

Abstract
Background and Purpose
Upon stimulation, neutrophils release their nuclear contents called neutrophil extracellular traps (NETs), which contain unfolded chromatin and lysosomal enzymes. NETs have been demonstrated to play a critical role in host defence, although the role of PGE2, a bioactive substance generated in inflammatory tissues, in the formation of NETs remains unclear.
Experimental Approach
The effects of PGE2, agonists and antagonists of its receptors, and modulators of the cAMP–PKA pathway on the formation of NETs were examined in vitro in isolated neutrophils and in vivo in a newly established mouse model.
Key Results
PGE2 inhibited PMA‐induced NET formation in vitro through EP2 and EP4 Gαs‐coupled receptors. Incubation with a cell‐permeable cAMP analogue, dibutyryl cAMP, or various inhibitors of a cAMP‐degrading enzyme, PDE, also suppressed NET formation. In the assay established here, where an agarose gel was s.c. implanted in mice and NET formation was detected on the surface of the gel, the extent of the NET formed was inhibited in agarose gels containing rolipram, a PDE4 inhibitor, and butaprost, an EP2 receptor agonist.
Conclusions and Implications
PGE2 inhibits NET formation through the production of cAMP. These findings will contribute to the development of novel treatments for NETosis‐related diseases.
Abbreviations
- BSA
bovine serum albumin
- cAMP
cyclic adenosine monophosphate
- db2cAMP
dibutyryl cyclic AMP
- MPO
myeloperoxidase
- NETs
neutrophil extracellular traps
- PAD
peptidylarginine deiminase
- PG
prostaglandin
- PGE2
prostaglandin E2
- PDE
phosphodiesterase
- PMA
phorbol 12‐myristate 13‐acetate
- PKA
protein kinase A
- PKC
protein kinase C
- ROS
reactive oxygen species
Tables of Links
| LIGANDS | |
| butaprost | L‐161982 |
| cAMP | L‐798106 |
| cilostazol | PF‐04418948 |
| Dibutyryl cAMP | PGE2 |
| H89 | rolipram |
| IBMX | SC‐51322 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a, 2013b).
Introduction
The innate immune system is the first line of host defence against infecting bacteria and neutrophils are the most abundant leukocyte involved in this response. They are recruited into inflammatory tissues after extravasation from blood vessels and eliminate bacteria by phagocytosis and the generation of reactive oxygen species (ROS). In addition, it has recently been demonstrated that neutrophils release their nuclear contents, including unfolded chromatins and lysosomal enzymes, which also play a critical role in killing bacteria by physically trapping them (Brinkmann et al., 2004). These released nuclear contents are called neutrophil extracellular traps (NETs), and the associated process is termed NETosis, to distinguish this type of neutrophil cell death mechanism from necrosis and apoptosis (Fuchs et al., 2007; Guimaraes‐Costa et al., 2009).
More recently, NETs have been demonstrated to have multiple, different functions (Brinkmann and Zychlinsky, 2012). For instance, NETs regulate thrombus formation through activation of platelet systems (Fuchs et al., 2010) and coagulation systems (Brinkmann and Zychlinsky, 2012), cancer metastasis by trapping cancer cells (Cools‐Lartigue et al., 2013) and the development of autoimmune diseases such as systemic lupus erythaematosus (Hakkim et al., 2010; Knight et al., 2013). Furthermore, NETs have been identified as the target antigens of anti‐citrullinated protein antibodies, a specific marker of rheumatoid arthritis (Khandpur et al., 2013), as many released proteins are citrullinated upon NETosis.
Investigations into the molecular mechanisms of NETosis have elucidated several key factors: firstly, PMA is often used to induce NETosis and reflects the importance of the activation of PKC in this phenomenon (Neeli and Radic, 2013). Secondly, ROS, downstream products of PKC activation, are critical for inducing NETosis because neutrophils of patients suffering from chronic granulomatous diseases, caused by impaired ROS production in neutrophils and macrophages, exhibit little if any NETosis (Fuchs et al., 2007). Thirdly, peptidylarginine deiminase 4 (PAD4), a Ca2+‐dependent enzyme that mediates protein citrullination, plays an important role in inducing NETosis as PAD4‐deficient mice show very little NETosis (Li et al., 2010). Because arginine, with a net positive charge, is changed into neutral citrulline by citrullination, the subsequent reduced positive charge of histones after citrullination weakens the tightly folded chromatin structure, enabling the apparent ‘NET’ formation (Wang et al., 2009). However, it remains unclear how PKC, ROS and PAD4 function at the molecular level at critical steps of NETosis, including the disappearance of the nuclear envelope, the swelling of the nucleus, the unfolding of chromatin, the disruption of organelles such as lysosomes and the release of cellular contents as a result of the disruption of the plasma membrane.
Cells in inflammatory tissues produce various bioactive substances, such as PGs, by the arachidonate cascade (Funk, 2001). Among these, PGE2 is considered a strong regulator of inflammation. PGE2's effects are bidirectional, namely, pro‐inflammatory on some occasions and anti‐inflammatory on others as its effects are mediated by four types of seven‐transmembrane receptor: EP1 coupled with Gαq, EP2 and EP4 coupled with Gαs and EP3 with Gαi (Sugimoto and Narumiya, 2007).
Here, we showed that PGE2 inhibited NETosis by inducing an increase in intracellular cAMP through its EP2 and EP4 receptors. We established a novel, evaluation assay for NETosis in vivo by the s.c. implantation of agarose gel in mice and showed that an inhibitor of PDE4, a major cAMP‐degrading enzyme expressed in neutrophils (Sousa et al., 2010) and an EP2 agonist butaprost strongly inhibited NETosis in vivo.
Methods
In vitro NETosis assay
Neutrophils were isolated from healthy human blood donors using Polymorphoprep (Alere Technologies AS, Jena, Germany) according to the manufacturer's instructions and as described previously (Nishioka et al., 1998). Briefly, 1 mL of the neutrophil of the Polymorphprep preparation was added to 2 mL of 75% PBS and centrifuged at 400× g for 10 min at 22°C. Pelleted cells were resuspended in 1 mL RPMI 1640 with 20 mg⋅mL−1 BSA. Cells were then incubated with 7 μg⋅mL−1 Hoechst 33342 and 50 nM mitotracker at 37°C for 30 min. After centrifugation at 120× g for 5 min at 4°C, pelleted cells were resuspended in 2 mL red blood cell lysis buffer (0.83 mg⋅mL−1 NH4Cl, 0.1 mg⋅mL−1 KHCO3 and 90 μg⋅mL−1 EDTA) and incubated at room temperature for 20 s. Then, the haemolysis was stopped by addition of 12 mL ice‐cold Hanks' balanced salt solution (Sigma). After centrifugation at 120× g for 5 min at 4°C, the pelleted cells were suspended in 1 mL RPMI 1640 with 20 mg⋅mL−1 BSA. More than 99% of cells (approximately 1–3 × 106/mL) in the final sample were granulocytes and used as ‘neutrophils’.
