

Abstract
Background and Purpose
In the RELAX‐AHF trial, a 48 h i.v. serelaxin infusion reduced systemic vascular resistance in patients with acute heart failure. Consistent with preclinical studies, serelaxin augments endothelial vasodilator function in rat mesenteric arteries. Little is known about the contribution of endothelium‐derived relaxing factors after a longer duration of continuous serelaxin treatment. Here we have assessed vascular reactivity and mechanistic pathways in mesenteric arteries and veins and the aorta after 48 or 72 h continuous i.v. infusion of serelaxin.
Experimental Approach
Male rats were infused with either placebo or serelaxin (13.3 μg·kg−1·h−1) via the jugular vein using osmotic minipumps. Vascular function was assessed using wire myography. Changes in gene and protein expression and 6‐keto PGF1α levels were determined by quantitative PCR, Western blot and ELISA respectively.
Key Results
Continuous i.v. serelaxin infusion augmented endothelium‐dependent relaxation in arteries (mesenteric and aorta) but not in mesenteric veins. In mesenteric arteries, 48 h i.v. serelaxin infusion increased basal NOS activity, associated with increased endothelial NOS (eNOS) expression. Interestingly, phosphorylated‐eNOSSer1177, eNOS and basal NOS activity were reduced in mesenteric arteries following 72 h serelaxin treatment. At 72 h, serelaxin treatment improved bradykinin‐mediated relaxation through COX2‐derived PGI2 production.
Conclusions and Implications
Continuous i.v. serelaxin infusion enhanced endothelial vasodilator function in arteries but not in veins. The underlying mediator at 48 h was NO but there was a transition to PGI2 by 72 h. Activation of the PGI2‐dependent pathway is key to the prolonged vascular response to serelaxin treatment.
Abbreviations
- BK
bradykinin
- EDH
endothelium‐derived hyperpolarization
- eNOS
endothelial NOS
- iNOS
inducible NOS
- KPSS
K+ physiological saline solution
- L‐NAME
N Ω‐nitro‐L‐arginine methyl ester hydrochloride
- pEC50
logarithm of the half maximal effective concentration
- Rmax
maximum relaxation
- RXFP1
relaxin/insulin‐like family peptide receptor 1
- SNP
sodium nitroprusside
Tables of Links
| TARGETS |
|---|
| GPCRs a |
| RXFP1, relaxin receptor |
| Enzymes b |
| eNOS |
| iNOS |
| COX‐1 |
| COX‐2 |
| Voltage‐gated ion channels c |
| KCa3.1 channels |
| KCa2.x channels |
| LIGANDS |
|---|
| Apamin |
| BK, bradykinin |
| L‐NAME |
| NO |
| PGI 2 |
| Serelaxin (relaxin 2) |
| TRAM‐34 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a b cAlexander et al., 2015a, 2015b, 2015c).
Introduction
The 6 kDa peptide hormone relaxin and its cognate receptor (RXFP1) are expressed in a variety of blood vessels, including mesenteric, renal, uterine arteries and the aorta (Novak et al., 2006; Vodstrcil et al., 2012; Jelinic et al., 2014), suggesting that relaxin has a role in modulating vascular tone. In small resistance arteries, this is mediated by endothelium‐derived hyperpolarization (EDH), nitric oxide (NO) and prostacyclin (PGI2). Intravenous infusion of serelaxin (the recombinant form of human relaxin‐2) in patients with acute heart failure reduced systemic vascular resistance, which is sustained once the infusion ceases (Ponikowski et al., 2014). Similarly, treatment of rodents with serelaxin decreases myogenic reactivity and enhances endothelium‐dependent relaxation in small renal and mesenteric arteries (Novak et al., 2002; Jelinic et al., 2014). A single i.v. tail vein injection of serelaxin enhances basal NOS activity and bradykinin (BK)‐evoked endothelium‐dependent relaxation in mesenteric arteries just 3 h after injection. This augmented vasodilation is mediated by intermediate‐conductance calcium‐activated potassium (KCa3.1) channel‐dependent EDH (Leo et al., 2014b). Interestingly, augmentation of BK‐mediated relaxation is sustained at least 24 h after a single i.v. injection of serelaxin. The mediator responsible for these changes is likely to be the vasodilator prostanoid PGI2 (Leo et al., 2014b).
