

Abstract
Background and Purpose
Although botulinum toxin type A (BoNT/A) is approved for chronic migraine treatment, its mechanism of action is still unknown. Dural neurogenic inflammation (DNI) commonly used to investigate migraine pathophysiology can be evoked by trigeminal pain. Here, we investigated the reactivity of cranial dura to trigeminal pain and the mechanism of BoNT/A action on DNI.
Experimental Approach
Because temporomandibular disorders are highly comorbid with migraine, we employed a rat model of inflammation induced by complete Freund's adjuvant, followed by treatment with BoNT/A injections or sumatriptan p.o. DNI was assessed by Evans blue‐plasma protein extravasation, cell histology and RIA for CGRP. BoNT/A enzymatic activity in dura was assessed by immunohistochemistry for cleaved synaptosomal‐associated protein 25 (SNAP‐25).
Key Results
BoNT/A and sumatriptan reduced the mechanical allodynia and DNI, evoked by complete Freund's adjuvant. BoNT/A prevented inflammatory cell infiltration and inhibited the increase of CGRP levels in dura. After peripheral application, BoNT/A‐cleaved SNAP‐25 colocalized with CGRP in intracranial dural nerve endings. Injection of the axonal transport blocker colchicine into the trigeminal ganglion prevented the formation of cleaved SNAP‐25 in dura.
Conclusions and Implications
Pericranially injected BoNT/A was taken up by local sensory nerve endings, axonally transported to the trigeminal ganglion and transcytosed to dural afferents. Colocalization of cleaved SNAP‐25 and the migraine mediator CGRP in dura suggests that BoNT/A may prevent DNI by suppressing transmission by CGRP. This might explain the effects of BoNT/A in temporomandibular joint inflammation and in migraine and some other headaches.
Abbreviations
- BoNT/A
botulinum toxin type A
- CFA
complete Freund's adjuvant
- DNI
dural neurogenic inflammation
- i.a.
intra‐articular
- i.g.
intraganglionic
- SNAP‐25
synaptosomal‐associated protein 25
- TMJ
temporomandibular joint
Tables of Links
| TARGETS |
|---|
| GPCRs |
| CGRP receptor |
| LIGANDS |
|---|
| CGRP |
| Colchicine |
| Sumatriptan |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Botulinum toxin type A (BoNT/A) blocks the vesicular release of neurotransmitters by proteolytic cleavage of a synaptic protein, synaptosomal‐associated protein 25 (SNAP‐25). SNAP‐25 is a part of the synaptic protein complex which is involved in Ca2+‐dependent exocytosis (Kalandakanond and Coffield, 2001; Blasi et al., 1993). This effect of BoNT/A at peripheral nerve endings is the basis of its therapeutic use in a range of neuromuscular (blepharospasm, focal dystonia and spasticity) and autonomic disorders (hyperhidrosis and bladder dysfunction) associated with neuronal over‐activity (Dressler, 2013). Based on large clinical studies, pericranially injected BoNT/A has also been approved for the treatment of chronic migraine (Diener et al., 2010). It is widely accepted that migraine headaches involve activation of trigeminal afferents innervating the meningeal blood vessels and dural neurogenic inflammation (DNI) (Moskowitz, 1990; Geppetti et al., 2012; Ramachandran and Yaksh, 2014). We have recently found that the activation of dural afferents, measured as plasma protein extravasation, can be evoked by extracranial pain in the trigeminal region (orofacial formalin‐evoked pain and infraorbital nerve constriction‐induced trigeminal neuropathy) (Filipović et al., 2012, 2014). The plasma protein extravasation induced by different types of pain was prevented by peripherally injected BoNT/A. The effect of BoNT/A in the cranial dura was associated with axonal transport of the toxin, because its effects were prevented by injection of colchicine directly into the trigeminal ganglion (Filipović et al., 2012).
In the present study, we investigated the effects of BoNT/A in a model of trigeminal pain induced by complete Freund's adjuvant (CFA) injection into the temporomandibular joint (TMJ), a common model of temporomandibular disorders (Harper et al., 2001; Villa et al., 2010). Temporomandibular disorders involve dysfunction of both the TMJ and masticatory muscles, leading to chronic pain (De Rossi et al., 2014). BoNT/A injections into masticatory muscles have been reported to reduce the tenderness and pain in patients suffering from temporomandibular disorders (Sunil Dutt et al., 2015). Severe forms of temporomandibular disorders are highly comorbid with primary headaches – up to 86% of patients suffer from migraine or other primary headaches (Bevilaqua Grossi et al., 2009; Franco et al., 2010). The underlying mechanism of the comorbidity is proposed to be related to extensive innervation of cranial dura by mandibular branch of trigeminal nerve (Schueler et al., 2013). So far, inflammation of the TMJ has been used preclinically to study the trigeminal sensitization associated with migraine (Villa et al., 2010; Thalakoti et al., 2007). CFA injection into the TMJ induces pain and inflammation leading to peripheral and central sensitization of trigeminal system (Villa et al., 2010). Similarly, by stimulating the TMJ with capsaicin, Thalakoti et al. (2007 found widespread peripheral sensitization in trigeminal ganglion cells. Accordingly, we hypothesized that TMJ pain might provide a suitable model to study trigeminal activation leading to DNI, as well as the mechanism of BoNT/A action in the trigeminovascular system, assumed to be involved in migraine and other headaches. In the TMJ inflammation model, apart from neurogenic plasma protein extravasation, we studied the effect of BoNT/A on CGRP, a neuropeptide considered the main mediator of trigeminal sensitization in migraine (Bigal et al., 2013).
Here, we have found that CFA‐evoked TMJ inflammation was accompanied by inflammatory changes in the cranial dura (plasma protein extravasation and inflammatory cell infiltration) and increased levels of CGRP. Additionally, following peripheral toxin injection, cleaved SNAP‐25, the product of BoNT/A enzymic activity, was colocalized with CGRP‐expressing dural afferents. BoNT/A prevented the CFA‐evoked dural inflammation and CGRP peptide increase in cranial dura.
Methods
Animal welfare and ethical statement
All animal care and experimental procedures were in accordance with the 2010/63/EU Directive on the protection of animals used for scientific purposes and the recommendations of International Association for the Study of Pain (Zimmerman, 1983) and were approved by the Ethical Committee of University of Zagreb School of Medicine (permit no. 07–76/2005–43). The experimental procedures used in the work described in this article were as humane as possible. All animal studies are described in compliance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010).
One hundred and five male Wistar rats (average weight 300–350 g; 3–3.5 months old; University of Zagreb School of Medicine, Croatia) were used in these experiments. Rats were kept under a constant 12 h/12 h light/dark cycle with free access to food and water. The animals were randomly allocated to different experimental treatments. The experimenter conducting the behavioural testing was unaware of the tretaments given to the animals.
CFA‐induced inflammatory pain in the TMJ
Animals were anesthetized with chloral hydrate (300 mg kg−1 i.p.). Injection into the TMJ was performed by inserting a 27 gauge needle medially through the skin below the inferior border of the zygomatic arch and superior to the mandibular condyl until it entered the joint capsule (Villa et al., 2010). Inflammation of the TMJ was elicited by injection of 50 μL of CFA into the left joint capsule. Control rats were injected intra‐articularly (i.a.) with saline (0.9% NaCl). Methylene blue was injected into a few animals, and the site of injection was examined in a preliminary experiment to confirm successful targeting of the TMJ.
