

Abstract
Background and Purpose
Sepsis is a life‐threatening clinical condition characterized by uncontrolled inflammatory responses and is a major cause of death in intensive care units. Histone deacetylase (HDAC) inhibitors have recently exhibited anti‐inflammatory properties. MAPK phosphatase (MKP) suppresses MAPK signalling, which plays an important role in inflammatory responses. The purpose of this study was to investigate the protective mechanisms of Compound 9a, a newly synthetized HDAC inhibitor, against septic injury.
Experimental Approach
The anti‐inflammatory properties of Compound 9a were assayed in LPS‐stimulated RAW264.7 cells. In vivo, polymicrobial sepsis was induced in C57BL/6 mice by caecal ligation and puncture (CLP). The mice were treated with Compound 9a (i.p., 10 mg∙kg−1) 2 h before and immediately after CLP.
Key Results
Compound 9a inhibited the increased production of TNF‐α, IL‐6 and NO in LPS‐stimulated RAW264.7 cells. In mice with CLP, Compound 9a improved survival rate, attenuated organ injuries and decreased serum TNF‐α and IL‐6 levels. CLP increased expression of toll‐like receptor 4, phosphorylated (p)‐p38, p‐JNK and p‐ERK proteins, which was attenuated by Compound 9a. Compound 9a decreased MKP‐1 association with HDAC1 and enhanced MKP‐1 acetylation and enhanced MKP‐1 association with p‐p38 and p‐ERK. Moreover, the inhibitory effects of Compound 9a on serum cytokine levels and phosphorylation of MAPK were abolished by MKP‐1 siRNA.
Conclusions and Implications
Our findings suggest that Compound 9a protected against septic injury by suppressing MAPK‐mediated inflammatory signalling.
Abbreviations
- Ac‐K
acetylated‐lysine
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BUN
blood urea nitrogen
- CLP
caecal ligation and puncture
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- MKP
MAPK phosphatase
- MTT
tetrazolium bromide
- SAHA
suberoylanilide hydroxamic acid
- TLR
toll‐like receptor
- TSA
trichostatin A
Tables of Links
| TARGETS |
|---|
| Enzymes a |
| HDAC1, histone deacetylase 1 |
| MAPK 1, ERK2 |
| MAPK 3, ERK1 |
| MAPK 8, JNK1 |
| MAPK 9, JNK2 |
| MAPK 14, p38α |
| Catalytic receptors b |
| TLR4 |
| LIGANDS |
|---|
| SAHA, vorinostat |
| TSA, trichostatin A |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a bAlexander et al., 2015a, 2015b).
Introduction
Sepsis is characterized by a systemic inflammatory response syndrome that results from a harmful host response to infection and is a major cause of morbidity and mortality in intensive care units. An uncontrolled hyper‐inflammatory response and inappropriate cytokine response during early sepsis have been proposed to cause multiple organ dysfunction syndrome during sepsis. Therefore, controlling inflammation during early sepsis may reduce organ injury and prevent death after septic insult. The liver is a major organ of the innate immune system and the inflammatory process in the liver contributes to diverse pathological events. Given the abundance of resident macrophages and the extensive endothelial surfaces in liver, it is highly likely that this organ plays a prominent role in the host immune response to infection (Wang and Chaudry, 1996). Moreover, the liver is believed to be a vulnerable organ responsible for the initiation of multiple organ dysfunction syndrome, which is the most lethal complication of sepsis.
Histone deacetylase (HDAC) tightly regulates gene expression through histone deacetylation. Because HDAC inhibitors can modulate gene expression, several HDAC inhibitors have been developed and are in clinical trials as novel anti‐cancer agents (Pan et al., 2007). Recent studies have found that HDAC inhibitors had pronounced anti‐inflammatory properties because they altered the acetylation status of non‐histone proteins, such as cell‐signalling proteins and transcription factors (Kim et al., 2001). Trichostatin A (TSA) significantly ameliorated histological glomerulonephritis and reduced inflammatory cytokines, such as IL‐6 and IFN‐γ in a model of systemic lupus erythematosus (Mishra et al., 2003). Suberoylanilide hydroxamic acid (SAHA) improved survival and attenuated pulmonary neutrophil infiltration in septic shock (Li et al., 2009). However, the molecular mechanisms underlying anti‐inflammatory effects of HDAC inhibitors during sepsis are not completely understood.
The MAPK family is a key component of inflammation and stress‐induced signal transduction cascades, which regulates a variety of cellular processes. The MAPK family includes p38, JNK and ERK; all of which are activated by phosphorylation (Chang and Karin, 2001). In earlier studies, JNK‐deficient mice had decreased serum levels of IL‐6 and IL‐1β in response to LPS. PD98059, an ERK‐specific inhibitor, reduced both activator protein 1 activity and expression of TNF mRNA in LPS‐stimulated macrophages (Lo et al., 2000). Furthermore, inhibiting MAPK signalling with tyrosine kinase inhibitors prevented LPS‐induced lethal toxicity (Novogrodsky et al., 1994). TSA inhibited JNK and p38 activation in LPS‐stimulated HUVECs (Hsu et al., 2011), and SAHA suppressed p38 and ERK activation in an endotoxemia model (Finkelstein et al., 2010). MAPK phosphatase (MKP) is a dual‐specificity phosphatase that inactivates MAPK signalling by dephosphorylation (Camps et al., 2000). MKP‐1 knockout mice with polymicrobial sepsis had more colony forming units in liver and increased mortality (Hammer et al., 2010).
The purpose of this study was to investigate the protective mechanisms of a newly synthetized HDAC inhibitor, Compound 9a, in sepsis, particularly with regard to MAPK signalling associated with inflammation.
Methods
Cell culture and treatment
RAW264.7 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin and maintained at 37°C in an atmosphere of 5% CO2. Six hours after incubation, the cells were starved with DMEM with 0.5% FBS. Eighteen hours after starvation, the cells were treated with vehicle (1% DMSO) or various concentrations (0.1, 0.5, 2.5 and 12.5 μM) of Compound 9a or SAHA. LPS (1 μg∙mL−1) was added 1 h after Compound 9a or SAHA treatment, and the culture media were harvested for assay.
Cell viability
RAW264.7 cells were seeded in 96‐well plates, incubated for 24 h in DMEM with 10% FBS and 1% penicillin/streptomycin and maintained at 37°C in an atmosphere of 5% CO2. The cells were treated with vehicle (1% DMSO) or various concentrations (0.1, 0.5, 2.5, 12.5 and 62.5 μM) of Compound 9a or SAHA. After 24 h, 100 μL of MTT solution (5 mg∙mL−1 in PBS) was added to each well, and the cells were incubated for 3 h. The medium was removed, and 100 μL of DMSO was added to each well to solubilize any deposited formazan. The OD of each well was measured at 450 nm with a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
NO and cytokine levels
To determine the level of NO production in media, equal volumes of culture medium and Griess reagent (1 part of 0.1% naphtylethylenediamine dihydrochloride in distilled water and 1 part of 1% sulfanilamide in 5% concentrated phosphoric acid) were mixed, and the absorbance of the mixture at 540 nm was measured with a microplate reader. TNF‐α and IL‐6 levels were determined with commercially available ELISA kits (BD Biosciences, San Jose, CA, USA) used according to the manufacturer's instructions.
Animals
All animal care and experimental procedures complied with the guidelines of the National Institutes of Health (NIH publication No. 86‐23, revised 1985) and were approved by the Sungkyunkwan University Animal Care Committee (SU12‐03), with adherence to the 3Rs (replacement, refinement and reduction). Reporting of the animal studies follows the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Male C57BL/6 mice weighing 23–25 g (8 weeks) were supplied by Orient Bio (Seongnam, Korea). The animals were housed in cages (4 per cage) located in temperature‐controlled rooms with a 12 h light–dark cycle and received water and food ad libitum.
