

Abstract
Background and Purpose
Doxorubicin is a powerful antineoplastic agent for treating a wide range of cancers. However, doxorubicin cardiotoxicity of the heart has largely limited its clinical use. All‐trans retinoic acid (ATRA) plays an important role in many cardiac biological processes, but its protective effects on doxorubicin‐induced cardiotoxicity remain unknown. Here, we studied the effect of ATRA on doxorubicin cardiotoxicity and the underlying mechanisms.
Experimental Approaches
Cellular viability assays, Western blotting and mitochondrial respiration analyses were employed to evaluate the cellular response to ATRA in H9c2 cells and primary cardiomyocytes. Quantitative PCR and gene knockdown were performed to investigate the underlying molecular mechanisms of ATRA's effects on doxorubicin cardiotoxicity.
Key Results
ATRA significantly inhibited doxorubicin‐induced apoptosis in H9c2 cells and primary cardiomyocytes. ATRA was more effective against doxorubicin cardiotoxicity than resveratrol and dexrazoxane. ATRA also suppressed reactive oxygen species generation and restored expression levels of mRNA and proteins in the phase II detoxifying enzyme system: nuclear factor‐E2‐related factor 2, manganese superoxide dismutase, haem oxygenase‐1, and mitochondrial function (mitochondrial membrane integrity, mitochondrial DNA copy numbers and mitochondrial respiration capacity, biogenesis and dynamics). Both a ERK1/2 inhibitor (U0126) and ERK2 siRNA, but not ERK1 siRNA, abolished the protective effect of ATRA against doxorubicin‐induced toxicity in H9c2 cells. Remarkably, ATRA did not compromise the anticancer efficacy of doxorubicin in gastric carcinoma cells.
Conclusions and Implications
ATRA protected cardiomyocytes against doxorubicin‐induced toxicity, by activating the ERK2 pathway, without compromising its anticancer efficacy. Therefore, ATRA is a promising candidate as a cardioprotective agent against doxorubicin cardiotoxicity.
Abbreviations
- ATRA
all‐trans retinoic acid
- Bcl‐2
B‐cell lymphoma 2
- Bcl‐xl
B‐cell lymphoma‐extra‐large
- BrdU
5‐bromo‐2′‐deoxyuridine
- Dox
doxorubicin
- Drp1
dynamin‐related protein 1
- FCCP
carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone
- HO1
haem oxygenase‐1
- Mfn1
mitofusin 1
- Mfn2
mitofusin 2
- MMP
mitochondrial membrane potential
- MnSOD
manganese superoxide dismutase
- MTT (3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide) Nrf2
nuclear factor‐E2‐related factor 2
- OCR
oxygen consumption rate
- OPA1
optic atrophy 1
- RARα
retinoic acid receptor α
- ROS
reactive oxygen species
- TFAM
mitochondrial transcription factor A
Tables of Links
| TARGETS | |
|---|---|
| Nuclear hormone receptors a | Enzymes b |
| PPARα | Caspase 3 |
| RARα | ERK1 |
| ERK2 | |
| HO1 | |
| JNK | |
| MEK | |
| SIRT3 (HDAC3) |
| LIGANDS | |
|---|---|
| ATRA | PD98059 |
| Bcl‐2 | Resveratrol |
| Dexrazoxane | SB203580 |
| Doxorubicin | SP600125 |
| LY294002 | U0126 |
| Wortmannin |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,2013b).
Introduction
Various types of human tumours can be treated with anthracyclines. Doxorubicin is one of the most widely used anthracyclines in chemopreventive applications. However, the cardiotoxicity of doxorubicin limits its clinical use (Minotti et al., 2004). An accumulation of doxorubicin‐induced oxidative stress leads to cardiac dysfunction, including cardiomyocyte death (Sterba et al., 2013). A decline in antioxidant enzyme activity (Gu et al., 2012) and mitochondrial dysfunction [energy metabolism, redox balance (Kuznetsov et al., 2011), decrease of mitochondrial biogenesis (Miyagawa et al., 2010) and dynamics (Kuzmicic et al., 2011)] have been reported in doxorubicin cardiotoxicity. ERK activation provides a possible mechanism for protection against doxorubicin‐induced cardiomyocyte damage (Izumi et al., 2006; Simoncikova et al., 2008). These observations suggest that some agents may protect against doxorubicin cardiotoxicity by inhibiting oxidative stress, restoring cardiac mitochondrial function or stimulating ERK activation.
Retinoic acid derivatives are essential for tissue homeostasis, cellular proliferation (Wiggert et al., 1978) and the prevention of various diseases, such as neurotoxicity (Cheng et al., 2013; Kim et al., 2013), hepatitis and hepatic ischaemia (Rao et al., 2010; Nagy, 2012). All‐trans retinoic acid (ATRA) protects cardiomyocytes from various stimuli by inhibiting oxidative stress, preventing cardiomyocyte apoptosis and attenuating p38 MAPK, JNK and NF‐κB activation (Choudhary et al., 2008; Lou et al., 2013; Nizamutdinova et al., 2013). However, the effect of ATRA on doxorubicin cardiotoxicity remains unknown. This study was aimed at evaluating the protective effects of ATRA against doxorubicin‐induced damage of cardiomyocytes and the potential involvement of the ERK signalling pathway in this protection.
Methods
Cell culture and treatment
H9c2 cells were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2 in DMEM supplemented with 10% FBS, penicillin (100 U·mL−1), and streptomycin (100 μg·mL−1). H9c2 cells were pretreated with different concentrations (5, 250, 1000 and 2000 nmol·L−1) of ATRA for 24 h and then were treated with 3 μmol·L−1 doxorubicin for another 24 h. In the experiments with inhibitors, H9c2 cells were pretreated for 1 h with U0126 (50 μmol·L−1), SB203580 (50 μmol·L−1), SP600125 (50 μmol·L−1), PD98059 (50 μmol·L−1), LY294002 (50 μmol·L−1) or wortmannin (1 μmol·L−1) before ATRA was added to the growth media.
Primary cultures of rat neonatal cardiomyocytes were prepared from the ventricles of 1‐ to 3‐day‐old Sprague–Dawley rat pups as previously described (Chlopcikova et al., 2001). Cardiomyocytes were seeded on six‐well plates overnight in DMEM with 10% FBS, penicillin (100 U·mL−1) and streptomycin (100 μg·mL−1). BrdU was used to inhibit the growth of cardiac fibroblasts. Primary cardiomyocytes were treated with different concentrations (0.1, 0.5, 1 or 2 μmol·L−1) of ATRA for 24 h and then were treated with doxorubicin.
Human gastric carcinoma SGC‐7901 and AGS cells were maintained at 37°C in RPMI 1640 medium with 10% FBS, penicillin (100 U·mL−1) and streptomycin (100 μg·mL−1). SGC‐7901 and AGS cells were pretreated with ATRA (2 μmol·L−1) for 24 h and then were treated with doxorubicin for 24 h.
MTT assay
For viability assays, the cells were incubated with MTT (5 μg·mL−1) for 1 h at 37°C, and then, the formazan precipitate was dissolved with dimethyl sulfoxide. The optical densities of the resulting formazan solutions were measured using a microplate reader (Thermo 1500) at 570 nm.
Detection of intracellular ROS
Intracellular reactive oxygen species (ROS) generation was analysed using the oxidation‐sensitive fluorescent dye carboxy‐H2DCF‐DA, which can be oxidized by ROS to emit green fluorescence. H9c2 cells were incubated with DCFH‐DA (1 μmol·L−1) at 37°C for 30 min and then were washed twice with PBS followed by image acquisition using an inverted florescence microscope (Olympus IX71).
Mitochondrial membrane potential evaluation
The mitochondrial membrane potential (MMP) in H9c2 cells was assessed using JC‐1 staining. H9c2 cells were incubated with JC‐1 (1 μmol·L−1) at 37°C for 30 min and then were washed with PBS followed by image acquisition using an inverted florescence microscope (Olympus IX71).
Real‐time PCR
Total RNA was extracted from the cells using TRIzol reagent (Roche, Basel, Switzerland) following the manufacturer's protocol. RT was performed using the PrimeScript RT‐PCR Kit followed by semi‐quantitative real‐time PCR using specific primers.
