

Abstract
Background and Purpose
There is emerging evidence suggesting that abnormal transport of amyloid‐β (Aβ) across the blood–brain barrier (BBB) is involved in diabetes‐associated cognitive decline. We investigated whether PPARγ agonists restore Aβ transport across the BBB and hippocampal plasticity in db/db mice.
Experimental Approach
Efflux and influx of Aβ across the BBB were determined by stereotaxic intra‐cerebral or i.a. infusion of [125I]‐Aβ1–40 respectively. Receptor for advanced glycation end products (RAGE) and low‐density lipoprotein receptor‐related protein 1 (LRP1), which are involved in Aβ influx and efflux, PPARγ and NF‐κB p65 at the BBB, as well as hippocampal Aβ, caspase‐3, Bax and Bcl‐2 were assayed by Western blot, immunohistochemistry and RT‐PCR. In vivo, hippocampal LTP was recorded, and Morris water maze and Y‐maze tasks were performed.
Key Results
Treatment with PPARγ agonists, rosiglitazone (0.8 mg·kg−1) and pioglitazone (9.0 mg·kg−1), for 6 weeks significantly increased Aβ efflux and decreased Aβ influx across the BBB in db/db mice. Concomitantly, they decreased hippocampal Aβ1–40 and Aβ1–42, suppressed neuronal apoptosis, as indicated by decreased caspase‐3 activity and increased ratio of Bcl‐2/Bax, and increased hippocampal plasticity, characterized by an enhanced in vivo LTP and better performance in behavioural tests. Furthermore, the PPARγ agonists induced the expression of LRP1 gene by activation of PPARγ and suppressed RAGE gene expression by inactivation of NF‐κB signalling at the BBB of db/db mice.
Conclusions and Implications
PPARγ agonists modify abnormal Aβ transport across the BBB and this is accompanied by amelioration of β‐amyloidosis and an improvement in hippocampal plasticity in diabetic mice.
Abbreviations
- AD
Alzheimer's disease
- Aβ
amyloid‐β
- AGEs
advanced glycation end products
- BBB
blood–brain barrier
- ID
injected dose
- LRP1
low‐density lipoprotein receptor related protein 1
- MWM
Morris water maze
- OCT
optimal cutting temperature
- PS
population spike
- RAGE
receptor for advanced glycation end products
- STZ
streptozotocin
- TCA
trichloroacetic acid
Tables of Links
| TARGETS | |
|---|---|
| Nuclear hormone receptors a | Other protein targets |
| PPARγ | Bax |
| Enzymes b | Bcl‐2 |
| Caspase 3 | FABP4 |
| RAGE |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,2013b).
Introduction
There is a mass of evidence suggesting that diabetes impacts cognitive function (Ott et al., 1999; Roberts et al., 2014), and epidemiological studies have also indicated that patients with diabetes are at a higher risk of developing Alzheimer's disease (AD) (Kopf and Frölich, 2009; Maher and Schubert, 2009; Morris et al., 2014). Cognitive deficits have also been reported in studies on rodent models of diabetes such as the db/db mice, which exhibit the characteristics of obesity, insulin resistance, hyperglycaemia and also have increased levels of corticosterone in the circulation due to a mutation that inactivates the leptin receptor (Hummel et al., 1996). These db/db mice have decreased levels of brain‐derived neurotrophic factor (BDNF) and increased inflammatory cytokines (IL‐1β, IL‐6 and TNF‐α) in the hippocampus, which cause impairments in hippocampus‐dependent spatial recognition memory, perforant path synaptic plasticity and adult neurogenesis (Stranahan et al., 2008; Hong et al., 2009; Dinel et al., 2011). Interestingly, there is accumulating evidence suggesting that diabetes affects the transport activity of the blood–brain barrier (BBB), which might be a causative factor in cognitive impairment and AD (Maeng et al., 2007; Huber, 2008; Hong et al., 2009; Liu et al., 2009). The BBB is primarily built up of brain endothelial cells, astrocyte end‐feet, pericytes, perivascular macrophages and a basal membrane. It is critical for maintaining brain function through regulating the transport of materials between the CNS and the peripheral circulation. An impairment of the influx and efflux transporters existing at the BBB may contribute to the pathogenesis of AD (Ueno et al., 2010).
Two major proteins, low‐density lipoprotein receptor‐related protein 1 (LRP1) and the receptor for advanced glycation end products (RAGE), are involved in receptor‐mediated amyloid β (Aβ) trafficking across the BBB (Deane et al., 2004; Zhang and Lee, 2011). RAGE, present at the lumina of the endothelial cytoplasm, is thought to transport blood Aβ into the brain, while LRP1, located at the ablumina of the endothelial cytoplasm, transports Aβ out of the brain (Shibata et al., 2000; Deane et al., 2003). These transporters at the BBB play a critical role in regulating Aβ homeostasis in the brain. We have previously shown that the up‐regulation of RAGE or down‐regulation of LRP1 at the BBB contribute to cerebral Aβ accumulation in streptozotocin (STZ)‐induced diabetic mice (Hong et al., 2009; Liu et al., 2009), which initiates the pathological cascades involved in the neuronal and neurovascular dysfunctions. Thus, rectifying the abnormal Aβ transport across the BBB is an important strategy for protecting neurons from damage caused by intracerebral Aβ accumulation under diabetic conditions. PPARs are ligand‐dependent transcription factors. PPARγ activation is known to modulate lipid and glucose metabolism and to increase insulin sensitization. Highly selective PPARγ agonists such as pioglitazone and rosiglitazone are currently approved for a p.o. monotherapy in patients with type 2 diabetes (Sood et al., 2000). Our recent research showed that pioglitazone reverses memory impairments in high‐fat diet plus STZ‐induced diabetic mice or STZ‐induced diabetic mice, and this involved inhibition of cerebral Aβ accumulation (Jiang et al., 2012; Liu et al., 2013). However, the mechanism by which these PPARγ agonists reduce cerebral Aβ is not well understood. Here, we investigated the effects of these PPARγ agonists on Aβ influx and efflux and hippocampal plasticity in db/db mice.
Methods
Animals and experimental protocol
Male C57BL/KsJ db/db (BKS. Cg‐m +/+ Leprdb/J) mice (approx. 6 weeks old, weighing 30–40 g; Model Animal Research Center of Nanjing University, China), and their age‐matched lean controls db/m mice were used for the experiments. The former strain of mice is obese and known to develop type 2 diabetes, followed by diabetic kidney disease (Zhang et al., 2013). The experimental procedures are in compliance with the European Communities Council Directive of 24 November 1986 (86/609/EEC), and the experimental protocol was approved by the Institutional Review Committee for the use of Animal Subjects of China Pharmaceutical University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). All animals were maintained in a well‐ventilated holding room at constant humidity of 55 ± 5% and temperature of 24 ± 1°C under a 12 h light/12 h dark cycle (lights on 07:00–19:00 h) with free access to water and standard laboratory chow.
db/db mice with similar degrees of body weight and hyperglycaemia were randomly divided into db/db vehicle (db/db + Veh), 0.8 mg·kg−1 rosiglitazone (db/db + Rsg 0.8) and 9.0 mg·kg−1 pioglitazone (db/db + Pio 9.0) groups. Age‐matched normal homozygote mice (db/m + Veh, n = 29) were administered the vehicle p.o. The total number of animals in each group was 29 to 31 mice. Rosiglitazone or pioglitazone were dissolved in 0.5% CMC‐Na. After intragastric administration of the drugs for 6 weeks, some of the mice from each group were used for stereotaxic intra‐cerebral infusion (n = 3–4 for each group) or i.a. infusion (n = 3–4 for each group) of [125I]‐Aβ1–40, others were assigned for in vivo LTP recording (n = 4–5 for each group). The rest of the mice were used for the behavioural tests. The expression of RAGE, LRP1, PPARγ and NF‐κB p65 was determined in the isolated brain capillaries and hippocampal Aβ, pro‐ and cleaved caspase‐3, Bax, Bcl‐2 and advanced glycation end products (AGEs) were also detected (n = 6–8 for each group). We also monitored the fasting blood glucose and insulin levels.