Experimental procedures
Caecal ligation and puncture (CLP) in rodents has become the most widely used model for experimental sepsis and is currently considered as the gold standard in sepsis research (Heuer et al., 2004). Thus, we used this model for studying the effects of Compound 9a on the underlying mechanisms of sepsis. Randomization was conducted by an individual other than the operator. The animals were selected randomly from the pool of all cages eligible for inclusion in the study and randomly divided into the experimental groups.
Polymicrobial sepsis was induced by CLP as described by Chaudry et al. (1979). Briefly, mice were anaesthetized with ketamine (100 mg∙kg−1; Yuhan Corporation, Seoul, Korea) and xylazine (10 mg∙kg−1; Boehringer Ingelheim, St. Joseph, MO, USA). After anaesthesia, the caecum was carefully exposed, ligated just distal to the ileocecal valve to avoid intestinal obstruction and punctured twice with a 20‐gauge needle. The punctured caecum was squeezed to expel fecal material and then returned to the abdominal cavity. The abdominal incision was closed with two layers of running suture. All animals were resuscitated with 1 mL normal saline injected s.c. immediately after the operation. Sham‐operated mice were subjected to laparotomy and intestinal manipulation; however, the caecum was neither ligated nor punctured.
In survival experiments, mice (10 mice per group) received Compound 9a (5, 10 and 20 mg∙kg−1), SAHA (50 mg∙kg−1) or 5% DMSO in saline (vehicle) administered i.p. 2 h before and immediately after CLP. The dose and time point of Compound 9a and SAHA administration were selected based on a previously published report (Li et al., 2010; Finkelstein et al., 2010) and our preliminary study. Mortality was recorded for up to 7 days after the procedure. To ensure that no late mortalities occurred, survivors were monitored for a further 3 weeks. On the basis of these survival assays, a dose of 10 mg∙kg−1 of Compound 9a was chosen for further biochemical studies. The animals were randomly assigned to four groups (each group, n = 6): (i) vehicle‐treated sham (sham), (ii) Compound 9a‐treated (10 mg∙kg−1) sham (9a), (iii) vehicle‐treated CLP (CLP) and (iv) Compound 9a‐treated (10 mg∙kg−1) CLP (CLP + 9a). Under anesthesia, blood samples (400 μL each) were collected from the inferior vena cava at various different timepoints after CLP. Liver tissue was isolated 3 h after CLP and immediately stored at −75°C until further biochemical assay.
Assessment of organ damage in CLP mice
Serum was separated by centrifugation at 10 000 × g for 10 min at 4°C. Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), creatinine, and LDH levels were measured with a Hitachi 7600 automatic analyzer (Hitachi, Tokyo, Japan).
Histological analysis
Twenty‐four hours after CLP, liver, lung, kidney and heart tissue samples were obtained for histological analysis. Each sample was fixed with 10% neutral‐buffered formalin. The samples were embedded in paraffin, sliced into 5 μm sections, and stained with haematoxylin and eosin. All histological analyses was conducted in a blinded manner. Histological changes were evaluated in random, non‐consecutive fields at ×200 magnification (Olympus BX51/Olympus DP71, Tokyo, Japan).
SiRNA gene silencing of MKP‐1
The siRNA for MKP‐1 and non‐specific control siRNA were purchased from Bioneer (Daejeon, Korea). MKP‐1 siRNA (30 μg) was diluted in 50 μL of sterile 10% glucose solution, and the volume was adjusted to 100 μL with RNase/DNase‐free water. In a separate tube, 14.4 μL of in vivo‐jetPEI (Polyplus‐transfection Inc., New York, USA) was diluted in 50 μL of sterile 10% glucose solution, and the volume was adjusted to 100 μL with RNase/DNase‐free water. The solutions were mixed and incubated for 15 min at room temperature to form the stable nucleic acids/in vivo‐jetPEI complex. MKP‐1 siRNAs or non‐specific control were injected via the tail vein twice in 2 days. Mice were subjected to CLP 2 days after the last siRNA injection. RAW264.7 cells were transfected with either siRNAs for MKP‐1 and non‐specific control using Lipofectamine® 2000 (Thermo Fisher Scientific Inc., Waltham, MA, USA). After 48 h of incubation, the cells were washed and then pretreated with or without Compound 9a (12.5 μM) for 1 h following LPS (1 μg∙mL−1). After incubation with LPS for 24 h, the culture media were harvested for further analysis. The siRNAs significantly decreased the levels of MKP‐1 protein expression in both in vivo and in vitro (Figure 8A, D).
Figure 8.
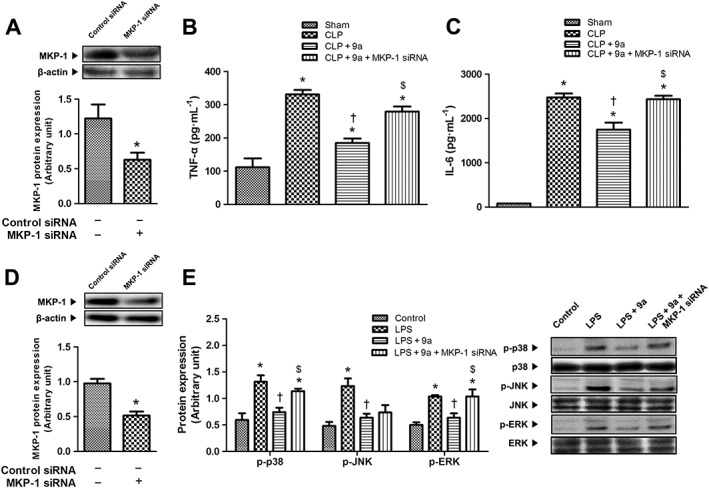
Effect of MKP‐1 siRNA on the inhibitory effect of Compound 9a on inflammation. Mice or RAW264.7 cells were transfected with control siRNA or MKP‐1 siRNA, 3 and 2 days before CLP or 48 h before LPS treatment (1 μg∙mL−1) respectively. Mice were injected i.p. with vehicle or 10 mg∙kg−1 Compound 9a 2 h before and immediately after CLP. RAW264.7 cell were treated with 12.5 μM Compound 9a 1 h before LPS treatment. Liver tissue or cells were collected 3 h after CLP or 24 h after LPS treatment respectively. MKP‐1 protein expression was measured to confirm the efficacy of MKP‐1 siRNA (A and D). The serum levels of TNF‐α (B) and IL‐6 (C) were measured. Phosphorylation of p38, JNK and ERK (E) was measured 24 h after LPS treatment. The results are presented as mean ± SEM of six mice per group or mean ± SEM of independent experiments. * P < 0.05, significantly different from control siRNA, sham or control. † P < 0.05, significantly different from CLP or LPS. $ P < 0.05, significantly different from CLP + 9a or LPS + 9a.
Protein extraction
Liver tissue was homogenized in PRO‐PREP™ Protein Extraction Solution (iNtRON Biotechnology, Seongnam, Korea) or NE‐PER® (Pierce Biotechnology, Rockford, IL) to extract total cell proteins and nuclear/cytosolic proteins, respectively, according to the manufacturer's instructions. Protein concentrations were determined with the bicinchoninic acid (BCA) Protein Assay kit (Pierce Biotechnology).
Western blot analysis
Protein samples (16–20 μg) were loaded on 7.5–15% polyacrylamide gels, separated by SDS‐PAGE and transferred to PVDF membranes using the Semi‐Dry Trans‐Blot Cell (Bio‐Rad Laboratories, Hercules, CA, USA). After the transfer, the membranes were blocked for 1 h with 5% skim milk powder in Tris‐buffered saline with 0.1% Tween‐20 at room temperature. The blots were incubated with primary antibodies overnight at 4°C and subsequently incubated with secondary antibodies for 1 h at room temperature. Bands were detected with an ECL detection system (iNtRON Biotechnology), according to the manufacturer's instructions. The intensities of immunoreactive bands were determined with Total‐Lab TL120 software (Nonlinear Dynamics, Newcastle, UK). Primary antibodies against mouse acetylated histone H3, histone H3, NF‐κB, IκB, phosphorylated (p)‐p38, p38, p‐JNK1/2, JNK1/2, p‐ERK1/2, ERK1/2 and HDAC1 (Cell Signaling Technology, Berverly, MA) and toll‐like receptor (TLR) 4, MKP‐1 and acetylated lysine (Ac‐K; Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used. The signals were standardized to β‐actin (Sigma Aldrich) to whole lysate and lamin B1 (Abcam, Cambridge, MA, USA) for the nuclear fraction.