In contrast to acute i.v. injection, s.c. infusion of serelaxin for 5 days increased endothelial vasodilator function in the mesenteric arteries but this effect was entirely mediated by NO (van Drongelen et al., 2013, 2011, 2012; Jelinic et al., 2014). Similarly, in small renal arteries, the same treatment elicits an endothelium‐derived NO‐dependent reduction in myogenic reactivity (Novak et al., 2002; Jeyabalan et al., 2003). Ex vivo studies on rat aortic rings treated in vitro with TNF‐α demonstrated that co‐incubation with serelaxin for 48 h reversed endothelial dysfunction and counteracts reactive oxygen species and arginase II generation caused by TNF‐α (Dschietzig et al., 2012). Serelaxin treatment also induces an increase in endothelial NOS (eNOS) phosphorylation and activity. However, it is unknown if in vivo serelaxin treatment produces similar effects in the aorta.
The beneficial effects of serelaxin treatment on vascular function are not observed in all systemic blood vessels. Thus, s.c. serelaxin infusion (3–5 days) has no effect on endothelial vasodilator function in mesenteric veins, saphenous arteries and veins (Li et al., 2005; Jelinic et al., 2014). Taken together, all these studies demonstrate that there is heterogeneity in response to serelaxin, dependent on the vascular bed‐studied. We have also demonstrated that acute i.v. serelaxin augments endothelial function in resistance arteries through a combination of NO, EDH and/or PGI2. However, no studies have investigated if the route of serelaxin treatment (s.c. vs. i.v.) and/or duration of treatment (hours vs. days) influences the vasodilator pathways that are stimulated to augment endothelial function and whether these effects vary between vascular beds.
A phase III clinical trial (RELAX‐AHF) demonstrated that a continuous 48 h i.v. infusion of serelaxin to acute heart failure patients improved dyspnoea relief, reduced in‐hospital worsening of acute heart failure and decreased cardiovascular and all‐cause mortality at 180 days (Teerlink et al., 2013). These effects of serelaxin were, in part attributed to the rapid and short‐term reduction in systemic vascular resistance, most likely to be due to arterial vasodilation (Ponikowski et al., 2014). The mechanism(s) of vasodilation evoked by 48 h or longer of serelaxin treatment via continuous i.v. infusion are not known as most preclinical studies treat animals with serelaxin via s.c. administration for 5 days. Therefore, the aims of the present study were to investigate changes in endothelial vasodilator function of systemic blood vessels following a 48 h continuous i.v. infusion of serelaxin and, importantly, to determine whether or not NO, EDH or PGI2 is involved at this critical time point. We also sought to investigate if i.v. serelaxin treatment beyond 48 h caused time‐dependent and region‐dependent changes in the contribution of endothelial‐derived factors.
Methods
Animal welfare and ethical statement
All animal care and experimental procedures complied with the National Health and Medical Research Council of Australia code of practice for the care and use of animals for scientific purposes and were approved by the Faculty of Science, The University of Melbourne Animal Experimentation Ethics Committee (AEC# 1212410.1). Animal studies are reported as recommended by the ARRIVE guidelines (McGrath and Lilley, 2015). All rats were maintained on an automated time cycle of 12 h light/dark at 20°C, with standard food pellets (Barastock, Vic., Australia) and water available ad libitum.
Serelaxin treatment in animals
Male Wistar rats (~300–350 g; Animal Resource Centre, Perth, WA, Australia) were randomly divided into three groups: (i) controls (consisting of placebo‐treated rats pooled from 48 and 72 h time points, n = 33), (ii) 48 h (n = 23) and (iii) 72 h (n = 22) serelaxin treatment. For continuous i.v. infusion of serelaxin, all animals were anaesthetised with 2% isoflurane (Univentor 400, Agnthos, AB, Lidingö, Sweden) in oxygen via inhalation. Once anaesthetised, rats were chronically instrumented with a silicone catheter (Cat No: 0007710, Bioscientific, Gymea, NSW, Australia) implanted in the right jugular vein with the tip located at the junction of the anterior vena cava and right atrium. The catheter was attached to a 3 day Alzet osmotic minipump (Model 1003D, Bioscientific) to infuse either serelaxin (13.3 μg·kg−1·h−1) or placebo (20 mmol·L−1 sodium acetate, pH 5.0) into the jugular vein. The dose of serelaxin (13.3 μg·kg−1·h−1) was equivalent to the standard infusion rate of 4 μg·h−1 (Jelinic et al., 2014). Either 48 or 72 h post‐onset of serelaxin infusion, blood samples were obtained from the left ventricle via cardiac puncture under 2% isoflurane anaesthesia. Plasma concentrations of serelaxin (n = 8–10 per group) were measured in duplicate using the Human Relaxin‐2 Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA) following the manufacturer's protocol. The limit of detection of detection was 15.6 pg·mL, and the intra‐ and inter‐coefficients of variation were 2.3–4.7% and 5.5–10.2% respectively.