Behavioural testing
The next day (24 h) following CFA injection, behavioural assessment of mechanical allodynia was performed using the von Frey monofilaments (Stoelting Co., Wood Dale, IL, USA) as previously described in detail (Filipović et al., 2012). Filaments produced a calibrated bending force of 0.16, 0.4, 0.6, 1, 2, 4, 6, 8 and 10. The rats were placed in a transparent plastic cage for 10 min to accommodate to the experimental environment until they assumed their normal sniffing/no locomotion position. For each session, a series of von Frey filaments were applied on the tested side of the face in ascending order, starting at 0.16 g, with three attempts until a defined behavioural response was elicited. Each time, the measurement started on the side contralateral to the CFA injection. A positive reaction was interpreted as defensive forepaw movement and/or escape/attack reaction after stimulation of whisker pad area with filaments. In total, the measurements were performed in three sessions in 10 min intervals. If no response was observed, we assigned 10 g as the withdrawal threshold, because the force exerted by thicker filaments (>10 g) was large enough to push the head of animals.
Pharmacological treatments
BoNT/A injections
The rats were anesthetized with chloral hydrate (300 mg kg−1 i.p.) (first anaesthetic) for different BoNT/A treatments, 3 days prior to the CFA (50 μL) injection in the left TMJ (performed under the second anaesthetic). For intra‐articular injections, BoNT/A in a dose of 5 U kg−1 and 20 μL volume was injected into the left TMJ capsule of anesthetized rats, as described earlier. For intraganglionic injections,: rats were injected with BoNT/A in a dose of 2 U kg−1 (2 μL volume) into the left trigeminal ganglion of anesthetized rats, as described previously (Matak et al., 2011; Filipović et al., 2012). In brief, a Hamilton syringe needle (Hamilton Microliter #701; Hamilton, Bonaduz, Switzerland) was inserted through the skin into the infraorbital foramen and advanced through the infraorbital canal and foramen rotundum into the trigeminal ganglion.
The multiple facial injections we made as follows: anesthetized rats were injected with BoNT/A at four sites: (i) bilaterally into the rat forehead above the orbital arch and (ii) bilaterally into the whisker pad. Five microlitre Injections (5μLper site) were administered using a Hamilton syringe. A total dose of 5 U kg−1 was employed and divided in four equal doses (1.25 U kg−1 per site).
Sumatriptan
A group of animals were given sumatriptan, p.o., 24 h after CFA injection into the TMJ. The p.o. dose of 175 μg kg−1 was calculated on the basis of previously used i.v. dose (50 μg kg−1) corrected for p.o. bioavailability (25–30%) in rats (Dallas et al., 1989; Schuh‐Hofer et al., 2003). Mechanical allodynia was measured, as described above, 2 h after the administration of sumatriptan.
Dural neurogenic plasma protein extravasation
Plasma protein extravasation, as an indicator of neurogenic inflammation, was measured 24 h after CFA injection. This was measured by injecting Evans blue dye which complexes to plasma proteins. Anaesthetized animals were perfused transcardially with 500 mL of saline 30 min after injection of 1 mL Evans blue solution (40 mg kg−1) into the tail vein. Supratentorial dura was dissected into the left (ipsilateral to CFA treatment) and right sides (contralateral to CFA) and weighed. Evans blue was extracted in formamide, and the absorbance of Evans blue was measured spectrophotometrically. The amounts of extravasated Evans blue were calculated using the standard concentration curve, as previously described in detail (Filipović et al., 2012).
RIA for CGRP
For the measurement of CGRP immunoreactivity with RIA, animals were injected with BoNT/A into the TMJ, as described above. One day after the induction of TMJ inflammation, animals were deeply anesthetized with chloral hydrate (300 mg kg−1 i.p.). Approximately 100 μL of CSF was withdrawn from cisterna magna using 27½ gauge syringe needle inserted percutaneously between the occipital bone and atlas. Only transparent CSF samples were taken for further analysis. The sample was rapidly frozen by immersing the sealed Eppendorf tube containing the CSF in liquid nitrogen and kept at −80°C. Immediately following the CSF sampling, anesthetized animals were killed by decapitation. Supratentorial dura, brainstem and trigeminal ganglion were quickly dissected, frozen in liquid nitrogen and kept at −80°C until further use. The frozen brainstem was placed in cryostat‐cooled environment (−25°C) for dissection of ipsilateral trigeminal nucleus caudalis without thawing. The nucleus was excised manually using a pre‐cooled microtome blade, scalpel and forceps. Dissected tissue was further kept at −80°C until homogenization.
Tissue samples were weighed and immediately homogenized with 1 mL distilled water and 20 μL of aprotinin solution (Trasylol, Bayer, Germany). Trigeminal ganglia and caudal nucleus samples were manually homogenized in a glass homogeniser, while dura was homogenized using a Polytron mechanical homogenizer. The samples were then centrifuged for 10 min at 8944 g, and the procedure was repeated with the resulting supernatant. Final supernatants were kept at −30°C until further analysis. CSF was directly used as a RIA sample without further preparation.
Radioimmunoassay was performed similarly as previously described (Németh et al., 1998; Pozsgai et al., 2012). In brief, samples or CGRP standards (Sigma) were diluted in buffer for RIA containing 1:120 000 anti‐CGRP polyclonal antibody (Sigma) and tracer containing radio‐iodinated CGRP standard. Diluted samples were incubated at 4°C for 48 h. Antigen‐bound and free CGRP peptides were then separated by adding 100 μL of distilled water with 10% activated charcoal, 2% dextran and 0.2% fat‐free milk powder. The samples were vortexed and centrifuged at 2010 g for 20 min. Levels of radioactivity of the pellets containing the free peptide and supernatant containing the antibody‐bound peptide were determined with a γ counter. Concentrations of CGRP (fmol mg−1 or fmol mL−1) in samples were calculated based on a standard concentration curve.
Histology and immunohistochemistry of the dura mater
In order to assess inflammatory cell infiltration in the dura mater by histology, animals were injected with BoNT/A (5 U kg−1) and CFA into the TMJ as described above. One day after CFA, the anaesthetized animals were perfused with saline and 250 mL of 4% paraformaldehyde in PBS. Ipsilateral and contralateral supratentorial dura were carefully dissected and placed in paraformaldehyde fixative containing 15% sucrose, followed by 30% sucrose in PBS on the next day. After 48 h, the samples were stored at −80°C until further use.
Histological study of the cranial dural tissue was performed using standard Giemsa staining. Bright field microphotographs were taken with Olympus BX‐51 microscope coupled with DP‐70 digital camera (Olympus, Tokyo, Japan) under constant condenser light intensity and camera exposition. The number of Giemsa‐stained cell profiles was automatically quantified in four to five non‐overlapping visual fields (obtained at 20× magnification) per single animal, using cellSens Dimension programme (Olympus) as previously described in detail (Filipović et al., 2014). Five animals per group were examined.