Data and statistical analysis
Design of this study conformed to the guidance on experimental design and analysis in pharmacology (Curtis et al., 2015). Results are presented as mean ± SEM. The survival data were analysed with the Kaplan–Meier curve and log‐rank test. All other data were analysed by the un‐paired Student's t‐test between two values and by one‐way or two‐way anova ,where more than two groups were compared. The Bonferroni test was used for post hoc comparisons. Differences between groups were considered statistically significant at P < 0.05.
Materials
Compound 9a, (E)‐N‐hydroxy‐4‐(2‐styrylthiazol‐4‐yl)butanamide was synthesized in the Organomedicinal Chemistry Laboratory of School of Pharmacy, Sungkyunkwan University. The chemical structure, detailed synthetic route and identification of Compound 9a are provided in Supporting Information Figure S1–S3. Compounds were dissolved in DMSO and stored at −20°C as small aliquots.
DMEM, PBS, penicillin/streptomycin (10 000 U∙mL−1 and 10 000 μg∙mL−1, respectively) and FBS were obtained from Gibco BRL, Life Technologies (Grand Island, NY). SAHA, LPS (Escherichia coli serotype O127:B8), tetrazolium bromide (MTT) and all other materials required for culturing cells were purchased from Sigma Aldrich (St. Louis, MO, USA). All other chemicals used in this study were of reagent grade.
Results
Compound 9a suppresses cytokine release in LPS‐stimulated RAW264.7 cells
To evaluate the anti‐inflammatory properties of Compound 9a, the levels of released cytokines were determined in LPS‐stimulated RAW264.7 cells. Compound 9a did not have any cytotoxicity on RAW264.7 cells at concentrations up to 12.5 μM, while SAHA showed cytotoxicity at 2.5 μM (Figure 1A). LPS markedly increased TNF‐α and IL‐6 release over control. These increases were suppressed by Compound 9a in a concentration‐dependent manner. SAHA also suppressed the increases in TNF‐α and IL‐6 release in a concentration‐dependent manner (Figure 1B, C). After cells were incubated with LPS for 24 h, NO release was also increased, relative to the the control value and this increase was attenuated by Compound 9a. SAHA showed less inhibitory effects on NO release than Compound 9a in LPS‐stimulated RAW264.7 cells (Figure 1D).
Figure 1.
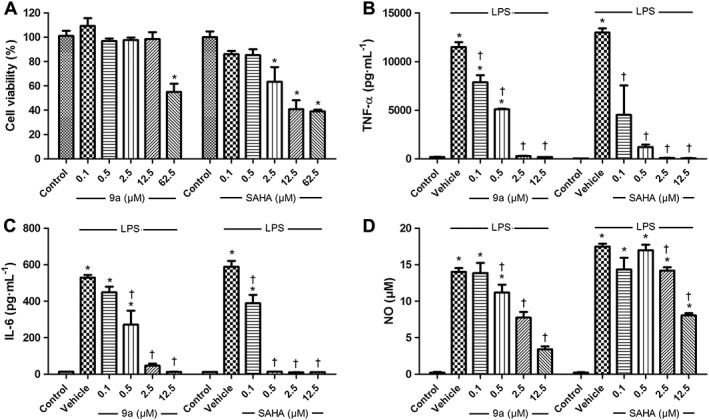
Effect of Compound 9a on the cytokine and NO release in LPS‐stimulated RAW264.7 cells. For the cellular toxicity assay, RAW264.7 cells were treated with vehicle, various concentrations of Compound 9a or suberoylanilide hydroxamic acid (SAHA) for 24 h (A). To measure cytokine release, RAW264.7 cells were treated with vehicle, various concentrations of Compound 9a or SAHA for 1 h before LPS (1 μg∙mL−1) stimulation. Twenty‐four hours after LPS stimulation, release of TNF‐α (B), IL‐6 (C) and NO (D) was measured. The results are presented as mean ± SEM of independent experiments. * P < 0.05, significantly different from control. † P < 0.05, significantly different from vehicle.
Compound 9a protects mice against sepsis‐induced mortality
In the untreated CLP group, the survival rate on the first day was 70% and stabilized at 30% on the third day after CLP. The survival rate of mice treated with 5 or 20 mg∙kg−1 Compound 9a plateaued on the third day but was not significantly different from the untreated group (P = 0.2942 and P = 0.1542, respectively). A log‐rank analysis of 7‐day survival demonstrated that only the dose of 10 mg∙kg−1 Compound 9a significantly improved survival compared with the untreated CLP group (Figure 2A). Moreover, Compound 9a (10 mg∙kg−1) was more effective in improving survival than a higher dose of SAHA (50 mg∙kg−1) in CLP mice (Figure 2B).
Figure 2.
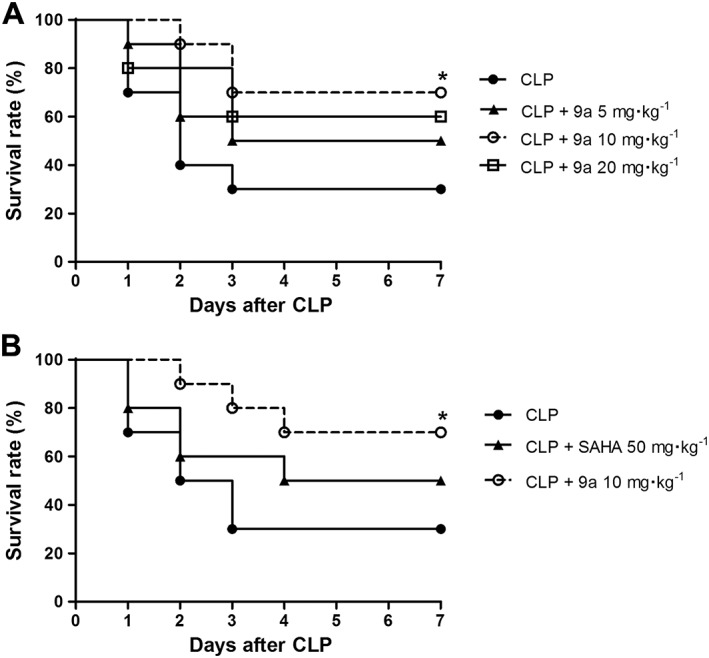
Effect of Compound 9a on mortality following CLP in mice. Mice were injected i.p. with vehicle, Compound 9a (5, 10 or 20 mg∙kg−1) or SAHA (50 mg∙kg−1) 2 h before and immediately after CLP (n = 10 per group). Animals were monitored for 7 days after CLP. * P < 0.05, significantly different from CLP.
Compound 9a increases acetylated‐histone H3 protein expression
To assess HDAC inhibition by Compound 9a in our in vivo system, we measured levels of the acetylated‐histone H3 protein. In the CLP group, acetylated‐histone H3 protein levels were decreased compared with the sham group and this decrease was attenuated by Compound 9a (Figure 3).
Figure 3.
