufacturer's recommendations and immunoprecipitated without (control) or with an anti‐FoxO1 or anti‐CREB antibody and normal goat immunoglobulin G (IgG). Following washing and elution, immunoprecipitates were heated overnight at 65°C to reverse cross‐linking of DNA and protein. DNA fragments were purified by phenol‐chloroform extraction and ethanol precipitation. The purified DNA was subjected to PCR amplification using the forward primer 5′‐CACCGGGCTTACGCAATTTT‐3′ and the reverse primer 5′‐ACGCTCACTGCAAGTCGTAT‐3′, which were specifically designed from the COX‐2 promoter region (−139 to +29). PCR fragments were analysed on 2% agarose in 1× TAE gel containing ethidium bromide.
Cell migration assay
HTSMCs were grown to confluence in 6‐well plates and starved by incubating them in serum‐free DMEM/F‐12 medium for 24 h. The monolayer cells were manually scratched with a pipette tip to create extended and definite scratches in the centre of the well with a bright and clear field. The detached cells were removed by washing the cells once with PBS. Serum‐free DMEM/F‐12 medium plus 1 μM hydroxyurea with or without S1P was added to each well as indicated after pretreatment with the inhibitors for 1 h or mevastatin for 24 h. Images of migratory cells from the scratched boundary were observed and recorded at 48 h with a digital camera and light microscope (Olympus, Japan). Number of migratory cells was counted from the resulting four phase images for each point and then averaged for each experimental condition. The images shown represent one of three individual experiments.
Animal care and experimental procedures
Male ICR mice aged 6–8 weeks were purchased from the National Laboratory Animal Centre (Taipei, Taiwan) and handled according to the guidelines of Animal Care Committee of Chang Gung University and NIH Guides for the Care and Use of Laboratory Animals. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 20 animals were used in the experiments described here ICR mice were pretreated with mevastatin (4 mg·kg−1 body weight) by i.p. injection for 24 h, and then anaesthetized by i.p. injection of 200 μL of pentobarbital sodium (5 mg·mL−1), placed individually on a board in a near vertical position and the tongues were withdrawn with a lined forceps. The depth of anaesthesia was evaluated by pinching the animal's paw with forceps and all efforts were made to minimize suffering. S1P (0.03 mg·kg−1 body weight) was placed posterior in the throat and aspirated into lungs. Control mice were administered sterile 0.1% BSA. Mice regained consciousness after 30 min. BAL fluid was obtained through a tracheal cannula using 1 mL aliquots of ice‐cold PBS solution. BAL fluid was centrifuged at 500× g at 4°C, and cell pellets were washed and re‐suspended in PBS. Leukocyte count was determined by a haemocytometer.
Analysis of data
All the data are expressed as the mean or mean ± SEM of five individual experiments performed in duplicate or triplicate. The significance of differences between two groups was determined by Student's paired two‐tailed t‐test for Western blot data. For all other statistical analyses and comparisons of multiple groups, a GraphPad Prism Program (GraphPad, San Diego, CA) using ANOVA followed by Tukey's post hoc test has been used. A P < 0.05 value was considered significant.
Materials
Anti‐COX‐2 antibody was from Abcam (ab62331) (Burlingame, CA). Anti‐p47phox (sc‐14015), anti‐Nox2 (sc‐20782), anti‐p110 (sc‐7189), anti‐Akt (sc‐8312), anti‐CREB2 (sc‐200), anti‐FoxO1 (sc‐374427), anti‐PPARγ (sc‐7273), anti‐β‐actin (sc‐47778) and anti‐lamin A (sc‐20680) antibodies were from Santa Cruz (Santa Cruz, CA). Anti‐mTOR (#2972), anti‐phospho‐mTOR (#5536), anti‐phospho‐Akt (#9271), anti‐phospho‐CREB (#9191) and anti‐phospho‐FoxO1 (#9461) antibodies were from Cell Signaling (Danver, MA). S1P and mevastatin were from Cayman (Ann Arbor, MI). NS‐398 (N‐[2‐(cyclohexyloxy)‐4‐nitrophenyl]‐methanesulfonamide), celecoxib, edaravone, apocynin (APO), diphenyleneiodonium chloride (DPI), LY294002, SH‐5 (D‐3‐deoxy‐2‐O‐methyl‐myo‐inositol 1‐[(R)‐2‐methoxy‐3‐(octadecyloxy)propyl hydrogen phosphate]), rapamycin, H89, AS1242586 (5‐amino‐7‐(cyclohexylamino)‐1‐ethyl‐6‐fluoro‐4‐oxo‐1,4‐dihydroquinoline‐3‐carboxylic acid) and GW9662 were from Biomol (Plymouth Meetings, PA). Luciferase assay kit was from Promega (Madison, WI). 2′,7′‐Dichlorodihydrofluorescein diacetate (DCFH‐DA) and 2′,7′‐bis‐(2‐carboxyethyl)‐5‐(and‐6)‐carboxyfluorescein, acetoxymethyl ester (BCECF/AM) were from Molecular Probes (Eugene, OR). SDS‐PA GE supplies were from MDBio Inc. (Taipei, Taiwan). CAY10444 (2‐undecyl‐thiazolidine‐4‐carboxylic acid), W123 (3‐(2‐(3‐hexylphenylamino)‐2‐oxoethylamino)propanoic acid) and all other reagents were from Sigma (St. Louis, MO).
Results
S1P induces COX‐2 expression via Nox2‐dependent ROS generation
S1P has been shown to regulate the development of hyperoxia‐induced lung injury via increased Nox‐dependent ROS generation (Harijith et al., 2013). We have demonstrated that S1P induced COX‐2 expression via S1P receptors in HTSMCs (Hsu et al., 2015). We found that increased the expression of COX‐2 by about onefold. Pretreatment with either a ROS scavenger (edaravone) or Nox inhibitors (apocynin and DPI) dose‐dependently attenuated the S1P‐induced increase in COX‐2 expression (Figure 1A). In addition, the increase in COX‐2 mRNA levels and promoter activity were also significantly attenuated by edaravone (mRNA, 75%; promoter, 40%), apocynin (mRNA 88%; promoter, 45%) or DPI (mRNA 85%; promoter, 44%) in HTSMCs (Figure 1B). We further observed that S1P markedly increased Nox activation and ROS generation, which were inhibited by apocynin, DPI, edaravone, W123 (S1P1 receptor antagonist) or CAY10444 (S1P3 receptor antagonist) (Figures 1C and D). Previous report indicates that activated Nox is a multimeric protein complex consisting of at least three cytosolic subunits of p47phox, p67phox and p40phox. Phosphorylation of p47phox leads to a conformational change allowing it to interact with p22phox (Lee et al., 2008). The p47phox organizes the translocation of other cytosolic factors; hence it is designated as an ‘organizer subunit’ (Lee et al., 2008). Moreover, we showed that transfection with siRNA of Nox2 or p47phox significantly inhibited S1P‐induced COX‐2 expression (Figure 1E). We also observed that transfection with Nox4 siRNA had no effect on S1P‐induced COX‐2 expression (data not shown). These data suggested that S1P increases COX‐2 expression through Nox2‐dependent ROS generation in HTSMCs.
Figure 1.