In the standard in vitro NETosis assay, NETosis was induced in 4 × 104 neutrophils suspended in 400 μL RPMI 1640 with 20 mg⋅mL−1 BSA in a coverglass chamber (Iwaki, Shizuoka, Japan; 5232–008; 5 × 5 mm per one chamber) by incubation with 50 nM PMA at 37°C for 90 min and stained with 100 nM sytox orange. In the analyses of cAMP‐regulating reagents, isolated neutrophils were pre‐incubated at 37°C for 15 min before PMA stimulation. NETosis was visually measured in more than 300 cells in five randomly selected fields by confocal microscopy, Leica TCS SP8 (Leica, Wetzler, Germany) (Figure 1A). Approximately 15% of neutrophils underwent NETosis after 90 min incubation with PMA (Figure 1B). However, because the rate of reaction varied, in vitro NETosis for experiments was evaluated between 75 and 105 min incubation (Figures 2, 3, 4).
Figure 1.
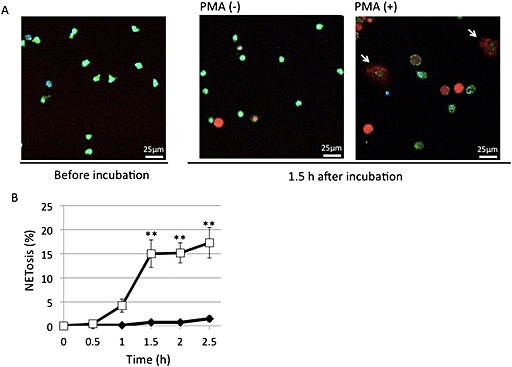
PMA‐induced NETosis in isolated neutrophils in vitro. After human peripheral neutrophils were incubated at 37°C for 1.5 h, without or with 50 nM PMA, they were stained with cell‐permeable DNA stain Hoechst 33342 (blue), mitochondrial stain mitotracker green FM (green) and cell‐impermeable DNA stain sytox orange (red) and analysed by confocal microscopy. (A) Typical photos with arrows indicating neutrophils undergoing NETosis. (B) Time‐dependent increase of cells undergoing NETosis. NETosis was visually measured in more than 300 cells in five randomly selected fields by confocal microscopy. Data are expressed as mean percentage of neutrophils undergoing NETosis per total number of cells ± SEM of five independent experiments. **P < 0.01 compared with the PMA (−) sample at each time point.
Figure 2.
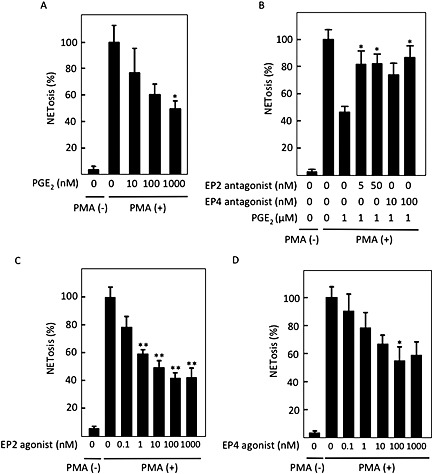
PGE2 inhibited PMA‐induced NETosis. After human peripheral neutrophils were incubated without or with 50 nM PMA in the presence of the indicated concentrations of (A) PGE2, (B) EP2 antagonist PF‐04418948 or EP4 antagonist L‐161982 with 1 μM PGE2, (C) EP2 agonist butaprost and (D) EP4 agonist CAY10598, cells undergoing NETosis were stained with cell‐impermeable DNA stain sytox orange. NETosis was visually measured in more than 300 cells in five randomly selected fields by confocal microscopy. Data shown are expressed as mean ± SEM of cells undergoing NETosis as a percentage of those stimulated with PMA alone in six (A) or five (B–D) independent experiments. *P < 0.05 and **P < 0.01 compared with NETosis with PMA alone for (A), (C) and (D) or that with PMA plus PGE2 for (B).
Figure 3.
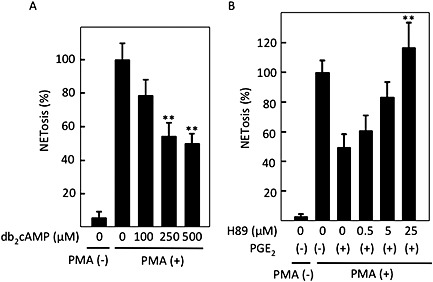
cAMP‐mediated inhibition of PMA‐induced NETosis. After human peripheral neutrophils were incubated without or with 50 nM PMA in the presence of the indicated concentrations of (A) cell‐permeable cAMP analogue, db2cAMP, and (B) PKA inhibitor H89 with 1 μM PGE2, cells undergoing NETosis were stained with cell‐impermeable DNA stain sytox orange. NETosis was visually measured in more than 300 cells in five randomly selected fields by confocal microscopy. Data shown are expressed as mean ± SEM of cells undergoing NETosis as a percentage of those stimulated with PMA alone in five independent experiments. **P < 0.01 compared with NETosis with PMA alone (A) and that with PMA plus PGE2 (B).
Figure 4.
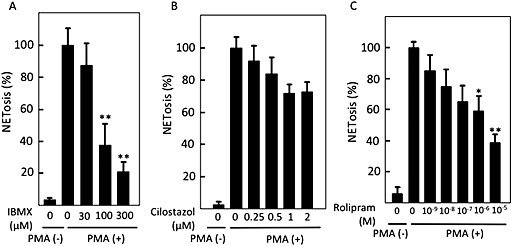
PDE inhibitors suppressed PMA‐induced NETosis. After human peripheral neutrophils were incubated without or with PMA in the presence of the indicated concentrations of (A) a pan‐PDE inhibitor, IBMX, (B) a PDE3 inhibitor, cilostazol, or (C) a PDE4 inhibitor, rolipram, cells undergoing NETosis were stained with cell‐impermeable DNA stain sytox orange. NETosis was visually measured in more than 300 cells in randomly selected five fields by confocal microscopy. Data shown are expressed as mean ± SEM of cells undergoing NETosis as a percentage of those stimulated with PMA alone in five independent experiments. *P < 0.05 and **P < 0.01 compared with NETosis with PMA alone.
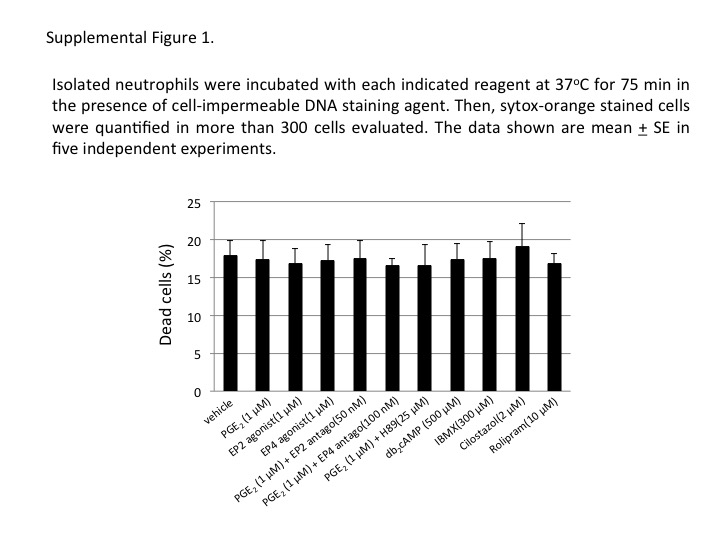
For the evaluation of the effects of cAMP‐regulating reagents on the neutrophil viability, isolated neutrophils were incubated with each indicated reagent at 37°C for 75 min in the presence of cell‐impermeable DNA staining agent, sytox orange. Then, sytox orange‐stained cells were quantified among more than 300 cells. cAMP‐regulating reagents used in this study did not affect cell viability of the neutrophils evaluated after 75 min incubation (Supporting Information Figure 1).