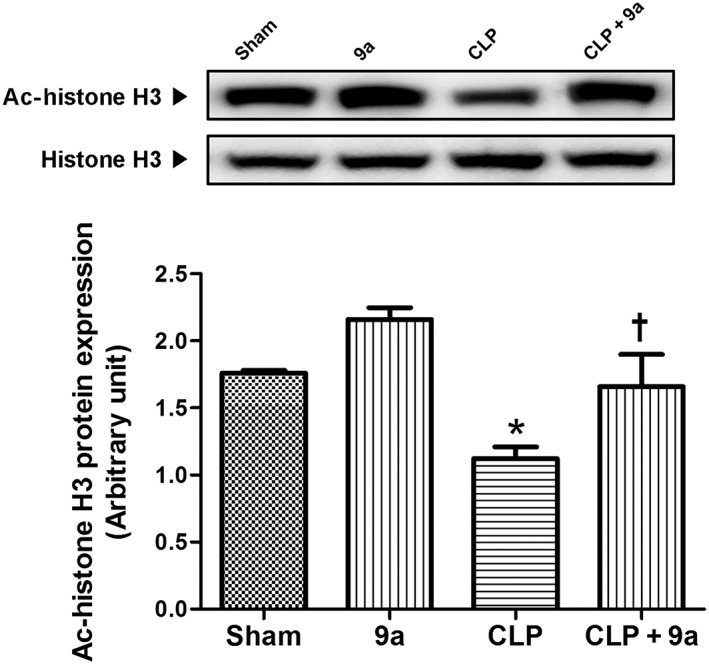
Effect of Compound 9a on hepatic levels of acetylated (Ac)‐histone H3 protein during sepsis. Mice were injected i.p. with vehicle or 10 mg∙kg−1 Compound 9a 2 h before and immediately after CLP. Liver tissue was collected 3 h after CLP. The results are presented as mean ± SEM of six mice per group. * P < 0.05, significantly different from sham. † P < 0.05, significantly different from CLP.
Compound 9a decreases CLP‐induced serum cytokine levels
In the sham group, serum TNF‐α levels remained constant at all times of sampling. In CLP mice, serum TNF‐α levels peaked at 3 h after CLP and gradually decreased until 24 h after CLP (Figure 4A). Serum IL‐6 levels in the sham group were similarly low and constant over the experimental period. The serum IL‐6 level started to increase 3 h after CLP, peaked at 6 h after CLP, gradually decreased until 12 h after CLP and remained constant until 24 h after CLP (Figure 4B). These changes in levels of both cytokines were attenuated by treatment of the CLP mice with Compound 9a (10 mg∙kg−1).
Figure 4.
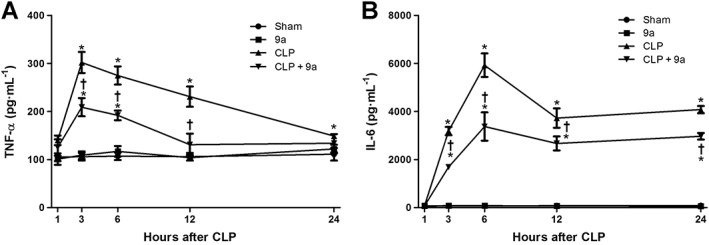
Effect of Compound 9a on serum cytokine levels during sepsis. Mice were injected i.p. with vehicle or 10 mg∙kg−1 Compound 9a 2 h before and immediately after CLP. The serum was collected 1, 3, 6, 12 and 24 h after CLP. Serum TNF‐α (A) and IL‐6 (B) levels were measured in indicated time. The results are presented as mean ± SEM of six mice per group. * P < 0.05 significantly different from sham. † P < 0.05, significantly different from CLP.
Compound 9a attenuates CLP‐induced organ injury
As shown in Figure 5A, serum ALT levels began to increase at 1 h after CLP, peaked at 12 h after CLP and then fell until 24 h after CLP. Serum AST levels began to increase at 1 h after CLP, peaked at 12 h after CLP and remained constant until 24 h after CLP. Serum creatinine levels began to increase at 1 h after CLP, peaked at 12 h after CLP and then gradually decreased until 24 h after CLP. Serum BUN levels began to increase at 6 h after CLP and peaked at 12 h after CLP. Serum LDH levels began to increase at 1 h after CLP and continued to increase until 12 h after CLP. Treatment with Compound 9a (10 mg∙kg−1) attenuated changes in serum ALT, AST, creatinine, BUN and LDH levels throughout the experiments. Histological evaluations revealed normal cell structures in the liver, lung, kidney and heart of sham‐operated mice (Figure 5B–E, respectively). Twenty‐four hours after CLP, significant histopathological changes (such as inflammatory cell infiltration and necrosis) were observed, and these pathological changes were attenuated by Compound 9a. Compound 9a alone did not induce any histological changes.
Figure 5.
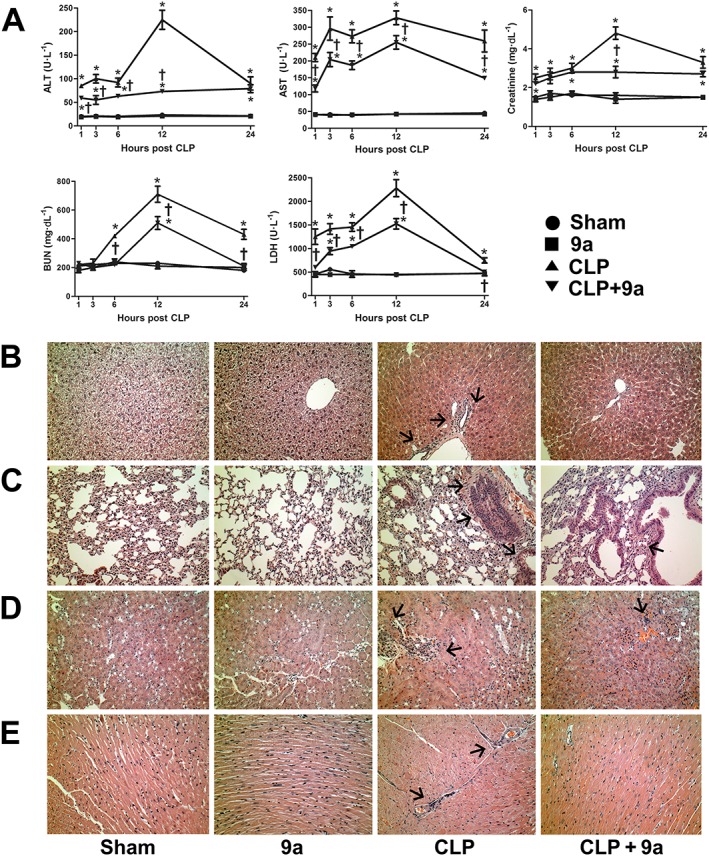
Effects of Compound 9a on organ injury during sepsis. Mice were injected i.p. with vehicle or 10 mg∙kg−1 Compound 9a 2 h before and immediately after CLP. Serum was collected 1, 3, 6, 12 and 24 h after CLP and serum ALT, AST, creatinine, BUN and LDH were measured (A). Liver (B), lung (C), kidney (D) and heart (E) tissue were collected 24 h after CLP. The results are presented as mean ± SEM of six mice per group. The arrows indicate inflammatory cell infiltration. * P < 0.05, significantly different from sham. † P < 0.05, significantly different from CLP.
Compound 9a decreases TLR4 signalling pathway
To determine the effect of Compound 9a on the TLR4 signalling pathway, we first determined the expression of TLR4 protein. CLP increased TLR4 protein expression to almost double that in the sham group, 3 h after CLP. These increases were attenuated by Compound 9a (Figure 6A). CLP increased translocation of NF‐κB to the nucleus, compared to that in the sham group. Cytosolic IκB protein expression significantly decreased to 81% of the sham group. Treatment with Compound 9a (10 mg∙kg−1) did not affect these changes induced by CLP (Figure 6B). Phosphorylation of p38, JNK and ERK increased compared with those in the sham group 3 h after CLP. However, these increases in protein phosphorylation were attenuated by treatment with Compound 9a (Figure 6C).
Figure 6.