Brain clearance studies
Brain interstitial fluid mediated clearance of [125I]‐Aβ1–40 was determined simultaneously with the reference marker [14C]‐inulin as has been described previously (Shibata et al., 2000; Hong et al., 2009). Briefly, a stainless steel guide cannula was stereotaxically inserted into the right caudate nucleus of anaesthetized mice (i.p. chloral hydrate, 350 mg·kg−1). The coordinates for the cannula were 1.9 mm lateral to bregma and 0.9 mm anterior, and 2.9 mm below the dural surface (Shibata et al., 2000; Cirrito et al., 2005). Mice were allowed to recover for 1 week after surgery. After this period, mice were re‐anaesthetized to conduct radiotracer studies. Tracer fluid (0.5 μl) containing 60 nM [125I]‐Aβ1–40 was injected over a period of 5 min, along with 12 nM of [14C]‐inulin, at 30 min after anaesthesia. After injecting these tracers, blood and brain samples were collected within 30 min and their radioactivity was analysed. Trichloroacetic acid (TCA) was added to the plasma and brain samples at a final concentration of 10% and then the samples were centrifuged at 4°C, 24 840 g for 10 min. A γ counter was used to analysis radioactivity in the supernatant and pellet fractions. Based on the TCA analysis, [125I]‐Aβ1–40 injected into the brain was more than 97% intact and several indices including %[125I]‐Aβ1–40 recovered in the brain, the percentage of BBB clearance and the percentage of injected dose (ID) mL‐1 plasma were determined. The percentage of radioactivity remaining in the brain after microinjection was determined as
and the percentage of BBB clearance of [125I]‐Aβ1–40 was calculated as
and then distribution of [125I]‐Aβ1–40 in the blood was expressed as % injected dose ml‐1 plasma
where N i is the radioactivity injected into the brain, and N b is the radioactivity remaining in the brain at the end of the experiment. In all calculations, cpm is counts per minute for TCA‐precipitated 125I radioactivity and dpm is disintegration per minute for [14C]‐inulin. Inulin, is a metabolically inert polar reference marker and is neither transported across the BBB nor retained by the brain.
Brain perfusion technique
This method was conducted to evaluate the influx of radiolabelled ligands across the BBB and has been described previously (Deane et al., 2003; Liu et al., 2009). Briefly, after the mice had been anaesthetized by an i.p. injection of chloral hydrate, the right common carotid artery of mice was connected to an extracorporeal perfusion circuit and perfused with oxygenated medium containing d‐glucose, electrolytes, dextran (formula weight 70 ,000, 48 g·L−1) and sheep red cells at 1 mL·min−1. Then 4.5 nmol·L−1 of [125I]‐Aβ1–40 was infused i.a. simultaneously with [14C]‐inulin at 0.1 mL·min−1. Electrolytes, pH, blood gases, arterial BP and perfusion pressure were kept within physiological range. Mice were killed, and their brains were used to analyse radioactivity at a preconcerted time (10 min). Trichloroacetic acid sedimentation was used to evaluate the integrity of 125I‐Aβ1–40. The permeability surface area product of the [125I]‐Aβ1–40 was determined with [14C]‐inulin, using the following calculation.
where T is the infusion time.
Isolation of brain microvessels
Brain microvessels were isolated and prepared as described previously (Singh et al., 2013). Mice were anaesthetized i.p. with chloral hydrate (350 mg·kg−1) and then decapitated, then pial vessels and the cerebral cortex were removed and chopped into pieces in an ice‐cold dish. These were then homogenized in a 5 mL handheld glass dounce tissue grinder with 3.5‐fold excess volume of ice‐cold buffer solution containing (in mM): sodium pyruvate (1), HEPES (15), glucose (10), NaHCO3 (25), KH2PO4 (1.2), CaCl2 (2.5), KCl (4.7), NaCl (130) and MgSO4 (1.2), at pH 7.4. The homogenate was suspended in an equal volume of 26% dextran (MW 64 000–76 000), spun at 5800 g for 15 min, and the pellet (brain vessels) was carefully separated from the supernatant. The pellet was then resuspended in an ice‐cold buffered solution, filtered using a 100 μm filter, and the filtrate passed through a 40 μm filter. The unfiltered microvessels were harvested by washing into a low binding tube and resuspended in an ice‐cold buffered solution. The microvessels were then washed three times with 1.5 mL ice‐cold buffered solution and then resuspended in an ice‐cold lysis buffer, sonicated, centrifuged at 20 000 g for 20 min, and supernatant was used for Western blot or RT‐PCR analysis.
Behavioural assessments
The Morris water maze (MWM) was used to assess spatial and long‐term memory and the Y‐maze test for the evaluation of working and short‐term memory. These behavioural tests were carried out as described previously (Tang et al., 2013; Tang et al., 2014). Furthermore, the general locomotor activity of the mice in this study was also assessed by use of an open field test, as described previously (Wang et al., 2014). More detailed descriptions of these behavioural tasks can be seen in the Supporting Information.
In vivo hippocampal LTP recording
Mice were anaesthetized with i.p. urethane (1.0 g·kg−1) and then immobilized on a stereotactic frame (SR‐5, Narishige, Japan) as described previously (Tang et al., 2014). A stimulating electrode and a recording glass pipette were inserted into the perforant path and dentate gyrus of the dorsal hippocampus respectively. Coordinates for the stimulating electrodes (tungsten with Teflon coating; Bilaney) were 3.0 mm from the midline, 3.8 mm caudal to bregma and 1.5 mm below the dural surface and for the recording electrode, 1.4 mm from the midline, 2 mm caudal to bregma and 1.5 mm below the dural surface. The responses evoked were filtered at 1/60 Hz and amplified, 100 μs pulses of 0.3 mA until the appearance of a population spike (PS). Input–output curves were firstly generated so as to confirm the maximal amplitude of the PS, and then the intensity of the stimulus was set at a level that evoked a PS between 55% and 65% of the maximum amplitude. The amplitude of the PS was collected and averaged every 5 min. High‐frequency stimulation, eight sessions of 400 μs stimuli at 400 Hz with a 10 s interval, was applied to induce LTP which was measured as normalized PS amplitude (%). All stimulation and recording were performed by an online computerized oscilloscope–stimulator and data analysis interface system. The percentage of the ratio of absolute PS amplitude to basal value was used to represent the PS amplitude.
Data analysis
Data shown are expressed as mean ± SEM. In particular, a repeated measure ANOVA with ‘group’ as the between‐subject factor and ‘days’ as the within‐subject factor were used to analysis group differences in the MWM escape latencies. The other data were analysed by a one‐way ANOVA followed by a Dunnett's post hoc analysis for multiple comparisons. The level of statistical significance was P < 0.05. All analyses were carried out using spss 20.0.