Vehicles were used as controls in each experiment. Most of the reagents except for butaprost were solubilized in ethanol as a vehicle. The final concentration of ethanol was no more than 0.5% in the in vitro NETosis assay, which had no effects on NET formation. Butaprost was solubilized in methylacetate. The final concentration of methylacetate was 0.1%, which had no effects on NET formation either. Each set of in vitro NET formation assays was performed in the presence of equal concentration of vehicle. Blood from three or four independent donors was used in each set of experiments.
In vivo NETosis assay
All studies involving animals were reported in accordance with the ARRIVE Guidelines for reporting animal experiments (Kilkenny et al., 2010; McGrath et al., 2010). Every effort was made to minimize the number of animals used and their suffering. An agarose gel (20 mg⋅mL−1 of Agarose S, 5 L × 5 W × 1 H mm), with or without 1 mM rolipram or 100 μM butaprost, was implanted s.c. and aseptically in the back of a C57BL/6J mouse (8–12 weeks, 17–22 g; Japan SLC Inc, Shizuoka, Japan) by a small skin incision (5 mm) after i.p. of sodium pentobarbital (40–50 mg/kg). After surgery, food and water were available ad libitum. Unless otherwise specified, the agarose gel was collected after 8 or 12 h and stained with sytox orange without fixation. NETosis on the visceral surface of the agarose gel was evaluated by confocal microscopy. To quantify NETosis, sytox orange‐positive areas in central nine squares were analysed with image j as shown in Figure 7A.
Figure 7.
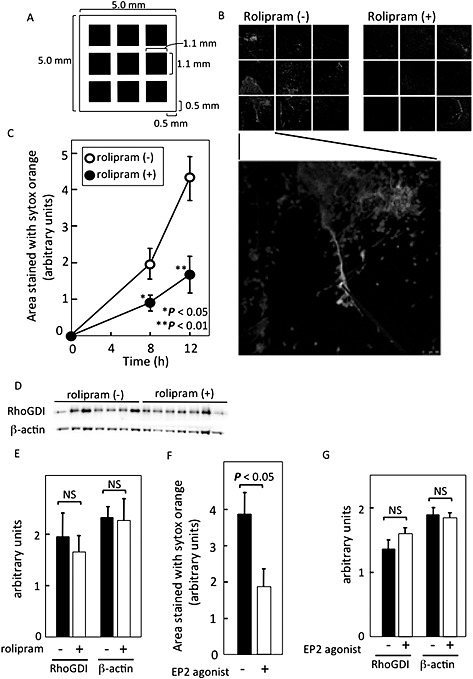
cAMP inhibited NETosis in vivo. (A) The quantification method is shown. An agarose gel implanted s.c. in a mouse was excised and stained with cell‐impermeable DNA stain sytox orange. Nine central photos were taken as indicated in the figure, followed by the measurement of the sytox orange‐positive areas by image j. (B) Typical photos of sytox orange‐stained agarose gels with or without 1 mM rolipram, a PDE4 inhibitor, after implantation for 12 h in the s.c. tissue of a mouse. (C) Quantification of sytox orange‐positive NET areas at indicated times. NET areas are expressed as mean ± SEM of 12 animals per group at 8 h and six animals per group at 12 h after implantation. (D, E) S.c. implanted gels with or without rolipram were collected after 8 h (n = 7 in each group) and the gel‐associated intracellular proteins, RhoGDI and β‐actin were analysed by immunoblotting (D) and quantified using image j (E). (F, G) Agarose gels, with or without the EP2 agonist, butaprost, were implanted s.c. in mice and collected after 12 h. They were immediately stained with sytox orange and quantified using image j as described above (each group n = 9) (F). The gel‐associated RhoGDI and β‐actin were analysed by immunoblotting (each group, n = 7) (G). The data are expressed as mean ± SEM. NS, not significant.
Evaluation of live neutrophils on the implanted gel was performed by measurement of intracellular proteins such as RhoGDI and β‐actin in separate sets of experiments. For this, gels were treated with 110 μL of Laemmli's SDS sample buffer (Laemmli, 1970) containing 0.5% cholic acid, and the eluates were analysed by probing with anti‐RhoGDI (BD Bioscience, San Jose, CA, USA) and anti‐β‐actin (Sigma) antibodies as indicators of live cells, followed by quantification with image j.
Immunohistochemistry
Agarose gels obtained in the in vivo NETosis assay were fixed by incubation with 4% paraformaldehyde at room temperature for 15 min, followed by treatment with 1 mg⋅mL−1 Triton X‐100 (Sigma, St Louis, MO, US) in PBS for 5 min. Gels were then incubated with anti‐Ly6G antibody at room temperature in PBS containing 1 mg⋅mL−1 BSA at room temperature for 1 h. After washing with PBS twice, gels were visualized with Alexa Fluor 488 goat anti‐rat IgG for 1 h at room temperature and analysed by confocal laser fluorescence microscopy (Leica TCS SP8). Immunostaining with MPO antibody was performed in the same way except for without treating gels with Triton X‐100 and using Alexa Fluor 488 goat anti‐rabbit IgG as a secondary antibody.
Measurement of ROS
Isolated neutrophils from healthy donors were incubated with 5 μM DHR123 (Cayman), a cell‐permeable fluorescent ROS indicator, and the cAMP‐regulating reagents indicated, such as PGE2, rolipram and IBMX at 37°C for 15 min, and then stimulated with 50 nM PMA at 37°C for 5 min. The samples were placed on ice and immediately analysed by flow cytometer, FC500 ( Beckman Coulter, Brea, Carifornia, US). Mean fluorescence intensity was analysed in three independent experiments with similar results.
Measurement of cAMP
Isolated neutrophils, 3 × 106 in 3 mL, were incubated for 30 min at 37°C with indicated reagents. Cells were collected by centrifugation and lysed with 150 μL 0.1 M HCl containing 1% Triton X‐100. After centrifugation, cAMP in the supernatant was quantified with the cAMP Complete elisa Kit (Enzo Life Science, Tokyo, Japan) according to the manufacturer's instructions.