The primer sequences were as follows:
Rat HO1:
-
(forward) 5′‐TGCTCGCATGAACACTCTG‐3′,
(reverse) 5′‐TCCTCTGTCAGCAGTGCCT‐3′;
Rat Nrf2:
-
(forward) 5′‐TTCCTCTGCTGCCATTAGTCAGTC‐3′,
(reverse) 5′‐GCTCTTCCATTTCCGAGTCACTG‐3′;
Rat MnSOD:
-
(forward) 5′‐TGGACAAACCTGAGCCCTAA‐3′,
(reverse) 5′‐GACCCAAAGTCACGCTTGATA‐3′;
Rat α‐tubulin:
-
(forward) 5′‐TGCTGCCATTGCCACCATCA‐3′,
(reverse) 5′‐CTCACCCTCACCCTCCACCG‐3′.
mtDNA content assay
Total DNA was extracted using the QIAamp DNA Mini Kit. Quantitative PCR was performed using 18S rRNA as an endogenous reference gene and a mitochondrial D‐loop as the target gene. Quantitative PCR reactions were performed using the SYBR Premix Ex Taq™, 0.5 μL of each primer (10 pmol·L−1) and 2.5 ng of template (DNA) or no template, with RNase‐free water added to a final volume of 10 μL. The cycling conditions were as follows: initial denaturation at 95°C for 5 min, followed by 40 cycles of 95°C for 30 s, 55°C for 30 s and 72°C for 20 s.
The following primers were used:
Rat mitochondrial D‐loop:
-
(forward) 5′‐CACCCAAGAACAGGGTTTGT‐3′,
(reverse) 5′‐TGGCCATGGGTATGTTGTTAA‐3′;
Rat 18S rRNA:
-
(forward) 5′‐TAGAGGGACAAGTGGCGTTC‐3′,
(reverse) 5′‐CGCTGAGCCAGTCAGTGT‐3′
The mtDNA content was determined as the ratio of the mitochondrial D‐loop to 18S rRNA. The results were presented as a percentage of the control.
Mitochondrial bioenergetics measurement
H9c2 cells were seeded in XF 24‐well microplates (Seahorse Bioscience, Billerica, MA, USA). Medium was changed to unbuffered DMEM after treatment, and the plate was incubated in a non‐CO2 incubator (37°C; 60 min) before running on the XF24 Analyzer (Seahorse Biosciences) to measure mitochondrial respiration. The final concentrations of the mitochondrial inhibitors were 10 μmol·L−1 antimycin A, 6 μmol·L−1 FCCP and 10 μmol·L−1 oligomycin. Briefly, four parameters of mitochondrial function were calculated from the bioenergetics profile: basal oxygen consumption rate (OCR), ATP‐inked OCR and proton leak OCR. After detection, the cell numbers were calculated, and OCR was adjusted accordingly.
Western blotting
The cell samples were extracted in lysis buffer containing protease and phosphatase inhibitors. The protein concentrations of the lysates were determined using the BCA™ protein assay kit. Samples containing approximately 20 μg of protein were separated by SDS‐PAGE and transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% non‐fat milk in TBST for 1 h at room temperature, incubated with the indicated antibodies overnight at 4°C and then incubated with the corresponding secondary antibodies for 1 h at room temperature. Western blots were developed using ECL and quantified by scanning densitometry.
siRNA transfection
siRNAs targeting ERK1 and ERK2 were used to silence rat ERK1 and ERK2 respectively. The siRNA transfections were performed with Lipofectamine 2000 according to the manufacturer's instructions. Briefly, 100 nmol of siRNA was mixed with 5 μL of Lipofectamine 2000. The complex was added to the H9c2 cells for 24 h before any other treatments were processed. A scrambled probe was used as a negative control.
The sequences for the siRNAs were as follows:
Rat ERK1:
-
(forward) 5′‐GGCCUCAAGUACAUACACUTT‐3′,
(reverse) 5′‐AGUGUAUGUACUUGAGGCCTT‐3′;
Rat ERK2:
-
(forward) 5′‐CCUGAGAGGAUUAAAGUAUTT‐3′,
(reverse) 5′‐AUACUUAAUCCUCUCAGGTT‐3′;
Rat: negative control:
-
(forward) 5′‐UUCUCCGAACGUGUCACGUTT‐3′,
(reverse) 5′‐ACGUGACACGUUCGGAGAATT‐3′.
Statistical analysis
Data are expressed as the mean ± SEM of at least five independent experiments. The protective effects and inhibition were assessed using graphpad prism 6 software (GraphPad, San Diego, CA, USA). Relative protein semi‐quantification was performed using quantity one software (Bio‐Rad, Hercules, CA, USA). The mitochondrial respiration capacity was assessed using seahorse xf24 extracellular flux analysis software (Seahorse Bioscience, Billerica, MA, USA). The gene relative expression ratio was calculated by the 2−△△CT method. Differences between groups were assessed by anova followed by Tukey's multiple comparison tests. A P value less than 0.05 was considered to be statistically significant.
Chemicals and reagents
ATRA, resveratrol, dexrazoxane, doxorubicin, SP600125, SB203580, U0126, 5‐bromo‐2′‐deoxyuridine (BrdU), 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide (MTT), carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone (FCCP) and antimycin A were obtained from Sigma (St Louis, MO, USA). 2′,7′‐Dichlorodihydrofluorescein diacetate (DCFH‐DA), DAPI and JC‐1 were purchased from Invitrogen (Carlsbad, CA, USA). Oligomycin and primary antibodies against B‐cell lymphoma 2 (Bcl‐2), B‐cell lymphoma‐extra‐large (Bcl‐xl), cleaved caspase‐3, PARP and phospho‐p44/42 MAPK (ERK1/2) (Thr202/Tyr204) were obtained from Cell Signaling Technology (Beverly, MA, USA); antibodies against ERK2, dynamin‐related protein 1 (Drp1), haem oxygenase‐1 (HO1), manganese superoxide dismutase (MnSOD), mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), nuclear factor‐E2‐related factor 2 (Nrf2), mitochondrial transcription factor A (TFAM), optic atrophy 1 (OPA1), α‐Tubulin and β‐actin were obtained from Santa Cruz Biotechnology (Heidelberg, Germany). Antibodies against PPARGC‐1α, OxPhos complex I subunit NDUFS3, OxPhos complex II 30 kDa subunit, OxPhos complex III subunit core 1, OxPhos complex IV subunit I and OxPhos Complex V subunit α were obtained from Invitrogen. Secondary HRP‐conjugated anti‐rabbit IgG (H + L), anti‐mouse (H + L) IgG, and anti‐goat (H + L) IgG antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA). Collagenases I and II were obtained from MP Biomedicals (Southern California, USA). Western lysis buffer was obtained from Beyotime Biotechnology (Shanghai, Jiangsu, China). The Pierce ECL western blotting substrate and BCA™ protein assay kit were purchased from Thermo Scientific (Rockford, USA). The QIAamp DNA Mini Kit was obtained from QIAGEN (Düsseldorf, Germany). TRIzol reagent was from Roche (Basel, Switzerland). The PrimeScript reverse transcription (RT)‐PCR Kit and SYBR premix Ex Taq II were obtained from Takara (Tokyo, Japan). The primers were from ShengGong (Shanghai, China). The siRNA constructs were from Gene Pharma (Shanghai, China).
Results
ATRA protects H9c2 cells against doxorubicin‐induced death
The viability of H9c2 cells was reduced after doxorubicin treatment. Pretreatment with ATRA prevented doxorubicin‐induced loss of cell viability in a concentration‐dependent manner. Moreover, ATRA treatment alone did not affect cell viability (Figure 1A). For subsequent experiments, 1 or 2 μmol·L−1 ARTA was used. Furthermore, doxorubicin induced a reduction in Bcl‐2 and Bcl‐xl expression and activation of caspase‐3 and PARP. However, ATRA pretreatment effectively inhibited these changes in apoptosis‐associated proteins in H9c2 cells (Figure 1B and C).
Figure 1.