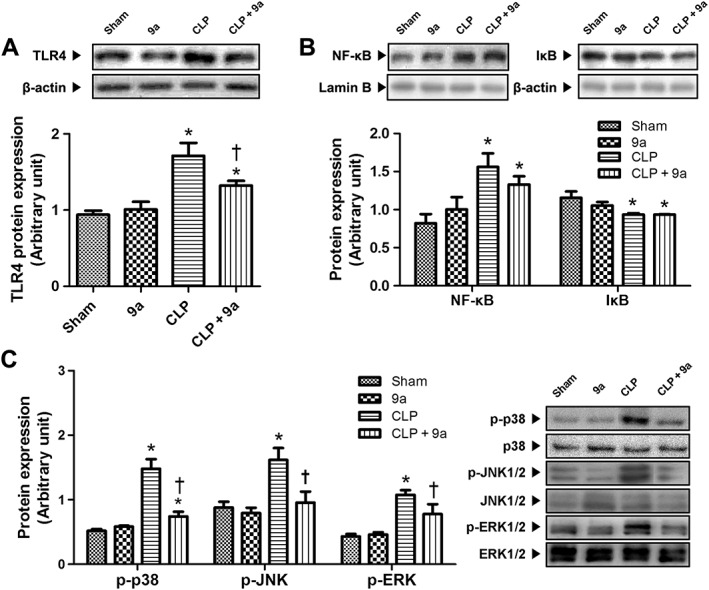
Effect of Compound 9a on TLR4 protein expression and signalling pathway during sepsis. Mice were injected i.p. with vehicle or 10 mg∙kg−1 Compound 9a 2 h before and immediately after CLP. The hepatic protein expressions of TLR4 (A), nuclear NF‐κB and cytosolic IκB (B) and MAPK (C) were measured 3 h after CLP. The results are presented as mean ± SEM of six mice per group. * P < 0.05, significantly different from sham. † P < 0.05, significantly different from CLP.
Compound 9a increases MKP‐1 acetylation and association with MAPK
To investigate the effect of Compound 9a on the phosphatase MKP‐1, we used immunoprecipitation assays. As shown in Figure 7A, the association of MKP‐1 with p‐p38 and p‐ERK decreased significantly 3 h after CLP, and the decrease was attenuated by Compound 9a. MKP‐1 association with p‐JNK tended to decrease 3 h after CLP and was not affected by Compound 9a. Levels of acetylated‐MKP‐1 protein tended to decline 3 h after CLP and was attenuated by Compound 9a (Figure 7B). MKP‐1 association with HDAC1, HDAC2 and HDAC3 increased significantly 3 h after CLP. Compound 9a attenuated the association of MKP‐1 with HDAC1 but not HDAC2 or HDAC3.
Figure 7.
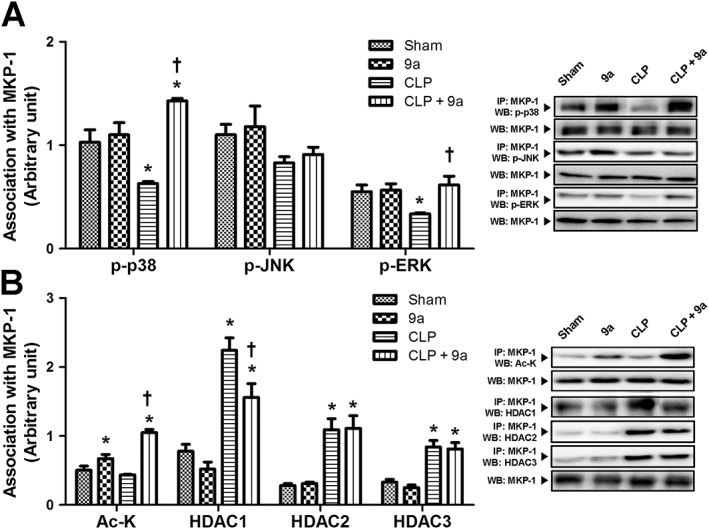
Effect of Compound 9a on MKP‐1 association with MAPK and acetylation during sepsis. Mice were injected i.p. with vehicle or 10 mg∙kg−1 Compound 9a 2 h before and immediately after CLP. Association of MKP‐1 with MAPK (A) and with Ac‐K and HDAC (B) were measured. The results are presented as mean ± SEM of six mice per group. Liver tissue was collected 3 h after CLP. * P < 0.05, significantly different from sham. † P < 0.05, significantly different from CLP.
MKP‐1 siRNA abolishes inhibitory effects of Compound 9a on inflammation
Involvement of MKP‐1 in the protective effects of Compound 9a against sepsis was further confirmed by MKP‐1 gene silencing methods. MKP‐1 siRNA significantly decreased MKP‐1 protein expression in both in vivo and in vitro (Figure 8A, D). The serum levels of TNF‐α and IL‐6 were increased, relative to those in the sham group 3 h after CLP and these increases were attenuated by Compound 9a. In mice treated with, MKP‐1 siRNA and CLP, this effect of Compound 9a was abolished (Figure 8B, C). Phosphorylation of p38 MAPK and ERK was double that in the control group, in LPS‐stimulated RAW264.7 cells, and these higher levels were attenuated by Compound 9a. MKP‐1 siRNA abolished the inhibitory activity of Compound 9a on phosphorylation of p38 MAPK and ERK. Increased phosphorylation of JNK, compared with that of the control group was also attenuated by Compound 9a in LPS‐stimulated RAW264.7 cells. However, the inhibitory activity of Compound 9a on phosphorylation of JNK was not abolished by MKP‐1 siRNA. (Figure 8E).
Discussion
HDAC is an important pharmacological target in cancer and neurodegenerative diseases, such as Parkinson's and Alzheimer's diseases, with over 80 clinical trials currently underway (Kazantsev and Thompson, 2008). Recently, HDAC inhibitors have been found to have anti‐inflammatory properties. The structure of hydroxamic acid enhances its HDAC inhibitory activity by coordinating a zinc ion in the active site of the enzyme. Therefore, most reported HDAC inhibitors are hydroxamic acid derivatives, such as SAHA and TSA (Suzuki and Miyata, 2005). SAHA reduced TNF‐α and IL‐1β mRNA levels in septic mice induced by LPS (Li et al., 2009), and TSA attenuated acute lung injury in polymicrobial sepsis (Zhang et al., 2010). Although HDAC inhibitors have various pharmacological effects, clinical experience with HDAC inhibitors remains limited because of their cytotoxicity (Subramanian et al., 2010). In a preliminary study, we screened several new synthetic HDAC inhibitors in vitro, focusing on both HDAC inhibitory activity and anti‐inflammatory properties. Out of 20 candidates, Compound 9a ((E)‐N‐hydroxy‐4‐(2‐styrylthiazol‐4‐yl)butanamide), a hydroxamic acid derivative, had significant HDAC inhibitory activity in cell‐based and enzyme‐based assays (data under review). Also, in the present study, Compound 9a exerted anti‐inflammatory effects in LPS‐stimulated RAW264.7 cells. Of particular interest was our finding that Compound 9a had lower cell toxicity than SAHA in RAW264.7 cells.
Histones are acetylated by histone acetyltransferase (HAT), which is necessary for gene transcription. HAT activity is balanced by HDAC, which removes the acetyl group from histones and impairs gene transcription (Egger et al., 2004). HDAC inhibitors enhance the acetylation of lysine residues on histones. SAHA increased histone H3 and H4 acetylation in myeloma cells (Jeong et al., 2014) and reversed the decrease in histone H3 acetylation induced by LPS in mice (Li et al., 2009). TSA treatment resulted in pronounced histone H4 hyperacetylation in breast carcinoma cell lines (Vigushin et al., 2001). In the liver, low oxygen decreased histone H3 acetylation to 50–75% of control levels in a model of anoxia in turtles (Krivoruchko and Storey, 2010). In the present study, levels of acetylated‐histone H3 protein decreased significantly in CLP animals, and Compound 9a increased hepatic levels of histone H3 acetylation.