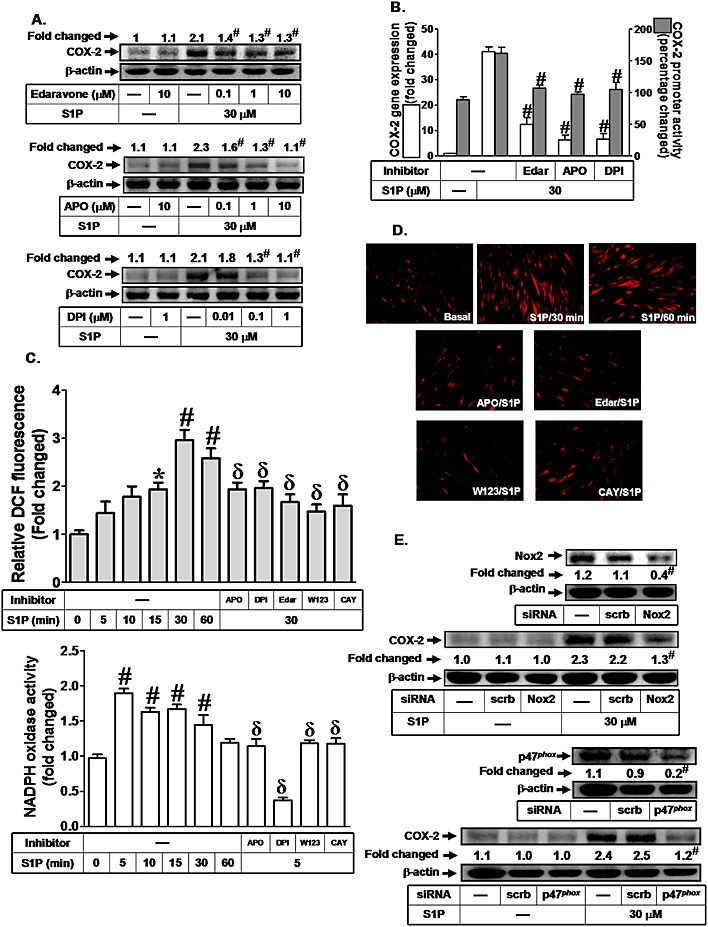
NADPH oxidase/ROS play key roles in S1P‐induced COX‐2 expression. (A) HTSMCs were pretreated with edaravone, apocynin (APO) or DPI for 2 h, and then incubated with S1P for 6 h. The COX‐2 protein expression was determined by Western blot. (B) Cells were pretreated with edaravone (Edar, 10 μM), apocynin (APO, 10 μM) or DPI (1 μM) for 2 h, and then incubated with S1P for 4 h or 1 h. The COX‐2 mRNA expression and promoter activity were determined by real‐time PCR and promoter assay respectively. (C) Cells were treated with S1P for the indicated time intervals or pretreated with edaravone (Edar, 10 μM), apocynin (10 μM) and DPI (1 μM) for 2 h, or W123 (10 μM) and CAY10444 (CAY, 10 μM) for 1 h, and then incubated with S1P for 30 min. The ROS generation was measured (upper panel). Cells were treated with S1P for the indicated time intervals or pretreated with apocynin (10 μM), DPI (1 μM) for 2 h, or W123 (10 μM), CAY10444 (CAY, 10 μM) for 1 h, and then incubated with S1P for 5 min. The NADPH oxidase activity was measured (lower panel). (D) Cells were incubated with S1P (30 μM) for the indicated time intervals or pretreated with apocynin or edaravone (Edar) for 2 h, or W123, CAY10444 (CAY) for 1 h and then incubated with S1P for 30 min. DHE fluorescence images were observed. Images shown are representative of three independent experiments with similar results. (E) Cells were transfected with siRNA of scrambled, Nox2 or p47phox, and then incubated with S1P for 6 h. The protein levels of Nox2, p47phox and COX‐2 were determined by Western blot. Data are expressed as mean (A and E) or mean ± SEM (B and C) of five independent experiments. *P < 0.05; # P < 0.01, as compared with the cells exposed to S1P alone (A and B), vehicle (C) or S1P + scrambled siRNA (E). δ P < 0.05, as compared with the cells exposed to S1P alone (C).
Participation of PI3K/Akt/mTOR in S1P‐induced COX‐2 expression
PI3K/Akt/mTOR activation by S1P leads to cell migration, differentiation or mineralization in various cell types (Kalhori et al., 2013; Matsuzaki et al., 2013). Therefore, we suggested that the PI3K/Akt/mTOR pathway is involved in S1P‐induced increase in COX‐2 expression in HTSMCs. As shown in Figure 2A, pretreatment with the inhibitor of PI3K (LY294002), Akt (SH‐5) or mTOR (rapamycin) significantly attenuated the S1P‐induced increase in COX‐2 expression. In addition, LY294002 (mRNA, 80%; promoter, 45%), SH‐5 (mRNA, 77%; promoter, 48%) and rapamycin (mRNA, 79%; promoter, 47%) also inhibited the S1P‐induced increase in COX‐2 mRNA expression and promoter activity (Figure 2B). Furthermore, we demonstrated that S1P time‐dependently stimulated Akt phosphorylation in HTSMCs, which was reduced by W123, CAY10444, LY294002 or SH‐5, but not rapamycin (Figure 2C). We noticed that S1P also stimulated mTOR phosphorylation, which was reduced by W123, CAY10444, LY294002, SH‐5 or rapamycin (Figure 2C). ROS have been shown to regulate Akt/mTOR activation induced by various stimuli (Chen et al., 2013; Dilshara et al., 2014). Here, we found that S1P‐induced mTOR phosphporylation was inhibited by edaravone, apocynin or DPI in HTSMCs (Figure 2C). We also demonstrated that ROS‐dependent mTOR phosphorylation was not mediated through Akt (Figure 2C). To confirm the roles of PI3K, Akt and mTOR in S1P‐induced increase in COX‐2 expression, as shown in Figure 2D, transfection with siRNA of p110, Akt or mTOR attenuated the S1P‐induced increase in COX‐2 protein levels in HTSMCs. These results suggest that S1P‐induced COX‐2 up‐regulation is mediated through PI3K/Akt‐ and Nox2/ROS‐dependent mTOR activation in HTSMCs.
Figure 2.
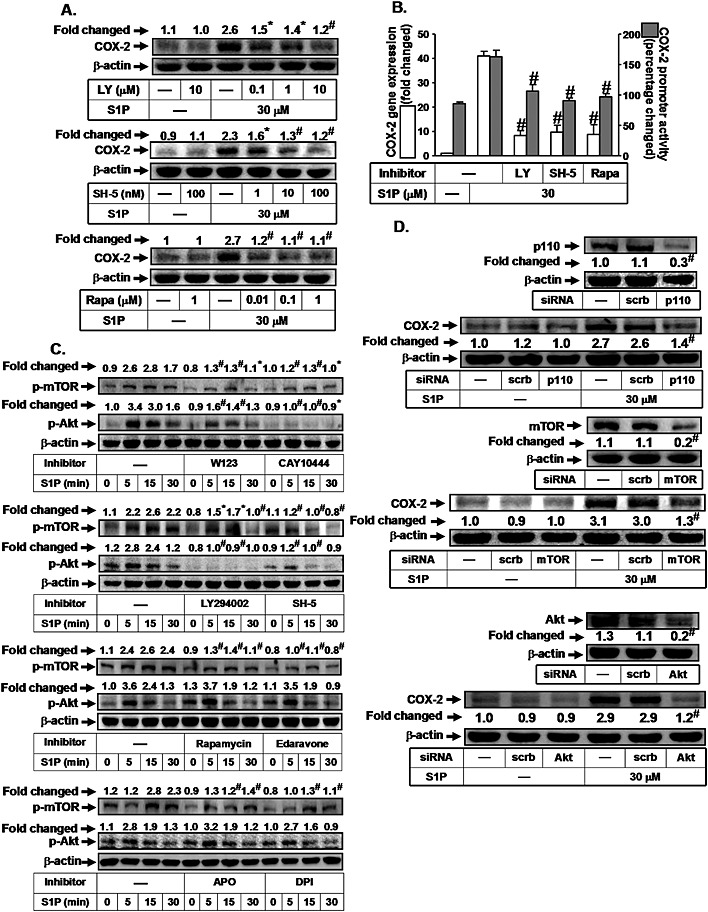
S1P induces COX‐2 expression via PI3K/Akt‐dependent mTOR. (A) HTSMCs were pretreated with LY294002 (LY), SH‐5 or rapamycin (Rapa) for 1 h, and then incubated with S1P for 6 h. The COX‐2 protein expression was determined by Western blot. (B) Cells were pretreated with LY294002 (LY, 10 μM), SH‐5 (100 nM) or Rapamycin (Rapa, 1 μM) for 1 h, and then incubated with S1P for 4 h or 1 h. The COX‐2 mRNA expression and promoter activity were determined by real‐time PCR and promoter assay, respectively. (C) Cells were pretreated without or with W123, CAY10444, LY294002, SH‐5 or rapamycin for 1 h or edaravone, apocynin or DPI for 2 h, and then incubated with S1P (30 μM) for the indicated time intervals. The levels of phospho‐mTOR and phospho‐Akt were determined by Western blot. (D) Cells were transfected with siRNA of scrambled, p110, mTOR or Akt, and then incubated with S1P for 6 h. The protein levels of p110, mTOR, Akt and COX‐2 were determined by Western blot. Data are expressed as mean (A, C and D) or mean ± SEM (B) of five independent experiments. *P < 0.05; # P < 0.01, as compared with the cells exposed to S1P alone or S1P + scrambled siRNA (D).