To investigate the possible spread of peripherally injected BoNT/A to dural afferents, animals were injected in the TMJ unilaterally with 5 or 15 U kg−1 BoNT/A, as described above. One group of animals was injected with 15 U kg−1 BoNT/A into the whisker pad. An additional group of animals was injected unilaterally with a total dose of 20 U kg−1 BoNT/A (7 U per 350 g rat) divided in four injection sites (1.75 U/20 μL per site) – (i) TMJ, (ii) whisker pad, (iii) medial (forehead) and (iv) lateral (temporal) cranial region. Six days after peripheral injection of BoNT/A, animals were anesthetized and perfused for immunohistochemistry with saline and paraformaldehyde fixative.
Dural samples were stained for cleaved SNAP‐25 using the free‐floating procedure as previously described (Matak et al., 2014). In brief, dissected dura was washed in PBS, blocked with 10% normal goat serum and incubated overnight at room temperature with 1:1600 anti‐BoNT/A‐cleaved SNAP‐25 antibody (provided by Ornella Rossetto, University of Padua, Italy) in PBS containing 1% goat serum. The antibody binds specifically to BoNT/A‐cleaved SNAP‐25 and not the intact SNAP‐25 (Matak et al., 2011). Next day, the samples were incubated with Alexa Fluor 555 anti‐rabbit secondary antibody. Stained dura was carefully spread on the glass slides and cover‐slipped with an anti‐fading agent. In animals injected at four different sites or only into the TMJ (5 U kg−1), additional labelling with rabbit anti‐CGRP antibody (1:5000, Sigma) was performed. In order to prevent a possible cross‐reactivity of cleaved SNAP‐25 with CGRP, a modified primary antibody elution procedure with pre‐heated acidic buffer (50°C, pH = 2, 25 mM glycine and 1% SDS) was performed, as described previously in detail (Matak et al., 2014). After the elution, the dural samples were stained with anti‐CGRP and Alexa Fluor 488 secondary antibody. The appearance of cleaved SNAP‐25 Alexa Fluor 555 stained fibre profiles, observed before and after antibody elution, was unchanged. Cross‐reactivity controls (omitted CGRP antibody) showed no Alexa Fluor 488 signal in association with cleaved SNAP‐25 fibers, as reported previously (Matak et al., 2014).
Investigation of the effect of the axonal transport inhibitor, colchicine, on antinociceptive activity and appearance of cleaved SNAP‐25 in dura mater following BoNT/A injection
By blocking the axonal transport within the trigeminal ganglion, we examined the involvement of the axonal traffic via the trigeminal nerve of BoNT/A for its antinociceptive activity and for the presence of cleaved SNAP‐25 in the dura mater. Anesthetized animals were injected in the TMJ with saline or BoNT/A (5 U kg−1). Immediately after TMJ injection, the animals were injected with 2 μL of saline or an equal volume of the axonal transport blocker colchicine (5 mM) into the trigeminal ganglion, percutaneously via the infraorbital canal as previously described (Filipović et al., 2012). Seven days after i.a. and i.g. treatments, the animals were treated with CFA, and the mechanical allodynia was measured after 24 h, as described above. Then, the animals were anaesthetized and perfused with saline and fixative, and the dural tissue was processed and stained for immunohistochemistry of BoNT/A‐cleaved SNAP‐25 as described above.
Data analysis
Results are presented as means ± SEM and analysed by one‐way ANOVA followed by the Newman–Keuls post hoc test. P < 0.05 was considered significant.
Materials
The suppliers of the reagents used were as follows: Evans blue (Merck KGaA, Darmstadt, Germany) reconstituted in 0.9% saline to obtain the required dose (40 mg kg−1); CFA cell suspension (Sigma, St. Louis, MO, USA); colchicine (Sigma, St. Louis, MO, USA); sumatriptan (Glaxo Wellcome, Taplow, UK) reconstituted in drinking water; and BoNT/A diluted in 0.9% saline (Botox®; Allergan Inc., Irvine, CA, USA). One unit (1 U) of BoNT/A preparation contains 48 pg of purified Clostridium botulinum neurotoxin type A complex.
Results
CFA‐evoked bilateral allodynia is reduced by i.a. and i.g. BoNT/A, and oral sumatriptan
Animals treated with CFA injected into the TMJ developed mechanical allodynia 24 h after the injection. Allodynia appeared bilaterally. Pre‐treatment with BoNT/A injected ipsilaterally to the CFA injection [both i.a. (5 U kg−1) and i.g. (2 U kg−1)] 3 days before CFA, reduced the mechanical allodynia bilaterally (P < 0.001). Similarly, 2 h after administration of sumatriptan (175 μg kg−1, p.o.), the mechanical allodynia was reduced bilaterally (P < 0.001). The differences between the BoNTA and sumatriptan treatments were not significant (Figure 1).
Figure 1.
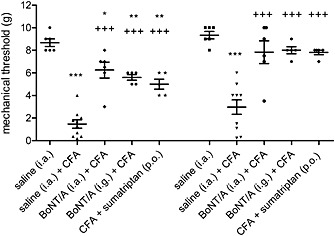
BoNT/A and sumatriptan effects on bilateral allodynia induced by unilateral TMJ inflammation. BoNT/A (5 U kg−1) was injected into the TMJ (5 U kg−1 i.a.) or trigeminal ganglion (2 U kg−1 i.g.) 3 days before CFA. Facial allodynia was measured with von Frey filaments 24 h after CFA injection into the TMJ. Sumatriptan (175 mg kg−1) was administered p.o. 24 h after CFA, and allodynia was measured 2 h after sumatriptan. Scatter plot represents data of individual animals, and horizontal lines and bars indicate mean ± SEM. n (animals per group) = 5–9. *P < 0.05, ** P < 0.01, ***P < 0.001, significantly different from saline control; +++ P < 0.001, significantly different from saline + CFA; one‐way ANOVA followed by Newman–Keuls post hoc test.
BoNT/A and sumatriptan reduce plasma protein extravasation in dura mater
Dural plasma protein extravasation was significantly increased bilaterally in CFA‐injected animals compared with control values (Figures 2, 3, 4). Plasma protein extravasation in the ipsilateral dura was double that on the contralateral side (P < 0.001, t‐test for dependent samples). BoNT/A injected both i.a. (5 U kg−1) and i.g. (2 U kg−1), as well as sumatriptan (175 μg kg−1 p.o.), reduced the ipsilateral dural plasma protein extravasation (Figure 2). In the contralateral side, none of the treatments affected the DNI.
Figure 2.
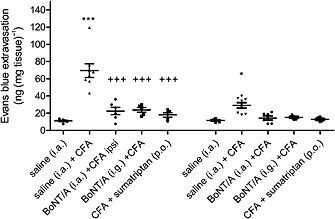
The effect of BoNT/A and sumatriptan on Evans blue/plasma protein extravasation in dura mater after TMJ inflammation. BoNT/A was injected into the TMJ (5 U kg−1 i.a.) or trigeminal ganglion (2 U kg−1 i.g.) 3 days before CFA. Sumatriptan (175 mg kg−1) was administered p.o. 24 h after CFA. Four days following BoNT/A or 2 h after sumatriptan rats were injected with Evans blue (i.v., 40 mg kg−1) and perfused with saline. Dura was collected for formamide extraction and spectrophotometric measurement of Evans blue dye which extravasates in complex with plasma proteins. Scatter plot represents data from individual animals, and horizontal lines and bars indicate mean ± SEM. n (animals per group) = 5–9. *P < 0.05, ***P < 0.001, significantly different from saline control; ++ P < 0.01; +++ P < 0.001, significantly different from saline + CFA; one‐way ANOVA followed by Newman–Keuls post hoc test.