Evaluation of adhesion of neutrophils in vitro
Human neutrophils (1.0 × 105⋅mL−1) were incubated for 15 min at 37°C with indicated reagents such as PGE2 and rolipram in a coverglass chamber (Iwaki, Tokyo), followed by addition of 5 nM PMA and then incubated for another 10 min. After being washed with PBS, cells adhering to the bottom of the dish were lysed with 100 μL of Laemmli's SDS‐containing sample buffer (Laemmli, 1970) with 0.5% cholate. Samples were analysed by immunoblotting with anti‐β‐actin antibody.
Statistics
The data are expressed as mean ± SEM of five or more experiments. The statistical analysis was performed using the JMP PRO 11 (Cary, North Carolina, US). With the time course experiments (Figure 1), data were analysed with the two‐way anova followed by Tukey's test. The data in Figures 2, 3, 4, 5 and Supporting Information Figures 1 and 3 were evaluated with the one‐way anova followed by Tukey's test. Differences between two sets of data (Figure 7) were evaluated by use of Student's unpaired t‐test. A P value of less than 0.05 was considered statistically significant.
Figure 5.
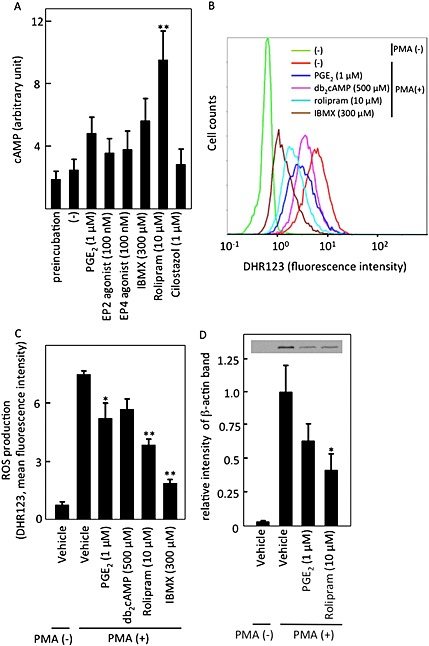
The effects of cAMP‐increasing reagents on cAMP and ROS levels in isolated neutrophils. (A) cAMP levels were quantified in isolated neutrophils after incubation with indicated reagents for 30 min as described in Methods. Data shown are expressed as mean ± SEM of cAMP levels (in arbitrary units) in five independent experiments. *P < 0.05 and **P < 0.01 compared with values obtained with vehicle. (B, C) Isolated neutrophils were incubated with 50 nM PMA in the presence of indicated reagents at 37°C for 5 min, and ROS generation was analysed by flow cytometry (B). Data are expressed as mean ± SEM of mean fluorescence intensity in five independent experiments (C). (D) The effects of cAMP‐increasing reagents on cell adhesion was examined by incubating isolated neutrophils with 5 nM PMA in the presence of the indicated reagents at 37°C for 10 min in a coverglass chamber. After washing with PBS once, the adhering cells were lysed, and β‐actin was analysed by immunoblotting, followed by quantification with image j. Data shown are expressed as mean ± SEM of relative intensity of β‐actin band in seven independent experiments. (C, D) *P < 0.05 and **P < 0.01 compared with values obtained with the PMA alone.
Materials
Cell‐permeable compound Hoechst 33342 and cell‐impermeable sytox orange used as DNA fluorescence stains were purchased from Dojindo (Kumamoto, Japan) and Life Technologies (Tokyo, Japan) respectively. Mitotracker was from Life Technologies. Agarose S and PGE2 were from Wako Chemicals (Osaka, Japan). A pan‐PDE inhibitor, IBMX (Talpain et al., 1995), a selective PDE3 inhibitor, cilostazol (Sudo et al., 2000), and a selective PDE4 inhibitor, rolipram (Talpain et al., 1995), were from Tocris (Bristol, UK), Sigma (St Louis, MO, USA) and Wako Chemicals respectively. An EP2 agonist, butaprost (Talpain et al., 1995), and an EP4 agonist, CAY10598 (Wang et al., 2014), were from Cayman (Ann Arbor, MI, USA). An EP1 antagonist, SC‐51322 (Tanaka et al., 1998), EP2 antagonist, PF‐04418948 (af Forselles et al., 2011), and an EP4 antagonist, L‐161982 (Cherukuri et al., 2007), were from Cayman. An EP3 antagonist, L‐798106 (Orie and Clapp, 2011), was from Sigma. A PKA inhibitor, H89 (Chijiwa et al., 1990), was from Cell Signaling (Danvers, MA, USA). Antibodies used were the following: rabbit polyclonal anti‐histone H3 and citrullinated histone H3 (Abcam, Cambridge, MA, USA), rabbit polyclonal anti‐myeloperoxidase (MPO) (Dako, Glostrup, Denmark) and anti‐Ly6G (Stirling et al., 2009) (BD Pharmingen, San Jose, CA, USA). Secondary antibodies used were Alexa Fluor 488 goat anti‐rat IgG and Alexa Fluor 488 goat anti‐rabbit IgG (both Life Technologies). All other substances, including PMA, dibutyryl cAMP (db2cAMP) and cilostazol, were from Sigma.
Ethical statement
This study was performed under approval of the Ethics Committee and the Animal Experiment Committee of Tohoku University, Sendai, Japan.
Results
PMA‐induced NETosis in vitro
Isolated neutrophils were incubated without or with PMA and stained with a cell‐impermeable DNA‐stain, sytox orange (red), a cell‐permeable DNA stain Hoechst 33342 (blue) and mitotracker (green) (Figure 1A). Cells undergoing NETosis were clearly observed as having NET‐like structures in red with a larger area than that of an intact neutrophil (Figure 1A). The induction of NETosis was time‐dependent (Figure 1B), and NETosis was induced in approximately 15% of neutrophils by 1.5 h. We generally analysed NETosis at 1.5 h after PMA stimulation. However, because the rate of reaction varied somewhat, NETosis was evaluated between 75 and 105 min in some experiments (Figures 2, 3, 4).
PGE2 inhibited NETosis through its stimulation of Gαs‐coupled, EP2 and EP4 receptors
We evaluated the effects of PGE2 on NETosis in neutrophils in the in vitro assay. As shown in Figure 2A, PGE2 inhibited PMA‐induced NETosis in a concentration‐dependent manner. The inhibition by PGE2 was partially reversed either by an EP2 antagonist, PF‐04418948, or by an EP4 inhibitor, L‐161982, to comparable degrees (Figure 2B), but not by an EP1 antagonist, SC‐51322, or an EP3 antagonist, L‐798106 (data not shown). Additionally, a specific EP2 agonist, butaprost, and an EP4 agonist, CAY10598, inhibited PMA‐induced NETosis in a concentration‐dependent manner (Figure 2C and D). Thus, NETosis was inhibited by PGE2, which was mediated comparably by both EP2 and EP4 receptors.