Materials
Rosiglitazone and pioglitazone were both purchased from Jiangsu Hengrui Medicine Co. Ltd. (Nanjing, China). Antibodies were obtained from different companies: anti‐Aβ1–40, anti‐Aβ1–42 and anti‐RAGE from Abcam Technology Co., Ltd. (Hong Kong); anti‐nuclear factor κB (NF‐κB) p65 and anti‐PPARγ from Cell Signaling Technology, Inc. (Boston, MA, USA); Anti‐LRP1 from Santa Cruz Biotechnology, Inc. (Heidelberg, Germany); anti‐histone H3, anti‐β‐actin and secondary antibodies from Bioworld Technology Co., Ltd. (Minneapolis, MN, USA). Streptavidin‐biotin complex immunohistochemistry kit was purchased from Boster Biotechnology Co., Ltd. (Wuhan, China). Mouse insulin and AGEs elisa kit were purchased from Cusabio Biotech Co., Ltd. (Wilmington, DE, USA). Coomassie (Bradford) protein assay kit and Glucose Oxidase Kit were purchased from Nanjing Jiancheng Biotech Institute (Nanjing, China). Nucleoprotein extraction kit was purchased from Sangon Biotech Co., Ltd. (Shanghai, China). Human Aβ1–40 protein fragment was purchased from Sigma‐Aldrich (USA). Na[125I] and [14C]‐inulin (5,000 Da, 1.5 μCi·mg−1) were purchased from Chengdu Gaotong Isotope Co., Ltd. (Chengdu, China) and American Radiolabeled Chemicals, Inc. (USA) respectively. Human [125I]‐Aβ1–40 was synthesized at Jiangnan Institute of Nuclear Medicine (Wuxi, China). More than 99% of [125I]‐Aβ1–40 (4,330 Da; 70 μCi·μg−1) radioactivity was present in the form of nonoxidized monomeric peptide. Aliquots of radiolabelled Aβ1–40 were kept at −20°C for a maximum of 4 weeks before use. All the other chemicals were of analytical grade and commercially available.
Results
PPARγ agonists increase [125I]‐Aβ1–40 efflux across the BBB in db/db mice
To observe the changes in Aβ efflux across the BBB in db/db mice, we carried out brain clearance studies. As shown in Figure 1, the efflux of [125I]‐Aβ1–40 from the brain at 30 min was markedly reduced in db/db mice compared with that of the control db/m group, as indicated by a significant decrease in the percentage of [125I]‐Aβ1–40 BBB clearance (P < 0.01; Figure 1A) and increase in the percentage of [125I]‐Aβ1–40 recovery in brain (P < 0.01; Figure 1B). Moreover, intact [125I]‐Aβ1–40 was shown to be present in plasma within 30 min of its administration to the brain and its level was markedly lowered in plasma (P < 0.01; Figure 1C) from the db/db mice compared with that of the db/m mice. Rosiglitazone or pioglitazone administration markedly increased the distribution of [125I]‐Aβ1–40 in plasma (both P < 0.05) and the percentage of [125I]‐Aβ1–40 BBB clearance (both P < 0.01), while the percentage of [125I]‐Aβ1–40 recovered in the brain significantly decreased (both P < 0.01).
Figure 1.
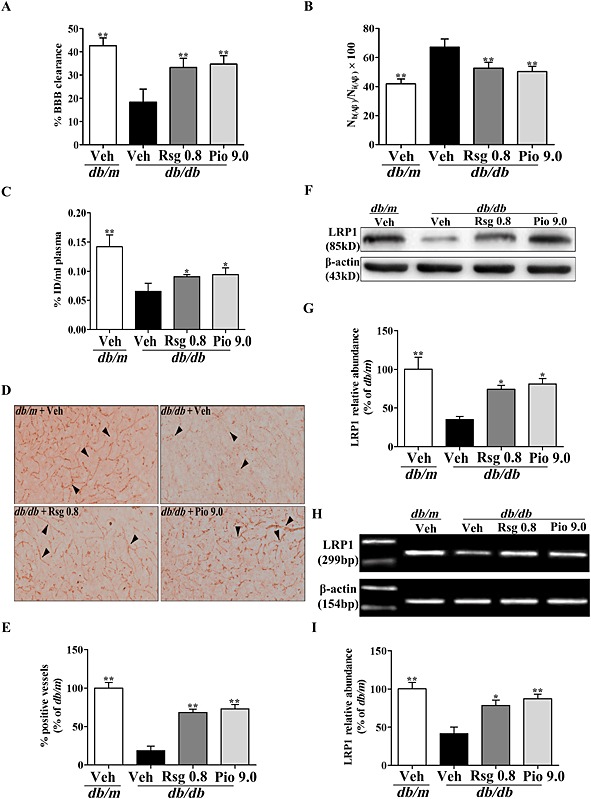
PPARγ agonists increase [125I]‐Aβ1‐40 efflux and LRP1 expression at the BBB in db/db mice. [125I]‐Aβ1‐40 and [14C]‐inulin were injected simultaneously, the percentage of [125I]‐Aβ1‐40 BBB clearance (A) and the levels of [125I]‐Aβ1‐40 in the brain (B) and plasma (C) were determined by γ counting after 30 min. (D) Representative immunohistochemical microphotographs of LRP1, (E) analysis for % of LRP1‐positive vessels (arrow, brain microvessels positive LRP1), (F) representative immunoblot detected by Western blot, (G) protein quantification, (H) LRP1 mRNA expression and (I) mRNA quantification of expressed as a percentage of the db/m are shown. Original magnification is 200×. Values shown are expressed as mean ± SEM; n = 3–4. *P < 0.05, **P < 0.01 versus db/db + Veh group.
PPARγ agonists increase LRP1 expression at the BBB in db/db mice
LRP1 has been shown to mediate the transcytosis of Aβ out of the brain across the BBB (Shibata et al., 2000; Hong et al., 2009). To further determine whether the increased Aβ efflux across the BBB induced by the PPARγ agonists results from the up‐regulation of LRP1 expression, we determined LRP1 expression at the BBB in db/db mice. Both vascular immunohistochemical staining and Western blot analysis of the isolated brain microvessels showed that LRP1 protein was significantly decreased in db/db mice treated with vehicle but markedly increased in db/db mice treated repeatedly with rosiglitazone or pioglitazone (both P < 0.05; Figure 1D‐G). As shown in Figure 1H, I, RT‐PCR analysis further indicated that the PPARγ agonists increased LRP1 protein by enhancing its mRNA transcription at the BBB in db/db mice.
PPARγ agonists decrease [125I]‐Aβ1–40 influx across the BBB in db/db mice
To determine whether PPARγ agonists affect Aβ influx across the BBB in db/db mice, we carried out brain perfusion experiments with [125I]‐Aβ1–40, evaluated as cerebrovascular permeability surface area product. As indicated in Figure 2A, the influx of circulating [125I]‐Aβ1–40 across the BBB was significantly increased in db/db mice relative to that of db/m mice (P < 0.01), whereas chronic treatment with rosiglitazone or pioglitazone markedly reduced the influx of peripheral [125I]‐Aβ1–40 across the BBB in db/db mice (P < 0.05 and P < 0.01 respectively).
Figure 2.
