

Abstract
In 2003, the first report was published that presented proof of principle for a novel class of FRET biosensors for use in living cells. This novel sensor class was built on the base of GPCRs, which represent an integral transmembrane receptor family passing the membrane seven times and are thus also called the 7TM receptor family. As an estimated number of 30% of all marketed drugs exert their effects by modulating GPCR function, these initial reports promised the gain of novel insights into receptor function. Such FRET sensors have slowly, but progressively, made their way into the standard toolbox for GPCR research as several groups are now reporting on the generation and use of these sensors. By now, FRET sensors have been reported for 18 different GPCRs, and more are expected to be added. These particular receptor sensors have been used to investigate receptor dynamics in living cells to evaluate ligand binding and ligand efficacy in real time, to study voltage and mechanosensitivity of GPCRs or to study the influence of receptor polymorphisms on receptor function in real‐time. In this review we will describe the different design principles of these GPCR‐based sensors and will summarize their current biological applications in living cells.
Abbreviations
- CFP
cyan fluorescent protein
- CLIP
mutant of SNAP preferring O2‐benzylcytosine
- FlAsH
fluorescein arsenical hairpin binder
- IL‐3
third intracellular loop
- ReAsH
resorufin arsenical hairpin binder
- SNAP
previously known as genetically modified AGT: O6‐alkylguanine‐DNA alkyltransferase
- TM
transmembrane
- YFP
yellow fluorescent protein
Tables of Links
| LIGANDS | |
| 5‐HT | Dopamine |
| ACh | Gallamine |
| Adenosine | Morphine |
| Atropine | Noradrenaline (NA) |
| Caffeine | Octopamine |
| Carbachol | Oxotremorine M |
| Clonidine | Yohimbine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Design principles of GPCR‐based FRET sensors
GPCRs comprise an important class of drug targets, as an estimated number of 30% of marketed drugs exert their effects by modulating this class of receptors (Howard et al., 2001). During the past decade, we have gained tremendous insights, due to crystal structural analysis, into the molecular mechanisms that lead to activation of these receptors (Venkatakrishnan et al. 2013). However, the molecular dynamics of these membrane receptors represent an important aspect of the signalling cascades that are triggered upon ligand binding or other possible stimuli. Rapid dynamic transitions between different receptor states appear to be important for receptor function, and ligand binding exhibits an allosteric coupling between the binding pocket and the cytoplasmic part of the receptor (Manglik et al. 2015). This dynamic coupling will determine if a ligand acts as an agonist, partial agonist or antagonist. Interestingly, for the fully active receptor state, a stabilizing role for G proteins or β‐arrestin has been described (Manglik et al., 2015). This opens the possibility that dynamic transitions might also play a role in ligand‐directed signalling bias between these pathways. Dynamic events are currently not resolved by crystal structural analysis. One way to investigate receptor dynamics in real time is represented by the use of FRET approaches to study GPCRs. The design of GPCR‐based FRET sensors follows the same basic principles as outlined for other protein classes in excellent reviews (Campbell, 2009; Miyawaki, 2011; Newman et al., 2011). However, a few special pitfalls for the design of such sensors have to be considered as this class of receptors is special due to its integral membrane structure, offering an N‐terminus, three extracellular and intracellular loops and a C‐terminus for insertion or addition of fluorescent tags (Lohse et al., 2012). Fluorescently‐labelled GPCRs were already used in the mid‐nineties to study conformational changes upon ligand binding (Gether et al., 1995) and were further developed into FRET‐based probes by using a combination with fluorescence quenchers as a second chromophore (Ghanouni et al., 2001). However, such early designed sensors were generally purified receptor proteins, which needed to be chemically labelled. Nonetheless, these sensors provided valuable evidence that the transmembrane regions and cytoplasmic loops of GPCRs undergo significant movements upon ligand binding of the receptor (Hoffmann et al., 2008). Our group utilized this information, but followed a different approach by using genetically encoded fluorescent proteins, which had become available for general use by the laboratory of Roger Y. Tsien (Heim and Tsien, 1996). With the intention of keeping this review focussed, we will concentrate on unimolecular FRET sensors which are suited for the analysis of protein conformational changes.
Basic requirements for FRET
FRET can occur between two fluorescent molecules, a donor and acceptor fluorophore. The donor molecule emits generally at the shorter wavelength, and FRET occurs between the excited state of the donor and the ground state or the acceptor molecule. For FRET to occur, the emission spectrum of the donor has to overlap with the excitation spectrum of the acceptor although no photon emission is involved. Because FRET involves a long‐range dipole–dipole interaction between both fluorophores, the efficiency of FRET is influenced by the relative distance and relative orientation of the dipoles (Jares‐Erijman and Jovin, 2003, 2006 and the references therein). For each fluorophore combination, a characteristic distance (termed Förster distance) defines half‐maximal energy transfer. Because the relative distance is effective with the six power (r−6), its effect is maximal at the Förster distance. Therefore, the distance dependency can be a challenge because an optimal distance between the two fluorophores needs to be achieved in order to be able to observe even small distance changes. Hence, once a FRET sensor has been successfully generated, the same modification sites might not work for a different fluorophore combination (personal unpublished experience). Furthermore, for non‐fluorescent protein fluorophores, the degree of labelling of the acceptor plays an important role because a ‘vacant’ donor would create a fluorescent signal, but no change in signal would be observed if only an unlabelled acceptor was close. In the best case, this would spoil the overall ‘dynamic range’ of the sensor and lead to smaller signal amplitudes. In the worst case, it would lead to false measurements, particularly in cases when quantitative FRET signals are desired. One also needs to be aware that such measurements represent ensemble measurements of receptors even when single cells are investigated. Hence, the observed signal will represent a sum of tens of thousands of receptors and therefore needs to be interpreted with appropriate caution when it comes to movements at molecular resolution. Several important issues need to be taken into account for signal correction, like fluorescence bleed‐through and wrong excitation (Vilardaga et al., 2003; Hoffmann et al., 2005). Without question, it is also necessary to study co‐expression of individually donor‐ or acceptor‐labelled receptors as control. Such control experiments are required to rule out the possibility that changes in FRET, which might occur by receptor dimer rearrangements, are observed and interpreted as movements of individual protomers (Hlavackova et al., 2012). A more detailed discussion of these issues or labelling protocols is beyond the scope of this review. As mentioned before and to avoid further discussion on the effects of relative protein expression on FRET signals in bimolecular sensors (Miyawaki and Niino, 2015), we will concentrate on unimolecular FRET sensors for GPCRs.
CFP–YFP‐based versus FlAsH–CFP‐based probe design
The cyan fluorescent protein (CFP) in combination with the yellow fluorescent protein (YFP) was used as the initial combination of fluorophores (Figure 1). These genetically encoded fluorophores were inserted into the third intracellular loop (IL‐3) of the α2A‐adrenoceptor or parathyroid hormone receptor (PTH‐receptor), and the second fluorescent protein was fused to the C‐terminus (Vilardaga et al., 2003). Because FRET is dependent on the distance and relative orientation of the fluorophores, a significant number of constructs needed to be designed to optimize the relative positioning of the two fluorophores. This was achieved by inserting CFP in different positions of IL‐3 as well as truncating the C‐terminus at various positions. These modifications might of course be problematic to the overall protein function, because the IL‐3 region is also a coupling region for G proteins (Oldham and Hamm, 2008) and the C‐terminus is often involved in correct receptor localization and interaction with many other proteins (Bockaert et al., 2004). Therefore, rigorous characterization of receptor functions is always required to verify that no vital receptor functions like ligand binding or receptor signalling have been altered (Vilardaga et al., 2003; Hoffmann et al., 2005; Maier‐Peuschel et al., 2010; Reiner et al., 2010; Xu et al., 2012). The generation of the α2A‐adrenoceptor and PTH‐receptor sensors allowed the study of receptor activation in living cells for the first time. Since then, sensors for several other GPCR family members have been generated and thus opened the possibility to study several phenomena of GPCR biology, which will be discussed later in special sections.