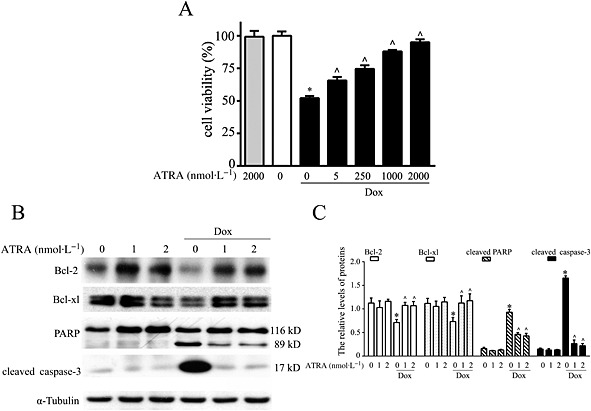
ATRA protects H9c2 cells from doxorubicin‐induced cell death. (A) ATRA inhibits doxorubicin‐induced viability loss in H9c2 cells (n = 5). (B) Representative images and (C) semi‐quantitative Western blot analysis of apoptosis‐associated proteins (Bcl‐2, Bcl‐xl, cleaved PARP and cleaved caspase‐3) (n = 5). Quantitative values are computed as the ratios of the density of the targeted protein to that of α‐tubulin. Data are expressed as the mean ± SEM and were analysed by anova. *P < 0.05 versus control and ^P < 0.05 versus Dox.
ATRA protects primary cardiomyocytes against doxorubicin‐induced cell death
Pretreatment with ATRA prevented doxorubicin‐induced viability loss in a concentration‐dependent manner (Figure 2A), and the reduction in Bcl‐2 and Bcl‐xl expression and caspase‐3 and PARP activation in primary cardiomyocytes (Figure 2B and C), findings that were similar to those in H9c2 cells.
Figure 2.
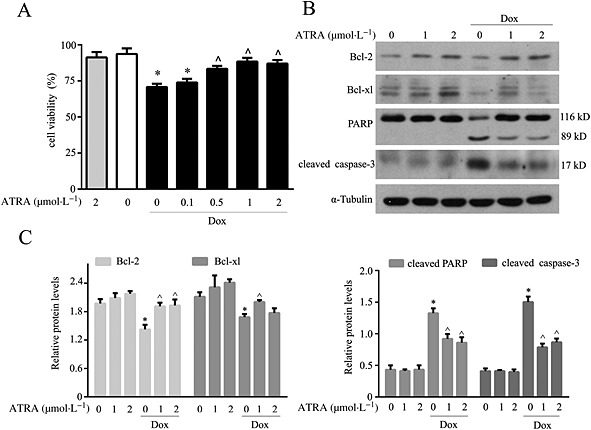
ATRA protects primary cardiomyocytes against doxorubicin‐induced cell death. (A) ATRA inhibits the doxorubicin‐induced viability loss in primary cardiomyocytes (n = 5). (B) Western blot analysis of apoptosis‐associated proteins (n = 5). (C) Quantitative results of apoptosis‐associated proteins (Bcl‐2, Bcl‐xl, cleaved PARP and cleaved caspase‐3). Quantitative values are computed as the ratios of density of the targeted protein to that of α‐tubulin. Data are expressed as the mean ± SEM and were analysed by anova. *P < 0.05 versus control and ^P < 0.05 versus Dox.
ATRA is more effective than resveratrol and dexrazoxane against doxorubicin‐induced cardiotoxicity
Both resveratrol and dexrazoxane showed significant protection against doxorubicin‐induced cardiotoxic effects only at a concentration of 100 μmol·L−1, while ATRA started to show protection at a concentration of 2 μmol·L−1, which is much lower than that used for resveratrol (100 μmol·L−1) and dexrazoxane (100 μmol·L−1) (Figure 3A). ATRA was more effective at scavenging ROS (Figure 3B) and inhibiting cell apoptosis (Figure 3C) than resveratrol and dexrazoxane at the same concentration (2 μmol·L−1). Moreover, 100 μmol·L−1 ATRA and dexrazoxane inhibited apoptosis better than resveratrol did at the same concentration (Figure 3D).
Figure 3.
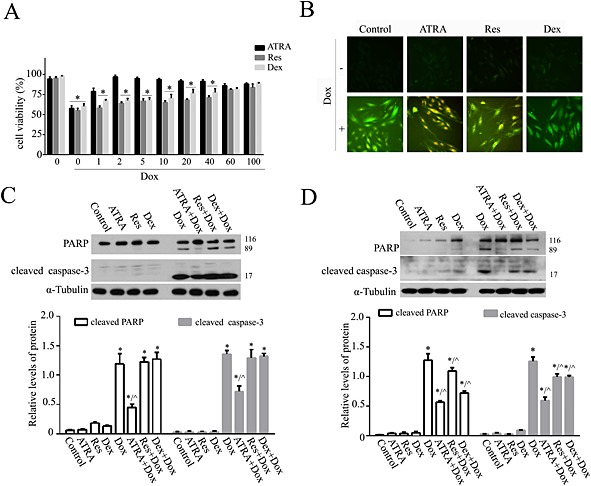
ATRA is more effective than resveratrol (Res) and dexrazoxane (Dex) against doxorubicin‐induced cardiotoxicity. (A) The differences among ATRA, resveratrol and dexrazoxane treatment against doxorubicin‐induced viability loss in H9c2 cells (n = 5). (B) DCFH‐DA (10 μmol·L−1) staining for the detection and evaluation of ROS. Representative images from each group (n = 6) are presented. Western blot analysis of the effects of (C) 2 and (D) 100 μmol·L−1 ATRA, resveratrol and dexrazoxane on doxorubicin‐induced apoptosis (cleaved PARP and cleaved caspase‐3) (n = 5). Above: representative images; below: semi‐quantitative analysis. Quantitative values are computed as the ratios of the density of the targeted protein to α‐tubulin. Data are expressed as the mean ± SEM and were analysed by anova. *P < 0.05 versus control and ^P < 0.05 versus Dox.
ATRA inhibits doxorubicin‐induced oxidative stress
Doxorubicin induced oxidative stress in H9c2 cells by provoking intracellular ROS generation (green fluorescence) and reducing the expression of phase II detoxifying enzymes (Nrf2, HO1 and MnSOD). Pretreatment with ATRA (1 and 2 μmol·L−1) abolished ROS generation (Figure 4A). Furthermore, ATRA induced an up‐regulation of Nrf2, HO 1 and MnSOD mRNA levels and protected against the doxorubicin‐induced decrease in Nrf2 and MnSOD mRNA levels (Figure 4B). ATRA prevented the decrease in the expression levels of the components associated with the phase II detoxifying enzyme system in doxorubicin‐treated H9c2 cells (Figure 4C and D).
Figure 4.
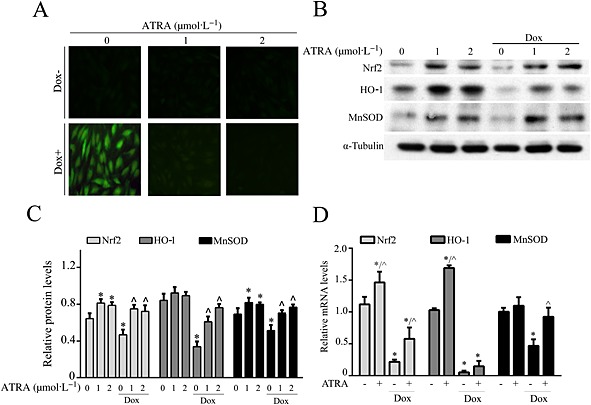
ATRA reduces oxidative stress in doxorubicin‐treated H9c2 cells. (A) DCFH‐DA staining for the detection and evaluation of ROS. Representative images from each group (n = 6) are presented. (B) Western blot analysis of phase II detoxifying enzymes (n = 5). (C) Semi‐quantitative analysis for (Western blot analysis) of phase II detoxifying enzymes (Nrf2, HO1 and MnSOD). (D) PCR analysis of the mRNA levels of phase II detoxifying enzymes in doxorubicin‐treated cells. Quantitative values are computed as the ratios of the density of the targeted protein to that of α‐tubulin. Except as noted, the concentrations of ATRA (1 or 2 μmol·L−1) and doxorubicin 3 μmol·L−1 respectively. Data are expressed as the mean ± SEM. Data were analysed by anova. *P < 0.05 versus control and ^P < 0.05 versus Dox.