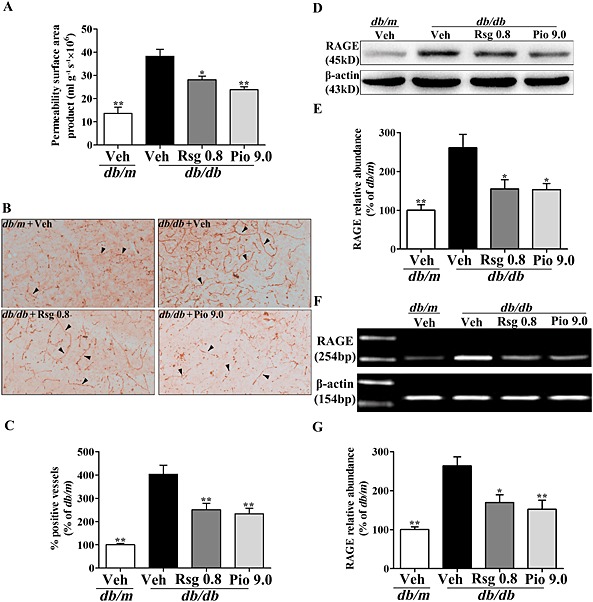
PPARγ agonists decrease [125I]‐Aβ1‐40 influx and RAGE expression at the BBB in db/db mice. (A) Cerebrovascular permeability surface area product was determined by γ counting after 10 min of [125I]‐Aβ1‐40 infusion. (B) Representative immunohistochemical microphotographs of RAGE, (C) analysis for % of RAGE‐positive vessels (arrow, brain microvessels positive RAGE), (D) representative immunoblot detected by Western blot, (E) protein quantification, (F) RAGE mRNA expression and (G) mRNA quantification of RAGE expressed as the percentage of the db/m are shown. Original magnification is 200×. Values shown are expressed as mean ± SEM; n = 3–4. *P < 0.05, **P < 0.01 versus db/db + Veh group.
PPARγ agonists decrease RAGE expression at the BBB in db/db mice
It has been confirmed that RAGE mediates Aβ influx across the BBB (Deane et al., 2003; Liu et al., 2009). To clarify whether the decreased Aβ influx across the BBB induced by PPARγ agonists can be attributed to the down‐regulation of RAGE protein, we evaluated the expression of RAGE protein at the BBB in db/db mice. Immunohistochemical staining showed that brain microvessels positive for RAGE staining were obviously increased in db/db mice treated with vehicle but significantly decreased in db/db mice treated with rosiglitazone or pioglitazone (Figure 2B, C). Western blot analysis of isolated brain microvessels also showed a significant reduction in the level of RAGE protein in db/db mice treated with rosiglitazone or pioglitazone (both P < 0.05; Figure 2D, E). As shown in Figure 2F, G, RT‐PCR analysis further revealed that the PPARγ agonists significantly decreased RAGE mRNA transcription at the BBB in db/db mice.
Effects of PPARγ agonists on PPARγ and NF‐κB p65 at the BBB in db/db mice
The next important question that we addressed is how PPARγ agonists regulate LRP1 or RAGE expression at the BBB. The binding site of PPARγ has been shown to be located in the promoter site of the adipocyte LRP1 gene, and the expression of functional LRP1 is promoted by PPARγ ligand‐induced up‐regulation of transcription (Gauthier et al., 2003). Interestingly, we found that PPARγ protein was markedly down‐regulated in the nuclei of brain microvessel, endothelial cells of db/db mice (P < 0.01; Figure 3A, B), and significantly elevated in those of db/db mice treated with PPARγ agonists (P < 0.05 and P < 0.01 respectively). Analysis of the putative promoter region of the RAGE gene has revealed the presence of potential NF‐κB binding sites (Li and Schmidt, 1997). Like peripheral tissue, NF‐κB signalling activation was observed at the BBB in db/db diabetic mice, as indicated by increased NF‐κB p65 in the nuclei of brain microvessel, endothelial cells (both P < 0.01; Figure 3A, C). More importantly, treatment with rosiglitazone or pioglitazone markedly reduced nuclear NF‐κB p65 in brain microvessel, endothelial cells, which suggests that PPARγ agonists suppress NF‐κB signalling at the BBB in diabetic mice.
Figure 3.
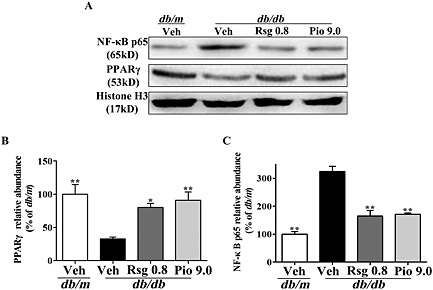
Effects of PPARγ agonists on PPARγ and NF‐κB p65 at the BBB in db/db mice. NF‐κB p65 or PPARγ (A) was detected by Western blot. Histone H3 was used as a loading control. Quantification of PPARγ (B) and NF‐κB p65 (C) are expressed as a percentage of the db/m. Values shown are expressed as mean ± SEM; n = 3. *P < 0.05, **P < 0.01 versus db/db + Veh group.
PPARγ agonists reduce Aβ levels and apoptosis‐related molecules in the brain of db/db mice
Next, in order to determine whether Aβ levels in the brain decrease when the Aβ efflux across the BBB is altered and the effects of the PPARγ agonists, Aβ levels in the hippocampus, a key brain region related to memory, were assayed. As expected, treatment with rosiglitazone or pioglitazone effectively and reversibly suppressed the increases in Aβ1–40 and Aβ1–42 (P < 0.05 and P < 0.01 respectively; Figure 4A‐C) in the hippocampus of db/db mice. Immunohistochemical staining also demonstrated similar results (Figure 4D‐G). As Aβ accumulation is likly to cause activation of apoptotic pathways in brain (Stadelmann et al., 1999; Awasthi et al., 2005; Rohn et al., 2008), we also investigated the expressions of apoptosis‐related molecules, caspase‐3, Bcl‐2 and Bax, as by Western blot analysis. As shown in the Supporting Information Fig. S1, rosiglitazone and pioglitazone treatment markedly suppressed caspase‐3 activation, Bax up‐regulation and Bcl‐2 down‐regulation (both P < 0.05) in the hippocampus.
Figure 4.
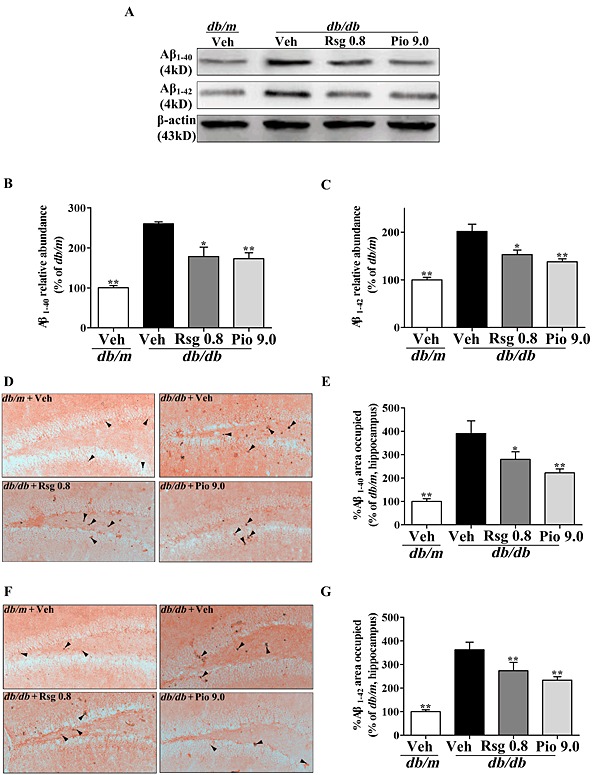
PPARγ agonists reduce Aβ levels in the brain of db/db mice. (A) Representative immunoblot of Aβ1‐40 and Aβ1‐42 detected by Western blot, quantification of Aβ1‐40 (B) and Aβ1‐42 (C) expressed as a percentage of the db/m are presented. Representative immunohistochemical microphotographs of Aβ1‐40 (D) and Aβ1‐42 (F), analysis for % of Aβ1‐40 (E) and Aβ1‐42 (G) area occupied (arrow, Aβ1‐40 or Aβ1‐42 positive area respectively) are shown. Original magnification is 200×. Values shown are expressed as mean ± SEM; n = 3. *P < 0.05, **P < 0.01 versus db/db + Veh group.