Intracellular cAMP inhibited NETosis
Because both EP2 and EP4 receptors are Gαs‐coupled receptors, the effects of PGE2 could be mediated by an increase in intracellular cAMP. As shown in Figure 3A, a cell‐permeable cAMP analogue, db2cAMP, inhibited PMA‐induced NETosis in a concentration‐dependent manner. Because most of the cAMP signal is mediated by PKA, we examined the effect of the PKA inhibitor, H89, on PMA‐induced NETosis. As shown in Figure 3B, H89 completely reversed the PGE2‐mediated inhibition of PMA‐induced NETosis. This suggests that intracellular cAMP inhibited NETosis through activation of PKA.
Intracellular cAMP is degraded by PDEs. IBMX, a strong pan‐PDE inhibitor, potently inhibited PMA‐induced NETosis in a concentration‐dependent manner (Figure 4A). Cilostazol, a selective PDE3 inhibitor, tended to reduce the PMA‐induced NETosis without statistical significance (Figure 4B), while rolipram, a selective PDE4 inhibitor, strongly affected PMA‐induced NETosis (Figure 4C). This again suggests the involvement of cAMP in the inhibition of neutrophil NETosis and that the contribution of PDE4 to the inhibition of NETosis is more dominant compared to PDE3.
cAMP inhibited ROS production and adhesion
We next evaluated the intracellular cAMP levels after the various treatments. As shown in Figure 5A, rolipram significantly increased the intracellular cAMP levels at 30 min after the treatment. Other reagents also tended to increase its levels, although the increases were not statistically significant. Nevertheless, the cAMP levels largely tended to correlate with the extent of the inhibition of NET formation.
Next, the production of ROS was measured at 5 min after PMA stimulation by flow cytometry (Figure 5B). As shown in Figure 5C, most of cAMP‐increasing reagents decreased ROS production, as shown previously (Liu and Simon, 1996), and this effect was inversely correlated with NET formation in the neutrophils. Thus, cAMP could reduce ROS production, which in turn inhibited NET formation. Furthermore, reagents that increased cAMP, such as PGE2 and rolipram, inhibited neutrophil adhesion in vitro (Figure 5D).
Establishment of an assay evaluating NETosis in vivo
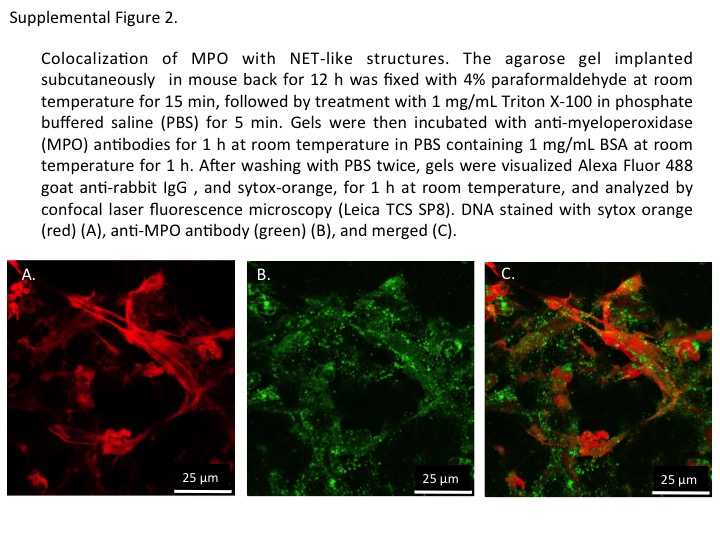
We next sought to clarify whether NETosis could be controlled by modulation of the cAMP system in vivo. For this purpose, we first established an in vivo NETosis assay. When an agarose gel (5 L × 5 W × 1 H mm) was implanted s.c. into the back of a mouse, cells migrated to it. The recruited cells stayed on the surface of the agarose gel and tended to form several clusters but barely entered the gel (Figure 6A). Almost all the cells were identified as neutrophils because they were stained by the mouse neutrophil‐specific marker, Ly6G (Stirling et al., 2009) (Figure 6C and D). The implanted gel was stained with cell‐permeable DNA stain Hoechst 33342, cell‐impermeable DNA indicator sytox orange and mitotracker, and the surface of the visceral side of the gel was observed by confocal laser microscopy (Figure 6E and F). The cells on the surface tended to form a few clusters. Most of the cells in some clusters did not undergo NETosis (Figure 6E), while those in other clusters exhibited NETosis exhibiting NET‐like structures that appeared pink when stained with both sytox orange and Hoechst 33342 (Figure 6F). The intensity of the fluorescence of the NET‐like structures was drastically diminished by incubation with DNase‐1 (Figure 6G). The gels were immunostained for MPO after fixation without detergent treatment. With this method, MPO became stained within sytox orange‐positive neutrophils that were considered to have a ruptured plasma membrane, but not in sytox orange‐negative neutrophils (Figure 6H). These results indicate that MPO in intact cells was not stained. By use of this method, MPO could be detected on the sytox orange‐stained NET‐like structures (Figure 6I–K), as shown previously (Kessenbrock et al., 2009). Supporting Information Figure 2 also clearly demonstrates the co‐localization of NET‐like structures and MPO. Thus, we concluded that this method could be used to evaluate NETosis in vivo.
Figure 6.
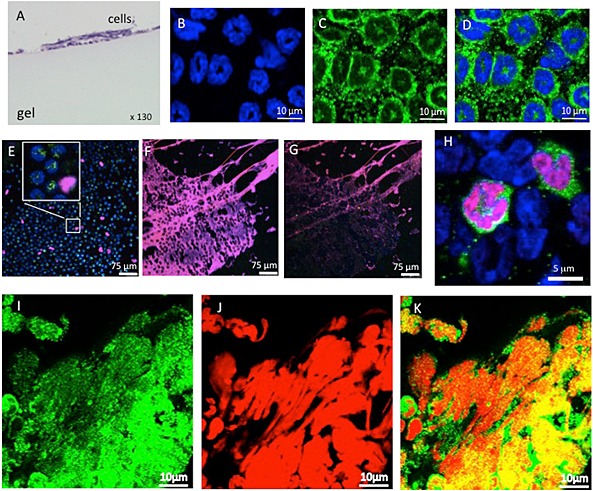
Establishment of an in vivo NETosis assay. An agarose gel was s.c. implanted in a C57BL/6J mouse, and cells on the gel surface were evaluated after 12 h. (A) Haematoxylin–eosin stain of a transverse section of the agarose gel. Data shown are representative of three independent experiments with a total of six mice. (B–D) The agarose gel was fixed with Triton X‐100‐containing buffer and stained with a cell‐permeable DNA dye, Hoechst 33342 (blue) (B), and an anti‐Ly6G antibody (green) (C) and then merged (D). Data shown are representative of two independent experiments with a total of five mice. (E, F) Non‐fixed live cells on the surface of the agarose gel were stained with Hoechst 33342 (blue), mitochondrial stain mitotracker green FM (green) and cell‐impermeable DNA stain sytox orange (red). Pink represents DNA stained with both Hoechst 33342 and sytox orange. The inset in (E) is a zoomed image. Data shown are representative of two independent experiments with a total of four mice. (G) Photo of the agarose gel shown in (F) after incubation with 200 U/mL DNase‐1 at 37°C for 1 h. Data shown are representative of two independent experiments with a total of four mice. (H) The gel was fixed without Triton X‐100 and stained with anti‐MPO antibody (green), cell‐impermeable sytox orange (red) and cell‐permeable Hoechst 33342 (blue). A merged photo of fields where cells did not undergo NETosis is shown and suggests that MPO in neutrophils with an intact plasma membrane was not stained. (I–K) Co‐localization of MPO with NET‐like structures. The agarose gel was fixed without Triton X‐100 and stained with anti‐MPO antibody (green) (I) and sytox orange (red) (J) and merged (K). Data shown in (H–K) are representative of three independent experiments with a total of seven mice.