Figure 1.
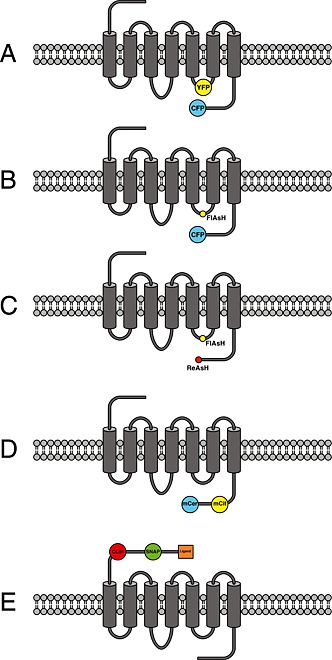
Schematic overview of different design principles of GPCR‐based FRET sensors: (A) Schematic representation of a GPCR modified with the cyan and yellow fluorescent protein in IL‐3 and at the C‐terminus (Vilardaga et al., 2003). (B) Same design principle as in (A), but insertion of the discontinuous CCPGCC amino acid sequence as the FlAsH‐binding motif into IL‐3 in combination with CFP fused to the C‐terminus of the receptor is used instead (Hoffmann et al., 2005). (C) Same design principle as in (A) or (B), but the fluorophore pair FlAsH–ReAsH was used in combination using the amino acid motifs CCPGCC and FLNCCPGCCMEP (Zürn et al., 2010). (D) Insertion of the FRET pair mCitrine–mCerulean, where both mCit and mCer are inserted into the C‐terminus of the receptor (Malik et al., 2013). (E) Insertion of SNAP‐ and CLIP‐tag into the N‐terminus of the receptor. SNAP‐ and CLIP‐tag can be labelled with dyes that are capable of performing FRET, and one fluorophore is additionally tagged with the respective receptor ligand (Masharina et al., 2012).
Initially, we suspected that the insertion of the large fluorescent proteins, consisting of 230 amino acids (27 kDa), might alter proper receptor function to an unacceptable degree. Therefore, we developed an alternative approach in parallel (Figure 1). We used a small cell‐permeant fluorescein‐derivative called FlAsH, which is an acronym for fluorescein arsenical hairpin binder (Griffin et al., 1998; Adams et al., 2002). In comparison with fluorescent proteins, this small soluble fluorophore binds specifically to a six amino acid sequence CCPGCC (one letter code) and is only 700 Da in size. Initial reports of problems using this fluorophore (Stroffekova et al., 2001) have long been solved (Martin et al., 2005; Hoffmann et al., 2010), and many groups have successfully used FlAsH in their research (Pomorski and Krezel, 2011). Because the excitation and emission properties of FlAsH are very close to those of YFP, one can use commercial filter pairs for CFP/YFP also for the combination of CFP/FlAsH. In our first study using FlAsH, we substituted the natural amino acid sequence within the IL‐3 of the adenosine A2A‐receptor with the specific amino acid binding sequence. We also created the same type of sensor using CFP/YFP and compared both approaches in a parallel fashion. Both approaches are schematically depicted in Figure 1. In the case of CFP/FlAsH, the aforementioned degree of labelling was investigated by optimizing the labelling protocols and parallel determination of FRET efficiency using different methods (Hoffmann et al. 2005; Hoffmann et al., 2010). No alteration in protein function was observed for the approach using CFP/FlAsH, whereas the approach using CFP/YFP led to a sensor that could not stimulate downstream signalling (Hoffmann et al., 2005). This disturbance in downstream signalling was found to be less pronounced for other GPCRs (Rochais et al. 2007). Both building principles led to receptors that could be stimulated with the endogenous agonist and showed similar kinetic properties of receptor response (Hoffmann et al., 2005). Interestingly, a fourfold increase in signal amplitude was observed when CFP/FlAsH was used instead of CFP/YFP (Hoffmann et al., 2005). The exact reason for this observation is still unknown, but most likely this results from a different relative orientation of the two fluorophores. Furthermore, because FlAsH is a bidentate fluorophore, it is tightly bound to the primary amino acid sequence (Madani et al., 2009), and relative changes in the orientation factors might contribute to the overall signal to a larger extent compared with CFP/YFP. This has been recently used by the group of Elliott Ross to optimize a sensor for the muscarinic M1 receptor using circular permutated versions of CFP (Chang and Ross, 2012). Such permutated versions of fluorescent proteins have the same spectral properties as the parent fluorescent protein, but the orientation of the dipole moment which is important for the FRET efficiency is different for each variant and thus can significantly influence sensor performance (Nagai et al., 2004). The above‐mentioned work by Chang and Ross is a nice example of how the use of fluorescent protein variants can significantly improve dynamic probe behaviour. A further indication of a particular role of the fluorophore orientation is the notion that an increase in the FRET ratio has only been reported for the combination of CFP/FlAsH (Nakanishi et al., 2006; Ziegler et al., 2011; Zhang et al., 2013). All currently reported intra‐molecular FRET sensors for GPCRs based on CFP/YFP have reported a decrease in the FRET ratio (Table 1), which would be consistent with an increase in fluorophore distance as seen in the crystal structures of the active β2‐adrenoceptor (Rasmussen et al., 2011a).
Table 1.