Recent studies have demonstrated that HDAC inhibitors effectively improved survival using in vivo models of CLP or LPS‐induced sepsis. Pre‐treatment and post‐treatment with HDAC inhibitors such as TSA and SAHA protected against septic injury (Zhang et al., 2010; Li et al., 2009). Although clinicians deal with treatment rather than prevention, there may be certain situations when individuals at risk could be identified for pre‐treatment, as protection against the effects of sepsis. Prevention is effective for children in Africa and hospitalized patients after serious surgical procedures with a high risk of sepsis (Pollach and Namboya, 2013; Sarangi et al., 1997). In our study, administering Compound 9a 2 h before and immediately after CLP improved the survival rate. In an attempt to widen the therapeutic window of Compound 9a, we tried delaying Compound 9a administration to after the onset of sepsis. Unfortunately, Compound 9a did not improve the survival rate when administered immediately after CLP or immediately and 24 h after CLP (data not shown).
The magnitude of the inflammatory response plays a critical role in the pathogenesis of sepsis. Furthermore, an excessive inflammatory response is closely related to mortality (Walley et al., 1996). In this study, Compound 9a reduced the systemic release of pro‐inflammatory cytokines, including TNF‐α and IL‐6, in polymicrobial sepsis. Excessive production of inflammatory cytokines results in capillary leakage, tissue injury and lethal organ failure. Multiple organ dysfunction is the critical cause of death in septic patients, and overall, mortality rates range from 30% to 100% depending on the number of damaged organs (Wu et al., 2008). In this study, the serum ALT, AST, creatinine, BUN and LDH levels peaked at 12 h after CLP. Treatment with Compound 9a not only decreased the serum levels of these markers of organ injury but also attenuated histopathological changes in liver. These results suggest that Compound 9a suppressed inflammatory cytokines, attenuated multiple organ dysfunction and improved survival during sepsis.
TLRs play a critical role in the pathogenesis of sepsis through recognition of microbes and are regarded as important upstream mediators that trigger excessive inflammatory response. Previous studies had revealed that up‐regulation of TLR4 protein expression plays a role in high mortality and pathophysiology of sepsis (Williams et al., 2003) and TLR4 activation facilitates the MAPK and NF‐κB pathways. Activation of the NF‐κB pathway results in translocation of NF‐κB to the nucleus and increased transcription of inflammatory genes, such as those for TNF‐α, IL‐1β and IL‐6. In septic patients, increased NF‐κB translocation to the nucleus was associated with higher mortality rate and worse clinical outcome (Abraham, 2003). In the present study, TLR4 protein expression significantly upregulated 3 h after CLP, which was attenuated by Compound 9a; however, Compound 9a did not affect the increased NF‐κB signalling pathway. There is now evidence that different acetylated isoforms of NF‐κB determine the activities of this transcription factor. Acetylation at Lys122 and Lys123 decreased NF‐κB DNA binding affinity (Huang et al., 2009), while acetylation at Lys221 enhanced the nuclear import of NF‐κB (Chen et al., 2002). The precise molecular mechanisms of the effects of HDAC inhibition on NF‐κB activity will require further study.
MAPK is a critical signalling molecule mediating inflammatory responses by regulating the expression of cytokines and inflammatory factors. MAPK is activated by sequential phosphorylation of its tyrosine and/or threonine residue (Raman et al., 2007). Previous studies have demonstrated that phosphorylation of p38, ERK and JNK regulated macrophage and T cell responses to traumatic injury. Furthermore, MAPK activation by phosphorylation contributes to sepsis‐induced organ injury (Yang et al., 2008). In a model of LPS‐induced endotoxemia, p‐p38, p‐ERK and p‐JNK expression significantly increased, and SAHA attenuated LPS‐induced increases in p‐p38 and p‐ERK, but not p‐JNK (Finkelstein et al., 2010). An HDAC inhibitor, KBH‐A42, inhibited cytokine production by suppressing phosphorylation of p38, but not ERK and JNK in an endotoxemia model (Kang et al., 2010). In the present study, p‐p38, p‐JNK and p‐ERK levels increased markedly after CLP and Compound 9a attenuated the increased MAPK phosphorylation. Compound 9a also inhibited phosphorylation of p38, JNK and ERK in LPS‐stimulated Kupffer cells (Supporting Information Figure S4). These results suggest that Compound 9a attenuated inflammatory responses by inhibiting MAPK phosphorylation.
MKP is a dual‐specificity phosphatase that inactivates MAPK by dephosphorylating tyrosine and threonine residues (Chi et al., 2006). To date, at least 10 MKPs have been identified in mammalian cells, with MKP‐1 as the archetype of the family. MAPK dephosphorylation by MKP‐1 is accompanied by the formation of a tight physical complex (Krivoruchko and Storey, 2010). Increased MKP‐1 levels coincide with p38, JNK and ERK inactivation in LPS‐treated peritoneal macrophages from mice (Zhao et al., 2006). In the present study, the association of MKP‐1 with p‐p38 and p‐ERK was significantly decreased after CLP, and this effect was attenuated by Compound 9a. The association of MKP‐1 with p‐JNK also tended to decrease after CLP, but Compound 9a had no effect. Our findings suggest that Compound 9a enhances the association of MAPK and MKP‐1.
MKP‐1 is regulated at both the transcriptional and post‐transcriptional levels (Wancket et al., 2012). In the present study, MKP‐1 protein expression did not decrease appreciably. This result might arise from the diverse regulation of MKP‐1 protein expression, including gene transcription and protein stability. MKP‐1 phosphorylation increases MKP‐1 stability (Brondello et al., 1999), and oxidization of the catalytic cysteine residues in MKP‐1 inhibits MKP‐1 activity (Kamata et al., 2005). Because it has roles in both MKP‐1 stability and activity, acetylation plays a key role in regulating MKP‐1. Recently, Cao et al. (2008) reported that acetylation of the K57 residue of MKP‐1 increased its interaction with p38. MKP‐1 acetylation is regulated by two enzymes, HDAC and HAT. Jeong et al. (2014) found that HDAC1, HDAC2 and HDAC3 deacetylated MKP‐1 in LPS‐treated RAW264.7 cells. In the present study, MKP‐1 acetylation tended to decrease after CLP, and this result was attenuated by Compound 9a. Furthermore, MKP‐1 interaction with HDAC1, but not HDAC2 or HDAC3, significantly increased after CLP and was attenuated by Compound 9a. To provide stronger evidence that Compound 9a really inhibited inflammation through MKP‐1, we used MKP‐1 gene silencing by siRNA. MKP‐1 siRNA abolished the inhibitory effect of Compound 9a on serum levels of TNF‐α and IL‐6 at 3 h after CLP. These data indicate that MKP‐1 is an essential factor for the protective effects of Compound 9a against septic injury. Moreover, MKP‐1 siRNA abolished the inhibitory activity of Compound 9a on phosphorylation of p38 MAPK and EKR in LPS‐stimulated RAW264.7 cells, but not that of JNK.
In conclusion, this study provides evidence of the anti‐inflammatory effect of HDAC inhibitors in sepsis, which is a novel strategy to treat sepsis. The current study demonstrated that Compound 9a inhibited MAPK‐mediated inflammatory responses and attenuated the subsequent organ damage. Moreover, we showed that Compound 9a enhanced the inhibitory effects of MKP‐1 on MAPK signalling through enhancing acetylation of MKP‐1. Therefore, Compound 9a appears to offer a novel therapeutic strategy for sepsis.