S1P induces FoxO1 activation and translocation via an mTOR‐dependent pathway
FoxO1 has been shown to be involved in the expression of various genes, such as HO‐1, catalase, sod2 and COX‐2 (Liu et al., 2013). Previously, it was demonstrated that insulin induces FoxO1 activation and translocation from the nucleus to the cytosol (Banerjee et al., 2010). Here, we investigated whether the S1P‐induced increase in COX‐2 expression was mediated through FoxO1. As shown in Figure 3A, S1P stimulated FoxO1 phosphorylation and translocation from the nucleus to the cytosol in a time‐dependent manner. We further investigated the relationship between NADPH oxidase/ROS, PI3K/Akt/mTOR and FoxO1 in response to S1P. As shown in Figure 3B, pretreatment with W123, CAY10444, edaravone, apocynin, DPI, LY294002, SH‐5 or rapamycin attenuated FoxO1 translocation from the nucleus to the cytosol. In addition, S1P‐stimulated FoxO1 phosphorylation was markedly inhibited by pretreatment with W123, CAY10444, apocynin, DPI, edaravone, rapamycin, LY294002 or SH‐5 (Figure 3C). Finally, we used a ChIP assay to determine whether S1P could stimulate the dissociation of FoxO1 from the COX‐2 promoter region. We showed that S1P stimulated FoxO1 dissociation in a time‐dependent manner (Figure 3D), which was reversed by W123, CAY10444, edaravone, apocynin, DPI, LY294002, SH‐5 or rapamycin (Figure 3E). These results demonstrate that S1P stimulates FoxO1 activation and translocation via PI3K/Akt‐ and Nox2/ROS‐dependent mTOR cascades.
Figure 3.
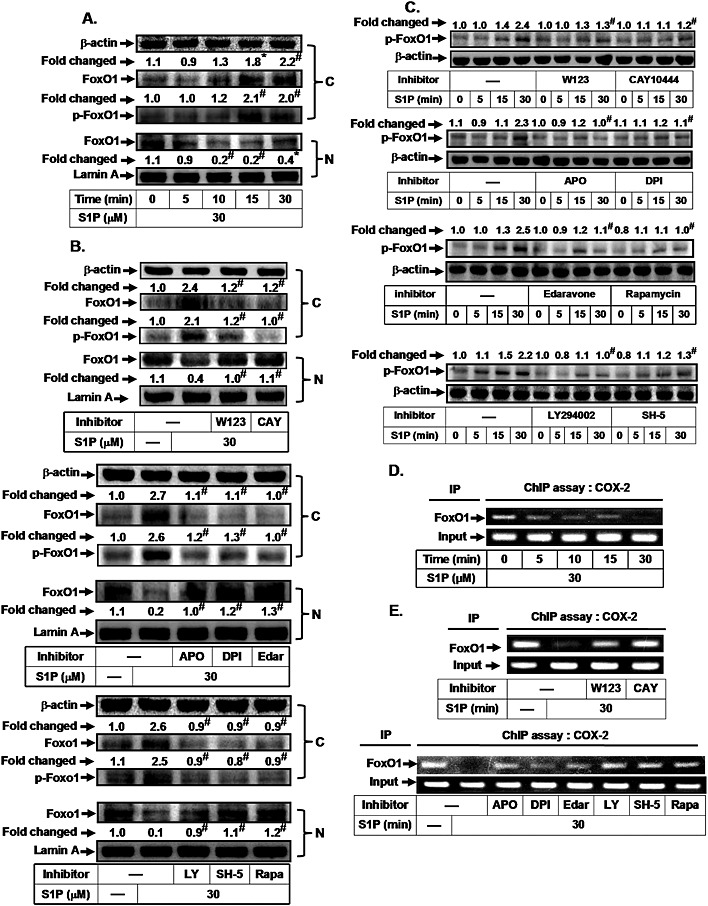
S1P induces FoxO1 activation and translocation via an mTOR‐dependent pathway. HTSMCs were (A) incubated with S1P (30 μM) for the indicated time intervals or (B) pretreated with W123, CAY10444 (CAY), apocynin (APO), DPI, edaravone (Edar), LY294002 (LY), SH‐5 or rapamycin (Rapa) for 1 h, and then incubated with S1P for 30 min. The cytosolic and nuclear fractions were prepared and subjected to Western blot analysis using an anti‐FoxO1 or anti‐phospho‐FoxO1 antibody. Lamin A and β‐actin were used as a marker protein for nuclear and cytosolic fractions, respectively. (C) Cells were pretreated without or with W123, CAY10444, apocynin, DPI, edaravone, rapamycin, LY294002 or SH‐5, and then incubated with S1P for the indicated time intervals. The levels of phospho‐FoxO1 were determined by Western blot. Cells were (D) incubated with S1P (30 μM) for the indicated time intervals or (E) pretreated with W123, CAY10444 (CAY), LY294002 (LY), SH‐5 and rapamycin (Rapa) for 1 h, or apocynin, DPI and edaravone (Edar) for 2 h, and then incubated with S1P for 30 min. The FoxO1 binding activities were analysed by a ChIP assay. Data are expressed as mean (A, B and C) of five independent experiments. *P < 0.05; # P < 0.01, as compared with the cells exposed to vehicle (A) or S1P alone (B and C).
The involvement of CREB in the up‐regulated expression of COX‐2 induced by S1P
CREB has been shown to play a critical role in the transcriptional activation of the COX‐2 gene in the early phase of adipogenesis (Fujimori et al., 2014). Therefore, we determined whether the S1P‐induced increased COX‐2 expression was mediated via activation of CREB in HTSMCs. As shown in Figure 4A, S1P‐induced COX‐2 expression was reduced by pretreatment with the inhibitor of PKA (H89) which is an upstream signalling molecule of CREB. In addition, pretreatment with H89 also diminished S1P‐induced COX‐2 mRNA levels (80%) and promoter activity (43%) (Figure 4B). To further confirm the role of CREB in S1P‐mediated COX‐2 promoter activity, a point‐mutated CREB COX‐2 promoter plasmid was used. As shown in Figure 4C, S1P‐stimulated COX‐2 promoter activity was prominently lost in HTSMCs transfected with the point‐mutated CREB COX‐2 promoter plasmid. To investigate whether S1P could stimulate CREB promoter activity, as shown in Figure 4D, S1P time‐dependently increased CREB promoter activity and this was attenuated by H89, W123 or CAY10444. We also found that S1P markedly stimulated CREB phosphorylation (Figure 4E) and translocation from the cytosol into the nucleus, which was inhibited by W123 or CAY10444 (Figure 4F). However, pretreatment with edaravone, apocynin, DPI, LY294002, SH‐5 or rapamycin had no effect on S1P‐mediated CREB phosphorylation and translocation (data not shown). To further confirm the role of CREB in S1P‐induced increased COX‐2 expression in HTSMCs, as shown in Figure 4G, transfection with CREB siRNA significantly reduced CREB protein expression, and then inhibited S1P‐induced COX‐2 expression in HTSMCs. Finally, we showed that S1P induced the binding of CREB to the COX‐2 promoter in a time‐dependent manner, which was reduced by W123, CAY10444, but not edaravone, apocynin, DPI, LY294002, SH‐5 or rapamycin (Figure 4H). Our results suggest that CREB acts as a crucial transcription factor in S1P‐induced increase in COX‐2 expression in HTSMCs.
Figure 4.