Resonance energy transfer biosensors for GPCR activation
| GPCR | Resonance technique | Comments | Reference |
|---|---|---|---|
| Receptor subtype | |||
| PTH receptor sensor and α2A‐adrenoceptor sensor with CFP/YFP | FRET | First sensor for GPCR activation based on FRET which was designed to study conformational changes in living cells | Vilardaga et al. 2003 |
| mGlu1 receptor CFP/YFP | FRET | Inter‐subunit FRET change between protomers, but no intra‐subunit FRET change | Tateyama et al. 2004 |
| A2A receptor sensor with FlAsH/CFP or CFP/YFP | FRET | First sensor using the combination FlAsH/CFP which showed no disturbance of G‐protein signalling | Hoffmann et al. 2005 |
| α2A adrenoceptor sensor with constitutive activity CFP/YFP | FRET | Inverse agonists exhibit signals opposite to that of agonists | Vilardaga et al. 2005 |
| B2 receptor sensor CFP/YFP | FRET | B2‐bradykinin receptor sensor responded to mechanosensitive stimuli | Chachisvilis et al., 2006 |
| α2A‐adrenoceptor sensor FlAsH/CFP | FRET | Partial and full agonists exhibit distinct receptor activation kinetics | Nikolaev et al. 2006 |
| β2‐adrenoceptor FlAsH/CFP | FRET | First sensor to observe an increase in FRET ratio upon agonist stimulation | Nakanishi et al. 2006 |
| β1‐adrenoceptor CFP/YFP | FRET | Clinically used antagonists exhibit different FRET signals for different polymorphic receptor variants | Rochais et al. 2007 |
| α2A‐adrenoceptor sensor FlAsH/CFP | FRET | Receptor crosstalk between α2A‐adrenergic receptor and μ‐opioid receptor shown by influence of the FRET signal | Vilardaga et al. 2008 |
| α2A‐adrenoceptor sensor FlAsH/CFP | FRET | Labelling with FlAsH in different positions reveals that partial agonists only induce conformational changes in some parts of the third intracellular loop | Zürn et al. 2009 |
| M1 receptor CFP/YFP | FRET | Complete kinetic analysis of the signalling cascade of the M1‐ACh‐receptor down to PLC | Jensen et al. 2009 |
| M2 receptor FlAsH/CFP | FRET | Allosteric modulation shown by alterations in the measured FRET signal | Maier‐Peuschel et al. 2010 |
| β2‐adrenoceptor CFP/YFP | FRET | Distinct conformational changes of adrenaline and noradrenaline | Reiner et al. 2010 |
| A2A receptor sensor with FlAsH/ReAsH | FRET | First receptor sensor that did not utilize a fluorescent protein, FRET efficiency determined by acceptor photobleaching | Zürn et al. 2010 |
| GABAB receptor Cerulean/YFP and M1 receptor Cerulean/YFP | FRET | Inter‐subunit FRET change between protomers, but no intra‐subunit FRET change; M1 receptor served as control | Matsushita et al. 2010 |
| M1, M3 and M5 receptor FlAsH/CFP | FRET | Comparison of different muscarinic receptor subtypes reveals differences in ligand‐dependent receptor kinetics | Ziegler et al. 2011 |
| B1 receptor FlAsH/CFP | FRET | Bradykinin‐dependent receptor activation is influenced by heterodimerization with carboxypeptidase M | Zhang et al. 2011 |
| M3 receptor FlAsH/CFP | FRET | M3 receptor and RASSL variant of M3 receptor exhibit similar conformational changes | Alvarez‐Curto et al. 2011 |
| β2‐adrenoceptor CFP/YFP | FRET | Receptor activation kinetics can be influenced by polymorphism | Ahles et al. 2011 |
| 5‐HT1B receptor Cerulean/Citrine | FRET | 5‐HT1B receptor sensor responded to mechanosensitive stimuli | Candelario and Chachisvilis, 2012 |
| AT1 receptor YFP/RLuc | BRET | First intra‐molecular GPCR based BRET sensor, allosteric modulation of protomers within a receptor dimer | Szalai et al. 2012 |
| M1 receptor CFP/YFP | FRET | Comparison of orthosteric and allosteric ligands | Markovic et al. 2012 |
| V2 receptor FlAsH/Lumi‐4 Tb | LRET | Structural information about biased agonism | Rahmeh et al. 2012 |
| mGlu1 receptor CFP/YFP | FRET | Inter‐subunit FRET change between protomers proceeds intra‐subunit FRET change | Hlavackova et al., 2012 |
| α2A‐adrenoceptor sensor with FlAsH/CFP | FRET | Non‐equilibrium activation demonstrates that G‐protein signalling and signal amplification are highly time‐dependent phenomena | Ambrosio and Lohse, 2012 |
| OX1 and OX2 receptors FlAsH/CFP | FRET | Slow activation kinetics observed as seen for the PTH receptor | Xu et al. 2012 |
| M3 receptor with constitutive activity FlAsH/CFP | FRET | Constitutive receptor activity increases affinity for ACh solely by increase in ligand off‐rate | Hoffmann et al. 2012 |
| M1 receptor FlAsH/CFP | FRET | Improved sensor performance by use of circular permutated versions of CFP | Chang and Ross, 2012 |
| M2 receptor FlAsH/CFP | FRET | Dualsteric receptor ligands exhibit distinct receptor conformations | Bock et al. 2012 |
| B1 receptor FlAsH/CFP and FlAsH/RLuc | FRET/BRET | Carboxypeptidase M influences bradykinin‐dependent receptor activation by allosteric mechanism | Zhang et al. 2013 |
| α2A‐adrenoceptor sensor with CFP/YFP | FRET | Receptor function is regulated by voltage | Rinne et al. 2013 |
| β1‐adrenoceptor CFP/YFP | FRET | Receptor sensor was used to monitor conformational changes evoked by human autoantibodies against β1‐AR in patients suffering from dilated cardiomyopathy | Bornholz et al. 2013 |
| M1 receptor CFP/YFP | FRET | Interaction of receptor and Gq‐protein stabilizes the active receptor state and delays receptor deactivation | Tateyama and Kubo, 2013b |
| M3 receptor and P2Y1 receptor CFP/YFP | FRET | Differentially influence of Gq‐protein on the active receptor state reveals receptor‐specific influence | Tateyama and Kubo, 2013a |
| M2 receptor FlAsH/CFP | FRET | Binding pose of dualsteric receptor ligands can dictate partial agonism | Bock et al. 2014 |
| A2A receptor sensor with FlAsH/CFP | FRET | Caffeine described as inverse agonist | Fernandez‐Duenas et al. 2014 |
| β1‐adrenoceptor CFP/YFP | FRET | Receptor polymorphism exhibits different receptor kinetics caused by receptor phosphorylation | Ahles et al. 2015 |
| Ghrelin receptor | LRET | First sensor using unnatural amino acid labelling incorporated by AMBER codon technology, purified receptors in lipid disks | Damian et al. 2015 |
This table summarizes all articles that we found performing a search for such biosensors listed in PubMed using the search GPCR and FRET (127 hits as of 1 September 2015), GPCR and BRET (106 hits as of 1 September 2015), GPCR and conformational sensor (9 hits as of 1 September 2015) as well as citations listed in Thomson Reuter science citation index to Vilardaga et al. 2003 or Hoffmann et al. 2005. LRET, luminescence resonance energy transfer.
FlAsH‐/ReAsH‐based probe design
To the best of our knowledge, currently, only a single report exists that describes the generation of a dually labelled GPCR in living cells using the combination of two small soluble fluorophores (Zürn et al. 2010). This sensor was published in the supplementary section of a report on the development of a dual labelling strategy in living cells and is based on the original construct A2A‐Flash3‐CFP (Hoffmann et al., 2005). To create this sensor, the C‐terminal CFP was replaced with a second binding motif for biarsenical dyes (Figure 1). Thus, this sensor contained two CCPGCC affinity‐sites with different affinities for FlAsH or its red resorufin‐based variant ReAsH. Selective labelling was successful and static FRET could be demonstrated by donor‐fluorescence recovery after acceptor bleaching (Zürn et al., 2010). However, dynamic changes upon ligand application could not be observed using this construct. This is a further indication for the need to individually optimize the sensor constructs in accordance with each labelling approach of choice.
Alternative probe design: GPCR‐based FRET sensors reporting on C‐terminal or N‐terminal movements
Although the recently reported X‐ray structures of active versus inactive GPCRs show a large movement of TM6, a smaller movement of TM5 and little changes in the resolved C‐terminal part of the receptors (Kruse et al., 2013), none of the above‐mentioned FRET sensors would currently be able to formally distinguish between movements within the IL‐3 region or the C‐terminus of the receptor. Thus, one publication with a different sensor design is of interest in this respect. The report by Granier et al. (2007 used a dual labelling strategy combining FlAsH and Alexa Fluor 568 and labelled the β2‐adrenoceptor at Cys265 underneath TM6, while the FlAsH‐binding motif was engineered at two different sites within the C‐terminus. The authors were able to monitor C‐terminal movements in two different positions of the receptor relative to the TM6 domain and therefore could investigate relative movements of the C‐terminus upon ligand application by combining two separately obtained datasets. However, this approach used purified receptors and thus does not necessarily report upon conformational changes occurring in living cells.