ATRA attenuates doxorubicin‐induced mitochondrial dysfunction
The mitochondrial membrane integrity in doxorubicin‐treated H9c2 cells was measured by JC‐1 staining. A change from red/orange to green fluorescence indicates mitochondrial membrane integrity loss, which leads to a decrease in the MMP. As shown in Figure 5A, pretreatment with ATRA attenuated the doxorubicin‐induced MMP decrease in H9c2 cells. The mtDNA content was quantified by real‐time PCR and measured as the ratio of D‐loop to 18S rRNA levels (Figure 5B). As shown in Figure 5C, ATRA pretreatment prevented the decrease in the ratio of mitochondrial D‐loop/18S rRNA and mitochondrial respiration capacity (basal respiration and ATP potential) in doxorubicin‐treated H9c2 cells. Doxorubicin also modified the expression of proteins associated with mitochondrial biogenesis and dynamics in H9c2 cells. Specifically, there were decreases in the expression of PPARGC‐1α, TFAM, Mfn1 and OPA1 (long and short isoforms) but no changes in the expression of Mfn2 and Drp1. There were different expression patterns of the respiratory chain complex proteins in doxorubicin‐treated H9c2 cells: a significant decrease in Complex I and Complex II, a slight decrease in Complex III and Complex V and a significant increase in Complex IV. Moreover, ATRA pretreatment inhibited all of these changes in doxorubicin‐treated H9c2 cells (Figure 5D and I).
Figure 5.
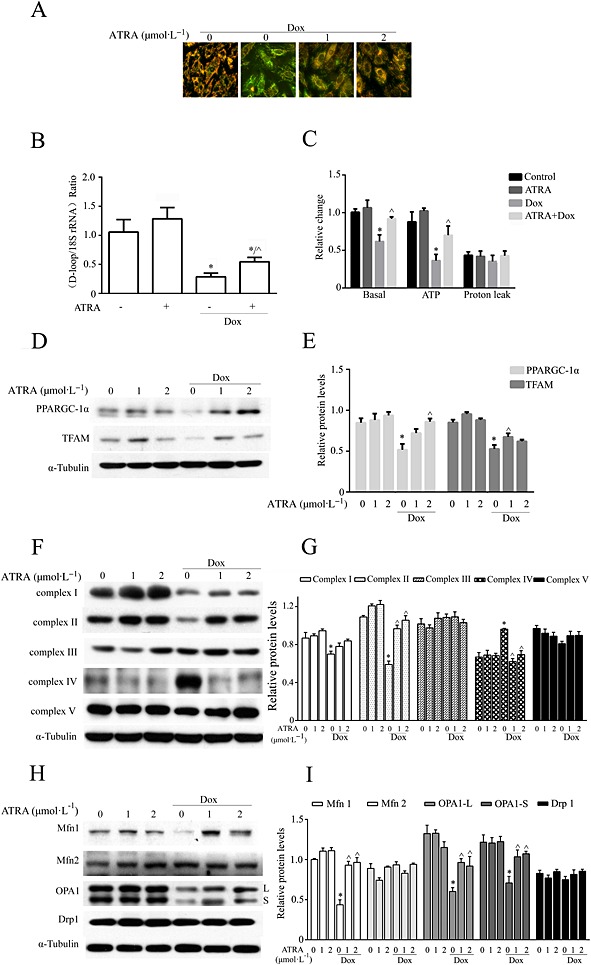
ATRA inhibits doxorubicin‐induced mitochondrial dysfunction in H9c2 cells. (A) JC‐1 staining for the MMP. Representative images from each group (n = 6) are presented. (B) Mitochondrial DNA copy number (n = 5). (C) Mitochondrial respiration capability (n = 5). Western blot analysis of (D) mitochondrial biogenesis‐associated proteins (PPARGC‐1α and TFAM), (F) mitochondrial respiratory chain complexes (Complexes I–V) and (H) mitochondrial dynamics‐associated proteins (Mfn1/2, OPA1 and Drp1). (E, G, I) Semi‐quantitative analysis of Western blots. Quantitative values are computed as the ratios of density of the targeted protein to that of α‐tubulin (n = 5). Except as noted, the concentrations of ATRA and Dox are 2 and 3 μmol·L−1 respectively. Data are expressed as the mean ± SEM and were analysed by anova. *P < 0.05 versus control and ^P < 0.05 versus Dox.
ERK signalling is involved in ATRA protection against doxorubicin‐induced cardiotoxicity
Various stimuli, such as cellular stresses and growth factors, can activate the MAPK family, which plays important roles in signal transduction. MAPK pathway inhibitors (U0126, PD98059 for ERK1/2, SB203580 for p38 and SP600125 for JNK) were used to determine which pathways are involved in ATRA cardioprotection. U0126, but not PD98059, SB203580 or SP600125, completely abolished the protective effect of ATRA against doxorubicin‐induced cardiotoxicity (Figure 6A). The Akt signalling pathway also plays an important role in doxorubicin‐induced cardiotoxicity. However, none of the Akt pathway inhibitors (LY294002 and wortmannin) abolished the ATRA protective effect against doxorubicin‐induced cardiotoxicity (Figure 6B). ATRA treatment evoked a marked up‐regulation of the phosphorylated ERK1/2 level in a time‐dependent manner, and the ERK phosphorylation levels were still significantly elevated at 24 h (Figure 6C‐i). The levels of ERK1/2 activated by ATRA were more significant than those of doxorubicin‐induced ERK1/2 activation in H9c2 cells. These results suggest that ATRA activates ERK1/2 to inhibit doxorubicin‐induced cell death signalling (Figure 6C‐ii). Moreover, U0126 almost completely blocked the ERK phosphorylation induced by ATRA (Figure 6C‐iii). Furthermore, U0126 abolished the cardioprotective effect of ATRA against doxorubicin‐induced apoptosis (caspase‐3 and PARP cleavage), oxidative stress (ROS generation), mitochondrial dysfunction (loss of mitochondrial membrane integrity) and ERK2 phosphorylation in H9c2 cells. U0126 significantly inhibited ATRA protection against doxorubicin cardiotoxicity (Figure 6D‐F). PD98059 significantly inhibited ERK1 and slightly inhibited ERK2 activation respectively. However, PD98059 did not abolish the protective effect of ATRA on cardiotoxicity (Figure 6G‐H). Taken together, these results show that ERK2 activation may play a key role in the cardioprotective effect of ATRA.
Figure 6.
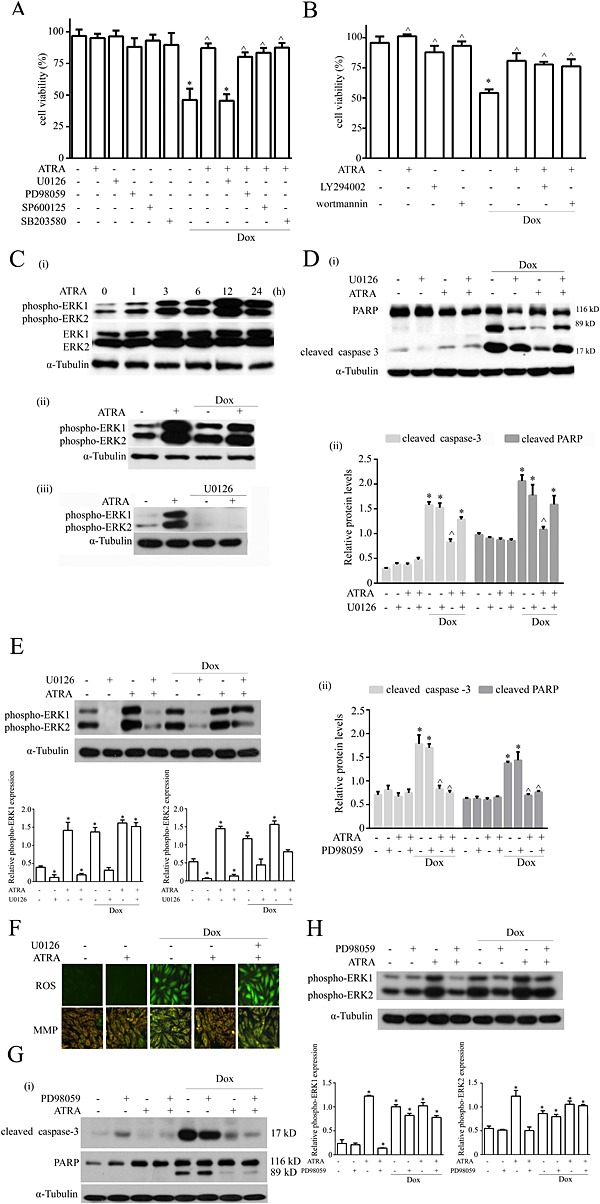
U0126 attenuates the cardioprotective effect of ATRA against doxorubicin‐induced damage in H9c2 cells. Effects of (A) MAPK and (B) Akt signalling pathway inhibitors on the protective effect of ATRA against doxorubicin‐induced cardiotoxicity (n = 5) (C) Western blotting for the effect of ATRA and U0126 on phosphorylated ERK1 and ERK2. (i) Doxorubicin and ATRA induced ERK1/2 activation in a time‐dependent manner. (ii) Effect of ATRA on ERK1/2 activation in doxorubicin‐induced cardiotoxicity. (iii) U0126 inhibited ATRA‐induced ERK1/2 activation. Western blotting for the inhibitory effects of (D) U0126 and (G) PD98059 on the protective effect of ATRA on doxorubicin‐induced cardiotoxicity in H9c2 cells, and quantitative analysis of apoptosis‐associated proteins (cleaved caspase‐3 and cleaved PARP). Quantitative values are computed as the ratios of the density of the targeted protein to that of α‐tubulin (n = 5). Western blotting for the effects of (E) U0126 and (H) PD98059 on phosphorylated ERK1 and ERK2 through inhibition of the ATRA effect on doxorubicin‐induced cardiotoxicity and semi‐quantitative analysis of ERK1 and ERK2. Quantitative values are computed as the ratios of the density of the targeted protein to that of α‐tubulin (n = 5). (F) ROS generation (DCFH‐DA staining) and the MMP (JC‐1 staining) abolished the protective effect of ATRA on doxorubicin‐induced cardiotoxicity in H9c2 cells. Representative images from each group (n = 6) are presented. Except as noted, the concentrations of ATRA and Dox were 2 and 3 μmol·L−1, respectively, the concentrations of inhibitors U0126, SB203580, SP600125, PD98059 and LY294002 were each 50 μmol·L−1, and the concentration of wortmannin was 1 μmol·L−1. Data are expressed as the mean ± SEM and were analysed by anova. *P < 0.05 versus control and ^P < 0.05 versus Dox.