Author contributions
S. J. Kim performed the research. S. J. Kim and S.‐M. Lee designed the research study. H. J. Park and Y. H. Jung contributed essential reagents or tools. S. J. Kim and H. J. Park analysed the data. S. J Kim and S.‐M. Lee wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 The synthetic routes of 9a. Reagents and conditions: (a) P2S5, THF, rt; (b) Br2, Et2O, 0°C then to room temperature (RT); (c) 12N HCl, CH3COOH, MeOH, 50°C then to 70°C; (d) MeOH, reflux; (e) 1N‐NaOH, MeOH, THF, RT; (f) EDCI, HOBt hydrate, NH2OH·HCl, TEA, CH3CN, DMF, RT. (E)‐3‐phenylprop‐2‐enethioamide (2) was prepared from thiation of cinnamide (1) under Lawesson's reagent. Diethyl 2‐acetylpentanedioate (3) was treated with bromine to afford acyl bromide, which was converted to methyl 6‐chloro‐5‐oxohexanoate (5) using 12NHCl and acetic acid. Next, (E)‐3‐phenylprop‐2‐enethioamide (2) was coupled with methyl 6‐chloro‐5‐oxohexanoate (5) to give (E)‐methyl 4‐(2‐styrylthiazol‐4‐yl)butanoate (6). Treatment of (E)‐methyl 4‐(2‐styrylthiazol‐4‐yl)butanoate (6) with NH2OH·HCl, EDCI, HOBt hydrate produced the compound 9a. Commercially available reagents were used without additional purification, unless otherwise stated. All reactions were performed under an inert atmosphere of nitrogen or argon. Nuclear magnetic resonance spectra (1H and 13C NMR) were recorded on a Bruker Unity 300 MHz and Varian Unit 500 MHz spectrometer for CD3OD solutions, and chemical shifts are reported as parts per million (ppm) relative to, respectively, residual CD3OD δH (3.31 ppm) and CD3OD δC (49.00 ppm) as internal standards. Resonance patterns are reported with the notations s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Coupling constants (J) are reported in hertz (Hz). Thin layer chromatography was carried out using plates coated with Kieselgel 60F254 (Merck). For flash column chromatography, E. Merck Kieselgel 60 (230–400 mesh) was used.
Figure S2 1H NMR spectrum of 9a. 1H NMR (500MHz,CD3OD): δ 7.62 (d, J= 5.6 Hz, 2H), 7.48–7.33 (m, J = 5H), 7.15 (s, 1H), 2.83–2.81 (t, J= 10 Hz, 2H), 2.19–2.17 (t, J= 10Hz, 2H), 2.07–2.05 (t, J= 10 Hz, 2H).
Figure S3 13C NMR spectrum of 9a. 13C NMR (125MHz, CD3OD): δ 172.64, 168.93, 158.27, 137.22, 136.11, 130.29, 130.13, 128.35, 121.95, 114.92, 33.25, 31.52, 26.49.
Figure S4 Effect of 9a on phosphorylation of MAPK in LPS‐stimulated Kupffer cells. Kupffer cells were isolated from SD rats (male; 160‐180 g) by differential centrifugation using Percoll. After the liver was digested with collagenase‐contained buffer, the suspension was filtered through cell strainer and cell fraction was obtained by centrifugation. The Kupffer cells fraction was collected, centrifuged at 1700 rpm for 5 min and suspended again in RPMI 1640 media. The Kupffer cells were incubated with 12.5 μM 9a 1 h before LPS treatment (10 ng·mL−1). The cell were harvested 24 h after LPS treatment. Phosphorylation of p38, JNK and ERK significantly increased to 3.5‐, 2.1‐ and 2.7‐fold than those of control group. The results are presented as mean ± SEM of independent experiments. Significantly different (*P<0.05) from control. Significantly different († P<0.05) from LPS.
Supporting info item
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST; 2012R1A5A2A28671860). So‐Jin Kim (2013034472; NRF‐2013H1A2A1034472) received ‘Global Ph.D. Fellowship Program’ support from the NRF funded by the MEST.
Kim, S.‐J. , Baek, K. S. , Park, H.‐J. , Jung, Y. H. , and Lee, S.‐M. (2016) Compound 9a, a novel synthetic histone deacetylase inhibitor, protects against septic injury in mice by suppressing MAPK signalling. British Journal of Pharmacology, 173: 1045–1057. doi: 10.1111/bph.13414.
References
- Abraham E (2003). Nuclear factor‐kappaB and its role in sepsis‐associated organ failure. J Infect Dis 187: S364–S369. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brondello JM, Pouyssegur J, McKenzie FR (1999). Reduced MAP kinase phosphatase‐1 degradation after p42/p44MAPK‐dependent phosphorylation. Science 286: 2514–2517. [DOI] [PubMed] [Google Scholar]
- Camps M, Nichols A, Arkinstall S (2000). Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J 14: 6–16. [PubMed] [Google Scholar]
- Cao W, Bao C, Padalko E, Lowenstein CJ (2008). Acetylation of mitogen‐activated protein kinase phosphatase‐1 inhibits toll‐like receptor signaling. J Exp Med 205: 1491–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Karin M (2001). Mammalian MAP kinase signalling cascades. Nature 410: 37–40. [DOI] [PubMed] [Google Scholar]
- Chaudry IH, Wichterman KA, Baue AE (1979). Effect of sepsis on tissue adenine nucleotide levels. Surgery 85: 205–211. [PubMed] [Google Scholar]
- Chen LF, Mu Y, Greene WC (2002). Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF‐kappaB. EMBO J 21: 6539–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, et al. (2006). Dynamic regulation of pro‐ and anti‐inflammatory cytokines by MAPK phosphatase 1 (MKP‐1) in innate immune responses. Proc Natl Acad Sci U S A 103: 2274–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA, et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger G, Liang G, Aparicio A, Jones PA (2004). Epigenetics in human disease and prospects for epigenetic therapy. Nature 429: 457–463. [DOI] [PubMed] [Google Scholar]
- Finkelstein RA, Li Y, Liu B, Shuja F, Fukudome E, Velmahos GC, et al. (2010). Treatment with histone deacetylase inhibitor attenuates MAP kinase mediated liver injury in a lethal model of septic shock. J Surg Res 163: 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer M, Echtenachter B, Weighardt H, Jozefowski K, Rose‐John S, Mannel DN, et al. (2010). Increased inflammation and lethality of Dusp1−/−mice in polymicrobial peritonitis models. Immunology 131: 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer JG, Bailey DL, Sharma GR, Zhang T, Ding C, Ford A, et al. (2004). Cecal ligation and puncture with total parenteral nutrition: a clinically relevant model of the metabolic, hormonal, and inflammatory dysfunction associated with critical illness. J Surg Res 121: 178–186. [DOI] [PubMed] [Google Scholar]
- Hsu YF, Sheu JR, Lin CH, Chen WC, Hsiao G, Ou G, et al. (2011). MAPK phosphatase‐1 contributes to trichostatin A inhibition of cyclooxygenase‐2 expression in human umbilical vascular endothelial cells exposed to lipopolysaccharide. Biochim Biophys Acta 1810: 1160–1169. [DOI] [PubMed] [Google Scholar]
- Huang B, Yang XD, Zhou MM, Ozato K, Chen LF (2009). Brd4 coactivates transcriptional activation of NF‐kappaB via specific binding to acetylated RelA. Mol Cell Biol 29: 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong Y, Du R, Zhu X, Yin S, Wang J, Cui H, et al. (2014). Histone deacetylase isoforms regulate innate immune responses by deacetylating mitogen‐activated protein kinase phosphatase‐1. J Leukoc Biol 95: 651–659. [DOI] [PubMed] [Google Scholar]
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M (2005). Reactive oxygen species promote TNFalpha‐induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120: 649–661. [DOI] [PubMed] [Google Scholar]
- Kang MR, Lee K, Kang JS, Lee CW, Lee KH, Kim JH, et al. (2010). KBH‐A42, a histone deacetylase inhibitor, inhibits the growth of doxorubicin‐resistant leukemia cells expressing P‐glycoprotein. Oncol Rep 23: 801–809. [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM (2008). Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov 7: 854–868. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, et al. (2001). Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med 7: 437–443. [DOI] [PubMed] [Google Scholar]