A very recent report described the design of several different β2‐adrenoceptors that report on G‐protein‐selective conformational changes (Malik et al., 2013). This group of investigators inserted two genetically encoded fluorophores (CFP and YFP) at different positions into the C‐terminus (Figure 1). In addition to these modifications, they also added a short amino acid sequence at the very tip of the receptor C‐terminus. This special sequence was derived from the C‐terminus of Gα‐subunits of G‐proteins and is different for each Gα‐subunit. This short sequence has been shown to determine specificity for receptor–G‐protein interactions (Conklin et al., 1996; Oldham and Hamm, 2008) and the recent X‐ray structure of the active β2‐adrenoceptors in complex with the Gs‐protein provided structural insights into how the corresponding G‐protein part inserts into the opening of TM5 and TM6 (Rasmussen et al., 2011b). Thus, by engineering a set of three individual receptor sensors, which were distinct by only a few amino acids of the corresponding Gi‐, Gs‐ or Gq‐protein‐derived sequence, the authors could investigate ligand‐dependent conformational changes and investigate G‐protein signalling selectivity of different receptor ligands (Malik et al., 2013). Depending on the individual ligands used, the relative opening of the TM5 and TM6 interface might be distinct, and the Gα‐specific sequence could insert into the receptor. This insertion would alter the relative orientation of the fluorophores inserted at the C‐terminus and thus report G‐protein‐selective conformational changes by a change in FRET signal (Malik et al., 2013). Although the reported signal changes were small, this approach is very promising because the measurements were done in whole cells and on large cell numbers. One current drawback was the measurement in cuvette systems, which did not permit testing of different ligands on the same cells. The second drawback might be that the tethered G‐protein sequence might influence or restrain the subset of receptor conformations that can be studied.
The latest FRET‐based GPCR probe development is distinct from all the above‐mentioned approaches because it does not monitor conformational changes upon receptor activation, but rather monitors ligand–receptor binding. This novel approach was developed by the group of Kai Johnsson (Masharina et al., 2012) for the GABAB receptor and utilizes a dual labelling strategy at the N‐terminus of the receptor (Fig. 1). It is based on the use of a SNAP‐ and CLIP‐tag which were derived from O6‐alkylguanine‐DNA alkyltransferase (Gautier et al., 2008). While the CLIP‐tag was labelled with an O2‐benzylcytosine‐coupled donor fluorophore, the SNAP‐tag was labelled with O6‐alkylguanine coupled to an acceptor fluorophore, which furthermore was linked to an antagonist for the respective GPCR. Thus, upon dual labelling, the antagonist binds to the receptor and restrains the N‐terminus in a closed position like a slingshot, and this correlates to a basal FRET signal. Upon agonist binding, the antagonist gets displaced from the binding pocket, and the restrained N‐terminus relaxes from the closed position which can be monitored by a change in FRET. Again, one needs to keep in mind the potential effect of the chosen antagonist on the receptor because it may influence what is observed. However, such constructs can probably be used to detect the release of endogenous agonists (Masharina et al., 2012) and could in principle be adapted to many other GPCRs and might open a novel area for GPCR research.
Biological applications of GPCR‐based FRET sensors
Because fluorescent labelling techniques for the detection of ligand binding to GPCRs (Ma et al. 2014; Ward and Milligan, 2014) and protocols or technical details of how such FRET experiments can be performed (Hoffmann et al., 2010; Vilardaga et al., 2013) have recently been reviewed, we will focus on the biological processes or phenomena that have been studied using unimolecular GPCR‐based FRET sensors. Table 1 represents a list of all original research articles we are aware of, which have developed or applied such GPCR‐based conformational resonance energy transfer sensors.
Kinetics of GPCR activation
The on‐rate of ligand binding to a given protein depends on the ligand concentration, and the on‐rate constant is expressed in M−1∙s−1, whereas the off‐rate is independent of ligand concentration, and the off‐rate constant is expressed in s−1. Thus, the conformational changes that are observed upon agonist binding to such GPCR‐based sensors (Figure 1) follow the same principle, and hence, the activation speed increases with ligand concentration. This can be seen in Figure 2, where an adenosine A2A receptor‐based FRET sensor was stimulated using different concentrations of adenosine. In the case of the adenosine receptor, the activation speed reaches a maximum of 60 ms (Hoffmann et al., 2005). Such experiments require high data sampling rates of 40 Hz or higher, meaning data points have to be collected every 25 ms or faster, and in turn illumination times will be extremely short. Such high sampling rates require FRET sensors with optimized FRET efficiencies and large FRET amplitudes because short illumination times will evoke only small FRET responses. Thus, the higher FRET amplitude observed for the combination of CFP/FlAsH is one good argument when kinetic experiments are intended, while a drawback of this combination is the faster bleaching of fluorescein compared with YFP (Hoffmann et al., 2005). Maximal activation kinetics for several receptors have now been reported and activation times for small diffusible endogenous ligands range from 40 to 100 ms, while peptide ligands were reported to act slower in the range of ~1 s (Lohse et al., 2012). These sensor constructs can also be used to generate concentration–response curves by analysing the maximal FRET response for each ligand concentration. An original experiment of this type is shown in Figure 2 for the muscarinic M3 receptor (Ziegler et al., 2011). Such sensors allowed comparison of ligand selectivity for different receptor family members similar to classical radioligand binding experiments. However, for the muscarinic receptor family, it was also reported that individual ligands like oxotremorine M exhibit kinetic differences for individual receptor subtypes (Ziegler et al., 2011). Interestingly, if concentration–response curves were analysed for different time points of receptor activation, non‐equilibrium receptor activation could be studied. Using the α2A‐adrenoceptor FlAsH /CFP based FRET sensor and noradrenaline as agonist, a time‐dependent shift in the potency of receptor activation was reported (Ambrosio and Lohse, 2012). This time‐dependent left shift of noradrenaline concentration–response curves was also observed downstream at the level of G‐protein activation. Such experiments revealed that receptor and G‐protein signalling are highly time‐dependent phenomena (Ambrosio and Lohse, 2012).
Figure 2.
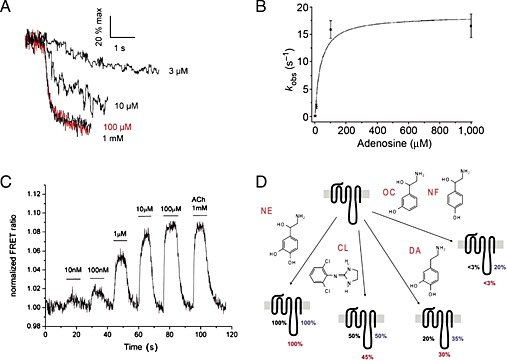
(A) Concentration‐dependent kinetics of adenosine‐mediated change in the FRET ratio. The recordings were expressed as % of the maximal response achieved with 1 mM adenosine, reproduced from Hoffmann et al., 2005. (B) Relationship between the adenosine concentration and kobs, the apparent rate constant of the change in the FRET ratio, reproduced from Hoffmann et al., 2005. (C) Concentration–response effects of ACh on the FRET response of the M3‐ACh‐receptor sensor, reproduced from Ziegler et al., 2011. (D) Differential effects of partial agonists in different receptor domains as observed for the α2a‐adrenoceptor, reproduced from Zürn et al., 2009.