ERK2 plays an important role in ATRA protection against doxorubicin‐induced cardiotoxicity
In addition, ERK1 and ERK2 siRNA were designed to confirm the results with U0126. The siRNAs were not toxic to H9c2 cells and showed efficient knockdown ability (Figure 7A and B). Knockdown of ERK1 or ERK2 partially or totally abolished the cardioprotection of ATRA against doxorubicin‐induced viability loss (Figure 7C), oxidative stress (ROS generation), mitochondrial dysfunction (loss of mitochondrial membrane integrity) (Figure 7D and E), and apoptosis (caspase‐3 and PARP cleavage) (Figure 7F and G) in H9c2 cells. Therefore, these results indicate that ERK, specifically ERK2, mediates the cardioprotective effect of ATRA against doxorubicin‐induced cardiotoxicity.
Figure 7.
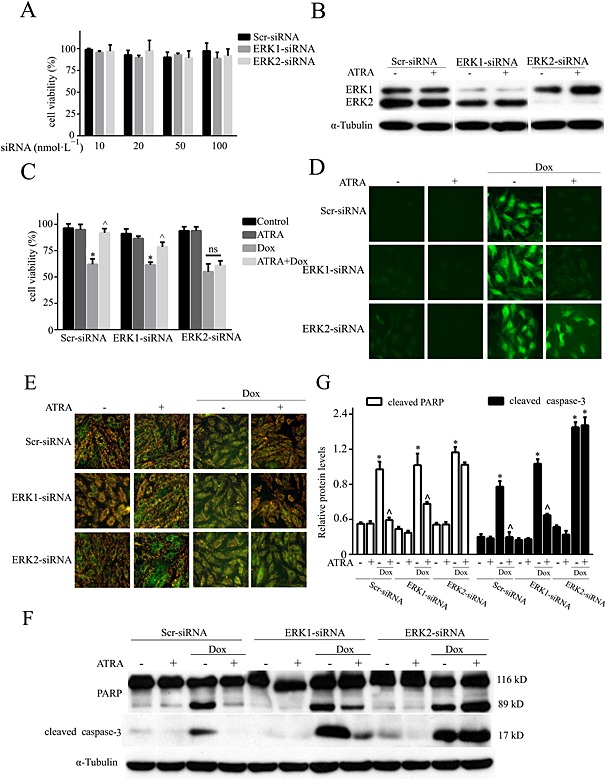
The role of ERK1 and ERK2 knockdown in the ATRA‐mediated protective effect on doxorubicin cardiotoxicity. (A) Toxicity of ERK1 and ERK2 siRNA in H9c2 cells; (B) silencing of ERK1 and ERK2 and ERK expression and activation under basal and ATRA‐stimulated conditions. Effects of ERK1 and ERK2 silencing on (C) cell viability, (D) ROS (DCFH‐DA staining), (E) MMP (JC‐1 staining) and (F) the levels of apoptosis‐associated proteins (Western blotting) in ATRA cardioprotection (n = 5). (G) Semi‐quantitative results for apoptosis‐associated proteins (cleaved PARP and cleaved caspase‐3). Quantitative values are computed as the ratios of the density of the targeted protein to that of α‐tubulin. Except as noted, the concentrations of ATRA and doxorubicin were 2 and 3 μmol·L−1 respectively. Data are expressed as the mean ± SEM and were analysed by anova. *P < 0.05 versus control and ^P < 0.05 versus Dox.
ATRA effects on doxorubicin‐induced cell death in gastric cancer cells
ATRA induced a loss of viability (Figure 8A) and the generation of ROS (Figure 8B) in both AGS and SGC‐7901 gastric cancer cells, although there were different responses to ERK, caspase‐3 and PARP activation in ATRA‐treated AGS and SGC‐7901 cells (Figure 8C and D). Moreover, ATRA did not compromise the oncological efficacy of doxorubicin in gastric carcinoma cells (Figure 8).
Figure 8.
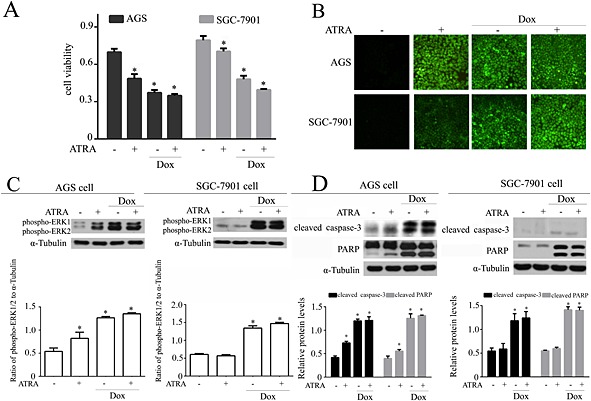
Effects of ATRA on doxorubicin‐induced cell death in gastric cancer cells. (A) ATRA‐induced viability loss in gastric cancer cells (n = 5). (B) ATRA‐induced ROS generation in gastric cancer cells. Representative images from each group (n = 6) are presented. Western blot and semi‐quantitative analysis for (C) ERK1/2 activation and (D) apoptosis‐associated proteins. Quantitative values are computed as the ratios of the density of the targeted protein to α‐tubulin (n = 5). Except as noted, the concentrations of ATRA and Dox were 2 and 3 μmol·L−1 respectively. Data are expressed as the mean ± SEM and were analysed by anova. *P < 0.05 versus control.
Discussion
The clinical efficacy of doxorubicin, one of the most effective anticancer drugs known, is severely limited for clinical application due to its concentration‐dependent cardiotoxicity (Minotti et al., 2004). The recent search for cardioprotective agents that can alleviate doxorubicin‐induced cardiotoxicity has only led to the identification of dexrazoxane as a possible candidate (Cvetkovic et al., 2005; Kane et al., 2008; Goey et al., 2010). However, its effectiveness is limited, and its long‐term benefits are unclear (Wiseman et al., 1998; Schlitt et al., 2014). The unique biological profile of ATRA has attracted considerable interest for many years, and it has been shown to have both neuro and hepatic protective effects (Nagy, 2012; Kim et al., 2013). In the present study it was revealed that ATRA can protect against doxorubicin‐induced cardiotoxicity. As shown in Figure 1, ATRA demonstrated strong cardioprotection in H9c2 cells. Although H9c2 cells are the typical in vitro cell model for cardiovascular disease research, particularly for doxorubicin‐induced cytotoxicity (Green et al., 2002; Chua et al., 2006; Li et al., 2006; Turakhia et al., 2007; Spagnuolo et al., 2011; Osman et al., 2013; Dong et al., 2014), there are some different characteristics between H9c2 cells and primary cardiomyocytes. Thus, we performed the same tests in primary cells and found that the effects of ATRA were in accordance with the conclusions obtained from the results in the from H9c2 cells (Figure 2).