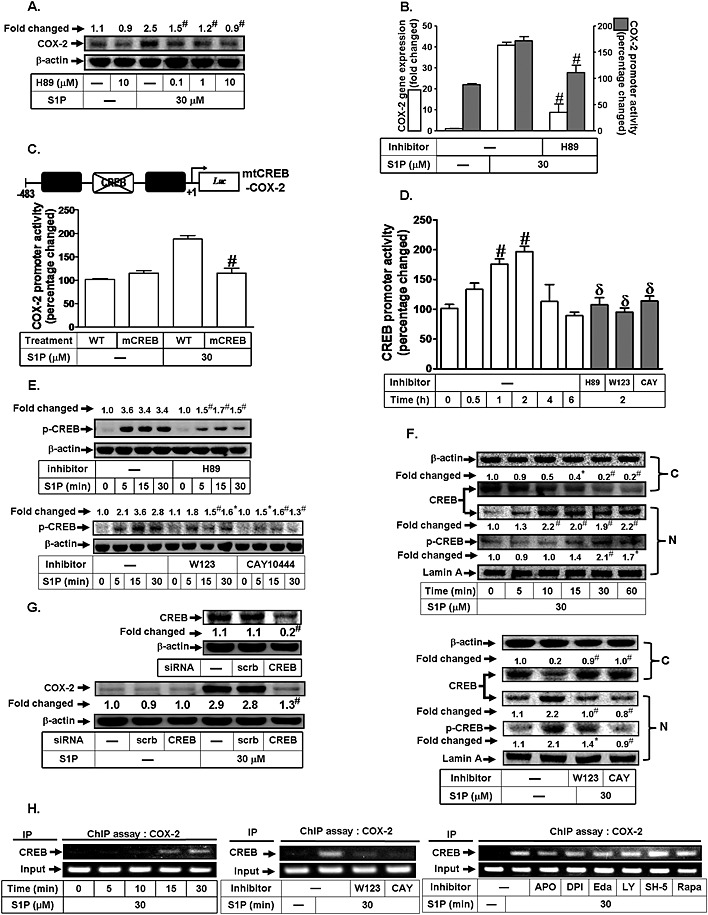
CREB is involved in S1P‐induced COX‐2 expression. (A) HTSMCs were pretreated with H89 for 1 h, and then incubated with S1P for 6 h. The COX‐2 protein expression was determined by Western blot. (B) Cells were pretreated with H89 (10 μM) for 1 h, and then incubated with S1P for 4 h or 1 h. The COX‐2 mRNA expression and promoter activity were determined by real‐time PCR and promoter assay, respectively. (C) Cells were transfected with wild‐type COX‐2 promoter reporter gene (WT‐COX‐2) or CREB‐mutated COX‐2 promoter reporter gene (mCREB‐COX‐2), and then incubated with or without S1P for 1 h. The COX‐2 promoter activity was determined. (D) Cells were treated with S1P for the indicated time intervals or pretreated with H89, W123 or CAY10444 (CAY) for 1 h, and then incubated with S1P for 2 h. The CREB promoter activity was measured. (E) Cells were pretreated without or with H89, W123 or CAY10444 for 1 h, and then incubated with S1P (30 μM) for the indicated time intervals. The levels of phospho‐CREB were determined by Western blot. (F) Cells were incubated with S1P (30 μM) for the indicated time intervals or pretreated with W123 or CAY10444 (CAY) for 1 h. The cytosolic and nuclear fractions were prepared and subjected to Western blot analysis using an anti‐CREB or anti‐phospho‐CREB antibody. Lamin A and β‐actin were used as a marker protein for nuclear and cytosolic fractions, respectively. (G) Cells were transfected with siRNA of scrambled or CREB, and then incubated with S1P for 6 h. The levels of CREB and COX‐2 were determined. (H) Cells were treated with S1P for the indicated time intervals or pretreated with W123 (10 μM), CAY10444 (CAY, 10 μM), LY294002 (LY, 10 μM), SH‐5 (10 nM) and rapamycin (Rapa, 1 μM) for 1 h, or apocynin (10 μM), DPI (1 μM) or Edaravone (Edar, 10 μM) for 2 h, and then incubated with S1P (30 μM) for 30 min. The CREB binding activities were analysed by a ChIP assay. Data are expressed as mean (A, E, F and G) or mean ± SEM (B–D) of five independent experiments. *P < 0.05; # P < 0.01, as compared with the cells exposed to S1P alone (A, B and E), vehicle (D and F) or S1P + scrambled siRNA (D). # P < 0.01, as compared with cells transfected with WT COX‐2 promoter stimulated by S1P (C). δ P < 0.05, as compared with the cells exposed to S1P for 2 h (D).
S1P induces cell migration via COX‐2‐dependent PGE2 production
S1P has been shown to induce the migration of renal mesangial cells via the production of COX‐2/PGE2 (Volzke et al., 2014). The migration of airway smooth muscle cells plays an important role in the development of various airway diseases. Therefore, we explored whether S1P could induce cell migration via COX‐2‐dependent PGE2 production in HTSMCs. As shown in Figure 5A, S1P induced an increase in cell migration, which was inhibited by pretreatment with the inhibitor of COX‐2 (CLC or NS‐398), suggesting that COX‐2 plays an essential role in S1P‐induced cell migration. ROS have been shown to induce cell migration induced by various stimuli (Bruder‐Nascimento et al., 2014; Kastl et al., 2014). In addition, the PI3K/Akt/mTOR pathway has been shown to be involved in cell migration induced by overexpression of the estrogen receptor (ER)α (Hou et al., 2014). Here, we found that pretreatment with W123, CAY10444, apocynin, DPI, edaravone, LY294002, SH‐5, rapamycin or H89 markedly attenuated cell migration induced by S1P (Figure 5B). We further determined whether S1P‐induced cell migration was mediated through PGE2. As shown in Figure 5C, PGE2 (30 μM) time‐dependently increased cell migration. However, S1P‐induced PGE2 generation was inhibited by apocynin, DPI, edaravone, LY294002, SH‐5, rapamycin or H89 (Figure 5D). Taken together, these results indicate that S1P induces cell migration via a COX‐2/PGE2‐dependent pathway.
Figure 5.
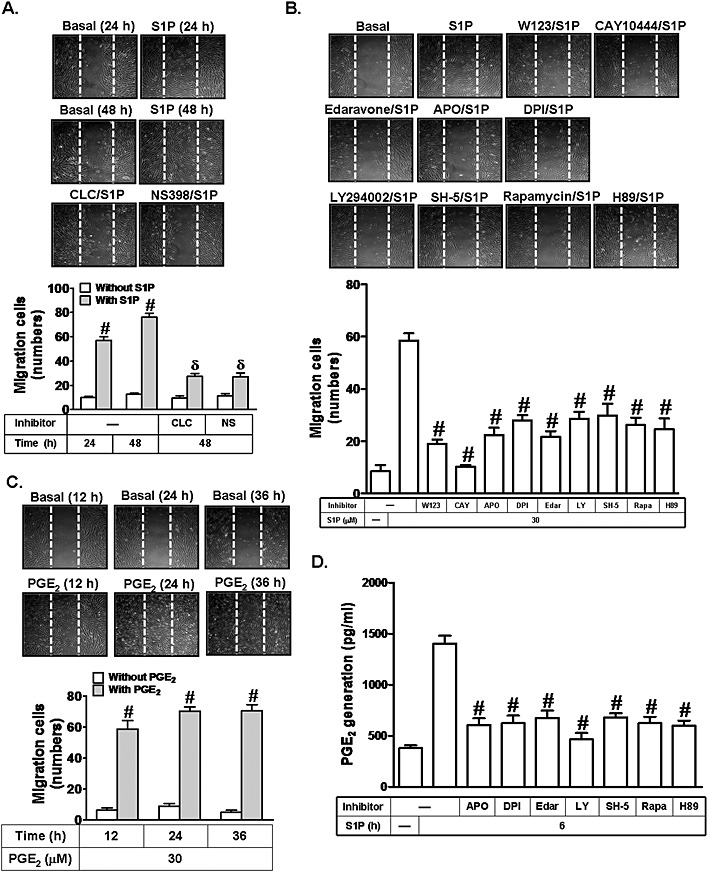
S1P induces COX‐2/PGE2‐dependent cell migration. (A) HTSMCs were treated with S1P (30 μM) for the indicated time intervals or pretreated without or with CLC (10 μM) or NS398 (NS, 10 μM) for 1 h, and then incubated with S1P (30 μM) for 48 h. Representative images are shown in the upper panel, and quantification of the images is shown in the lower panel. (B) Cells were pretreated without or with W123, CAY10444 (CAY), LY294002 (LY), SH‐5, rapamycin (Rapa), H89, edaravone (Edar), apocynin (APO) or DPI, and then incubated with S1P (30 μM) for 48 h. (C) Cells were treated with PGE2 (30 μM) for the indicated time intervals. Images shown are representative of three independent experiments with similar results. (D) Cells were pretreated with LY294002 (LY), SH‐5, rapamycin (Rapa), H89, edaravone (Edar), apocynin or DPI, and then incubated with S1P for 6 h. The release of PGE2 was measured. Data are expressed as mean ± SEM of five independent experiments. # P < 0.01, as compared with the cells exposed to S1P alone (B and D) or vehicle (A and C). δ P < 0.01, as compared with the cells exposed to S1P for 48 h (A).
Mevastatin inhibits S1P‐induced COX‐2/PGE2‐dependent cell migration via PPARγ receptors
Mevastatin has been reported to alleviate inflammatory responses by reducing the secretion of inflammatory cytokines and expression of adhesion molecules (Akasaki et al., 2009; Helbing et al., 2010). In addition, statins have been shown to play a protective role in coronary artery disease by inducing the expression of PPARγ (Pucci et al., 2011). Thus, we investigated whether mevastatin could inhibit S1P‐induced cell migration by inhibiting the generation of COX‐2/PGE2. As shown in Figures 6A and B, mevastatin significantly reduced S1P‐stimulated cell migration and PGE2 generation. In addition, pretreatment with GW9662 (a potent antagonist of PPARγ) reversed mevastatin‐reduced cell migration and PGE2 generation (Figures 6A and B). We also observed that mevastatin markedly inhibited S1P‐induced increased COX‐2 expression, which was reversed by GW9662 (Figure 6C, upper panel). To further confirm the role of PPARγ in mevastatin‐reduced S1P‐induced increase in COX‐2 expression, a PPARγ agonist (rosiglitazone) was used. In this study, pretreatment with rosiglitazone inhibited S1P‐induced increased COX‐2 expression, which was reversed by GW9662 (Figure 6C, lower panel). Moreover, transfection with PPARγ siRNA significantly reduced PPARγ protein expression, and then reversed the mevastatin‐reduced COX‐2 expression in S1P‐stimulated HTSMCs (Figure 6D). We also demonstrated that mevastatin inhibited S1P‐stimulated FoxO1 and CREB translocation (Figure 6E). Ultimately, we investigated whether mevastatin could mediate the interaction of CREB and FoxO1 with the COX‐2 promoter region. As shown in Figure 6F, mevastatin inhibited the S1P‐induced FoxO1 dissociation and CREB association with the COX‐2 promoter region, and these effects were reversed by GW9662. Taken together, these results show that mevastatin inhibits S1P‐induced COX‐2/PGE2‐dependent cell migration via PPARγ.