Partial and inverse agonism
At the time when this class of FRET‐based sensors was developed, the molecular details of partial agonistic behaviour or inverse agonism were only poorly understood. Therefore, GPCR‐based FRET sensors and kinetic analysis of receptor stimulation were used to investigate these phenomena. We are still far from a molecular understanding of partial or inverse agonism, but we are currently gaining more insights by crystal structural analysis of receptor ligand complexes (Katritch et al., 2009) and dynamic measurements (Vilardaga et al., 2005; Nikolaev et al., 2006; Zürn et al., 2009; Hoffmann et al., 2012; Bock et al., 2014). Partial agonists were found to induce smaller and most often slower FRET changes when compared with the reference ligand (Nikolaev et al., 2006). The initial study was performed using a α2A‐adrenoceptor FlAsH /CFP based FRET sensor and noradrenaline as reference agonist. However, because only one single receptor construct was used, limited information about partial agonism could be derived at the molecular level. A further study using three individual α2A‐adrenoceptor FlAsH /CFP based FRET sensors each with different labelling sites for FlAsH within IL‐3 (Figure 1), enabled more detailed information on partial agonism to be gained. As shown in Figure 2, all three constructs were exposed to several structurally related ligands. While noradrenaline induced conformational changes in all receptor constructs, the partial agonists clonidine, dopamine, octopamine and norphenephrine all induced distinct conformational changes with IL‐3 that was characteristic for each ligand (Zürn et al., 2009). This study provided evidence for the existence of different conformational changes in living cells. Furthermore, the α2A‐adrenoceptor FRET sensors also helped to investigate the phenomenon of inverse agonism. Inverse agonism is characterized by a decrease in basal receptor activity, which is thought to occur by a limited proportion of receptors being in an active state without agonist being present and can be enhanced by point mutations (Samama et al., 1993). Therefore, a constitutively active version of the initial α2A‐adrenoceptor CFP /YFP based FRET sensors as depicted in Figure 1 was developed by insertion of point mutations beneath TM6 and then exposed to yohimbine, a known inverse agonist of this receptor. It was observed that the FRET signal evoked by yohimbine was opposite to the signal induced by noradrenaline and occurred with slower kinetics (Vilardaga et al., 2005). The slower kinetic and opposite FRET signal might be a special feature for inverse agonism, because both observations were confirmed for atropine at a constitutively active version of the muscarinic M3 receptor (Hoffmann et al., 2012). Because constitutive activity is also frequently accompanied by higher agonist affinity, the authors studied the influence of constitutive activity on receptor activation and deactivation, as indicator for ligand on‐ and off‐rates. While the maximal receptor activation kinetics, as indicator for ligand on‐rates, were not altered, receptor deactivation was significantly prolonged (Hoffmann et al., 2012). This would indicate that at least for the muscarinic M3 receptor, the increase in agonist affinity by constitutive receptor activity was almost completely due to a decrease in ligand off‐rate. Very recently, it was demonstrated using the original construct A2A receptor‐Flash3‐CFP and experimental models of Parkinson's disease that caffeine, a legal stimulant consumed every day by millions of people, can act as an inverse agonist (Fernandez‐Duenas et al., 2014) at this receptor. Again, the change in FRET signal was opposed to that of the endogenous agonist adenosine. Once this inverse agonistic activity has been confirmed by others, this would be an interesting starting point to develop novel treatments for this neurological disorder.
Allosteric modulation of GPCRs
Allosteric modulation of GPCRs is a rapidly developing research field and holds great promise for drug development (Wootten et al., 2013). Testing for allosteric modulation is often a great challenge because the effect of the allosteric modulator may be observed for the probe used for screening, but may not influence the endogenous ligand and it is intended to influence in a patient. In this respect, it would be advantageous to be able to study the influence of the allosteric modulator directly using the endogenous ligand as the probe. Two recent publications used GPCR‐based FRET sensors to study allosteric modulators (Maier‐Peuschel et al., 2010; Markovic et al., 2012). The first study used the muscarinic M2 receptor and created a CFP‐based/FlAsH‐based sensor as schematically shown in Figure 1, while the second study used the muscarinic M1 receptor and created a CFP‐based/YFP‐based sensor as schematically shown in Figure 1. For the M2 receptor, it was demonstrated that ACh and carbachol could induce a rapid and concentration‐dependent decrease in FRET, while the allosteric modulator gallamine did not cause a change in FRET signal (Maier‐Peuschel et al., 2010). However, as shown in Figure 3, gallamine was able to reverse the signal change induced by ACh. The signal change occurred much faster than could be explained by agonist dissociation. This observation was interpreted as further support of the allosteric modulatory effect by gallamine, which interestingly was different for ACh and carbachol, demonstrating the potential of this technology to investigate the probe‐dependent effect of allosteric modulators. For the M1 receptor, it was demonstrated that allosteric agonists can be detected using such sensor constructs (Markovic et al., 2012). Allosteric modulation of protein function can also occur by interacting proteins. This was recently demonstrated using a B1 (kinin 1B) receptor‐based FRET sensor. Employing such a sensor, the group of Randal A. Skidgel could demonstrate positive allosteric modulation of the B1 receptor by carboxypeptidase M (Zhang et al. 2011 and 2013). Two further studies also reported that the Gαq‐protein could stabilize the high‐affinity state of the M1 and M3 receptors but not the P2Y1 receptor (Tateyama and Kubo, 2013a, 2013b). These studies demonstrate the possible use of such sensors to investigate signalling complexes.
Figure 3.
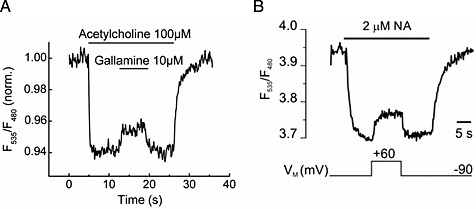
(A) Effects of gallamine on the FRET signals of a M2 receptor sensor expressed in HEK293 cells. Gallamine 10 μM reversed the signal induced by 100 μM ACh, reproduced from Maier‐Peuschel et al., 2010. (B) Voltage‐sensitivity of an α2A‐adrenoceptor. The biosensor was activated with 2 μM NA at −90 mV, reflected as a decrease in the FRET signal. Subsequent depolarization to +60 mV deactivates α2A‐adrenoceptor‐cam, indicated by a rise in the FRET ratio. Reproduced from Rinne et al., 2013.
Receptor dimers and higher order complexes of GPCRs
Oligomerization of GPCRs is a hot topic in GPCR research. The approaches and methods that are currently used to study GPCR dimerization have recently been covered in an up‐to‐date article as a result of an intense discussion at a GPCR workshop (Vischer et al., 2015). This area of research is the classic domain of bimolecular FRET sensors to study protein–protein interactions. Therefore, in this review, we will only cover work that has been carried out using unimolecular FRET sensors. We and others have also utilized this approach to study GPCR dimers and their activation. Using the before‐mentioned α2A‐adrenoceptor FlAsH‐ /CFP‐based FRET sensors (Figure 1), a modulatory effect of morphine via the μ‐opioid receptor could be shown (Vilardaga et al., 2008). This crosstalk was dependent upon co‐transfection of the μ‐opioid receptor and was due to a direct modulation at the level of the receptor itself. Furthermore, it could be demonstrated that morphine application led to a modulation of downstream signalling of the α2A‐adrenoceptor (Vilardaga et al., 2008). Several more studies investigated dimeric receptor complexes and their activation mode. It was reported that intermolecular FRET changes between both protomers of a dimer were clearly detectable using the metabotropic glutamate receptor‐1α (Tateyama et al., 2004), while intra‐molecular FRET changes within a receptor protomer could not be detected. Similarly, ligand‐induced subunit rearrangements of the GABAB receptor could only be detected when inter‐molecular rearrangement was investigated but not visible when intra‐molecular FRET changes were assessed (Matsushita et al., 2010). This absence of intra‐molecular FRET changes in class C GPCRs is not a general rule, because we could detect both inter‐ and intra‐molecular FRET changes using the mGlu1 receptor (Hlavackova et al., 2012). It could be demonstrated that the inter‐molecular FRET change occurred faster compared with the intra‐molecular signal change. Hence, a movement within the two protomers of a dimer might be required to allow activation of individual protomers, but this is currently speculation because receptor ensembles were measured and no single molecules.