It is essential to identify agents that can protect against doxorubicin‐induced cardiomyocyte apoptosis but without compromising its chemopreventive effects. Compared with that of resveratrol and dexrazoxane, the cardioprotective effect of ATRA against doxorubicin was mostly achieved at a much lower effective concentration. Although resveratrol can enhance the anticancer activity of doxorubicin and exert cardioprotective effects (Zhang et al., 2011; Xu et al., 2012), the efficacy of the inhibition of apoptosis and ROS generation was much lower than that of ATRA.
Previous research has demonstrated that the induction of oxidative stress, depletion of antioxidants and reduction of phase II detoxifying enzymes in response to doxorubicin have been implicated in many tissues such as the heart, kidney, liver and brain (Pal et al., 2012; Sterba et al., 2013; El‐Moselhy et al., 2014). Nrf2 is responsible for regulating the antioxidant response element (ARE)‐driven expression of genes encoding most antioxidant and phase II detoxification enzymes, such as glutamate–cysteine ligase, glutathione S‐transferase and HO1 (Bryan et al., 2013). In the present work, ATRA reduced doxorubicin‐induced oxidative stress by preventing ROS generation and activating the antioxidant defence system (HO1 and MnSOD). Consistent with our findings, the elevation of Nrf2, HO1 and SOD levels was shown to alleviate cardiotoxicity (Yu et al., 2013; Zhao et al., 2015). It is interesting that ATRA interferes with the recruitment of Nrf2 to the ARE through RARα and by reducing Nrf2 activity to increase the susceptibility to carcinogens in cancer cells (Wang et al., 2007), suggesting that ATRA may be able to evoke cardioprotection simultaneously with chemosensitization in anticancer therapy. The MEK/ERK signalling pathway can activate the expression of Nrf2 activity and nuclear translocation, and MEK/ERK inhibitors block the up‐regulation of Nrf2 expression (Zipper et al., 2003; Yuan et al., 2006; Copple, 2012; Bryan et al., 2013; Richter et al., 2014). Combining ATRA with doxorubicin activated the ERK pathway and induced Nrf2 expression in the present work, and ERK activation was shown to be important for the induction of the phase II detoxification enzyme involved in ATRA's cardioprotective effect.
Mitochondrial dysfunction is involved in cardiac energy deficits, which contribute to heart failure (Rosca et al., 2013). The present work demonstrated that doxorubicinin impaired the MMP, caused mtDNA lesions, had a deleterious effect on the respiratory chain and the expression of mitochondrial biogenesis, as well as decreasing protein dynamics. Hence, the restoration of mitochondrial biogenesis has been shown to contribute to the attenuation of doxorubicin‐induced cardiotoxicity (Miyagawa et al., 2010). Mitochondrial dynamics has emerged as an important active mechanism that affects normal mitochondrial function in heart failure (Marin‐Garcia et al., 2013). Aerobic exercise training and resveratrol have been shown to increase Mfn1/2 in doxorubicin‐treated mice (Dolinsky et al., 2013). Consistent with our findings, mitochondrial function can be improved by ATRA and its derivatives (Berdanier et al., 2001; Chiu et al., 2008; Choudhary et al., 2008; Lin et al., 2008; Klepinin et al., 2014). SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics to protect cells from doxorubicin‐mediated cell death (Samant et al., 2014). SIRT3 is likely to affect a specific response to ATRA (Krupkova et al., 2014). This possibility is interesting and warrants further investigation regarding ATRA‐regulated OPA1 deacetylation in our work. In previous studies, vitamin A was found to enhance the transcription of ATPase 6 (Berdanier et al., 2001). Furthermore, 9‐cis retinoic acid induced the expression of retinoid X receptor localized to the mitochondria and significantly increased the expression of ND1, ND6 and complex I RNA (Lin et al., 2008). However, effects of ATRA on mitochondrial biogenesis and mitochondrial dynamics through RARα in cardiomyocytes have not yet been reported.
There is accumulating evidence that the MAPK superfamily plays an important role in protecting against cardiomyocyte death (Adderley et al., 1999; Zhu et al., 1999; Khan et al., 2006). ERK is particularly implicated in the biological processes of cardiomyocytes, and ERK inhibition exacerbates cardiomyocyte death and heart function impairments (Purcell et al., 2007). ERK activation in doxorubicin‐induced cardiotoxicity offers cardioprotection against apoptosis (Fryer et al., 2001; Su et al., 2006; Xiang et al., 2009). In contrast, the inhibition of ERK activation aggravates oxidative stress‐induced cardiomyocyte apoptosis (Adderley and Fitzgerald, 1999). PD98059 mediates its inhibitory properties by binding to the ERK‐specific MAPK MEK, thereby preventing the phosphorylation of ERK1/2 (p44/p42 MAPK) by MEK1/2(Alessi et al., 1995). PD98059 binds to the inactive forms of ERK1 and prevents activation by upstream activators such as ATRA in our study. U0126 can inhibit both ERK1 and ERK2, while PD98059 inhibits ERK1 more potently than ERK2 (Crews et al., 1992; Favata et al., 1998). U0126 can inhibit both ERK1 and ERK2, while PD98059 inhibits ERK1 more potently than ERK2. PD98059 did not inhibit ERK2 completely, so PD98059 failed to abolish the protective effects of ATRA. Therefore, in our study, U0126 and siRNA inhibited ERK2 and totally abolished ATRA‐mediated cardioprotection in H9c2 cells. Doxorubicin has been shown to activate ERK1/2 to induce cell death previously (Cagnol et al., 2010). In our studies, ATRA‐activated ERK provided a self‐protection mechanism. A difference in the extent of ATRA‐induced ERK activation between cardiomyocytes and gastric cancer cells may be the core reason why ATRA acts differently to prevent doxorubicin‐induced cell death.
ATRA exhibits not only anticancer effects (Mangiarotti et al., 1998; Liu et al., 2003; Rosenberg et al., 2008; Long et al., 2014) but also acts as a new class of cardioprotective agent (Bernard et al., 2011; Guleria et al., 2011; Kim et al., 2013). The results of this study also raise the possibility that ATRA may have therapeutic potential to protect the heart against the toxicity of cancer chemotherapies, suggesting that these compounds, like vincristine (Chatterjee et al., 2008), deserve further extensive pharmacology and preclinical investigations.
Taken together, the present work showed that ATRA has more potential as an effective agent against doxorubicin‐induced cardiotoxicity than dexrazoxane and resveratrol. Moreover, ERK2 activation may mediate the cardioprotective effect of ATRA against doxorubicin‐induced cardiotoxicity by attenuating oxidative stress and restoring mitochondrial function.
Author contributions
L. Y. and C. L. performed the research. L. Y., C. L. and J. L. designed the research study. X. W., C. C. and W. S. contributed essential reagents or tools. L. Y. analysed the data. L. Y., C. L. and J. L. wrote the paper.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was partially supported by the National Basic Research Program (No.2015CB553602) and the National “Twelfth Five‐Year” Plan for Science & Technology Support (2012BAH30F03).
Yang, L. , Luo, C. , Chen, C. , Wang, X. , Shi, W. , and Liu, J. (2016) All‐trans retinoic acid protects against doxorubicin‐induced cardiotoxicity by activating the ERK2 signalling pathway. British Journal of Pharmacology, 173: 357–371. doi: 10.1111/bph.13377.