Effects of PPARγ agonists on hippocampal LTP in db/db mice
Aβ accumulation can also damage synaptic plasticity; thus, we recorded the hippocampal LTP, a well‐established indicator of synaptic plasticity closely related to memory. The LTP recording revealed that hippocampal LTP was modestly inhibited in db/db mice (130.90 ± 3.17%) after the tetanic stimulation persisting for more than 60 min compared with the db/m group (222.22 ± 7.46%, P < 0.01; Figure 5A, B), and markedly increased in db/db mice treated with rosiglitazone or pioglitazone (156.97 ± 6.21% and 173.66 ± 10.31%, P < 0.05 and P < 0.01 respectively).
Figure 5.
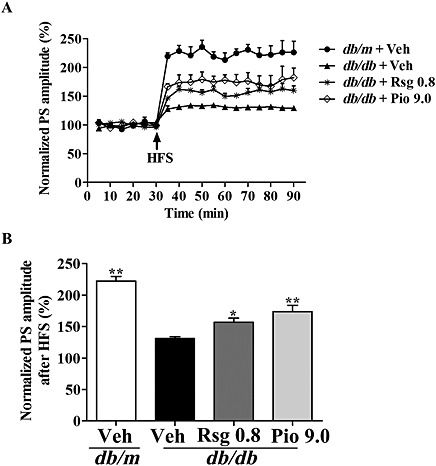
Effects of PPARγ agonists on hippocampal LTP in db/db mice. High‐frequency stimulation (HFS) was given at the end of 30 min baseline recording of PS. The time course of PS after HFS (A) and PS at 60 min post‐HFS is summarized in (B). Values shown are expressed as mean ± SEM; n = 4–5. *P < 0.05, **P < 0.01 versus db/db + Veh group.
PPARγ agonists ameliorate memory impairment in db/db mice
Hippocampal LTP should be in line with the enhanced hippocampus‐dependent learning and memory by the behavioural tests. In a non‐spatial visible‐platform variant of the MWM, mice showed similar escape latency indicating no significant difference in motivation and vision in each group (four trials per mouse per day for 2 days, effect of day, P < 0.01; effect of group, P > 0.05; effect of group‐by‐day interaction, P > 0.05; Figure 6A). In the spatial hidden‐platform variant, compared with the db/m group, db/db mice had a significantly increased escape latency (P < 0.05; Figure 6B). Interestingly, rosiglitazone or pioglitazone treatment significantly decreased escape latency (four trials per mouse per day for 3 days, effect of day, P < 0.01; effect of group, P < 0.01; effect of group‐by‐day interaction, P > 0.05). In the probe trial, db/db mice showed a significant decrease in the percentage of total time in the target quadrant (one trial per mouse for the last day, P < 0.01; Figure 6C, D), the number of target crossings (one trial per mouse for the last day, P < 0.01; Figure 6E) compared with the db/m group. In contrast, db/db mice treated with rosiglitazone or pioglitazone displayed significant increases in these indices compared with those of db/db group. The Y‐maze test also indicated that rosiglitazone or pioglitazone significantly increased the number of correct choices and decreased the latency to enter the shock‐free compartment in db/db mice (Figure 6F, G). For the open field test, there was no significant difference in the total distance travelled by each group (Supporting Information Fig. S2A). No significant effect of rosiglitazone or pioglitazone alone on memory was observed in age‐matched normal homozygote mice (data not shown). These data indicate that PPARγ agonists ameliorate memory impairments in db/db mice.
Figure 6.
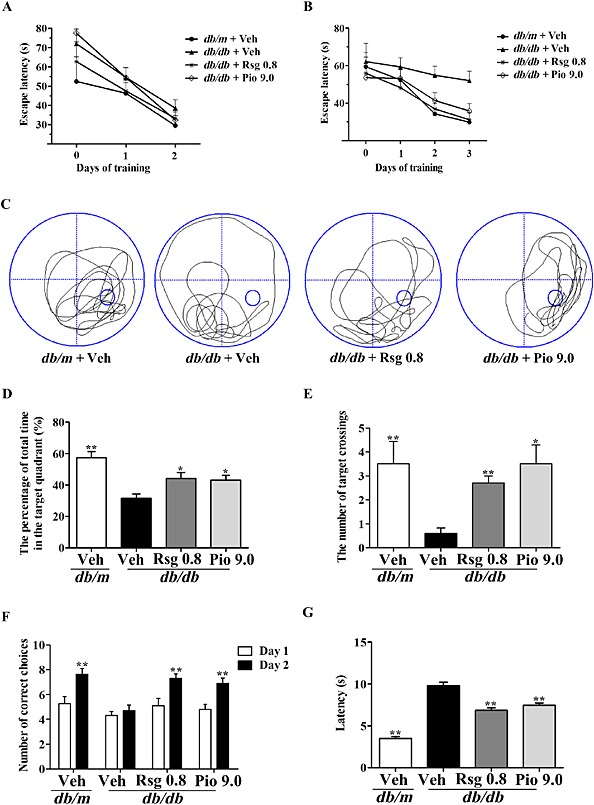
PPARγ agonists ameliorate memory impairment in db/db mice. In the MWM task, day 0 indicates performance on the first trial, and subsequent points represent average of all daily trials. (A) No differences were found in the escape latency among all groups during the 2‐day visible platform test. (B) Changes in escape latency to reach the hidden platform during the 3‐day acquisition trials, (C) representative swim paths during the spatial probe test, (D) the percentage of total time in the target quadrant, (E) the number of target crossings during the probe trial test, (F) the number of correct choices on days 1–2 and (G) the latency to enter the shock‐free compartment on day 2 in the Y‐maze test. Values shown are expressed as mean ± SEM; n = 8–10. *P < 0.05, **P < 0.01 versus db/db + Veh group.
Effects of PPARγ agonists on brain AGEs, fasting serum glucose and insulin levels in db/db mice
As shown in Table 1, Rosiglitazone or pioglitazone pronouncedly ameliorated hyperglycaemia (both P < 0.05), hyperinsulinaemia (P < 0.01 and P < 0.05 respectively) and inhibited the generation of AGEs in both the hippocampus and cortex (P < 0.01 and P < 0.05 respectively), but did not affect body weights.
Table 1.