- Krivoruchko A, Storey KB (2010). Epigenetics in anoxia tolerance: a role for histone deacetylases. Mol Cell Biochem 342: 151–161. [DOI] [PubMed] [Google Scholar]
- Li Y, Liu B, Fukudome EY, Kochanek AR, Finkelstein RA, Chong W, et al. (2010). Surviving lethal septic shock without fluid resuscitation in a rodent model. Surgery 148: 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, et al. (2009). Protective effect of suberoylanilide hydroxamic acid against LPS‐induced septic shock in rodents. Shock 32: 517–523. [DOI] [PubMed] [Google Scholar]
- Lo CJ, Chiu KC, Fu M, Chu A, Helton S (2000). Fish oil modulates macrophage P44/P42 mitogen‐activated protein kinase activity induced by lipopolysaccharide. JPEN J Parenter Enteral Nutr 24: 159–163. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS (2003). Histone deacetylase inhibitors modulate renal disease in the MRL‐lpr/lpr mouse. J Clin Invest 111: 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novogrodsky A, Vanichkin A, Patya M, Gazit A, Osherov N, Levitzki A (1994). Prevention of lipopolysaccharide‐induced lethal toxicity by tyrosine kinase inhibitors. Science 264: 1319–1322. [DOI] [PubMed] [Google Scholar]
- Pan LN, Lu J, Huang B (2007). HDAC inhibitors: a potential new category of anti‐tumor agents. Cell Mol Immunol 4: 337–343. [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SPH, Buneman OP et al, NC‐IUPHAR (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollach G, Namboya F (2013). Preventing intensive care admissions for sepsis in tropical Africa (PICASTA): an extension of the international pediatric global sepsis initiative: an African perspective. Pediatr Crit Care Med 14: 561–570. [DOI] [PubMed] [Google Scholar]
- Raman M, Chen W, Cobb MH (2007). Differential regulation and properties of MAPKs. Oncogene 26: 3100–3112. [DOI] [PubMed] [Google Scholar]
- Sarangi J, Coleby M, Trivella M, Reilly S (1997). Prevention of post splenectomy sepsis: a population based approach. J Public Health Med 19: 208–212. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Bates SE, Wright JJ, Espinoza‐Delgado I, Piekarz RL (2010). Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals 3: 2751–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Miyata N (2005). Non‐hydroxamate histone deacetylase inhibitors. Curr Med Chem 12: 2867–2880. [DOI] [PubMed] [Google Scholar]
- Vigushin DM, Ali S, Pace PE, Mirsaidi N, Ito K, Adcock I, et al. (2001). Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo . Clin Cancer Res 7: 971–976. [PubMed] [Google Scholar]
- Walley KR, Lukacs NW, Standiford TJ, Strieter RM, Kunkel SL (1996). Balance of inflammatory cytokines related to severity and mortality of murine sepsis. Infect Immun 64: 4733–4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wancket LM, Frazier WJ, Liu Y (2012). Mitogen‐activated protein kinase phosphatase (MKP)‐1 in immunology, physiology, and disease. Life Sci 90: 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Chaudry IH (1996). Mechanism of hepatocellular dysfunction during hyperdynamic sepsis. Am J Physiol 270: R927–R938. [DOI] [PubMed] [Google Scholar]
- Williams DL, Ha T, Li C, Kalbfleisch JH, Schweitzer J, Vogt W, et al. (2003). Modulation of tissue Toll‐like receptor 2 and 4 during the early phases of polymicrobial sepsis correlates with mortality. Crit Care Med 31: 1808–1818. [DOI] [PubMed] [Google Scholar]
- Wu JY, Tsou MY, Chen TH, Chen SJ, Tsao CM, Wu CC (2008). Therapeutic effects of melatonin on peritonitis‐induced septic shock with multiple organ dysfunction syndrome in rats. J Pineal Res 45: 106–116. [DOI] [PubMed] [Google Scholar]
- Yang M, Wu J, Martin CM, Kvietys PR, Rui T (2008). Important role of p38 MAP kinase/NF‐kappaB signaling pathway in the sepsis‐induced conversion of cardiac myocytes to a proinflammatory phenotype. Am J Physiol Heart Circ Physiol 294: H994–1001. [DOI] [PubMed] [Google Scholar]
- Zhang L, Jin S, Wang C, Jiang R, Wan J (2010). Histone deacetylase inhibitors attenuate acute lung injury during cecal ligation and puncture‐induced polymicrobial sepsis. World J Surg 34: 1676–1683. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Wang X, Nelin LD, Yao Y, Matta R, Manson ME, et al. (2006). MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med 203: 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The synthetic routes of 9a. Reagents and conditions: (a) P2S5, THF, rt; (b) Br2, Et2O, 0°C then to room temperature (RT); (c) 12N HCl, CH3COOH, MeOH, 50°C then to 70°C; (d) MeOH, reflux; (e) 1N‐NaOH, MeOH, THF, RT; (f) EDCI, HOBt hydrate, NH2OH·HCl, TEA, CH3CN, DMF, RT. (E)‐3‐phenylprop‐2‐enethioamide (2) was prepared from thiation of cinnamide (1) under Lawesson's reagent. Diethyl 2‐acetylpentanedioate (3) was treated with bromine to afford acyl bromide, which was converted to methyl 6‐chloro‐5‐oxohexanoate (5) using 12NHCl and acetic acid. Next, (E)‐3‐phenylprop‐2‐enethioamide (2) was coupled with methyl 6‐chloro‐5‐oxohexanoate (5) to give (E)‐methyl 4‐(2‐styrylthiazol‐4‐yl)butanoate (6). Treatment of (E)‐methyl 4‐(2‐styrylthiazol‐4‐yl)butanoate (6) with NH2OH·HCl, EDCI, HOBt hydrate produced the compound 9a. Commercially available reagents were used without additional purification, unless otherwise stated. All reactions were performed under an inert atmosphere of nitrogen or argon. Nuclear magnetic resonance spectra (1H and 13C NMR) were recorded on a Bruker Unity 300 MHz and Varian Unit 500 MHz spectrometer for CD3OD solutions, and chemical shifts are reported as parts per million (ppm) relative to, respectively, residual CD3OD δH (3.31 ppm) and CD3OD δC (49.00 ppm) as internal standards. Resonance patterns are reported with the notations s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Coupling constants (J) are reported in hertz (Hz). Thin layer chromatography was carried out using plates coated with Kieselgel 60F254 (Merck). For flash column chromatography, E. Merck Kieselgel 60 (230–400 mesh) was used.
Figure S2 1H NMR spectrum of 9a. 1H NMR (500MHz,CD3OD): δ 7.62 (d, J= 5.6 Hz, 2H), 7.48–7.33 (m, J = 5H), 7.15 (s, 1H), 2.83–2.81 (t, J= 10 Hz, 2H), 2.19–2.17 (t, J= 10Hz, 2H), 2.07–2.05 (t, J= 10 Hz, 2H).
Figure S3 13C NMR spectrum of 9a. 13C NMR (125MHz, CD3OD): δ 172.64, 168.93, 158.27, 137.22, 136.11, 130.29, 130.13, 128.35, 121.95, 114.92, 33.25, 31.52, 26.49.
Figure S4 Effect of 9a on phosphorylation of MAPK in LPS‐stimulated Kupffer cells. Kupffer cells were isolated from SD rats (male; 160‐180 g) by differential centrifugation using Percoll. After the liver was digested with collagenase‐contained buffer, the suspension was filtered through cell strainer and cell fraction was obtained by centrifugation. The Kupffer cells fraction was collected, centrifuged at 1700 rpm for 5 min and suspended again in RPMI 1640 media. The Kupffer cells were incubated with 12.5 μM 9a 1 h before LPS treatment (10 ng·mL−1). The cell were harvested 24 h after LPS treatment. Phosphorylation of p38, JNK and ERK significantly increased to 3.5‐, 2.1‐ and 2.7‐fold than those of control group. The results are presented as mean ± SEM of independent experiments. Significantly different (*P<0.05) from control. Significantly different († P<0.05) from LPS.
Supporting info item