A PDE4 inhibitor, rolipram, and an EP2 agonist, butaprost, inhibited NETosis in vivo
We quantified NETosis by measuring sytox orange‐stained areas in the central nine squares of a gel, as shown in Figure 7A. Typical photos are shown in Figure 7B. NETosis areas increased in a time‐dependent manner, and the PDE4 inhibitor, rolipram, significantly inhibited NETosis in vivo (Figure 7C). Rolipram did not affect the amounts of live cells associated with the gels at 8 h after implantation in mice, as deduced from the similar amounts of cytosolic protein RhoGDI (Boulter and Garcia‐Mata, 2010) and cytoskeletal protein β‐actin associated with the implanted gels in the presence and absence of rolipram (Figure 7D and E). Furthermore, the areas showing NETs were reduced on the EP2 agonist, butaprost‐containing gels, compared with those on the control gels (Figure 7F), while butaprost did not affect the amount of RhoGDI or β‐actin associated with the implanted gels (Figure 7G). We then examined the effects of rolipram and butaprost on the gel‐associated RhoGDI or β‐actin 4 h after implantation, when NETosis was barely visible on the gels. The amount RhoGDI or β‐actin was similar irrespective of the absence or presence of rolipram or butaprost (Supporting Information Figure 3), suggesting again that NETosis per se, but not recruitment and adhesion of neutrophils to the gels, was inhibited by a treatment that increased intracellular cAMP in the in vivo assay. Taken together, the data suggest that PGE2 inhibits NETosis by increasing intracellular cAMP both in vitro and in vivo.
Discussion
In this study, we demonstrated that PGE2 inhibits PMA‐induced NETosis in isolated neutrophils and that this effect is mediated through its Gαs‐coupled receptors, EP2 and EP4, resulting in increased intracellular cAMP. We subsequently demonstrated that PDE4 inhibitor, rolipram and EP2 agonist butaprost efficiently suppressed NETosis in a newly established in vivo assay.
The efficiency of NET formation in vitro appears to vary among different studies. One paper reported that NETosis was induced in approximately 40% of neutrophils by incubation with 50 nM PMA for 2 h (Brinkmann et al., 2013). This NETosis rate is more than twice that obtained in the present study (~15%) after a similar stimulus. However, their evaluation method differed from ours. Namely, they counted cells undergoing NETosis based on nuclear enlargement as detected by an image analyser, whereas we counted cells exhibiting apparent NET‐like formation by direct visual observation. Thus, the rate of NETosis could vary, depending on the methodology and the definition used. Nevertheless, in our study, approximately 15% of neutrophils stably exhibited NETosis after stimulation with 50 nM PMA for 90 min, as determined by visual observation: we used this method as our in vitro NETosis model throughout this study.
PGE2 is produced by various types of cells in inflammatory tissues and regulates many immune responses in a manner that is dependent on the particular disease: for example, PGE2 positively enhances the immune response in a cerebral infarction model (Ikeda‐Matsuo et al., 2006), while it suppresses the immune response in a colitis model (Sasaki et al., 2000). Negative regulation by PGE2 could, in part, be mediated by inhibition of NET formation by neutrophils, as shown here. Generally, the cellular functions of PGE2 are diverse because it can stimulate four types of receptor that are coupled to different G proteins, namely, Gαs, Gαi and Gαq.
In this study, we showed that selective EP2, as well as EP4, agonists comparably inhibited NETosis, while both EP2 and EP4 antagonists reversed the PGE2‐mediated inhibition of PMA‐induced NETosis to a similar extent, indicating that among the PGE2 receptors, EP2 and EP4 receptors play major roles in the regulation of neutrophil NETosis. Because both EP2 and EP4 receptors are coupled to Gαs, this suggests that cAMP is the second messenger downstream of PGE2 in neutrophils. Indeed, we showed that the cAMP levels in neutrophils after 30 min incubation with various cAMP‐inducing reagents were largely correlated with the inhibition of NET formation, which also supports a role for cAMP in the regulation of the formation of NET.
We subsequently demonstrated a critical role for cAMP in the inhibition of NETosis by showing that the cell‐permeable cAMP analogue, db2cAMP, as well as PDE inhibitors, inhibited NETosis. Because a PKA inhibitor reversed the PGE2‐induced inhibition of PMA‐induced NETosis, activation of PKA could be downstream of cAMP. Activation of PKA has been demonstrated to suppress an agonist‐induced increase in intracellular calcium ions (Anderson et al., 1998) and ROS production (Talpain et al., 1995), both of which are critical for inducing NETosis (Fuchs et al., 2007; Neeli and Radic, 2013). In this study, we also showed that the ROS levels in neutrophils after PMA stimulation in the presence of cAMP‐modulating reagents strongly correlated with the inhibition of NET formation, suggesting that the function of the cAMP–PKA pathway could, at least partly, be mediated by the regulation of ROS production. This is consistent with results from a recent study showing that Bordetella pertussis adenylate cyclase toxin inhibits NETosis through the production of cAMP and reduction of ROS (Eby et al., 2014). Thus, irrespective of intrinsic and extrinsic factors, an increase in cAMP in neutrophils could inhibit NETosis.
It is difficult to evaluate NETosis quantitatively in vivo, and only a few assays have been used: for example, NETosis has been qualitatively observed by intravital microscopy and quantified by measuring circulating DNA–MPO complexes in a ventilation‐induced lung injury model (Rossaint et al., 2014). In a transfusion‐related acute lung injury model, DNA–MPO in plasma was also used as a measure of NETosis (Caudrillier et al., 2012). However, DNA–MPO in plasma may not directly indicate the amount of NETosis. More recently, neutrophils undergoing NETosis have been directly observed in capillaries of liver sinusoids (McDonald et al., 2012; Tanaka et al., 2014), postcapillary venules of the caecum (Tanaka et al., 2014) and pulmonary capillaries (Tanaka et al., 2014) in an LPS‐induced sepsis model. However, the intensities of the inflammation observed in these models varied, and the quantification of cells undergoing NETosis was difficult. In contrast, the assay established here is very simple and easy to perform. We found that cells did not enter the gel, and those detected on the surface of the gel at 12 h were exclusively neutrophils. We clearly demonstrated that NETosis effectively occurred on the surface of the gel by showing (i) NET‐like structures stained with the cell‐impermeable DNA stain sytox orange; (ii) degradation of the NET‐like structure by DNase‐1; and (iii) co‐localization of MPO with the NET‐like structures. This in vivo NETosis assay could be adapted as a useful tool for in vivo pharmacological evaluations by possible inducers and inhibitors of NET formation, as shown, for example, for rolipram and butaprost in this study.