Figure 3.
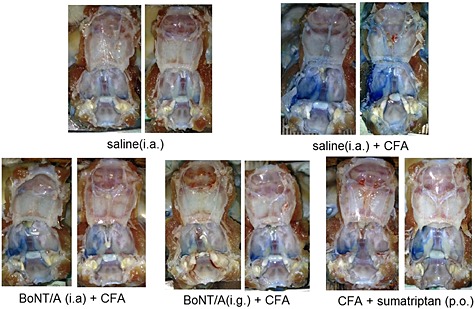
Neurogenic plasma protein extravasation in dura is reduced by i.a./i.g. BoNT/A and p.o. sumatriptan – photographs of open cranial cavities. Left side: TMJ was injected with CFA 1 day before animal perfusion with saline. BoNT/A was injected into the TMJ (5 U kg−1 i.a.) or trigeminal ganglion (2 U kg−1 i.g.) 3 days before CFA. Sumatriptan (175 mg kg−1) was administered p.o. 24 h after CFA. Four days following BoNT/A or 2 h after sumatriptan rats were intravenously injected with Evans blue (40 mg kg−1) and perfused with saline. Photographs were taken upon the perfusion with saline and the removal of brain tissue.
Figure 4.
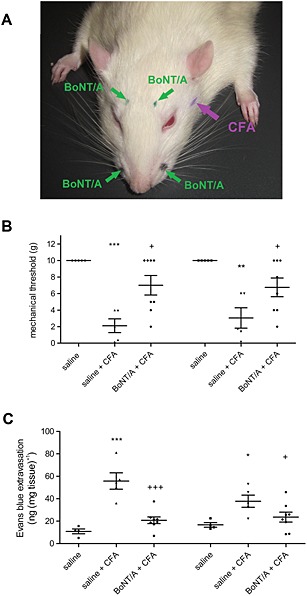
The effect of BoNT/A injection outside the TMJ on mechanical allodynia and dural Evans blue/plasma protein extravasation. BoNT/A (total dose 5 U kg−1) was injected at four sites (bilateral forehead and bilateral whisker pad injections) (A). Three days after BoNT/A rats were injected with CFA into the TMJ. Facial allodynia was measured with von Frey filaments 24 h after CFA injection. After behavioural measurement, rats were injected with Evans blue (i.v., 40 mg kg−1) and perfused with saline. Dura was harvested for formamide extraction and spectrophotometric measurement of Evans blue dye extravasated in complex with plasma proteins. (A) Sites of BoNT/A bilateral injections and position of TMJ to be injected with CFA. (B) The effect of BoNT/A on mechanical thresholds measured by von Frey filaments (mechanical allodynia). (C) The effect of bilateral Evans blue/plasma protein extravasation in the cranial dura. Scatter plot represents individual animal values, and horizontal lines and bars indicate mean ± SEM. n (animals per group) = 5–8. *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from saline control; + P < 0.05, significantly different from saline + CFA; +++ P < 0.001, significantly different from saline + CFA; one‐way ANOVA followed by Newman–Keuls post hoc test.
In a separate experiment, we employed four BoNT/A low‐dose bilateral injections into the face of the rats (Figure 4). As observed with the single BoNT/A injection into the TMJ, four injections outside of TMJ prevented both bilateral allodynia and the CFA‐evoked plasma protein extravasation (Figure 4).
TMJ inflammation induces dural tissue infiltration with inflammatory cells, which is prevented by BoNT/A
Histological staining of the dural tissue of CFA‐treated rats demonstrated an elevated number of automatically counted, Giemsa‐positive, cell nuclei, compared with those in saline‐treated animals (P < 0,001), indicating an inflammatory cell infiltration. Inflammatory cells present in CFA‐injected animals (not present in saline control) were identified by an experienced pathologist, as lymphocytes, monocytes and plasma cells, as previously found in a model of trigeminal neuropathy (Filipović et al., 2014). The lack of polymorphonuclear neutrophils in dura suggests the presence of a sterile inflammation. BoNT/A prevented the increased number of Giemsa positive profiles evoked by i.a. CFA (Figure 5).
Figure 5.
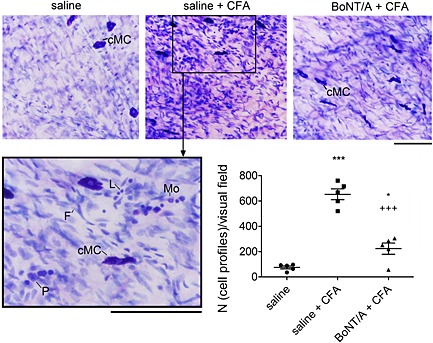
The effect of BoNT/A injection into the TMJ on inflammatory cell infiltration in dura mater in CFA‐treated rats. The 5 U kg−1 BoNT/A or saline were injected into the TMJ 3 days before the induction of TMJ inflammation by CFA. Histological staining of ipsilateral cranial dural tissue was performed using Giemsa staining. Number of Giemsa‐stained cell profiles was automatically quantified by CellSens Dimension visualizing programme (Olympus). Each data value represents mean of 4–5 visual fields per single animal. L, lymphocyte; Mo, monocyte; P, plasma cell, cMC, constitutive mast cell; F, fibrocyte. Scale bars = 100 μm. Scatter plot represents individual animal values, and horizontal lines and bars indicate mean ± SEM. n (animals per group) = 5. *P < 0.05, significantly different from saline control; ***P < 0.001, significantly different from saline control; +++ P < 0.001, significantly different from saline + CFA; one‐way ANOVA followed by Newman–Keuls post hoc test.
TMJ inflammation induces up‐regulation of CGRP in dura and TNC, which is reduced by i.a. BoNT/A
Following CFA‐induced TMJ inflammation, CGRP expression was significantly increased in dura mater and ipsilateral caudal trigeminal nuclei. BoNT/A injected into the TMJ prevented the CGRP increase in dura mater. The effect of BoNT/A on CGRP expression in trigeminal nuclei was not significant. CGRP concentration was not significantly altered in trigeminal ganglion and CSF (Figure 6).
Figure 6.