Effects of PPARγ agonists on brain AGEs, serum glucose and insulin levels in db/db mice
| Groups | Serum Glucose (mmol·L−1) | Serum insulin (mIU·L−1) | AGEs levels (μg·g−1 protein) | |
|---|---|---|---|---|
| Hippocampus | Cortex | |||
| db/m + Veh | 8.33 ± 1.47 ** | 8.63 ± 1.01 ** | 35.27 ± 3.15 ** | 46.31 ± 4.64 ** |
| db/db + Veh | 31.67 ± 1.83 | 45.70 ± 2.44 | 86.87 ± 6.14 | 97.61 ± 6.26 |
| db/db + Rsg 0.8 | 23.99 ± 1.85 * | 36.50 ± 2.10 ** | 64.15 ± 5.27 * | 75.96 ± 5.08 * |
| db/db + Pio 9.0 | 22.88 ± 2.78 * | 38.15 ± 1.88 * | 51.69 ± 4.51 ** | 66.27 ± 6.45 ** |
Values shown are expressed as mean ± SEM; n = 5–10.
P < 0.05.
P < 0.01 versus db/db + Veh group.
Discussion and conclusion
We initially demonstrated that PPARγ agonists increase LRP1 expression and Aβ transport from the brain into the bloodstream by stimulating PPARγ, and significantly decrease RAGE expression and Aβ transport from the bloodstream into the brain by inhibiting NF‐κB activation at the BBB in db/db mice. The bidirectional regulation of Aβ transport across the BBB was correlated with a decrease in Aβ accumulation and amelioration of apoptotic responses, as the PPARγ agonists suppressed caspase‐3 activation and increased the ratio of Bcl‐2/Bax in the hippocampus, which is associated with short‐ and long‐term memory. More importantly, the PPARγ agonists effectively improved hippocampus plasticity, as they enhanced LTP and improved learning and memory, reflected by an improved performance in the MWM and Y‐maze tasks. In addition, the PPARγ agonists also significantly inhibited the formation of cerebral AGEs and improved insulin resistance, as they decreased both serum glucose and insulin in the db/db mice.
The vascular hypothesis on cognitive dysfunction associated with diabetes proposes that functional or structural changes of the BBB, which result in the accumulation of numerous neurotoxic and vasculotoxic macromolecules in the brain or a reduction in cerebral blood flow and hypoxia, can induce neurodegeneration (Huber, 2008; Kodl and Seaquist, 2008; Bell et al., 2010). Specifically, abnormalities in the BBB decrease Aβ efflux from the brain into the bloodstream (Deane et al., 2004) and may increase the influx of peripheral Aβ into the brain (Atwood et al., 2002; Eisele et al., 2010), elevating brain Aβ levels. These changes occur in insulin‐deficient animals (Liu et al., 2006; Liu et al., 2008; Hong et al., 2009; Liu et al., 2009). Interestingly, db/db mice with hyperinsulinaemia also showed a similar alteration in Aβ influx and efflux across the BBB. An increased expression of RAGE expression in cerebral vessels, neurons and glial cells has been shown to be associated with diabetes‐related cognitive impairment (Toth et al., 2006), suggesting that RAGE has a crucial role in the development of cerebral dysfunction (Yan et al., 1997). RAGE, a member of the immunoglobulin superfamily, is a multiligand receptor that binds to a range of molecules including AGEs, Aβ and the S100/calgranulin family of pro‐inflammatory mediators (Hofmann et al., 1999; Stern et al., 2002). The most prominent among these is Aβ, which plays a key role in AD pathology. RAGE is associated with chronic diabetes‐related damage in the kidneys, retina, peripheral nerves and blood vessels (Kim et al., 2005). RAGE gene promoter contains NF‐κB transcription elements, which interact between cell‐surface RAGE and its ligand setting up a positive feedback mechanism, which can rapidly accelerate disease progression (Li and Schmidt, 1997). In our study in db/db mice, AGEs and Aβ significantly accumulated in the brain, and NF‐κB p65 translocated into the nucleus of brain microvessel endothelial cells, subsequently stimulating the expression of RAGE gene and the transport of peripheral Aβ into the brain. PPARγ plays a key role in maintaining cerebrovascular health. Its genetic inactivation in mice causes increased reactive oxygen species generation, endothelial dysfunction and hypertrophy of cerebral arterioles (Beyer et al., 2008; Ketsawatsomkron et al., 2010). Overexpression of PPARγ or its activation with ligands can decrease the expression and activity of MMP and adhesion molecules in the vasculature (Keen et al., 2004; Ramirez et al., 2008; Huang et al., 2009), thereby maintaining levels of tight junction proteins and preventing leukocyte–endothelial interactions (Fiala et al., 2002). Evidence is emerging suggesting that the PPARs interact with NF‐κB to decrease its activity, which may suppress gene expression (Ricote et al., 1998; Zipser et al., 2007). In the present study, PPARγ stimulation by its agonists led to decreases in NF‐κB activity, RAGE expression and Aβ influx at the BBB in db/db mice. LRP1 also binds to a broad range of ligands, including α2‐macroglobulin, apoE, amyloid precursor protein (APP) and Aβ (Zlokovic, 2004). It has been demonstrated that functional LRP1 expression is regulated by PPARγ ligands, which involves ligand‐induced up‐regulation of a transcription factor via the activation of PPARγ‐RXRα heterodimers that bind to a newly identified PPRE in the promoter region of the LRP1 gene (Gauthier et al., 2003). As expected, pioglitazone or rosiglitazone stimulated PPARγ and increased its localization in the nucleus of the isolated brain microvessel cells, consequently, LRP1 expression and its associated Aβ efflux through the BBB were significantly increased in db/db mice. The neurotoxicity of Aβ, especially Aβ1–42, a more neurotoxic Aβ species, has been well documented (Lai et al., 2014). It was observed in db/db mice that hippocampal Aβ accumulation was accompanied by apoptotic responses, and the PPARγ agonists effectively suppressed apoptosis, as indicated by decreased caspase‐3 activity and an increased ratio of Bcl‐2/Bax in the hippocampus. Many studies have demonstrated that diabetes induces cognitive impairments. In particular, the hippocampus is susceptible to the negative consequences of the diabetic condition. Stimulating PPARγ in diabetes can reverse behavioural impairments and increase LTP at the perforant path–dentate gyrus synapses. This restorative effect on hippocampal plasticity is involved in the amelioration of cerebral β‐amyloidosis, because there are no differences in the glucose or insulin levels in the hippocampus of insulin‐resistant and insulin‐deficient animals (Stranahan et al., 2008). In addition, chronic treatment with a PPARγ agonist also showed significant attenuation of hyperglycaemia and hyperinsulinaemia in db/db mice; thus, these peripheral effects should contribute somewhat to the reduction in RAGE‐mediated Aβ influx at the BBB because the formation of AGEs is also reduced. Furthermore, db/db mice are also characterized by an increase in body weight, leading to abnormal lipid metabolism and insulin resistance and an increase in the incidence of cognitive impairment or AD (Kanoski and Davidson, 2011). It has been shown that PPARγ agonists improve lipid utilization by multiple pathways in adipose tissue, inducing an increase in the levels of lipid metabolism‐associated genes, such as lipoprotein lipase and fatty acid binding protein 4 (FABP4; Kawai et al., 2010). These changes in gene expression promote the incorporation of free fatty acids into adipose tissue, which lowers serum free fatty acids levels and increases lipid deposits in the liver and skeletal muscle. Thus, PPARγ agonists improve the memory impairment in db/db mice in both a direct and indirect way.
Taken together, our results show that PPARγ agonists not only increase LRP1‐mediated Aβ efflux but also decrease RAGE‐mediated Aβ influx across the BBB in diabetic mice, and this bidirectional effect resulted in a marked decrease in hippocampal Aβ and improvements in synaptic plasticity. Our findings suggest that PPARγ agonists can be promoted for potential therapeutic use for the prevention and treatment of diabetes‐associated cognitive decline, which has important implications for patients with type 2 diabetes.