In the development of our model, we reasoned that because neutrophils try to wrap large foreign bodies with decondensed chromatin released by NETosis (Branzk et al., 2014), an agarose gel would be similarly recognized as a huge foreign body by neutrophils, and NETosis would thus occur on its surface. Interestingly, NETs were observed to form in clusters but did not diffusely cover the whole surface of the gel. Furthermore, most of the neutrophils that accumulated on the gels underwent NETosis in some cluster areas, while almost no cells undergoing NETosis were observed in other cluster areas, suggesting the NETs recruited and induced the NETosis of other neighbouring neutrophils. Possible factors that recruit and/or induce NETosis may be present in NETs and would be worthy of future investigations.
Among the several PDEs identified so far, it has been demonstrated that PDE4 plays a major role in neutrophil inflammation and apoptosis (Sousa et al., 2010). Therefore, it was not surprising that the PDE4 inhibitor, rolipram, had more marked effects on NETosis than a PDE3 inhibitor, cilostazol. Using the in vivo NETosis assay established here, we examined the effect of increased cAMP on NETosis with a rolipram‐containing gel. The extent of NETs, evaluated as sytox orange‐positive areas, on the rolipram‐containing gel surface was found to be approximately half of those of the control (Figure 7C). The inhibition of NET formation by rolipram was unlikely to be due to the suppression of the recruitment/adhesion of neutrophils to the gels because similar amounts of a cytosolic protein, RhoGDI, and also a cytoskeletal protein, β‐actin, were detected on the gels irrespective of the absence or presence of rolipram (Figure 7D and E, and Supporting Information Figure 3). These data differed from those obtained in the in vitro experiments, which showed that rolipram inhibited isolated neutrophil adhesion (Figure 5D). The precise reason for this variation is unclear, but it could be due to the specific experimental conditions used.
In addition to rolipram, we showed that an EP2 agonist inhibited NETosis in vivo (Figure 7F and G, and Supporting Information Figure 3). Thus, these data strongly suggest that intracellular cAMP inhibits NET formation in vivo. We also tried to evaluate the effect of a EP4 agonist in vivo: preliminary experiments showed that it did not significantly inhibit NET formation (data not shown), although it inhibited NET formation in vitro (Figure 2D). The precise reasons for this discrepancy remain unknown. However, it might possibly be due to the lack of an effective release of EP4 agonist from gels, or due to species differences, because human neutrophils were used in the in vitro assay (Figure 2D) while mice were used in the in vivo experiments.
Because some PDE inhibitors, such as cilostazol, are currently used in a clinical setting, our results suggest that PDE inhibitors, especially PDE4 inhibitors, could be useful for NETosis‐related diseases such as systemic lupus erythaematosus (Hakkim et al., 2010; Knight et al., 2013).
In summary, in the present study we have demonstrated, using in vitro assays and a newly developed in vivo model of NETosis, that PGE2, a critical regulator of inflammation, inhibits NETosis by activation of the cAMP–PKA pathway through the activation of its Gαs‐coupled receptors, EP2 and EP4. Elucidation of the signalling pathways of NETosis, including the data presented here, could contribute to the development of novel treatment strategies for NETosis‐related diseases such as systemic lupus erythaematosus.
Author contributions
H. H. was responsible for the conception and design of the experiments. K. S. performed most of experiments with the support of R. S. and T. K.. T. H. and N. S. performed experiments for Figure 5. D‐A. T. contributed to the in vivo experiments for Figure 7. Y. A. performed the pathological analysis of the in vivo experiments. K. S., R. S., T. K. and H. H. analysed and discussed the data. K. S. and H. H. drafted the paper, and R. S. and T. K. commented on the manuscript. All authors approved the final version of the manuscript.
Conflict of interest
None.
Supporting information
Figure S1 Isolated neutrophils were incubated with each indicated reagent at 37°C for 75 min in the presence of cell‐impermeable DNA staining agent, sytox orange. Then, sytox orange‐stained cells were quantified in more than 300 cells evaluated. The data shown are mean ± SEM in five independent experiments.
Figure S2 Co‐localization of myeloperoxidase (MPO) with NET‐like structures. The agarose gel implanted s.c. in the mouse back for 12 h was fixed with 4% paraformaldehyde at room temperature for 15 min, followed by treatment with 1 mg⋅mL−1 Triton X‐100 in PBS for 5 min. Gels were then incubated with anti‐MPO antibodies for 1 h at room temperature in PBS containing 1 mg⋅mL−1 BSA. After being washed with PBS twice, gels were visualized using Alexa Fluor 488 goat anti‐rabbit lgG, and sytox orange, for 1 h at room temperature, and analysed by confocal laser fluorescence microscopy (Leica TCS SP8). DNA stained with sytox orange (red) (A), anti‐MPO antibody (green) (B) and merged (C).
Figure S3 Gels, s.c. implanted, containing vehicle, rolipram and the EP2 agonist butaprost were collected after 4 h (n = 6 in each group), and the gel‐associated intracellular proteins, RhoGDI and β‐actin, were analysed by immunoblotting. The data shown are mean ± SEM.
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Acknowledgements
This work was in part supported by a JSPS KAKENHI grant (number 25670136). It was also supported by the Good Practice (GP) Program, ‘Nurturing MD researchers competitive in the world arena, Tohoku University School of Medicine’ by the Ministry of Education, Culture, Sports, Science, and Technology, Japan to K. S., T. H. and N. S. We thank Drs. Satoshi Yamada and Hiroshi Nakase (The Department of Gastroenterology and Hepatology, Graduate School of medicine, Kyoto University, Kyoto, Japan) and Dr. Hidenori Arai (The National Center for Geriatrics and Gerontology, Obu, Japan) for valuable suggestions for statistical analyses.
Shishikura, K. , Horiuchi, T. , Sakata, N. , Trinh, D.‐A. , Shirakawa, R. , Kimura, T. , Asada, Y. , and Horiuchi, H. (2016) Prostaglandin E2 inhibits neutrophil extracellular trap formation through production of cyclic AMP. British Journal of Pharmacology, 173: 319–331. doi: 10.1111/bph.13373.