Figure 6.
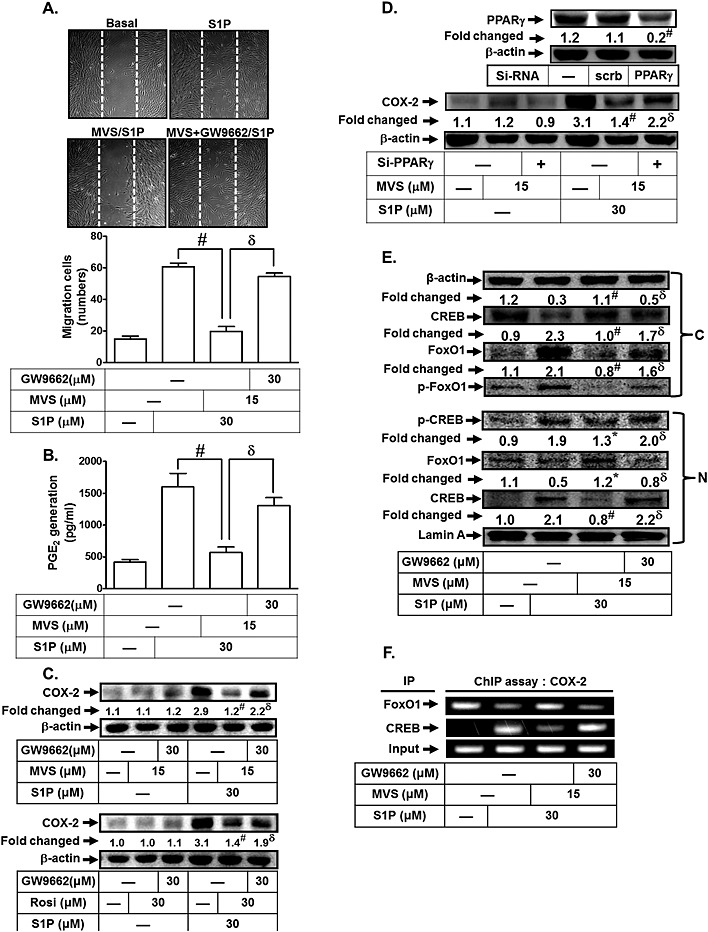
Mevastatin (MVS) attenuates S1P‐promoted cell migration via PPARγ. (A, B) HTSMCs were pretreated with GW9662 (30 μM) for 1 h, and then treated with mevastatin (15 μM) for 24 h, followed by incubation with S1P (30 μM) for 48 h (cell migration) or 6 h (PGE2 generation). (A) Representative images of cell migration are shown in the upper panel, and quantification of the images is shown in the lower panel. (B) The PGE2 levels were measured. (C) Cells were pretreated with GW9662 (30 μM) for 1 h, and then treated with mevastatin (15 μM) or Rosiglitazone (Rosi) (30 μM) for 24 h, followed by incubation with S1P (30 μM) for 6 h. The levels of COX‐2 were determined by Western blot. (D) Cells were transfected with siRNA or scrambled of PPARγ, pretreated with mevastatin for 24 h and then incubated with S1P for 6 h. The levels of PPARγ and COX‐2 protein were determined by Western blot. (E) Cells were pretreated with GW9662 (30 μM) for 1 h, and then treated with mevastatin (15 μM) for 24 h, followed by incubation with S1P (30 μM) for 30 min. The cytosolic and nuclear fractions were prepared and subjected to Western blot analysis using an anti‐CREB, anti‐FoxO1, anti‐phospho‐CREB or anti‐phospho‐FoxO1 antibody. (F) Cells were pretreated with GW9662 (30 μM) for 1 h, and then treated with mevastatin (15 μM) for 24 h, followed by incubation with S1P (30 μM) for 30 min. The CREB and FoxO1 binding activities were analysed by a ChIP assay. Data are expressed as mean ± SEM (A and B) or mean (C, D and E) of five independent experiments. *P < 0.05; # P < 0.01, as compared with the cells exposed to S1P alone (A, B, C and E) or S1P + scrambled siRNA (D). δ P < 0.05, as compared with the cells exposed to S1P + mevastatin (A, B, C and E), S1P + Rosi (C), or S1P + mevastatin + scrambled siRNA (D).
Mevastatin attenuates S1P‐evoked airway inflammation and leukocyte infiltration in vivo
Moreover, we determined whether S1P could induce airway inflammation in vivo. In an in vivo study, mice were intratracheally administered S1P. As shown in Figure 7A, S1P caused a pulmonary haematoma, which was inhibited by pretreatment with mevastatin. In addition, BAL fluid was acquired, and airway tissues were homogenized to extract proteins. As shown in Figure 7B, S1P significantly enhanced the number of leukocytes (eosinophils and neutrophils) in BAL fluid, an effect attenuated by pretreatment with mevastatin. The effect of mevastatin on COX‐2 protein expression was also confirmed by using mice as an animal model (Figure 7C). These data suggest that pretreatment with mevastatin inhibits S1P‐induced airway inflammatory responses both in vitro and in vivo.
Figure 7.
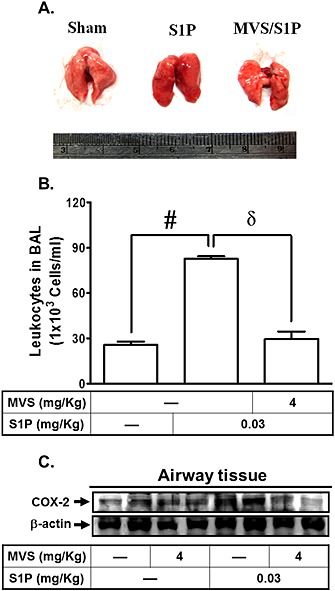
Mevastatin (MVS) attenuates S1P‐induced airway inflammatory responses in vivo. (A) Mice were pretreated with mevastatin or vehicle (Sham), i.p., for 24 h, and then administered mevastatin with or without S1P (S1P), i.t., for 24 h. Changes in the appearance of the lung were observed. (B) BAL fluid was acquired, and the number of leukocytes was counted with a haemocytometer. (C) The proteins were extracted from airway tissues, and the protein levels of COX‐2 and β‐actin were determined by Western blot. Data are expressed as mean ± SEM of five mice in each group. # P < 0.01, as compared with mice injected with vehicle. δ P < 0.05, as compared with the cells exposed to S1P alone.
Discussion and conclusions
COX‐2 plays a pivotal role in the production of pro‐inflammatory eicosanoids. Moreover, COX‐2 is thought to be an important mediator in airway inflammatory responses. Several lines of evidence indicate that high levels of PGE2, synthesized by COX‐2, are also involved in inflammatory responses (Lee et al., 2010). COX‐2 inhibitors, such as etodolac, weaken the cough reflex and inflammatory responses in the airways of asthmatic patients, suggesting that COX‐2 plays a critical role in airway diseases (Ishiura et al., 2009). S1P plays an important role in allergic responses, including asthma and anaphylaxis (Ammit et al., 2001). Several reports indicate that statins possess anti‐inflammatory and anti‐oxidant properties in vitro and in vivo (Akasaki et al., 2009; Li et al., 2012; Shao et al., 2012; Wu et al., 2013). Statins have been shown to inhibit oxLDL‐induced COX‐2 expression (Shao et al., 2012). However, the molecular mechanisms underlying the inhibitory effect of mevastatin on the increase in COX‐2/PGE2 induced in HTSMCs remain unclear.
In this study, as depicted in Figure 8, S1P markedly increased Nox/ROS production and COX‐2/PGE2‐dependent cell migration. Pretreatment with mevastatin inhibited S1P‐induced increased COX‐2 expression, PGE2 release, cell migration, and FoxO1 and CREB activation and translocation. The observed suppression of S1P‐induced COX‐2 expression and PGE2 release by mevastatin was attenuated by GW9662 or transfection with PPARγ siRNA in HTSMCs (Figure 6). The in vivo results also indicate that mevastatin blocks the S1P‐induced pulmonary haematoma (Figure 7); the S1P‐enhanced leukocyte count in BAL and COX‐2 expression was attenuated in mevastatin‐pretreated mice.