References
- Adderley SR, Fitzgerald DJ (1999). Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2‐mediated induction of cyclooxygenase‐2. J Biol Chem 274: 5038–5046. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR (1995). PD 098059 is a specific inhibitor of the activation of mitogen‐activated protein kinase kinase in vitro and in vivo . J Biol Chem 270: 27489–27494. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. (2013a). The concise guide to PHARMACOLOGY 2013/14: nuclear hormone receptors. Br J Pharmacol 170: 1652–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. (2013b). The concise guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdanier CD, Everts HB, Hermoyian C, Mathews CE (2001). Role of vitamin A in mitochondrial gene expression. Diabetes Res Clin Pract 54: S11–S27. [DOI] [PubMed] [Google Scholar]
- Bernard Y, Ribeiro N, Thuaud F, Turkeri G, Dirr R, Boulberdaa M, et al. (2011). Flavaglines alleviate doxorubicin cardiotoxicity: implication of Hsp27. PLoS One 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan HK, Olayanju A, Goldring CE, Park BK (2013). The Nrf2 cell defence pathway: Keap1‐dependent and ‐independent mechanisms of regulation. Biochem Pharmacol 85: 705–717. [DOI] [PubMed] [Google Scholar]
- Cagnol S, Chambard JC (2010). ERK and cell death: mechanisms of ERK‐induced cell death–apoptosis, autophagy and senescence. FEBS J 277: 2–21. [DOI] [PubMed] [Google Scholar]
- Chatterjee K, Zhang J, Tao R, Honbo N, Karliner JS (2008). Vincristine attenuates doxorubicin cardiotoxicity. Biochem Biophys Res Commun 373: 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Martinez AA, Morado J, Scofield V, Roberts JL, Maffi SK (2013). Retinoic acid protects against proteasome inhibition associated cell death in SH‐SY5Y cells via the AKT pathway. Neurochem Int 62: 31–42. [DOI] [PubMed] [Google Scholar]
- Chiu HJ, Fischman DA, Hammerling U (2008). Vitamin A depletion causes oxidative stress, mitochondrial dysfunction, and PARP‐1‐dependent energy deprivation. FASEB J 22: 3878–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlopcikova S, Psotova J, Miketova P (2001). Neonatal rat cardiomyocytes – a model for the study of morphological, biochemical and electrophysiological characteristics of the heart. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 145: 49–55. [PubMed] [Google Scholar]
- Choudhary R, Baker KM, Pan J (2008). All‐trans retinoic acid prevents angiotensin II‐ and mechanical stretch‐induced reactive oxygen species generation and cardiomyocyte apoptosis. J Cell Physiol 215: 172–181. [DOI] [PubMed] [Google Scholar]
- Chua CC, Liu X, Gao J, Hamdy RC, Chua BH (2006). Multiple actions of pifithrin‐alpha on doxorubicin‐induced apoptosis in rat myoblastic H9c2 cells. Am J Physiol Heart Circ Physiol 290: H2606–H2613. [DOI] [PubMed] [Google Scholar]
- Copple IM (2012). The Keap1‐Nrf2 cell defense pathway – a promising therapeutic target? Adv Pharmacol 63: 43–79. [DOI] [PubMed] [Google Scholar]
- Crews CM, Alessandrini A, Erikson RL (1992). The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science 258: 478–480. [DOI] [PubMed] [Google Scholar]
- Cvetkovic RS, Scott LJ (2005). Dexrazoxane: a review of its use for cardioprotection during anthracycline chemotherapy. Drugs 65: 1005–1024. [DOI] [PubMed] [Google Scholar]
- Dolinsky VW, Rogan KJ, Sung MM, Zordoky BN, Haykowsky MJ, Young ME, et al. (2013). Both aerobic exercise and resveratrol supplementation attenuate doxorubicin‐induced cardiac injury in mice. Am J Physiol Endocrinol Metab 305: E243–E253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Q, Chen L, Lu Q, Sharma S, Li L, Morimoto S, et al. (2014). Quercetin attenuates doxorubicin cardiotoxicity by modulating Bmi‐1 expression. Br J Pharmacol 171: 4440–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Moselhy MA, El‐Sheikh AA (2014). Protective mechanisms of atorvastatin against doxorubicin‐induced hepato–renal toxicity. Biomed Pharmacother 68: 101–110. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, et al. (1998). Identification of a novel inhibitor of mitogen‐activated protein kinase kinase. J Biol Chem 273: 18623–18632. [DOI] [PubMed] [Google Scholar]
- Fryer RM, Pratt PF, Hsu AK, Gross GJ (2001). Differential activation of extracellular signal regulated kinase isoforms in preconditioning and opioid‐induced cardioprotection. J Pharmacol Exp Ther 296: 642–649. [PubMed] [Google Scholar]
- Goey AK, Schellens JH, Beijnen JH, Huitema AD (2010). Dexrazoxane in anthracycline induced cardiotoxicity and extravasation. Ned Tijdschr Geneeskd 154: A1155. [PubMed] [Google Scholar]
- Green PS, Leeuwenburgh C (2002). Mitochondrial dysfunction is an early indicator of doxorubicin‐induced apoptosis. Biochim Biophys Acta 9: 94–101. [DOI] [PubMed] [Google Scholar]
- Gu J, Song ZP, Gui DM, Hu W, Chen YG, Zhang DD (2012). Resveratrol attenuates doxorubicin‐induced cardiomyocyte apoptosis in lymphoma nude mice by heme oxygenase‐1 induction. Cardiovasc Toxicol 12: 341–349. [DOI] [PubMed] [Google Scholar]
- Guleria RS, Choudhary R, Tanaka T, Baker KM, Pan J (2011). Retinoic acid receptor‐mediated signaling protects cardiomyocytes from hyperglycemia induced apoptosis: role of the renin‐angiotensin system. J Cell Physiol 226: 1292–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi M, Masaki M, Hiramoto Y, Sugiyama S, Kuroda T, Terai K, et al. (2006). Cross‐talk between bone morphogenetic protein 2 and leukemia inhibitory factor through ERK 1/2 and Smad1 in protection against doxorubicin‐induced injury of cardiomyocytes. J Mol Cell Cardiol 40: 224–233. [DOI] [PubMed] [Google Scholar]
- Kane RC, McGuinn WD Jr, Dagher R, Justice R, Pazdur R (2008). Dexrazoxane (Totect): FDA review and approval for the treatment of accidental extravasation following intravenous anthracycline chemotherapy. Oncologist 13: 445–450. [DOI] [PubMed] [Google Scholar]
- Khan M, Varadharaj S, Ganesan LP, Shobha JC, Naidu MU, Parinandi NL, et al. (2006). C‐phycocyanin protects against ischemia‐reperfusion injury of heart through involvement of p38 MAPK and ERK signaling. Am J Physiol Heart Circ Physiol 290: H2136–H2145. [DOI] [PubMed] [Google Scholar]
- Kim JH, Yu KS, Jeong JH, Lee NS, Lee JH, Jeong YG, et al. (2013). All‐trans‐retinoic acid rescues neurons after global ischemia by attenuating neuroinflammatory reactions. Neurochem Res 38: 2604–2615. [DOI] [PubMed] [Google Scholar]
- Klepinin A, Chekulayev V, Timohhina N, Shevchuk I, Tepp K, Kaldma A, et al. (2014). Comparative analysis of some aspects of mitochondrial metabolism in differentiated and undifferentiated neuroblastoma cells. J Bioenerg Biomembr 46: 17–31. [DOI] [PubMed] [Google Scholar]
- Krupkova M, Liska F, Sedova L, Krenova D, Kren V, Seda O (2014). Pharmacogenomic analysis of retinoic‐acid induced dyslipidemia in congenic rat model. Lipids Health Dis 13: 13–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmicic J, Del Campo A, Lopez‐Crisosto C, Morales PE, Pennanen C, Bravo‐Sagua R, et al. (2011). Mitochondrial dynamics: a potential new therapeutic target for heart failure. Rev Esp Cardiol 64: 916–923. [DOI] [PubMed] [Google Scholar]
- Kuznetsov AV, Margreiter R, Amberger A, Saks V, Grimm M (2011). Changes in mitochondrial redox state, membrane potential and calcium precede mitochondrial dysfunction in doxorubicin‐induced cell death. Biochim Biophys Acta 6: 21. [DOI] [PubMed] [Google Scholar]
- Li K, Sung RY, Huang WZ, Yang M, Pong NH, Lee SM, et al. (2006). Thrombopoietin protects against in vitro and in vivo cardiotoxicity induced by doxorubicin. Circulation 113: 2211–2220. [DOI] [PubMed] [Google Scholar]
- Lin Y‐W, Lien L‐M, Yeh T‐S, Wu H‐M, Liu Y‐L, Hsieh R‐H (2008). 9‐cis retinoic acid induces retinoid X receptor localized to the mitochondria for mediation of mitochondrial transcription. Biochem Biophys Res Commun 377: 351–354. [DOI] [PubMed] [Google Scholar]