References
- af Forselles KJ, Root J, Clarke T, Davey D, Aughton K, Dack K, et al (2011). In vitro and in vivo characterization of PF‐04418948, a novel, potent and selective prostaglandin EP(2) receptor antagonist. Br J Pharmacol 164: 1847–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R, Goolam Mahomed A, Theron AJ, Ramafi G, Feldman C (1998). Effect of rolipram and dibutyryl cyclic AMP on resequestration of cytosolic calcium in FMLP‐activated human neutrophils. Br J Pharmacol 124: 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter E, Garcia‐Mata R (2010). RhoGDI: a rheostat for the Rho switch. Small GTPases 1: 65–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al (2014). Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol 15: 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V, Zychlinsky A (2012). Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol 198: 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al (2004). Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Goosmann C, Kühn LI, Zychlinsky A (2013). Automatic quantification of in vitro NET formation. Front Immunol 3: 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al (2012). Platelets induce neutrophil extracellular traps in transfusion‐related acute lung injury. J Clin Invest 122: 2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherukuri DP, Chen XB, Goulet AC, Young RN, Han Y, Heimark RL, et al (2007). The EP4 receptor antagonist, L‐161,982, blocks prostaglandin E2‐induced signal transduction and cell proliferation in HCA‐7 colon cancer cells. Exp Cell Res 313: 2969–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, et al (1990). Inhibition of forskolin‐induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP‐dependent protein kinase, N‐[2‐(p‐bromocinnamylamino)ethyl]‐5‐isoquinolinesulfonamide (H‐89), of PC12D pheochromocytoma cells. J Biol Chem 265: 5267–5272. [PubMed] [Google Scholar]
- Cools‐Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B et al (2013). Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest 123: 3446–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eby JC, Gray MC, Hewlett EL (2014). Cyclic AMP‐mediated suppression of neutrophil extracellular trap formation and apoptosis by the Bordetella pertussis adenylate cyclase toxin. Infect Immun 82: 5256–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al (2007). Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176: 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al (2010). Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 107: 15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CD (2001). Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294: 1871–1875. [DOI] [PubMed] [Google Scholar]
- Guimaraes‐Costa AB, Nascimento MT, Froment GS, Soares RP, Morgado FN, Conceicao‐Silva F, et al (2009). Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc Natl Acad Sci U S A 106: 6748–6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al (2010). Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A 107: 9813–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda‐Matsuo Y, Ota A, Fukada T, Uematsu S, Akira S, Sasaki Y (2006). Microsomal prostaglandin E synthase‐1 is a critical factor of stroke‐reperfusion injury. Proc Natl Acad Sci U S A 103: 11790–11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al (2009). Netting neutrophils in autoimmune small‐vessel vasculitis. Nat Med 15: 623–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandpur R, Carmona‐Rivera C, Vivekanandan‐Giri A, Gizinski A, Yalavarthi S, Knight JS, et al (2013). NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 5 178ra140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW . (2010). Animal research: reporting in vivo experiments: the ARRIVE Guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight JS, Zhao W, Luo W, Subramanian V, O'Dell AA, Yalavarthi S, et al (2013). Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest 123: 2981–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y (2010). PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 207: 1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Simon MI (1996). Regulation by cAMP‐dependent protein kinease of a G‐protein‐mediated phospholipase C. Nature 382: 83–87. [DOI] [PubMed] [Google Scholar]
- McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P (2012). Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 12: 324–333. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL (2010). Guidelines for reporting experiments involving animals: the ARRIVE Guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeli I, Radic M (2013). Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front Immunol 4: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka H, Horiuchi H, Arai H, Kita T (1998). Lysophosphatidylcholine generates superoxide anions through activation of phosphatidylinositol 3‐kinase in human neutrophils. FEBS Lett 441: 63–66. [DOI] [PubMed] [Google Scholar]
- Orie NN, Clapp LH (2011). Role of prostanoid IP and EP receptors in mediating vasorelaxant responses to PGI2 analogues in rat tail artery: evidence for Gi/o modulation via EP3 receptors. Eur J Pharmacol 654: 258–265. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucleic Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossaint J, Herter JM, van Aken H, Napirei M, Doring Y, Weber C, et al (2014). Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap‐mediated sterile inflammation. Blood 123: 2573–2584. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Hirata I, Maemura K, Hamamoto N, Murano M, Toshina K, et al (2000). Prostaglandin E2 inhibits lesion formation in dextran sodium sulphate‐induced colitis in rats and reduces the levels of mucosal inflammatory cytokines. Scand J Immunol 51: 23–28. [DOI] [PubMed] [Google Scholar]
- Sousa LP, Lopes F, Silva DM, Tavares LP, Vieira AT, Rezende BM, et al (2010). PDE4 inhibition drives resolution of neutrophilic inflammation by inducing apoptosis in a PKA‐PI3K/Akt‐dependent and NF‐kappaB‐independent manner. J Leukoc Biol 87: 895–904. [DOI] [PubMed] [Google Scholar]
- Stirling DP, Liu S, Kubes P, Yong VW (2009). Depletion of Ly6G/Gr‐1 leukocytes after spinal cord injury in mice alters wound healing and worsens neurological outcome. J Neurosci 29: 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo T, Tachibana K, Toga K, Tochizawa S, Inoue Y, Kimura Y, et al (2000). Potent effects of novel anti‐platelet aggregatory cilostamide analogues on recombinant cyclic nucleotide phosphodiesterase isozyme activity. Biochem Pharmacol 59: 347–356. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Narumiya S (2007). Prostaglandin E receptors. J Biol Chem 282: 11613–11617. [DOI] [PubMed] [Google Scholar]
- Talpain E, Armstrong RA, Coleman RA, Vardey CJ (1995). Characterization of the PGE receptor subtype mediating inhibition of superoxide production in human neutrophils. Br J Pharmacol 114: 1459–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Shibuya I, Kabashima N, Ueta Y, Yamashita H (1998). Inhibition of voltage‐dependent calcium channels by prostaglandin E2 in rat melanotrophs. Endocrinology 139: 4801–4810. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Koike Y, Shimura T, Okigami M, Ide S, Toiyama Y, et al (2014). In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PLoS One 9 e111888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Lu X, Peng K, Du Y, Zhou SF, Zhang A, et al (2014). Prostaglandin E‐prostanoid4 receptor mediates angiotensin II‐induced (pro)renin receptor expression in the rat renal medulla. Hypertension 64: 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al (2009). Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 184: 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Isolated neutrophils were incubated with each indicated reagent at 37°C for 75 min in the presence of cell‐impermeable DNA staining agent, sytox orange. Then, sytox orange‐stained cells were quantified in more than 300 cells evaluated. The data shown are mean ± SEM in five independent experiments.
Figure S2 Co‐localization of myeloperoxidase (MPO) with NET‐like structures. The agarose gel implanted s.c. in the mouse back for 12 h was fixed with 4% paraformaldehyde at room temperature for 15 min, followed by treatment with 1 mg⋅mL−1 Triton X‐100 in PBS for 5 min. Gels were then incubated with anti‐MPO antibodies for 1 h at room temperature in PBS containing 1 mg⋅mL−1 BSA. After being washed with PBS twice, gels were visualized using Alexa Fluor 488 goat anti‐rabbit lgG, and sytox orange, for 1 h at room temperature, and analysed by confocal laser fluorescence microscopy (Leica TCS SP8). DNA stained with sytox orange (red) (A), anti‐MPO antibody (green) (B) and merged (C).
Figure S3 Gels, s.c. implanted, containing vehicle, rolipram and the EP2 agonist butaprost were collected after 4 h (n = 6 in each group), and the gel‐associated intracellular proteins, RhoGDI and β‐actin, were analysed by immunoblotting. The data shown are mean ± SEM.
Supporting info item
Supporting info item
Supporting info item