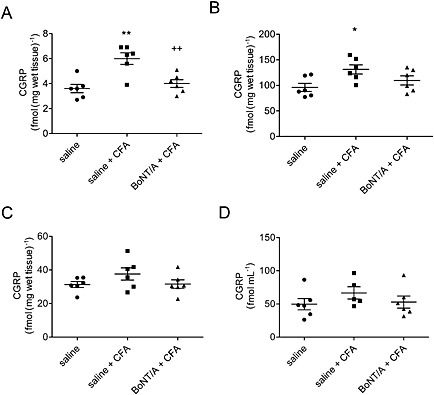
BoNT/A effect on concentration of CGRP protein in dura, trigeminal nucleus caudalis, trigeminal ganglion and CSF in CFA‐treated rats. The 5 U kg−1 BoNT/A was injected into the TMJ 3 days before the CFA treatment. Tissues were collected 1 day post‐CFA, and the CGRP concentration was analysed by RIA. (A) Dura mater; (B) ipsilateral trigeminal nucleus caudalis; (C) ipsilateral trigeminal ganglion; and (D) CSF. Scatter plot represents individual animal values, and horizontal lines and bars indicate mean ± SEM. n (animals per group) = 6. *P < 0.05, significantly different from saline control; **P < 0.01, significantly different from saline control; ++ P < 0.01, significantly different from saline + CFA; one‐way ANOVA followed by Newman–Keuls post hoc test.
Cleaved SNAP‐25 colocalizes with CGRP‐expressing afferents of the dura mater after BoNT/A peripheral treatment
In animals injected peripherally with BoNT/A, we observed the presence of cleaved SNAP‐25 in the injected‐side lateral and parietal dura near the dural blood vessels after BoNT/A multiple (Figure 7) and single injections into TMJ and whisker pad (not shown). Cleaved SNAP‐25 was also visible in non‐vascular areas of dura. All examined fibers containing SNAP‐25 co‐expressed bright granular immunoreactivity for CGRP (Figure 7). Contralateral dura was devoid of cleaved SNAP‐25, ruling out possible systemic BoNT/A diffusion (Figure 7). Complete colocalization of CGRP and cleaved SNAP‐25 was also visible after the single injection of BoNT/A (5 U kg−1) into the TMJ (not shown).
Figure 7.
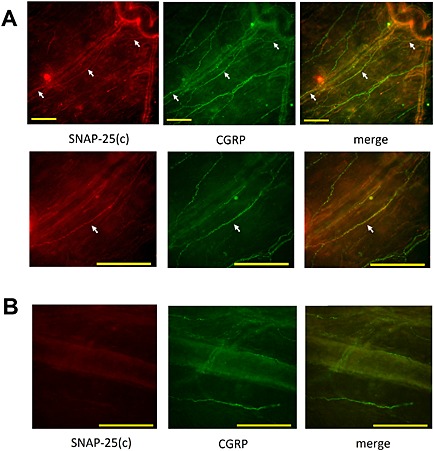
Colocalization of truncated SNAP‐25 and CGRP in ipsilateral cranial dura after BoNT/A injection in the periphery. BoNT/A 20 U kg−1 total dose was injected into four different sites (TMJ, whisker pad, and frontal and temporal regions; 1.75 U/20 μL per site) on the left side of the head. Animals were perfused for immunohistochemistry 6 days later. (A) Upper panel: lower magnification fluorescent microphotograph shows the course of a single‐cleaved SNAP‐25 [SNAP‐25(c)]‐immunoreactive fibre (arrows, red immunofluorescence) in the vicinity of dural blood vessels, which colocalizes with CGRP (green fibers). Lower panel: higher magnification image of the middle part of cleaved SNAP‐25‐immunoreactive fibre, which colocalizes with granular CGRP immunofluorescence. (B) Microphotograph of contralateral side dura of the same animal without detectable cleaved SNAP‐25 in CGRP‐expressing afferents. The images are representative of the data obtained from four animals. Scale bars = 100 μm.
Anti‐nociceptive activity and enzymatic activity of BoNT/A in dura mater are axonal transport‐dependent
The anti‐nociceptive actions of BoNT/A on CFA‐induced pain was prevented by the axonal transport blocker colchicine injected into the trigeminal ganglion (Figure 8). This is in line with previous findings that BoNT/A antinociceptive activity is dependent on axonal transport (Filipović et al., 2012). Similarly, cleaved SNAP‐25 was no longer found in dura after treatment with colchicine (Figure 8). This finding suggests that, after local peripheral injection of 5 U kg−1 in the facial area, BoNT/A is axonally transported to the ipsilateral dural primary afferents by microtubule‐dependent mechanism through the ganglion.
Figure 8.
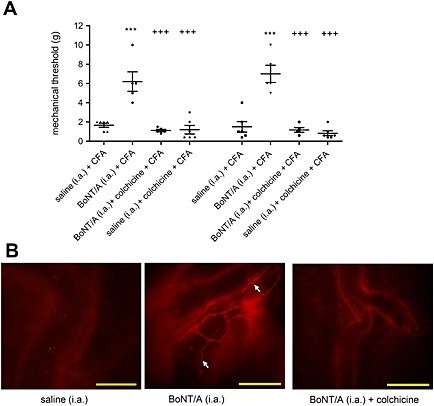
BoNT/A antinociceptive activity and occurrence of cleaved SNAP‐25 in dura mater is dependent on axonal transport. (A) Preventive effect of i.a. BoNT/A (5 U kg−1) on mechanical allodynia evoked by CFA injection into the TMJ is prevented by colchicine (5 mM) injection into the trigeminal ganglion. Scatter plot represents individual animal values, and horizontal lines and bars indicate mean ± SEM. n (animals per group) = 5–6. ***P < 0.001, significantly different from saline i.a. + CFA; +++ P < 0.001, significantly different from BoNT/A i.a. + CFA; one‐way ANOVA followed by Newman–Keuls post hoc test. (B) Colchicine prevented the occurrence of cleaved SNAP‐25 immunofluorescence in dura mater. The image is representative of the data obtained from four animals per group. Scale bar = 100 μm.
Discussion
By studying the TMJ inflammation evoked by CFA, we found that the pain in this experimental condition was accompanied by DNI. The neurogenic plasma protein leakage in cranial dural tissue was accompanied by increased inflammatory cell infiltration and up‐regulation of CGRP peptide levels (Figures 2, 3, 4, 6).
Along with the present report, we have also shown that other painful stimuli in the trigeminal region (formalin‐induced or experimental neuropathic pain) were accompanied by DNI (Filipović et al., 2012, 2014). These observations demonstrate the occurrence of DNI in experimental trigeminal pain. To study migraine, other authors induced DNI more ‘artificially’ by different chemical or electrical stimuli (Markowitz et al., 1987; Buzzi and Moskowitz, 1990; O'Shaughnessy and Connor, 1994; Arulmani et al., 2006; Nelson et al., 2010; Akerman et al., 2013). Current opinion suggests that the migraine headache involves CNS dysfunction, accompanied by activation of the trigeminovascular system (Williamson and Hargreaves, 2001), and release of vasoactive peptides which induce DNI (Markowitz et al., 1987). This is not limited to experimental animals because it was clinically observed that DNI accompanies migraine and cluster headache attacks (Göbel et al., 2000; Knotkova and Pappagallo 2007). Thus, ongoing pain in the TMJ area, as well as other orofacial pain models, can be employed to study the sensitization of trigeminal dural afferents, assumed to be present in migraine and other headaches. In the present experiments, peripherally injected BoNT/A reduced mechanical allodynia and inflammatory changes in the cranial dura [plasma protein extravasation and cellular inflammatory response (Figures 1, 2)]. The similar effects of BoNT/A injections given directly into the ganglion suggests that BoNT/A action is primarily associated with the trigeminal system (Figures 1, 2).