Modulatory effects of transient protein modifications and ligand‐independent receptor modulation
Polymorphic variants of proteins have been described for several GPCRs. In this respect, it is interesting to see that such different variants can behave differently upon receptor activation. This was investigated for the polymorphic variants of the β‐adrenoceptor family (Ahles et al., 2011 and 2015). The kinetic response pattern was different for the Arg16‐Gly or Gln27‐Glu variants of the β2‐adrenoceptor. This was not evident when a single stimulus was compared, but became apparent when each variant was exposed to multiple repetitive stimuli. This difference in receptor activation was shown to depend on receptor phosphorylation and β‐arrestin2 interaction because the effect disappeared upon saponin treatment for cell permeabilization (Ahles et al., 2011). A similar effect was recently observed for the polymorphic variants of the β1‐adrenoceptor (Ahles et al., 2015).
Two recent studies have demonstrated the influence of other stimuli upon receptor activation using such GPCR‐based FRET sensors as depicted in Figure 1. The first study could demonstrate the influence of 15 mM benzoyl alcohol, a known membrane fluidity enhancer, upon 5‐HT1B receptor activation (Candelario and Chachisvilis, 2012). This led to direct 5‐HT1B receptor activation in a similar manner as controlled by the endogenous ligand 5‐HT. The second study investigated the influence of membrane voltage upon adrenoceptor function (Rinne et al., 2013) and found a direct modulation of active receptor states by voltage. The study used the previously described α2A‐CFP‐based/YFP‐based FRET sensors (Vilardga et al., 2003). A clear change in FRET ratio could be observed when the membrane potential was changed from −90 to +60 mV and strikingly resembles changes that were observed upon allosteric modulation by ligands at the muscarinic M2 receptor (Figure 3). The molecular mechanism leading to voltage‐dependent conformational changes and receptor activation are currently unknown.
Conclusion and future directions
The generation of GPCR‐based FRET sensors is currently an individual process and still needs to be individually optimized. However, we think the examples of versatile applications (Figure 4) compiled for these sensors in this review article clearly demonstrate that the added work of designing such sensors is outweighed by the added value of information obtained by their ability to investigate GPCR pharmacology and biology. As we discover new signalling modalities of GPCRs, like signalling from within the cell (Irannejad et al., 2013; Lohse and Hofmann, 2015), we also face novel challenges for labelling strategies. Some interesting developments are seen by the incorporation of unnatural amino acids into proteins that offer unique chemistry for functional coupling in living cells and allow a site‐selective labelling in cells (Haney et al., 2015). Such approaches allow the use of brighter organic fluorophores that were already used in vitro in single‐molecule FRET experiments (Tyagi and Lemke, 2015). Hence, such fluorophores should be bright enough to be resolved in subcellular compartments of living cells, although the total protein number within such compartments would probably be small. Another major challenge will be to adopt such RET sensors for use in in vivo situations. This will require joint efforts from several different scientific disciplines, and a detailed discussion has recently been published (van Unen et al., 2015). Hence, much remains to be done, but the improvements in our knowledge and understanding of these fascinating class of proteins will reward investigators for their efforts.
Figure 4.
Schematic overview of current research applications for GPCR‐based FRET sensors.
Conflict of interest
The University of Würzburg does hold a patent on this technology: WO2004057333 A1.
Acknowledgements
This work was supported by the DFG “Sonderforschungsbereich/Transregio 166: High‐end light microscopy elucidates receptor function ‐ ReceptorLight” (project C2; CH) and Marie Curie Initial Training Networks (ITN) “WntsApp” (CH).
Stumpf, A. D. , and Hoffmann, C. (2016) Optical probes based on G protein‐coupled receptors – added work or added value?. British Journal of Pharmacology, 173: 255–266. doi: 10.1111/bph.13382.
References
- Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, et al (2002). New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J Am Chem Soc 124: 6063–6076. [DOI] [PubMed] [Google Scholar]
- Ahles A, Rochais F, Frambach T, Bunemann M, Engelhardt S (2011). A polymorphism‐specific "Memory" mechanism in the {beta}2‐adrenergic receptor. Sci Signal 4: ra53. [DOI] [PubMed] [Google Scholar]
- Ahles A, Rodewald F, Rochais F, Bünemann M, Engelhardt S (2015). Interhelical interaction and receptor phosphorylation regulate the activation kinetics of different human beta1‐adrenoceptor variants. J Biol Chem 290: 1760–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al (2013). The Concise Guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Curto E, Prihandoko R, Tautermann CS, Zwier JM, Pediani JD, Lohse MJ, et al (2011). Developing chemical genetic approaches to explore G protein‐coupled receptor function: validation of the use of a receptor activated solely by synthetic ligand (RASSL). Mol Pharmacol 80: 1033–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosio M, Lohse MJ (2012). Nonequilibrium activation of a G‐protein‐coupled receptor. Mol Pharmacol 81: 770–777. [DOI] [PubMed] [Google Scholar]
- Bock A, Merten N, Schrage R, Dallanoce C, Bätz J, Klöckner J, et al (2012). The allosteric vestibule of a seven transmembrane helical receptor controls G‐protein coupling. Nat Commun 3: 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock A, Chirinda B, Krebs F, Messerer R, Bätz J, Muth M, et al (2014). Dynamic ligand binding dictates partial agonism at a G protein‐coupled receptor. Nat Chem Biol 10: 18–20. [DOI] [PubMed] [Google Scholar]
- Bockaert J, Fagni L, Dumuis A, Marin P (2004). GPCR interacting proteins (GIP). Pharmacol Ther 103: 203–221. [DOI] [PubMed] [Google Scholar]
- Bornholz B, Weidtkamp‐Peters S, Schmitmeier S, Seidel CA, Herda LR, Felix SB, et al (2013). Impact of human autoantibodies on beta1‐adrenergic receptor conformation, activity, and internalization. Cardiovasc Res 97: 472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RE (2009). Fluorescent‐protein‐based biosensors: modulation of energy transfer as a design principle. Anal Chem 81: 5972–5979. [DOI] [PubMed] [Google Scholar]
- Candelario J, Chachisvilis M (2012). Mechanical stress stimulates conformational changes in 5‐hydroxytryptamine receptor 1B in bone cells. Cell Mol Biol 5: 277–286. [Google Scholar]
- Chachisvilis M, Zhang YL, Frangos JA (2006). G protein‐coupled receptors sense fluid shear stress in endothelial cells. Proc Natl Acad Sci U S A 103: 15463–15468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Ross EM (2012). Activation biosensor for G protein‐coupled receptors: a FRET‐based m1 muscarinic activation sensor that regulates G(q). PLoS One 7: e45651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin BR, Herzmark P, Ishida S, Voyno‐Yasenetskaya TA, Sun Y, Farfel Z, et al (1996). Carboxyl‐terminal mutations of Gq alpha and Gs alpha that alter the fidelity of receptor activation. Mol Pharmacol 50: 885–890. [PubMed] [Google Scholar]