Figure 8.
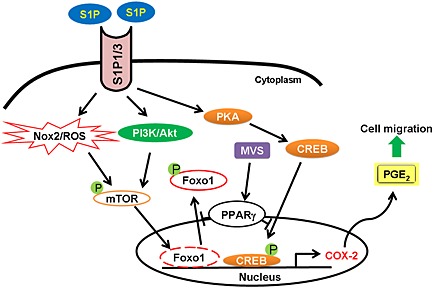
Schematic diagram illustrating the proposed signalling pathways involved in S1P‐induced COX‐2 expression and cell migration in HTSMCs. S1P induces COX‐2/PGE2‐dependent cell migration via Nox2/ROS‐ and PI3K/Akt‐dependent mTOR/FoxO1 and CREB phosphorylation. Moreover, mevastatin (MVS) inhibits S1P‐induced FoxO1 and CREB phosphorylation and translocation, and then suppresses cell migration via PPARγ.
Several lines of evidence indicate that S1P‐induced diverse biological effects are mediated through S1P receptors (Aarthi et al., 2011). These various effects are partly elicited by the binding of S1P to a family of five GPCRs (S1P receptors), termed S1P1‐5. S1P1, S1P2 and S1P3 receptors have been shown to be expressed on various cell types, including airway smooth muscle cells (Ishii et al., 2004; Brinkmann, 2007). Indeed, we found that S1P1, S1P2 and S1P3 receptors are expressed on HTSMCs (Hsu et al., 2015). Moreover, we demonstrated that the inhibition of S1P1 and S1P3 receptors significantly attenuated S1P‐induced increased COX‐2 expression in HTSMCs (Hsu et al., 2015). Thus, we proposed that S1P1 and S1P3 receptors play key roles in S1P‐induced increase in COX‐2 expression in these cells.
Our report also indicated that cells and tissues are routinely subjected to sublethal doses of various oxidants, either exogenously through environmental exposure or endogenously through inflammatory processes (Lee and Yang, 2012). ROS are intracellularly generated from several sources, including mitochondrial respiration, cytochrome P450, the Nox system, xanthine/xanthine oxidase and metabolism of arachidonic acid (Lee and Yang, 2012). In in vitro experiments using human lung microvascular endothelial cells, it was shown that exogenous S1P stimulated intracellular ROS generation (Harijith et al., 2013). S1P also caused Nox activation and intracellular H2O2 generation in NIH3T3 fibroblasts (Catarzi et al., 2011). Moreover, in HTSMCs, our data, for the first time, show a novel role of Nox in the S1P‐induced ROS production and the resulting increase in the induction of COX‐2/PGE2. Here, we believed that S1P‐induced ROS generation was, at least in part, due to Nox activation in HTSMCs. In the future, we will investigate whether S1P‐induced ROS production can be mediated via other sources, such as xanthine oxidase or mitochondrial respiration in these cells.
The PI3Ks are a conserved family of signal transduction enzymes that are involved in cellular activation, inflammatory responses, chemotaxis and apoptosis. PI3K/Akt have been shown to be downstream components of ROS activated by different stimuli in various cell types (Lee and Yang, 2012; Yang et al., 2012). However, we showed that, in HTSMCs, S1P stimulated Akt activation in a Nox/ROS‐independent manner. mTOR, initially known as the mammalian target of rapamycin, integrates the input from upstream pathways, including insulin, growth factors and amino acids (Oya, 2010). mTOR also senses cellular nutrients, oxygen and energy levels. In addition, the PI3K/Akt/mTOR signalling pathway plays a central role in cell proliferation, growth and survival under physiological conditions (Oya, 2010). Here, we showed that S1P‐induced increased COX‐2 expression was reduced via the inhibition of mTOR. However, we also found that PI3K/Akt and Nox/ROS play key roles in mediating mTOR phosphorylation in these cells. Thus, S1P stimulated mTOR activation via the PI3K/Akt‐ and Nox/ROS‐dependent pathways.
FoxO transcription factors are involved in several physiological and pathological processes, including ageing, cancer and neurological diseases (Tikhanovich et al., 2013). There are four isoforms, FoxO1, FoxO3, FoxO4 and FoxO6 in mammals. Among the FoxO family members, FoxO1 has been shown to be involved in various cellular functions, such as gluconeogenesis, cell cycle and autophagy (Tikhanovich et al., 2013). We noticed that FoxO1 can be induced by IL‐1β at early treatment and mediates the downstream expression of genes such as COX‐2 (Lappas, 2013). In the presence of insulin and insulin‐like growth factor, the PI3K/Akt signalling pathway is activated, and the protein kinases such as Akt directly phosphorylate FoxO factors (Webb and Brunet, 2014). FoxO1 is phosphorylated by Akt at Thr24, Ser256 and Ser319 and translocated from the nucleus to the cytoplasm and degradation (Banerjee et al., 2010). In this study, we found that S1P induced PI3K/Akt/mTOR‐dependent FoxO1 phosphorylation, and the phosphorylated FoxO1 was translocated from the nucleus to the cytoplasm in HTSMCs. In addition, Nox/ROS also play key roles in regulating FoxO1 phosphorylation. These findings were similar to those from a previous study showing that insulin induces FoxO1 phosphorylation and translocation from the nucleus to the cytosol (Banerjee et al., 2010). In this situation, FoxO1 dissociated from the promoter of the target gene an enabled the gene to be expressed. In this study, we found that S1P induced FoxO1 translocation and dissociation from the COX‐2 promoter region via PI3K/Akt‐ and Nox/ROS‐dependent mTOR activation. However, CREB, a cellular transcription factor, also binds to certain DNA sequences (CRE, cAMP response elements) and thereby regulates their downstream expressions. CREB has been shown to play a critical role in the transcriptional activation of the COX‐2 gene in the early phase of adipogenesis (Fujimori et al., 2014). Here, we showed that S1P also induced the expression of CREB associated with the COX‐2 promoter to facilitate the COX‐2 expression, which was reduced by inhibiting CREB in HTSMCs (Figure 4). Choi et al. have also indicated that luteolin induces PI3K/Akt‐dependent CREB activation (Choi, 2011). Lee et al. (2011) also showed that raloxifene had an inhibitory effect on LPS‐induced NO production in RAW264.7 cells and that this was mediated through a ROS/p38 MAPK/CREB pathway via the up‐regulation of HO‐1, independently of oestrogen receptors. However, our results indicated that S1P‐induced CREB activation and translocation from the cytosol into the nucleus were not mediated through PI3K/Akt‐ and Nox/ROS‐dependent mTOR activation.
The migration of airway smooth muscle cells plays an important role in the development of various airway diseases. S1P has been shown to induce the migration of renal mesangial cells via COX‐2/PGE2 production (Volzke et al., 2014). Results from a previous study indicated that angiotensin II induces vascular smooth muscle cell migration via Nox1‐dependent ROS generation (Bruder‐Nascimento et al., 2014). In addition, Yang et al. (2015) demonstrated that the transcription factor SOX2 promotes the migration and invasion of laryngeal cancer cells by the induction of MMP‐2 via the PI3K/Akt/mTOR pathway. In this study, we established that S1P induced COX‐2/PGE2‐dependent cell migration via Nox2/ROS‐ and PI3K/Akt‐dependent mTOR/FoxO1 and CREB phosphorylation in HTSMCs.
Statins are now recognized as powerful anti‐inflammatory agents that exert beneficial effects beyond low‐density lipoprotein cholesterol reduction (Lefer, 2002). However, the effect of statins on airway diseases is still unclear. The use of statins in the treatment of asthma patients is still controversial. Ostroukhova et al. (2009) demonstrated that statins could promote allergic responses in asthma. However, results from other studies suggested that treatment with statins can increase lung function, enhance the ability of anti‐inflammatory drugs, and improve the symptoms of asthma (Maneechotesuwan et al., 2010; Braganza et al., 2011). In addition, simvastatin and mevastatin have been shown to reduce airway inflammation, epithelial injury, cytokine secretion and airway hyperresponsiveness via PPARγ in asthma (Pucci et al., 2011; Osmak, 2012). Moreover, statins have been shown to inhibit oxidant enzyme activity, such as that of reduced Nox and myeloperoxidase and up‐regulate the activity of antioxidant enzymes such as catalase and paraoxonase (Davignon et al., 2004). Zhu et al. (2012) demonstrated that the reduction in mucus secretion induced by rosuvastatin in airway remodelling and asthma was mediated through an attenuation of GABA A receptor‐mediated activity. On the other hand, mevastatin also reduced S1P‐induced FoxO1, CREB phosphorylation and translocation in HTSMCs (Figure 6). These mevastatin‐induced responses were reversed by GW9662 and PPARγ knockdown. These findings were consistent with the results observed in our in vivo animal experiments. S1P‐induced pulmonary haematoma was attenuated by mevastatin treatment and both the number of leukocytes and expression of COX‐2 were also reduced by mevastatin. Similar to our results, simvastatin has been shown to attenuate the OVA‐induced airway responsiveness and eosinophilia in BAL fluid (Liu et al., 2014). The results of the present study suggest that mevastatin attenuates the airway inflammatory response and reduces leukocyte (eosinophil and neutrophil) infiltration in animals in vivo. Mevastatin also attenuates S1P‐induced COX‐2/PGE2‐dependent cell migration via PPARγ in HTSMCs. In the future, we will determine which type of leukocyte (eosinophil or neutrophil) is attenuated by mevastatin in HTSMCs.