The recommended protocol for BoNT/A application in chronic migraine consists of multiple injections to 31 head and neck sites (Diener et al., 2010). A similar protocol is difficult to replicate in rats because of the smaller cranial dimensions. Thus, we injected BoNT/A bilaterally to the rat forehead region overlying the frontal bone (innervated by V1 ophthalmic trigeminal branch) and whisker pad (V2 maxillary branch). Such BoNT/A injections at four sites were effective in preventing CFA‐evoked pain and DNI similarly to the single BoNT/A injection into the TMJ (Figure 4). This demonstrates that the effects of BoNT/A on allodynia and DNI are not primarily mediated by its direct peripheral effect on CFA‐stimulated neurons.
Plasma protein extravasation in cranial dura is a useful marker of trigeminal activation, often employed in preclinical screening of antimigraine drugs (Markowitz et al., 1987; Buzzi and Moskowitz, 1990; O'Shaughnessy and Connor, 1994; Arulmani et al., 2006; Nelson et al., 2010; Akerman et al., 2013). DNI consists of two main components: vasodilation, which is mediated by CGRP, and plasma protein extravasation, which is mediated by substance P. Blocking only the substance P transmission by NK1 receptor antagonists did not reduce migraine symptoms, suggesting that CGRP transmission might play a more important role in the pathophysiology of migraine (Williamson and Hargreaves 2001; Peroutka, 2005). Thus, we investigated the possibility that the antimigraine actions of BoNT/A were associated with prevention of CGRP transmission. Here, we have found that peripherally injected BoNT/A prevented the CFA‐induced increase in CGRP levels in the cranial dura (Figure 6). Interestingly, in chronic migraine patients responsive to BoNT/A, the pretreatment CGRP plasma levels were increased in comparison with those in BoNT/A non‐responsive patients (Cernuda‐Morollón et al., 2014). After the treatment, BoNT/A normalized the elevated CGRP plasma levels (Cernuda‐Morollón et al., 2015). The authors posited that BoNT/A inhibits the release of CGRP from peripheral trigeminal neurons and, consequently, reduces the CGRP‐mediated trigeminal sensitization in migraine (Cernuda‐Morollón et al., 2015).
Because the anti‐migraine effect of BoNT/A is difficult to explain by its local action on peripheral, extracranial sensory nerves endings, it was suggested that BoNT/A exhibits its actions in pain and migraine by reaching dural trigeminal afferents (Matak and Lacković, 2014; Ramachandran and Yaksh, 2014). Previously, we reported that the effects of BoNT/A on trigeminal neuropathic pain and resulting DNI was prevented by colchicine injected into the ganglion, indicative of axonal transport of this toxin (Filipović et al., 2012). After BoNT/A peripheral injection, we detected cleaved SNAP‐25 in the cranial dura mater (Figures 7, 8). Moreover, cleaved SNAP‐25 and CGRP were colocalized in the ipsilateral dura (Figure 7). Peripherally administered BoNT/A may prevent the SNAP‐25‐mediated release of CGRP in cranial meninges and consequent CGRP effects presumably involved in migraine pathophysiology (Williamson and Hargreaves, 2001; Durham, 2008; Geppetti et al., 2012; Karsan and Goadsby, 2015).
It was recently found that BoNT/A reduces the mechanical sensitivity of extracranially projecting collaterals of dural afferents which exit the cranium through the skull bone sutures in rats (Burstein et al., 2014). However, in our experiments, BoNT/A effects on dura mater were present even if the toxin was administered away from cranial sutures (TMJ and whisker pad). Additionally, blockade of the axonal transport of the toxin by direct i.g. colchicine prevented the formation of cleaved SNAP‐25 in the dura (Figure 8) and other effects of BoNT/A on DNI (Filipović et al., 2012). Colchicine action is limited to the injection site (Kreutzberg, 1969; Cangiano and Fried, 1977) and, therefore any possible BoNT/A axonal traffic to the dura via extracranial collaterals of dural afferents should not be prevented by administration of colchicine into the ganglion. These observations do not support an important contribution of BoNT/A local activity on extracranially projecting dural afferent collaterals.
The question arising from the present experiments is how BoNT/A crosses from the trigeminal extracranial nerves to trigeminal nerve endings in dura. Dura and extracranial trigeminal regions are innervated by separate sensory neurons (Larrier and Lee, 2003; Shimizu et al., 2012). Therefore, the transcytosis of BoNT/A from the extracranial sensory neurons to neurons that innervate dura seems the most logical explanation for the occurrence of cleaved SNAP‐25 in dura mater after facial injection of the toxin. Up to now, transcytosis of BoNT/A between different neurons has been demonstrated directly inside the retina and brain (Restani et al., 2011, 2012). In the trigeminal region, BoNT/A transcytosis within the trigeminal ganglion after its peripheral injection has been suggested by Kitamura et al. (2009. The authors investigated the effect of BoNT/A on vesicular neurotransmitter release in trigeminal neurons acutely isolated from neuropathic rats subjected to infraorbital nerve constriction injury. BoNT/A injected into the rat face induced a profound reduction of vesicular neurotransmitter release in all neurons isolated from the ganglion. They assumed that, in order to induce a widespread effect, BoNT/A was transcytosed within the ganglion (Kitamura et al., 2009). In the trigeminal ganglion, facially injected BoNT/A reduced the expression of TRPV1 channels in neurons projecting to the dura mater (Shimizu et al., 2012). These authors proposed that the effects of BoNT/A were mediated by transcytosis of the toxin, within trigeminal gangliam from extracranially projecting neurons to neurons that innervate the dura (Shimizu et al., 2012). The exact place and mechanism of such putative transcytosis remain to be elucidated. It is likely to occur within the trigeminal ganglion itself (Shimizu et al., 2012), although transcytosis in the trigeminal sensory nuclei cannot be excluded (Ramachandran and Yaksh, 2014) (Figure 9).
Figure 9.
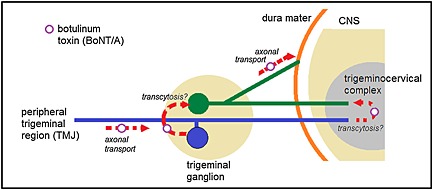
Possible sites of action of axonally transported BoNT/A in migraine and other headaches. Following injection in peripheral trigeminal region, BoNT/A is taken up by the extracranial trigeminal afferent (blue pseudounipolar neuron) and retrogradely transported to trigeminal ganglion. BoNT/A is then transcytosed to meningeal afferent (green pseudounipolar neuron) and anterogradely transported to dura mater where it inhibits neuropeptide release. Alternatively, the transcytosis can take place in the trigeminocervical complex.
The conventional antimigraine drug sumatriptan, an agonist of 5‐HT1B/D receptors, reduced the pain supersensitivity and dural plasma protein extravasation in CFA‐induced TMJ inflammation, as well (Figures 1, 2). Sumatriptan prevents the evoked release of CGRP and substance P in vitro and ex vivo (Buzzi and Moskowitz, 1990; Durham and Russo, 1999). Furthermore, sumatriptan reduces elevated CGRP concentrations in blood and saliva during migraine attacks (Goadsby et al., 1990; Bellamy et al., 2006). CGRP antagonists are reported to reduce the symptoms of acute migraine attacks (Edvinsson and Warfvinge, 2013). Antibodies against CGRP and CGRP receptors might also be effective as a prophylactic chronic migraine treatment (Edvinsson, 2015).