- Damian M, Mary S, Maingot M, M'Kadmi C, Gagne D, Leyris JP, et al (2015). Ghrelin receptor conformational dynamics regulate the transition from a preassembled to an active receptor: Gq complex. Proc Natl Acad Sci U S A 112: 1601–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Duenas V, Gomez‐Soler M, Lopez‐Cano M, Taura JJ, Ledent C, Watanabe M, et al (2014). Uncovering caffeine's adenosine A2A receptor inverse agonism in experimental parkinsonism. ACS Chem Biol 9: 2496–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier A, Juillerat A, Heinis C, Correa IR Jr, Kindermann M, Beaufils F, et al (2008). An engineered protein tag for multiprotein labeling in living cells. Chem Biol 15: 128–136. [DOI] [PubMed] [Google Scholar]
- Gether U, Lin S, Kobilka BK (1995). Fluorescent labeling of purified beta 2 adrenergic receptor. Evidence for ligand‐specific conformational changes. J Biol Chem 270: 28268–28275. [DOI] [PubMed] [Google Scholar]
- Ghanouni P, Steenhuis JJ, Farrens DL, Kobilka BK (2001). Agonist‐induced conformational changes in the G‐protein‐coupling domain of the beta 2 adrenergic receptor. Proc Natl Acad Sci U S A 98: 5997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S, Kim S, Shafer AM, Ratnala VR, Fung JJ, Zare RN, et al (2007). Structure and conformational changes in the C‐terminal domain of the beta2‐adrenoceptor: insights from fluorescence resonance energy transfer studies. J Biol Chem 282: 13895–13905. [DOI] [PubMed] [Google Scholar]
- Griffin BA, Adams SR, Tsien RY (1998). Specific covalent labeling of recombinant protein molecules inside live cells. Science 281: 269–272. [DOI] [PubMed] [Google Scholar]
- Haney CM, Wissner RF, Petersson EJ (2015). Multiply labeling proteins for studies of folding and stability. Curr Opin Chem Biol 28: 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim R, Tsien RY (1996). Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol 6: 178–182. [DOI] [PubMed] [Google Scholar]
- Hlavackova V, Zabel U, Frankova D, Bätz J, Hoffmann C, Prezeau L, et al (2012). Sequential inter‐ and intrasubunit rearrangements during activation of dimeric metabotropic glutamate receptor 1. Sci Signal 5: ra59. [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Gaietta G, Bünemann M, Adams SR, Oberdorff‐Maass S, Behr B, et al (2005). A FlAsH‐based FRET approach to determine G protein‐coupled receptor activation in living cells. Nat Methods 2: 171–176. [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Gaietta G, Zürn A, Adams SR, Terrillon S, Ellisman MH, et al (2010). Fluorescent labeling of tetracysteine‐tagged proteins in intact cells. Nat Protoc 5: 1666–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Nuber S, Zabel U, Ziegler N, Winkler C, Hein P, et al (2012). Comparison of the activation kinetics of the m3 acetylcholine receptor and a constitutively active mutant receptor in living cells. Mol Pharmacol 82: 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Zürn A, Bünemann M, Lohse MJ (2008). Conformational changes in G‐protein‐coupled receptors‐the quest for functionally selective conformations is open. Br J Pharmacol 153: S358–S366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard AD, McAllister G, Feighner SD, Liu Q, Nargund RP, Van der Ploeg LH, et al (2001). Orphan G‐protein‐coupled receptors and natural ligand discovery. Trends Pharmacol Sci 22: 132–140. [DOI] [PubMed] [Google Scholar]
- Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, et al (2013). Conformational biosensors reveal GPCR signalling from endosomes. Nature 495: 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jares‐Erijman EA, Jovin TM (2003). FRET imaging. Nat Biotechnol 21: 1387–1395. [DOI] [PubMed] [Google Scholar]
- Jares‐Erijman EA, Jovin TM (2006). Imaging molecular interactions in living cells by FRET microscopy. Curr Opin Chem Biol 10: 409–416. [DOI] [PubMed] [Google Scholar]
- Jensen JB, Lyssand JS, Hague C, Hille B (2009). Fluorescence changes reveal kinetic steps of muscarinic receptor‐mediated modulation of phosphoinositides and Kv7.2/7.3 K+ channels. J Gen Physiol 133: 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Reynolds KA, Cherezov V, Hanson MA, Roth CB, Yeager M, et al (2009). Analysis of full and partial agonists binding to beta2‐adrenergic receptor suggests a role of transmembrane helix V in agonist‐specific conformational changes. J Mol Recognit 22: 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, et al (2013). Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504: 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Nuber S, Hoffmann C (2012). Fluorescence/bioluminescence resonance energy transfer techniques to study G‐protein‐coupled receptor activation and signaling. Pharmacol Rev 64: 299–336. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Hofmann KP (2015). Spatial and temporal aspects of signaling by G‐protein‐coupled receptors. Mol Pharmacol 88: 572–578. [DOI] [PubMed] [Google Scholar]
- Ma Z, Du L, Li M (2014). Toward fluorescent probes for G‐protein‐coupled receptors (GPCRs). J Med Chem 57: 8187–8203. [DOI] [PubMed] [Google Scholar]
- Madani F, Lind J, Damberg P, Adams SR, Tsien RY, Graslund AO (2009). Hairpin structure of a biarsenical‐tetracysteine motif determined by NMR spectroscopy. J Am Chem Soc 131: 4613–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier‐Peuschel M, Frölich N, Dees C, Hommers LG, Hoffmann C, Nikolaev VO, et al (2010). A fluorescence resonance energy transfer‐based M2 muscarinic receptor sensor reveals rapid kinetics of allosteric modulation. J Biol Chem 285: 8793–8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik RU, Ritt M, Devree BT, Neubig RR, Sunahara RK, Sivaramakrishnan S (2013). Detection of G protein‐selective G protein‐coupled receptor (GPCR) conformations in live cells. J Biol Chem 288: 17167–17178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, et al (2015). Structural insights into the dynamic process of beta2‐adrenergic receptor signaling. Cell 161: 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic D, Holdich J, Al‐Sabah S, Mistry R, Krasel C, Mahaut‐Smith MP, et al (2012). FRET‐based detection of M(1) muscarinic acetylcholine receptor activation by orthosteric and allosteric agonists. PLoS One 7: e29946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Giepmans BN, Adams SR, Tsien RY (2005). Mammalian cell‐based optimization of the biarsenical‐binding tetracysteine motif for improved fluorescence and affinity. Nat Biotechnol 23: 1308–1314. [DOI] [PubMed] [Google Scholar]
- Masharina A, Reymond L, Maurel D, Umezawa K, Johnsson K (2012). A fluorescent sensor for GABA and synthetic GABA(B) receptor ligands. J Am Chem Soc 134: 19026–19034. [DOI] [PubMed] [Google Scholar]
- Matsushita S, Nakata H, Kubo Y, Tateyama M (2010). Ligand‐induced rearrangements of the GABA(B) receptor revealed by fluorescence resonance energy transfer. J Biol Chem 285: 10291–10299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A (2011). Development of probes for cellular functions using fluorescent proteins and fluorescence resonance energy transfer. Annu Rev Biochem 80: 357–373. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Niino Y (2015). Molecular spies for bioimaging‐fluorescent protein‐based probes. Mol Cell 58: 632–643. [DOI] [PubMed] [Google Scholar]
- Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A (2004). Expanded dynamic range of fluorescent indicators for Ca2+ by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci U S A 101: 10554–10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi J, Takarada T, Yunoki S, Kikuchi Y, Maeda M (2006). FRET‐based monitoring of conformational change of the beta2 adrenergic receptor in living cells. Biochem Biophys Res Commun 343: 1191–1196. [DOI] [PubMed] [Google Scholar]
- Newman RH, Fosbrink MD, Zhang J (2011). Genetically encodable fluorescent biosensors for tracking signaling dynamics in living cells. Chem Rev 111: 3614–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev VO, Hoffmann C, Bünemann M, Lohse MJ, Vilardaga JP (2006). Molecular basis of partial agonism at the neurotransmitter {alpha}2A‐adrenergic receptor and Gi‐protein heterotrimer. J Biol Chem 281: 24506–24511. [DOI] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE (2008). Heterotrimeric G protein activation by G‐protein‐coupled receptors. Nat Rev Mol Cell Biol 9: 60–71. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ; NC‐IUPHAR. (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 42: D1098‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomorski A, Krezel A (2011). Exploration of biarsenical chemistry—challenges in protein research. Chembiochem 12: 1152–1167. [DOI] [PubMed] [Google Scholar]
- Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, et al (2012). Structural insights into biased G protein‐coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci U S A 109: 6733–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, et al (2011a). Structure of a nanobody‐stabilized active state of the beta(2) adrenoceptor. Nature 469: 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al (2011b). Crystal structure of the beta2 adrenergic receptor‐Gs protein complex. Nature 477: 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner S, Ambrosio M, Hoffmann C, Lohse MJ (2010). Differential signaling of the endogenous agonists at the {beta}2‐adrenergic receptor. J Biol Chem 285: 36188–36198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne A, Birk A, Bünemann M (2013). Voltage regulates adrenergic receptor function. Proc Natl Acad Sci U S A 110: 1536–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochais F, Vilardaga JP, Nikolaev VO, Bünemann M, Lohse MJ, Engelhardt S (2007). Real‐time optical recording of beta1‐adrenergic receptor activation reveals supersensitivity of the Arg389 variant to carvedilol. J Clin Invest 117: 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ (1993). A mutation‐induced activated state of the beta 2‐adrenergic receptor. Extending the ternary complex model. J Biol Chem 268: 4625–4636. [PubMed] [Google Scholar]
- Stroffekova K, Proenza C, Beam KG (2001). The protein‐labeling reagent FLASH‐EDT2 binds not only to CCXXCC motifs but also non‐specifically to endogenous cysteine‐rich proteins. Pflugers Arch 442: 859–866. [DOI] [PubMed] [Google Scholar]
- Szalai B, Barkai L, Turu G, Szidonya L, Varnai P, Hunyady L (2012). Allosteric interactions within the AT(1) angiotensin receptor homodimer: role of the conserved DRY motif. Biochem Pharmacol 84: 477–485. [DOI] [PubMed] [Google Scholar]
- Tateyama M, Abe H, Nakata H, Saito O, Kubo Y (2004). Ligand‐induced rearrangement of the dimeric metabotropic glutamate receptor 1alpha. Nat Struct Mol Biol 11: 637–642. [DOI] [PubMed] [Google Scholar]
- Tateyama M, Kubo Y (2013a). Analyses of the effects of Gq protein on the activated states of the muscarinic M3 receptor and the purinergic P2Y1 receptor. Physiol Rep 1: e00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateyama M, Kubo Y (2013b). Binding of Gq protein stabilizes the activated state of the muscarinic receptor type 1. Neuropharmacology 65: 173–181. [DOI] [PubMed] [Google Scholar]
- Tyagi S, Lemke EA (2015). Single‐molecule FRET and crosslinking studies in structural biology enabled by noncanonical amino acids. Curr Opin Struct Biol 32: 66–73. [DOI] [PubMed] [Google Scholar]
- van Unen J, Woolard J, Rinken A, Hoffmann C, Hill SJ, Goedhart J, et al (2015). A perspective on studying G‐protein‐coupled receptor signaling with resonance energy transfer biosensors in living organisms. Mol Pharmacol 88: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM (2013). Molecular signatures of G‐protein‐coupled receptors. Nature 494: 185–194. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Bünemann M, Krasel C, Castro M, Lohse MJ (2003). Measurement of the millisecond activation switch of G protein‐coupled receptors in living cells. Nat Biotechnol 21: 807–812. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Nikolaev VO, Lorenz K, Ferrandon S, Zhuang Z, Lohse MJ (2008). Conformational cross‐talk between alpha2A‐adrenergic and mu‐opioid receptors controls cell signaling. Nat Chem Biol 4: 126–131. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Romero G, Feinstein TN, Wehbi VL (2013). Kinetics and dynamics in the G protein‐coupled receptor signaling cascade. Methods Enzymol 522: 337–363. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Steinmeyer R, Harms GS, Lohse MJ (2005). Molecular basis of inverse agonism in a G protein‐coupled receptor. Nat Chem Biol 1: 25–28. [DOI] [PubMed] [Google Scholar]
- Vischer HF, Castro M, Pin JP (2015). G protein‐coupled receptor multimers: a question still open despite the use of novel approaches. Mol Pharmacol 88: 561–571. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Milligan G (2014). Structural and biophysical characterisation of G protein‐coupled receptor ligand binding using resonance energy transfer and fluorescent labelling techniques. Biochim Biophys Acta 1838: 3–14. [DOI] [PubMed] [Google Scholar]
- Wootten D, Christopoulos A, Sexton PM (2013). Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov 12: 630–644. [DOI] [PubMed] [Google Scholar]
- Xu TR, Ward RJ, Pediani JD, Milligan G (2012). Intramolecular fluorescence resonance energy transfer (FRET) sensors of the orexin OX1 and OX2 receptors identify slow kinetics of agonist activation. J Biol Chem 287: 14937–14949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Tan F, Brovkovych V, Zhang Y, Skidgel RA (2011). Cross‐talk between carboxypeptidase M and the kinin B1 receptor mediates a new mode of G protein‐coupled receptor signaling. J Biol Chem 286: 18547–18561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Tan F, Skidgel RA (2013). Carboxypeptidase M is a positive allosteric modulator of the kinin B1 receptor. J Biol Chem 288: 33226–33240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler N, Bätz J, Zabel U, Lohse MJ, Hoffmann C (2011). FRET‐based sensors for the human M(1)‐, M(3)‐, and M(5)‐acetylcholine receptors. Bioorg Med Chem 19: 1048–1054. [DOI] [PubMed] [Google Scholar]
- Zürn A, Klenk C, Zabel U, Reiner S, Lohse MJ, Hoffmann C (2010). Site‐specific, orthogonal labeling of proteins in intact cells with two small biarsenical fluorophores. Bioconjug Chem 21: 853–859. [DOI] [PubMed] [Google Scholar]
- Zürn A, Zabel U, Vilardaga JP, Schindelin H, Lohse MJ, Hoffmann C (2009). Fluorescence resonance energy transfer analysis of {alpha}2a‐adrenergic receptor activation reveals distinct agonist‐specific conformational changes. Mol Pharmacol 75: 534–541. [DOI] [PubMed] [Google Scholar]