In summary, as depicted in Figure 8, our data indicate that S1P induces COX‐2/PGE2‐dependent cell migration via Nox2/ROS‐ and PI3K/Akt‐dependent mTOR/FoxO1 and CREB phosphorylation, in HTSMCs. Moreover, mevastatin inhibits S1P‐induced FoxO1 and CREB activation and translocation, and then suppresses cell migration via PPARγ.
Author contributions
CKH, CCL, LDH and CMY developed and designed experiments, and analysed and interpreted data. CKH, CCL and LDH conducted the experiments and collected data.
This article was drafted by CKH, CCL, LDH and CMY. All authors have read and approved the final version of this manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors thank Dr. Chen‐Yu Wang for proof reading the manuscript. This work was supported by MOST 104‐2320‐B‐182‐010 and MOST 104‐2320‐B‐182A‐003‐MY3 from the Ministry of Science and Technology, Taiwan; EMRPD1E1641 from the Ministry of Education, Taiwan and CMRPD1C0103, CMRPD1B0383, CMRPD1C0563, CMRPD1B0332, CMRPG3C1303 and CMRPG3B1093 from Chang Gung Medical Research Foundation, Taiwan.
Hsu, C.‐K. , Lin, C.‐C. , Hsiao, L.‐D. , and Yang, C.‐M. (2015) Mevastatin ameliorates sphingosine 1‐phosphate‐induced COX‐2/PGE2‐dependent cell migration via FoxO1 and CREB phosphorylation and translocation. British Journal of Pharmacology, 172: 5360–5376. doi: 10.1111/bph.13326.
References
- Aarthi JJ, Darendeliler MA, Pushparaj PN (2011). Dissecting the role of the S1P/S1PR axis in health and disease. J Dent Res 90: 841–854. [DOI] [PubMed] [Google Scholar]
- Akasaki Y, Matsuda S, Nakayama K, Fukagawa S, Miura H, Iwamoto Y (2009). Mevastatin reduces cartilage degradation in rabbit experimental osteoarthritis through inhibition of synovial inflammation. Osteoarthritis Cartilage 17: 235–243. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013b). The Concise Guide to PHARMACOLOGY 2013/14: nuclear hormone receptors. Br J Pharmacol 170: 1632–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013c). The Concise Guide to PHARMACOLOGY 2013/14:enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammit AJ, Hastie AT, Edsall LC, Hoffman RK, Amrani Y, Krymskaya VP et al. (2001). Sphingosine 1‐phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodelling in asthma. FASEB J 15: 1212–1214. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Meyer K, Mazumdar B, Ray RB, Ray R (2010). Hepatitis C virus differentially modulates activation of forkhead transcription factors and insulin‐induced metabolic gene expression. J Virol 84: 5936–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braganza G, Chaudhuri R, McSharry C, Weir CJ, Donnelly I, Jolly L et al. (2011). Effects of short‐term treatment with atorvastatin in smokers with asthma—a randomized controlled trial. BMC Pulm Med 11: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V (2007). Sphingosine 1‐phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther 115: 84–105. [DOI] [PubMed] [Google Scholar]
- Bruder‐Nascimento T, Chinnasamy P, Riascos‐Bernal DF, Cau SB, Callera GE, Touyz RM et al. (2014). Angiotensin II induces Fat1 expression/activation and vascular smooth muscle cell migration via Nox1‐dependent reactive oxygen species generation. J Mol Cell Cardiol 66: 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catarzi S, Romagnoli C, Marcucci G, Favilli F, Iantomasi T, Vincenzini MT (2011). Redox regulation of ERK1/2 activation induced by sphingosine 1‐phosphate in fibroblasts: involvement of NADPH oxidase and platelet‐derived growth factor receptor. Biochim Biophys Acta 1810: 446–456. [DOI] [PubMed] [Google Scholar]
- Chen S, Ren Q, Zhang J, Ye Y, Zhang Z, Xu Y et al. (2013). N‐acetyl‐L‐cysteine protects against cadmium‐induced neuronal apoptosis by inhibiting ROS‐dependent activation of Akt/mTOR pathway in mouse brain. Neuropathol Appl Neurobiol 40: 759–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi EM (2011). Luteolin protects osteoblastic MC3T3‐E1 cells from antimycin A‐induced cytotoxicity through the improved mitochondrial function and activation of PI3K/Akt/CREB. Toxicol In Vitro 25: 1671–1679. [DOI] [PubMed] [Google Scholar]
- Davignon J, Jacob RF, Mason RP (2004). The antioxidant effects of statins. Coron Artery Dis 15: 251–258. [DOI] [PubMed] [Google Scholar]
- Dilshara MG, Lee KT, Choi YH, Moon DO, Lee HJ, Yun SG et al. (2014). Potential chemoprevention of LPS‐stimulated nitric oxide and prostaglandin E2 production by α‐l‐rhamnopyranosyl‐(1‐‐>6)‐β‐d‐glucopyranosyl‐3‐indolecarbonate in BV2 microglial cells through suppression of the ROS/PI3K/Akt/NF‐κB pathway. Neurochem Int 67: 39–45. [DOI] [PubMed] [Google Scholar]
- Esposito E, Rinaldi B, Mazzon E, Donniacuo M, Impellizzeri D, Paterniti I et al. (2012). Anti‐inflammatory effect of simvastatin in an experimental model of spinal cord trauma: involvement of PPAR‐α. J Neuroinflammation 9: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori K, Yano M, Miyake H, Kimura H (2014). Termination mechanism of CREB‐dependent activation of COX‐2 expression in early phase of adipogenesis. Mol Cell Endocrinol 384: 12–22. [DOI] [PubMed] [Google Scholar]
- Harijith A, Pendyala S, Reddy NM, Bai T, Usatyuk PV, Berdyshev E et al. (2013). Sphingosine kinase 1 deficiency confers protection against hyperoxia‐induced bronchopulmonary dysplasia in a murine model: role of S1P signaling and Nox proteins. Am J Pathol 183: 1169–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbing T, Rothweiler R, Heinke J, Goetz L, Diehl P, Zirlik A et al. (2010). BMPER is upregulated by statins and modulates endothelial inflammation by intercellular adhesion molecule‐1. Arterioscler Thromb Vasc Biol 30: 554–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Zhao M, Wang T, Zhang G (2014). Upregulation of estrogen receptor mediates migration, invasion and proliferation of endometrial carcinoma cells by regulating the PI3K/AKT/mTOR pathway. Oncol Rep 31: 1175–1182. [DOI] [PubMed] [Google Scholar]
- Hsu CK, Lee IT, Lin CC, Hsiao LD, Yang CM (2014). Nox2/ROS‐dependent human antigen R translocation contributes to TNF‐α‐induced SOCS‐3 expression in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 306: L521–L533. [DOI] [PubMed] [Google Scholar]
- Hsu CK, Lee IT, Lin CC, Hsiao LD, Yang CM (2015). Sphingosine‐1‐phosphate mediates COX‐2 expression and PGE2/IL‐6 secretion via c‐Src‐dependent AP‐1 activation. J Cell Physiol 230: 702–715. [DOI] [PubMed] [Google Scholar]
- Ishii I, Fukushima N, Ye X, Chun J (2004). Lysophospholipid receptors: signaling and biology. Annu Rev Biochem 73: 321–354. [DOI] [PubMed] [Google Scholar]
- Ishiura Y, Fujimura M, Yamamoto H, Ishiguro T, Ohkura N, Myou S (2009). COX‐2 inhibition attenuates cough reflex sensitivity to inhaled capsaicin in patients with asthma. J Investig Allergol Clin Immunol 19: 370–374. [PubMed] [Google Scholar]
- Kalhori V, Kemppainen K, Asghar MY, Bergelin N, Jaakkola P, Tornquist K (2013). Sphingosine‐1‐phosphate as a regulator of hypoxia‐induced factor‐1α in thyroid follicular carcinoma cells. PLoS One 8: e66189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastl L, Sauer SW, Ruppert T, Beissbarth T, Becker MS, Süss D et al. (2014). TNF‐α mediates mitochondrial uncoupling and enhances ROS‐dependent cell migration via NF‐κB activation in liver cells. FEBS Lett 588: 175–183. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan A, van Nimwegen M, Leman K, Hoogsteden HC, Lambrecht BN (2013). Topical treatment targeting sphingosine‐1‐phosphate and sphingosine lyase abrogates experimental allergic rhinitis in a murine model. Allergy 68: 204–212. [DOI] [PubMed] [Google Scholar]