- Liu H, Zang C, Fenner MH, Possinger K, Elstner E (2003). PPARgamma ligands and ATRA inhibit the invasion of human breast cancer cells in vitro. Breast Cancer Res Treat 79: 63–74. [DOI] [PubMed] [Google Scholar]
- Long ZJ, Hu Y, Li XD, He Y, Xiao RZ, Fang ZG, et al. (2014). ATO/ATRA/anthracycline‐chemotherapy sequential consolidation achieves long‐term efficacy in primary acute promyelocytic leukemia. PLoS One 9: e104610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou S, Zhong L, Yang X, Xue T, Gai R, Zhu D, et al. (2013). Efficacy of all‐trans retinoid acid in preventing nickel induced cardiotoxicity in myocardial cells of rats. Food Chem Toxicol 51: 251–258. [DOI] [PubMed] [Google Scholar]
- Mangiarotti R, Danova M, Alberici R, Pellicciari C (1998). All‐trans retinoic acid (ATRA)‐induced apoptosis is preceded by G1 arrest in human MCF‐7 breast cancer cells. Br J Cancer 77: 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin‐Garcia J, Akhmedov AT, Moe GW (2013). Mitochondria in heart failure: the emerging role of mitochondrial dynamics. Heart Fail Rev 18: 439–456. [DOI] [PubMed] [Google Scholar]
- Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L (2004). Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 56: 185–229. [DOI] [PubMed] [Google Scholar]
- Miyagawa K, Emoto N, Widyantoro B, Nakayama K, Yagi K, Rikitake Y, et al. (2010). Attenuation of Doxorubicin‐induced cardiomyopathy by endothelin‐converting enzyme‐1 ablation through prevention of mitochondrial biogenesis impairment. Hypertension 55: 738–746. [DOI] [PubMed] [Google Scholar]
- Nagy L (2012). Would eating carrots protect your liver? A new role involving NKT cells for retinoic acid in hepatitis. Eur J Immunol 42: 1677–1680. [DOI] [PubMed] [Google Scholar]
- Nizamutdinova IT, Guleria RS, Singh AB, Kendall JA Jr, Baker KM, Pan J (2013). Retinoic acid protects cardiomyocytes from high glucose‐induced apoptosis through inhibition of NF‐kappaB signaling pathway. J Cell Physiol 228: 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman AM, Al‐Harthi SE, Alarabi OM, Elshal MF, Ramadan WS, Alaama MN, et al. (2013). Chemosensetizing and cardioprotective effects of resveratrol in doxorubicin‐treated animals. Cancer Cell Int 13: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Ahir M, Sil PC (2012). Doxorubicin‐induced neurotoxicity is attenuated by a 43‐kD protein from the leaves of Cajanus indicus L. via NF‐kappaB and mitochondria dependent pathways. Free Radic Res 46: 785–798. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell NH, Wilkins BJ, York A, Saba‐El‐Leil MK, Meloche S, Robbins J, et al. (2007). Genetic inhibition of cardiac ERK1/2 promotes stress‐induced apoptosis and heart failure but has no effect on hypertrophy in vivo . Proc Natl Acad Sci U S A 104: 14074–14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao J, Zhang C, Wang P, Lu L, Zhang F (2010). All‐trans retinoic acid alleviates hepatic ischemia/reperfusion injury by enhancing manganese superoxide dismutase in rats. Biol Pharm Bull 33: 869–875. [DOI] [PubMed] [Google Scholar]
- Richter M, Winkel AF, Schummer D, Gerlitz M, de Hoop M, Brunner B, et al. (2014). Pau d'arco activates Nrf2‐dependent gene expression via the MEK/ERK‐pathway. J Toxicol Sci 39: 353–361. [DOI] [PubMed] [Google Scholar]
- Rosca MG, Hoppel CL (2013). Mitochondrial dysfunction in heart failure. Heart Fail Rev 18: 607–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg PB, Mielke MM, Tschanz J, Cook L, Corcoran C, Hayden KM, et al. (2008). Effects of cardiovascular medications on rate of functional decline in Alzheimer disease . Am J Geriatr Psychiatry 16: 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, et al. (2014). SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol 34: 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitt A, Jordan K, Vordermark D, Schwamborn J, Langer T, Thomssen C (2014). Cardiotoxicity and oncological treatments. Dtsch Arztebl Int 111: 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoncikova P, Ravingerova T, Barancik M (2008). The effect of chronic doxorubicin treatment on mitogen‐activated protein kinases and heat stress proteins in rat hearts. Physiol Res 57: S97–S102. [DOI] [PubMed] [Google Scholar]
- Spagnuolo RD, Recalcati S, Tacchini L, Cairo G (2011). Role of hypoxia‐inducible factors in the dexrazoxane‐mediated protection of cardiomyocytes from doxorubicin‐induced toxicity. Br J Pharmacol 163: 299–312. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sterba M, Popelova O, Vavrova A, Jirkovsky E, Kovarikova P, Gersl V, et al. (2013). Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal 18: 899–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su HF, Samsamshariat A, Fu J, Shan YX, Chen YH, Piomelli D, et al. (2006). Oleylethanolamide activates Ras‐Erk pathway and improves myocardial function in doxorubicin‐induced heart failure. Endocrinology 147: 827–834. [DOI] [PubMed] [Google Scholar]
- Turakhia S, Venkatakrishnan CD, Dunsmore K, Wong H, Kuppusamy P, Zweier JL, et al. (2007). Doxorubicin‐induced cardiotoxicity: direct correlation of cardiac fibroblast and H9c2 cell survival and aconitase activity with heat shock protein 27. Am J Physiol Heart Circ Physiol 293: H3111–H3121. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Hayes JD, Henderson CJ, Wolf CR (2007). Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc Natl Acad Sci U S A 104: 19589–19594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggert B, Bergsma DR, Helmsen R, Chader GJ (1978). Vitamin‐a receptors – retinoic acid binding in ocular tissues. Biochem J 169: 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman LR, Spencer CM (1998). Dexrazoxane. A review of its use as a cardioprotective agent in patients receiving anthracycline‐based chemotherapy. Drugs 56: 385–403. [DOI] [PubMed] [Google Scholar]
- Xiang P, Deng HY, Li K, Huang GY, Chen Y, Tu L, et al. (2009). Dexrazoxane protects against doxorubicin‐induced cardiomyopathy: upregulation of Akt and Erk phosphorylation in a rat model. Cancer Chemother Pharmacol 63: 343–349. [DOI] [PubMed] [Google Scholar]
- Xu X, Chen K, Kobayashi S, Timm D, Liang Q (2012). Resveratrol attenuates doxorubicin‐induced cardiomyocyte death via inhibition of p70 S6 kinase 1‐mediated autophagy. J Pharmacol Exp Ther 341: 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Cui L, Zhang Z, Zhao Q, Li S (2013). α‐Linolenic acid attenuates doxorubicin‐induced cardiotoxicity in rats through suppression of oxidative stress and apoptosis. Acta Biochim Biophys Sin 45: 817–826. [DOI] [PubMed] [Google Scholar]
- Yuan X, Xu C, Pan Z, Keum YS, Kim JH, Shen G, et al. (2006). Butylated hydroxyanisole regulates ARE‐mediated gene expression via Nrf2 coupled with ERK and JNK signaling pathway in HepG2 cells. Mol Carcinog 45: 841–850. [DOI] [PubMed] [Google Scholar]
- Zhang C, Feng Y, Qu S, Wei X, Zhu H, Luo Q, et al. (2011). Resveratrol attenuates doxorubicin‐induced cardiomyocyte apoptosis in mice through SIRT1‐mediated deacetylation of p53. Cardiovasc Res 90: 538–545. [DOI] [PubMed] [Google Scholar]
- Zhao M, Guo H, Chen J, Wang J, Huang H, Zheng S, et al. (2015). 5‐aminolevulinic acid combined with sodium ferrous citrate ameliorates H2O2‐induced cardiomyocyte hypertrophy via activation of the MAPK/Nrf2/HO‐1 pathway. Am J Physiol Cell Physiol 308: 4. [DOI] [PubMed] [Google Scholar]
- Zhu W, Zou Y, Aikawa R, Harada K, Kudoh S, Uozumi H, et al. (1999). MAPK superfamily plays an important role in daunomycin‐induced apoptosis of cardiac myocytes. Circulation 100: 2100–2107. [DOI] [PubMed] [Google Scholar]
- Zipper LM, Mulcahy RT (2003). Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicol Sci 73: 124–134. [DOI] [PubMed] [Google Scholar]