In conclusion, as demonstrated here, BoNT/A might have beneficial effect on experimental TMJ pain and the accompanying dural inflammation. The effects of BoNT/A in the cranial dura could be reconstructed as follows: after peripheral injection, BoNT/A is taken up by sensory nerve endings and axonally transported to trigeminal ganglion. After transcytosis, the toxin reaches dural nerve endings containing CGRP and suppresses the CGRP‐mediated sensitization of the trigeminovascular system and DNI. At present, this seems as the most convincing hypothesis of the action of BoNT/A in migraine and other headaches.
Acknowledgements
The work was supported by grants from Croatian Ministry of Science, Education and Sport (no. 108‐1080003‐0001 awarded to Z. L.), Croatian National Science Foundation (no. O‐1259‐2015 awarded to Z. L.), Hungarian National Brain Research Program (SROP4.2.2.A‐11/1/KONV‐2012‐0024 and the KTIA NAP_13‐1‐2013‐0001 awarded to Z. H.) and National Brain Research Program B (Chronic Pain Research Group; KTIA_NAP_13–2014‐0022; awarded to Z. L., 888819). We thank Mrs. Teréz Bagoly for her excellent technical contribution to the CGRP RIA and Prof Vladimir Trkulja for the suggestions regarding the presentation of results. Antibody to cleaved SNAP‐25 was kindly provided by Assistant Prof Ornella Rossetto (University of Padua, Italy). The histological analysis was performed by pathologist Prof Mara Dominis.
Author contributions
Z. L., B. F. and I. M. conceived and designed the study. Z. L., B. F., I. M. and Z. H analysed and interpreted the data. Z. L., B. F., I. M. and Z. H. drafted the manuscript. All authors have approved the final version of the manuscript. All authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflicts of interest
The authors declare no conflict of interest.
Lacković, Z. , Filipović, B. , Matak, I. , and Helyes, Z. (2016) Activity of botulinum toxin type A in cranial dura: implications for treatment of migraine and other headaches. British Journal of Pharmacology, 173: 279–291. doi: 10.1111/bph.13366.
References
- Akerman S, Holland PR, Hoffmann J (2013). Pearls and pitfalls in experimental in vivo models of migraine: dural trigeminovascular nociception. Cephalalgia 33: 577–592. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. (2013). The concise guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arulmani U, Gupta S, VanDenBrink AM, Centurión D, Villalón CM, Saxena PR (2006). Experimental migraine models and their relevance in migraine therapy. Cephalalgia 26: 642–659. [DOI] [PubMed] [Google Scholar]
- Bellamy JL, Cady RK, Durham PL (2006). Salivary levels of CGRP and VIP in rhinosinusitis and migraine patients. Headache 46: 24–33. [DOI] [PubMed] [Google Scholar]
- Bevilaqua Grossi D, Lipton RB, Bigal ME (2009). Temporomandibular disorders and migraine chronification. Curr Pain Headache Rep 13: 314–318. [DOI] [PubMed] [Google Scholar]
- Bigal ME, Walter S, Rapoport AM (2013). Calcitonin gene‐related peptide (CGRP) and migraine current understanding and state of development. Headache 53: 1230–1244. [DOI] [PubMed] [Google Scholar]
- Blasi J, Chapman ER, Link E, Binz T, Yamasaki S, de Camilli P, et al. (1993). Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP‐25. Nature 365: 160–163. [DOI] [PubMed] [Google Scholar]
- Burstein R, Zhang X, Levy D, Aoki KR, Brin MF (2014). Selective inhibition of meningeal nociceptors by botulinum neurotoxin type A: therapeutic implications for migraine and other pains. Cephalalgia 34: 853–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzi MG, Moskowitz MA (1990). The antimigraine drug, sumatriptan (GR43175), selectively blocks neurogenic plasma extravasation from blood vessels in dura mater. Br J Pharmacol 99: 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cangiano A, Fried JA (1977). The production of denervation‐like changes in rat muscle by colchicine, without interference with axonal transport or muscle activity. J Physiol 265: 63–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cernuda‐Morollón E, Martínez‐Camblor P, Ramón C, Larrosa D, Serrano‐Pertierra E, Pascual J (2014). CGRP and VIP levels as predictors of efficacy of onabotulinumtoxin type A in chronic migraine. Headache 54: 987–995. [DOI] [PubMed] [Google Scholar]
- Cernuda‐Morollón E, Ramón C, Martínez‐Camblor P, Serrano‐Pertierra E, Larrosa D, Pascual J (2015). OnabotulinumtoxinA decreases interictal CGRP plasma levels in chronic migraine patients. Pain 156: 820–824. [DOI] [PubMed] [Google Scholar]
- Dallas FA, Dixon CM, McCulloch RJ, Saynor DA (1989). The kinetics of 14C‐GR43175 in rat and dog. Cephalalgia 9: 53–56. [DOI] [PubMed] [Google Scholar]
- de Rossi SS, Greenberg MS, Liu F, Steinkeler A (2014). Temporomandibular disorders: evaluation and management. Med Clin North Am 98: 1353–1384. [DOI] [PubMed] [Google Scholar]
- Diener HC, Dodick DW, Aurora SK, Turkel CC, Degryse RE, Lipton RB, et al. (2010). PREEMPT 2 Chronic Migraine Study Group. OnabotulinumtoxinA for treatment of chronic migraine: results from the double‐blind, randomized, placebo‐controlled phase of the PREEMPT 2 trial. Cephalalgia 30: 804–814. [DOI] [PubMed] [Google Scholar]
- Dressler D (2013). Botulinum toxin therapy: its use for neurological disorders of the autonomic nervous system. J Neurol 260: 701–713. [DOI] [PubMed] [Google Scholar]
- Durham PL (2008). Inhibition of calcitonin gene‐related peptide function: a promising strategy for treating migraine. Headache 48: 1269–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham PL, Russo AF (1999). Regulation of calcitonin gene‐related peptide secretion by a serotonergic antimigraine drug. J Neurosci 19: 3423–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L (2015). CGRP receptor antagonists and antibodies against CGRP and its receptor in migraine treatment. Br J Clin Pharmacol . doi:10.1111/bcp.12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L, Warfvinge K (2013). CGRP receptor antagonism and migraine therapy. Curr Protein Pept Sci 14: 386–392. [DOI] [PubMed] [Google Scholar]
- Filipović B, Matak I, Bach‐Rojecky L, Lacković Z (2012). Central action of peripherally applied botulinum toxin type A on pain and dural protein extravasation in rat model of trigeminal neuropathy. PLoS One 7: e29803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipović B, Matak I, Lacković Z (2014). Dural neurogenic inflammation induced by neuropathic pain is specific to cranial region. J. Neural Transm 121: 555–563. [DOI] [PubMed] [Google Scholar]
- Franco AL, Gonçalves DA, Castanharo SM, Speciali JG, Bigal ME, Camparis CM (2010). Migraine is the most prevalent primary headache in individuals with temporomandibular disorders. J Orofac Pain 24: 287–292. [PubMed] [Google Scholar]