- Lappas M (2013). Forkhead box O1 (FOXO1) in pregnant human myometrial cells: a role as a pro‐inflammatory mediator in human parturition. J Reprod Immunol 99: 24–32. [DOI] [PubMed] [Google Scholar]
- Lee IT, Lee CW, Tung WH, Wang SW, Lin CC, Shu JC et al. (2010). Cooperation of TLR2 with MyD88, PI3K, and Rac1 in lipoteichoic acid‐induced cPLA2/COX‐2‐dependent airway inflammatory responses. Am J Pathol 176: 1671–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IT, Wang SW, Lee CW, Chang CC, Lin CC, Luo SF et al. (2008). Lipoteichoic acid induces HO‐1 expression via the TLR2/MyD88/c‐Src/NADPH oxidase pathway and Nrf2 in human tracheal smooth muscle cells. J Immunol 181: 5098–5110. [DOI] [PubMed] [Google Scholar]
- Lee IT, Yang CM (2012). Role of NADPH oxidase/ROS in pro‐inflammatory mediators‐induced airway and pulmonary diseases. Biochem Pharmacol 84: 581–590. [DOI] [PubMed] [Google Scholar]
- Lee SA, Kim EY, Jeon WK, Woo CH, Choe J, Han S et al. (2011). The inhibitory effect of raloxifene on lipopolysaccharide‐induced nitric oxide production in RAW264.7 cells is mediated through a ROS/p38 MAPK/CREB pathway to the up‐regulation of heme oxygenase‐1 independent of estrogen receptor. Biochimie 93: 168–174. [DOI] [PubMed] [Google Scholar]
- Lee YS, Lee SJ, Seo KW, Bae JU, Park SY, Kim CD (2013). Homocysteine induces COX‐2 expression in macrophages through ROS generated by NMDA receptor‐calcium signaling pathways. Free Radic Res 47: 422–431. [DOI] [PubMed] [Google Scholar]
- Lefer DJ (2002). Statins as potent antiinflammatory drugs. Circulation 106: 2041–2042. [DOI] [PubMed] [Google Scholar]
- Li M, Liu Y, Shi H, Zhang Y, Wang G, Xu J et al. (2012). Statins inhibit pulmonary artery smooth muscle cell proliferation by upregulation of HO‐1 and p21WAF1. Naunyn Schmiedebergs Arch Pharmacol 385: 961–968. [DOI] [PubMed] [Google Scholar]
- Lin CC, Lee IT, Wu WL, Lin WN, Yang CM (2012). Adenosine triphosphate regulates NADPH oxidase activity leading to hydrogen peroxide production and COX‐2/PGE2 expression in A549 cells. Am J Physiol Lung Cell Mol Physiol 303: L401–L412. [DOI] [PubMed] [Google Scholar]
- Lin CC, Lee IT, Yang YL, Lee CW, Kou YR, Yang CM (2010). Induction of COX‐2/PGE2/IL‐6 is crucial for cigarette smoke extract‐induced airway inflammation: role of TLR4‐dependent NADPH oxidase activation. Free Radic Biol Med 48: 240–254. [DOI] [PubMed] [Google Scholar]
- Liu JN, Suh DH, Yang EM, Lee SI, Park HS, Shin YS (2014). Attenuation of airway inflammation by simvastatin and the implications for asthma treatment: is the jury still out? Exp Mol Med 46: e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Cui Y, Li M, Xu H, Zuo J, Fang F et al. (2013). Cobalt protoporphyrin induces HO‐1 expression mediated partially by FOXO1 and reduces mitochondria‐derived reactive oxygen species production. PLoS One 8: e80521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maneechotesuwan K, Ekjiratrakul W, Kasetsinsombat K, Wongkajornsilp A, Barnes PJ (2010). Statins enhance the anti‐inflammatory effects of inhaled corticosteroids in asthmatic patients through increased induction of indoleamine 2, 3‐dioxygenase. J Allergy Clin Immunol 126: 754–762. [DOI] [PubMed] [Google Scholar]
- Matsuzaki E, Hiratsuka S, Hamachi T, Takahashi‐Yanaga F, Hashimoto Y, Higashi K et al. (2013). Sphingosine‐1‐phosphate promotes the nuclear translocation of β‐catenin and thereby induces osteoprotegerin gene expression in osteoblast‐like cell lines. Bone 55: 315–324. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodai A, Machida T, Izumi S, Hamaya Y, Kohno T, Igarashi Y et al. (2007). Sphingosine 1‐phosphate induces cyclooxygenase‐2 via Ca2+‐dependent, but MAPK‐independent mechanism in rat vascular smooth muscle cells. Life Sci 80: 1768–1776. [DOI] [PubMed] [Google Scholar]
- Osmak M (2012). Statins and cancer: current and future prospects. Cancer Lett 324: 1–12. [DOI] [PubMed] [Google Scholar]
- Ostroukhova M, Kouides RW, Friedman E (2009). The effect of statin therapy on allergic patients with asthma. Ann Allergy Asthma Immunol 103: 463–468. [DOI] [PubMed] [Google Scholar]
- Oya M (2010). mTOR inhibitors. Nihon Rinsho 68: 1067–1071. [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MM, Oskeritzian CA, Falanga YT, Hoffman RK, Amrani Y, Krymskaya VP et al. (2013). A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell‐dependent murine model of allergic asthma. J Allergy Clin Immunol 131: 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucci A, Formato L, Muscio M, Brscic E, Pizzimenti S, Ferroni F et al. (2011). PPARγ in coronary atherosclerosis: in vivo expression pattern and correlations with hyperlipidemic status and statin treatment. Atherosclerosis 218: 479–485. [DOI] [PubMed] [Google Scholar]
- Ryan JJ, Spiegel S (2008). The role of sphingosine‐1‐phosphate and its receptors in asthma. Drug News Perspect 21: 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Q, Shen LH, Hu LH, Pu J, Jing Q, He B (2012). Atorvastatin suppresses inflammatory response induced by oxLDL through inhibition of ERK phosphorylation, IκBα degradation, and COX‐2 expression in murine macrophages. J Cell Biochem 113: 611–618. [DOI] [PubMed] [Google Scholar]
- Tikhanovich I, Cox J, Weinman SA (2013). Forkhead box class O transcription factors in liver function and disease. J Gastroenterol Hepatol 28 (Suppl 1): 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo BT, Morton D Jr, Komaragiri S, Millena AC, Leath C, Khan SA (2013). TGF‐β effects on prostate cancer cell migration and invasion are mediated by PGE2 through activation of PI3K/AKT/mTOR pathway. Endocrinology 154: 1768–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volzke A, Koch A, Meyer Zu HD, Huwiler A, Pfeilschifter J (2014). Sphingosine 1‐phosphate (S1P) induces COX‐2 expression and PGE2 formation via S1P receptor 2 in renal mesangial cells. Biochim Biophys Acta 1841: 11–21. [DOI] [PubMed] [Google Scholar]
- Webb AE, Brunet A (2014). FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci 39: 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Tian S, Zhou H, Wu Y (2013). Statins protect human endothelial cells from TNF‐α induced inflammation via ERK5 activation. Biochem Pharmacol 85: 1753–1760. [DOI] [PubMed] [Google Scholar]
- Yang CM, Lin CC, Lee IT, Lin YH, Yang CM, Chen WJ et al. (2012). Japanese encephalitis virus induces matrix metalloproteinase‐9 expression via a ROS/c‐Src/PDGFR/PI3K/Akt/MAPKs‐dependent AP‐1 pathway in rat brain astrocytes. J Neuroinflammation 9: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Hui L, Wang Y, Yang H, Jiang X (2015). SOX2 promotes the migration and invasion of laryngeal cancer cells by induction of MMP‐2 via the PI3K/Akt/mTOR pathway. Oncol Rep 34: 2627–2635. [DOI] [PubMed] [Google Scholar]
- Zhu T, Zhang W, Wang DX, Huang NW, Bo H, Deng W et al. (2012). Rosuvastatin attenuates mucus secretion in a murine model of chronic asthma by inhibiting the γ‐aminobutyric acid type A receptor. Chin Med J (Engl) 125: 1457–1464. [PubMed] [Google Scholar]