Author contributions
H. H. designed the experiments and wrote the manuscript. W. H., C. F., Z. K. L. and T. S. S. performed the experiments. L. Y., M. M. X. and L. J. M. performed the isotope experiments. H. M. and S. H. B. analysed the data and revised the manuscript. All the authors agreed that the final approval of the version to be published and ensured questions relating to the accuracy or integrity of any part of the work were appropriately investigated and resolved.
Conflict of interest
The authors declare that there is no duality of interest associated with this manuscript.
Supporting information
Figure S1 Effects of PPARγ agonists on the levels of apoptosis‐related molecules including caspase‐3, Bcl‐2 and Bax in hippocampus of db/db mice. (A) Pro‐caspase‐3, cleaved caspase‐3, Bcl‐2 and Bax were determined in hippocampus by Western blot using respective antibodies. (B) Caspase‐3 activation is expressed as the ratio of caspase‐3 fragment to pro‐caspase‐3, (C) Bcl‐2 and (D) Bax were expressed as the ratio (in percentage) of the db/m. Values shown are expressed as mean ± SEM; n = 3. *P<0.05, **P<0.01 versus db/db+Veh group.
Figure S2 Effects of PPARγ agonists on the general locomotor activity in db/db mice. (A) The total distance travelled in the open field test is shown. Values shown are expressed as mean ± SEM; n = 8–10.
Supporting info item
Acknowledgements
We acknowledge the technical assistance of Dr Feng Hao of XBL‐China and Prof. Yu Hui Xin of Jiangnan Institute of Nuclear Medicine. This work was supported by grants from the National Natural Science Foundation of China (81273497), the Program for Changjiang Scholars and Innovative Research Team in University (IRT1193), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Wang, H. , Chen, F. , Zhong, K. L. , Tang, S. S. , Hu, M. , Long, Y. , Miao, M. X. , Liao, J. M. , Sun, H. B. , and Hong, H. (2016) PPARγ agonists regulate bidirectional transport of amyloid‐β across the blood–brain barrier and hippocampus plasticity in db/db mice. British Journal of Pharmacology, 173: 372–385. doi: 10.1111/bph.13378.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The concise guide to PHARMACOLOGY 2013/14: nuclear hormone receptors. Br J Pharmacol 170: 1652–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The concise guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood CS, Bishop GM, Perry G, Smith MA (2002). Amyloid‐beta: a vascular sealant that protects against hemorrhage? J Neurosci Res 70: 356. [DOI] [PubMed] [Google Scholar]
- Awasthi A, Matsunaga Y, Yamada T (2005). Amyloid‐beta causes apoptosis of neuronal cells via caspase cascade, which can be prevented by amyloid‐beta‐derived short peptides. Exp Neurol 196: 282–289. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Saqare AP, Singh I, LaRue B, Deane R et al (2010). Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68: 409–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer AM, Baumbach GL, Halabi CM, Modrick ML, Lynch CM, Gerhold TD et al (2008). Interference with PPARγ signaling causes cerebral vascular dysfunction, hypertrophy and remodeling. Hypertension 51: 867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB et al (2005). P‐glycoprotein deficiency at the blood‐brain barrier increases amyloid‐beta deposition in an Alzheimer disease mouse model. J Clin Invest 115: 3285–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E et al (2003). RAGE mediates amyloid‐β peptide transport across the blood–brain barrier and accumulation in brain. Nat Med 9: 907–913. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K et al (2004). LRP/amyloid beta‐peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43: 333–344. [DOI] [PubMed] [Google Scholar]
- Dinel AL, André C, Aubert A, Ferreira G, Layé S, Castanon N (2011). Cognitive and emotional alterations are related to hippocampal inflammation in a mouse model of metabolic syndrome. PLoS One 6: e24325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Oberm Obermüller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H et al (2010). Peripherally applied Abeta‐containing inoculates induce cerebral beta‐amyloidosis. Science 330: 980–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC et al (2002). Cyclooxygenase‐2‐positive macrophages infiltrate the Alzheimer's disease brain and damage the blood‐brain barrier. Eur J Clin Invest 32: 360–371. [DOI] [PubMed] [Google Scholar]
- Gauthier A, Vassiliou G, Benoist F, McPherson R (2003). Adipocyte low density lipoprotein receptor‐related protein gene expression and function is regulated by peroxisome proliferator‐activated receptor gamma. J Biol Chem 278: 11945–11953. [DOI] [PubMed] [Google Scholar]
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y et al (1999). RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97: 889–901. [DOI] [PubMed] [Google Scholar]
- Hong H, Liu LP, Liao JM, Wang TS, Ye FY, Wu J et al (2009). Downregulation of LPR1 at the blood–brain barrier in streptozotocin‐induced diabetic mice. Neuropharmacology 56: 1054–1059. [DOI] [PubMed] [Google Scholar]
- Huang W, Eum SY, András IE, Hennig B, Toborek M (2009). PPARα and PPARγ attenuate HIV induced dysregulation of tight junction proteins by modulations of matrix metalloproteinase and proteasome activities. FASEB J 23: 1596–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber JD (2008). Diabetes, cognitive function, and the blood–brain barrier. Curr Pharm Des 14: 1594–1600. [DOI] [PubMed] [Google Scholar]
- Hummel KP, Dickie MM, Coleman DL (1996). Diabetes, a new mutation in the mouse. Science 153: 1127–1128. [DOI] [PubMed] [Google Scholar]
- Jiang LY, Tang SS, Wang XY, Liu LP, Long Y, Hu M et al (2012). PPARγ agonist pioglitazone reverses memory impairment and biochemical changes in a mouse model of type 2 diabetes mellitus. CNS Neurosci Ther 18: 659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanoski SE, Davidson TL (2011). Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav 103: 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai M, Sousa KM, MacDougald OA, Rosen CJ (2010). The many facets of PPAR gamma: novel insights for the skeleton. Am J Physiol Endocrinol Metab 299: E3–E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen HL, Ryan MJ, Beyer A, Mathur S, Scheetz TE, Gackle BD et al (2004). Gene expression profiling of potential PPARγ target genes in mouse aorta. Physiol Genomics 18: 33–42. [DOI] [PubMed] [Google Scholar]
- Ketsawatsomkron P, Pelham CJ, Groh S, Keen HL, Faraci FM, Sigmund CD (2010). Does peroxisome proliferator‐activated receptor‐γ (PPARγ) protect from hypertension directly through effects in the vasculature? J Biol Chem 285: 9311–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, NC3Rs Reporting Guidelines Working Group (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Hudson BI, Moser B, Guo J, Rong LL, Lu Y et al (2005). Receptor for advanced glycation end products and its ligands: a journey from the complications of diabetes to its pathogenesis. Ann N Y Acad Sci 1043: 553–561. [DOI] [PubMed] [Google Scholar]
- Kodl CT, Seaquist ER (2008). Cognitive dysfunction and diabetes mellitus. Endocr Rev 29: 494–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf D, Frölich L (2009). Risk of incident Alzheimer's disease in diabetic patients: a systematic review of prospective trials. J Alzheimers Dis 16: 677–685. [DOI] [PubMed] [Google Scholar]