- Geppetti P, Rossi E, Chiarugi A, Benemei S (2012). Antidromic vasodilatation and the migraine mechanism. J. Headache Pain 13: 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goadsby PJ, Edvinsson L, Ekman R (1990). Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 28: 183–187. [DOI] [PubMed] [Google Scholar]
- Göbel H, Czech N, Heinze‐Kuhn K, Heinze A, Brenner W, Muhle C, et al. (2000). Evidence of regional plasma protein extravagation in cluster headache using Tc‐99 m albumin SPECT. Cephalalgia 20: 287. [Google Scholar]
- Harper RP, Kerins CA, McIntosh JE, Spears R, Bellinger LL (2001). Modulation of the inflammatory response in the rat TMJ with increasing doses of complete Freund's adjuvant. Osteoarthritis Cartilage 9: 619–624. [DOI] [PubMed] [Google Scholar]
- Kalandakanond S, Coffield JA (2001). Cleavage of SNAP‐25 by botulinum toxin type A requires receptor‐mediated endocytosis, pH‐dependent translocation, and zinc. J. Pharmacol. Exp. Ther. 296: 980–986. [PubMed] [Google Scholar]
- Karsan N, Goadsby PJ (2015). Calcitonin gene‐related peptide and migraine. Curr Opin Neurol 28: 250–254. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. PLoS Biol 8: e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Matsuka Y, Spigelman I, Ishihara Y, Yamamoto Y, Sonoyama W, et al. (2009). Botulinum toxin type a (150 kDa) decreases exaggerated neurotransmitter release from trigeminal ganglion neurons and relieves neuropathy behaviors induced by infraorbital nerve constriction. Neuroscience 159: 1422–1429. [DOI] [PubMed] [Google Scholar]
- Knotkova H, Pappagallo M (2007). Imaging intracranial plasma extravasation in a migraine patient: a case report. Pain Med 8: 383–387. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW (1969). Neuronal dynamics and axonal flow. IV. Blockage of intra‐axonal enzyme transport by colchicine. Proc Natl Acad Sci U S A 62: 722–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrier D, Lee A (2003). Anatomy of headache and facial pain. Otolaryngol Clin North Am 36: 1041–1053. [DOI] [PubMed] [Google Scholar]
- Markowitz S, Saito K, Moskowitz MA (1987). Neurogenically mediated leakage of plasma protein occurs from blood vessels in dura mater but not brain. J Neurosci 7: 4129–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matak I, Bach‐Rojecky L, Filipović B, Lacković Z (2011). Behavioral and immunohistochemical evidence for central antinociceptive activity of botulinum toxin A. Neuroscience 186: 201–207. [DOI] [PubMed] [Google Scholar]
- Matak I, Lacković Z (2014). Botulinum toxin A, brain and pain. Prog Neurobiol 119–120: 39–59. [DOI] [PubMed] [Google Scholar]
- Matak I, Rossetto O, Lacković Z (2014). Botulinum toxin type A selectivity for certain types of pain is associated with capsaicin‐sensitive neurons. Pain 155: 1516–1526. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA (1990). Basic mechanisms in vascular headache. Neurol Clin 8: 801–815. [PubMed] [Google Scholar]
- Nelson DL, Phebus LA, Johnson KW, Wainscott DB, Cohen ML, Calligaro DO, et al. (2010). Preclinical pharmacological profile of the selective 5‐HT1F receptor agonist lasmiditan. Cephalalgia 30: 1159–1169. [DOI] [PubMed] [Google Scholar]
- Németh J, Görcs T, Helyes Z, Oroszi G, Kocsy T, Pintér E, et al. (1998). Development of a new sensitive CGRP radioimmunoassay for neuropharmacological research. Neurobiology (Bp). 6: 473–475. [PubMed] [Google Scholar]
- O'Shaughnessy CT, Connor HE (1994). Investigation of the role of tachykinin NK1, NK2 receptors and CGRP receptors in neurogenic plasma extravasation in rat dura mater. Eur J Pharmacol 263: 193–198. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC‐IUPHAR(2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peroutka SJ (2005). Neurogenic inflammation and migraine: implications for the therapeutics. Mol Interv 5: 304–311. [DOI] [PubMed] [Google Scholar]
- Pozsgai G, Hajna Z, Bagoly T, Boros M, Kemény Á, Materazzi S, et al. (2012). The role of transient receptor potential ankyrin 1 (TRPA1) receptor activation in hydrogen‐sulphide‐induced CGRP‐release and vasodilation. Eur J Pharmacol 689: 56–64. [DOI] [PubMed] [Google Scholar]
- Ramachandran R, Yaksh TL (2014). Therapeutic use of botulinum toxin in migraine: mechanisms of action. Br J Pharmacol 171: 4177–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restani L, Antonucci F, Gianfranceschi L, Rossi C, Rossetto O, Caleo M (2011). Evidence for anterograde transport and transcytosis of botulinum neurotoxin A (BoNT/A). J Neurosci 31: 15650–15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restani L, Novelli E, Bottari D, Leone P, Barone I, Galli‐Resta L, et al. (2012). Botulinum neurotoxin A impairs neurotransmission following retrograde transynaptic transport. Traffic 13: 1083–1089. [DOI] [PubMed] [Google Scholar]
- Schueler M, Messlinger K, Dux M, Neuhuber WL, de Col R (2013). Extracranial projections of meningeal afferents and their impact on meningeal nociception and headache. Pain 154: 1622–1631. [DOI] [PubMed] [Google Scholar]
- Schuh‐Hofer S, Boehnke C, Reuter U, Siekmann W, Lindauer U, Arnold G, et al. (2003). A fluorescence‐based method to assess plasma protein extravasation in rat dura mater using confocal laser scanning microscopy. Brain Res Brain Res Protoc 12: 77–82. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Shibata M, Toriumi H, Iwashita T, Funakubo M, Sato H, et al. (2012). Reduction of TRPV1 expression in the trigeminal system by botulinum neurotoxin type‐A. Neurobiol Dis 48: 367–378. [DOI] [PubMed] [Google Scholar]
- Sunil Dutt C, Ramnani P, Thakur D, Pandit M (2015). Botulinum toxin in the treatment of muscle specific Oro‐facial pain: a literature review. J Maxillofac Oral Surg 14: 171–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalakoti S, Patil VV, Damodaram S, Vause CV, Langford LE, Freeman SE, et al. (2007). Neuron‐glia signaling in trigeminal ganglion: implications for migraine pathology. Headache 47: 1008–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa G, Ceruti S, Zanardelli M, Magni G, Jasmin L, Ohara PT, et al. (2010). Temporomandibular joint inflammation activates glial and immune cells in both the trigeminal ganglia and in the spinal trigeminal nucleus. Mol Pain 6: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson DJ, Hargreaves RJ (2001). Neurogenic inflammation in the context of migraine. Microsc Res Tech 53: 167–178. [DOI] [PubMed] [Google Scholar]
- Zimmerman M (1983). Ethical guidelines for investigations of experimental pain in conscious animals. Pain 16: 109–110. [DOI] [PubMed] [Google Scholar]