- Lai J, Hu M, Wang H, Hu M, Long Y, Miao MX et al (2014). Montelukast targeting the cysteinyl leukotriene receptor 1 ameliorates Aβ1‐42‐induced memory impairment and neuroinflammatory and apoptotic responses in mice. Neuropharmacology 79: 707–714. [DOI] [PubMed] [Google Scholar]
- Li J, Schmidt AM (1997). Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem 272: 16498–16506. [DOI] [PubMed] [Google Scholar]
- Liu H, Xu X, Yang Z, Deng Y, Liu X, Xie L (2006). Impaired function and expression of P‐glycoprotein in blood–brain barrier of streptozotocin‐induced diabetic rats. Brain Res 1123: 245–252. [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu H, Yang J, Liu X, Lu S, Wen T et al (2008). Increased amyloid beta‐peptide (1–40) level in brain of streptozotocin‐induced diabetic rats. Neuroscience 153: 796–802. [DOI] [PubMed] [Google Scholar]
- Liu LP, Hong H, Liao JM, Wang TS, Wu J, Chen SS et al (2009). Upregulation of RAGE at the blood–brain barrier in streptozotocin‐induced diabetic mice. Synapse 63: 636–642. [DOI] [PubMed] [Google Scholar]
- Liu LP, Yan TH, Jiang LY, Hu W, Hu M, Wang C et al (2013). Pioglitazone ameliorates memory deficits in streptozotocin‐induced diabetic mice by reducing brain β‐amyloid through PPARγ activation. Acta Pharmacol Sin 34: 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeng HJ, Kim MH, Jin HE, Shin SM, Tsuruo T, Kim SG et al (2007). Functional induction of P‐glycoprotein in the blood–brain barrier of streptozotocin‐induced diabetic rats: evidence for the involvement of nuclear factor‐kappaB, a nitrosative stress‐sensitive transcription factor, in the regulation. Drug Metab Dispos 35: 1996–2005. [DOI] [PubMed] [Google Scholar]
- Maher PA, Schubert DR (2009). Metabolic links between diabetes and Alzheimer's disease. Expert Rev Neurother 9: 617–630. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JK, Vidoni ED, Honea RA, Burns JM (2014). Impaired glycemia and Alzheimer's disease. Neurobiol Aging 35: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM (1999). Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 53: 1937–1942. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP et al (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez SH, Heilman D, Morsey B, Potula R, Haorah J, Persidsky Y (2008). Activation of peroxisome proliferator‐activated receptor γ (PPARγ) suppresses Rho GTPases in human brain microvascular endothelial cells and inhibits adhesion and transendothelial migration of HIV‐1 infected monocytes. J Immunol 180: 1854–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK (1998). The peroxisome proliferator‐activated receptor‐γ is a negative regulator of macrophage activation. Nature 391: 79–82. [DOI] [PubMed] [Google Scholar]
- Roberts RO, Knopman DS, Przybelski SA, Mielke MM, Kantarci K, Preboske GM et al (2014). Association of type 2 diabetes with brain atrophy and cognitive impairment. Neurology 82: 1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT, Vyas V, Hernandez‐Estrada T, Nichol KE, Christie LA, Head E (2008). Lack of pathology in a triple transgenic mouse model of Alzheimer's disease after overexpression of the anti‐apoptotic protein Bcl‐2. J Neurosci 28: 3051–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B et al (2000). Clearance of Alzheimer's amyloid‐ss(1–40) peptide from brain by LDL receptor‐related protein‐1 at the blood–brain barrier. J Clin Invest 106: 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh I, Sagare AP, Coma M, Perlmutter D, Gelein R, Bell RD et al (2013). Low levels of copper disrupt brain amyloid‐β homeostasis by altering its production and clearance. Proc Natl Acad Sci U S A 110: 14771–14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood V, Colleran K, Burge MR (2000). Thiazolidinediones: a comparative review of approved uses. Diabetes Technol Ther 2: 429–440. [DOI] [PubMed] [Google Scholar]
- Stadelmann C, Deckwerth TL, Srunivasan A, Bancher C, Brück W, Jellinger K et al (1999). Activation of caspase‐3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease: evidence for apoptotic cell death. Am J Pathol 155: 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern D, Yan SD, Yan SF, Schmidt AM (2002). Receptor for advanced glycation endproducts: A multiligand receptor magnifying cell stress in diverse pathologic settings. Adv Drug Deliv Rev 54: 1615–1625. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP (2008). Diabetes impairs hippocampal function through glucocorticoid‐mediated effects on new and mature neurons. Nat Neurosci 11: 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SS, Wang XY, Hong H, Long Y, Li YQ, Xiang GQ et al (2013). Leukotriene D4 induces cognitive impairment through enhancement of CysLT1R‐mediated amyloid‐β generation in mice. Neuropharmacology 65: 182–192. [DOI] [PubMed] [Google Scholar]
- Tang SS, Hong H, Chen L, Mei ZL, Ji MJ, Xiang GQ et al (2014). Involvement of cysteinyl leukotriene receptor 1 in Aβ1–42‐induced neurotoxicity in vitro and in vivo . Neurobiol Aging 35: 590–599. [DOI] [PubMed] [Google Scholar]
- Toth C, Schmidt AM, Tuor UI, Francis G, Foniok T, Brussee V et al (2006). Diabetes, leukoencephalopathy and rage. Neurobiol Dis 23: 445–461. [DOI] [PubMed] [Google Scholar]
- Ueno M, Nakagawa T, Wu B, Onodera M, Huang CL, Kusaka T et al (2010). Transporters in the brain endothelial barrier. Curr Med Chem 17: 1125–1138. [DOI] [PubMed] [Google Scholar]
- Wang H, Mei ZL, Zhong KL, Hu M, Long Y, Miao MX et al (2014). Pretreatment with antiasthmatic drug ibudilast ameliorates Aβ1–42‐induced memory impairment and neurotoxicity in mice. Pharmacol Biochem Behav 124: 373–379. [DOI] [PubMed] [Google Scholar]
- Yan SD, Stern D, Schmidt AM (1997). What's the RAGE? The receptor for advanced glycation end products (RAGE) and the dark side of glucose. Eur J Clin Invest 27: 179–181. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lee DH (2011). Sink hypothesis and therapeutic strategies for attenuating Abeta levels. Neuroscientist 17: 163–173. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li R, Shi W, Liang X, Liu S, Ye Z et al (2013). NFAT2 inhibitor ameliorates diabetic nephropathy and podocyte injury in db/db mice. Br J Pharmacol 170: 426–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipser BD, Johanson CE, Gonzalez L, Berzin TM, Tavares R, Hulette CM et al (2007). Microvascular injury and blood–brain barrier leakage in Alzheimer's disease. Neurobiol Aging 28: 977–986. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV (2004). Clearing amyloid through the blood–brain barrier. J Eurochem 89: 807–811. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of PPARγ agonists on the levels of apoptosis‐related molecules including caspase‐3, Bcl‐2 and Bax in hippocampus of db/db mice. (A) Pro‐caspase‐3, cleaved caspase‐3, Bcl‐2 and Bax were determined in hippocampus by Western blot using respective antibodies. (B) Caspase‐3 activation is expressed as the ratio of caspase‐3 fragment to pro‐caspase‐3, (C) Bcl‐2 and (D) Bax were expressed as the ratio (in percentage) of the db/m. Values shown are expressed as mean ± SEM; n = 3. *P<0.05, **P<0.01 versus db/db+Veh group.
Figure S2 Effects of PPARγ agonists on the general locomotor activity in db/db mice. (A) The total distance travelled in the open field test is shown. Values shown are expressed as mean ± SEM; n = 8–10.
Supporting info item