Isolation of mesenteric arteries, veins and aortae
Following blood collection, the animals were killed via cervical dislocation under anaesthesia. The mesenteric arcade and aorta were isolated and immediately placed in ice cold Krebs bicarbonate solution (mmol·L−1: 120 NaCl, 5 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 11.1 D‐glucose and 2.5 CaCl2). Small mesenteric arteries and veins (third‐order branch of the superior mesenteric artery, internal diameter ~300 μm) and abdominal aortae were cleared of fat and loose connective tissue, cut into rings of 2 mm in length and mounted on a Mulvany–Halpern wire myograph (model 610 M, Danish Myo Technology, Aarhus, Denmark). The remaining arteries were snap frozen in liquid nitrogen and stored at −80°C for further analysis. After the mesenteric arteries and veins were mounted on the myograph, they were allowed to stabilize at zero tension for 15 min before normalization, as described previously (Leo et al., 2011c, 2012; Jelinic et al., 2014). All experiments were performed at 37°C in the presence of 95% O2 and 5% CO2.
Assessment of vascular reactivity
Vascular reactivity experiment was performed as previously described (Leo et al., 2012, 2014b) with the following modifications. Briefly, vessels were maximally contracted with high K+ physiological saline solution (KPSS, 100 mmol·L−1), and the integrity of the endothelium was determined. This was achieved by pre‐contracting the blood vessels to 70–80% KPSS using phenylephrine (0.1–3 μmol·L−1). ACh‐induced (10 μmol·L−1) relaxation was >95% of the pre‐contracted tone. To assess endothelial and vascular smooth muscle function, blood vessels were pre‐contracted to a similar level (70–80% of KPSS contraction) using phenylephrine (0.1–3 μmol·L−1), followed by cumulative concentration–response curves to the endothelium‐dependent agonists, BK (0.1 nmol·L−1 to 1 μmol·L−1, for mesenteric arteries and veins) or ACh (1 nmol·L−1 to 10 μmol·L−1, for aorta), and the endothelium‐independent agonist, sodium nitroprusside (SNP, 0.01 nmol·L−1 to 10 μmol·L−1). Previous studies from our laboratory (Jelinic et al., 2014; Leo et al., 2014b) demonstrated that serelaxin treatment selectively enhances BK‐evoked but not ACh‐evoked endothelium‐dependent relaxation in the mesenteric arteries. Thus, in the current study, we did not assess ACh‐induced relaxation in the mesenteric arteries. Furthermore, our preliminary experiments revealed that BK failed to evoke endothelium‐dependent in the abdominal aorta relaxation (data not shown); thus, ACh was used to evaluate endothelial function. In addition, responses to BK or ACh were examined after 20 min incubation with different combinations of pharmacological blockers including L‐NAME (200 μmol·L−1), the COX inhibitor indomethacin (Indo, 1 μmol·L−1), selective COX‐1 inhibitor SC560 (1 μmol·L−1), selective COX‐2 inhibitor NS398 (1 μmol·L−1), the KCa3.1 channel inhibitor TRAM34 (1 μmol·L−1), the inhibitor of small‐conductance calcium‐activated potassium (KCa2.x) channels, apamin (1 μmol·L−1) and the prostacyclin synthase (PGI2S) inhibitor U51605 (1 μmol·L−1).
Assessment of basal NOS activity
In a separate set of experiments, the effects of serelaxin treatment on basal NO activity were examined through the addition of L‐NAME (200 μmol·L−1) in endothelium‐intact rings sub‐maximally contracted with phenylephrine (10–100 nmol·L−1) to ~20% of KPSS contraction. Under these conditions, a contractile response to L‐NAME was considered to reflect the level of basal NOS activity (Leo et al., 2011b).
Western blotting
Western blots were performed as described previously (Leo et al., 2011a) with the following modifications. Frozen endothelium‐intact mesenteric arteries or aortae were placed in a pre‐chilled Wig‐L‐Bug® capsule with a metal ball and pulverized with a Digital Wig‐L‐Bug amalgamator (Dentsply Ltd., York, PA, USA). The samples were dissolved in 300 μL of ice‐cold lysis buffer (100 mmol·L−1 NaCl, 10 mmol·L−1 Tris, 2 mmol·L−1 EDTA, 0.5% w/v sodium deoxycholate, 1% v/v Triton X‐100, pH 7.4) with protease and phosphatase inhibitor cocktails according to the manufacturers' instructions (Roche, Sydney, NSW, Australia). Total protein concentration of the samples was quantified using a BCA protein assay kit (ThermoScientific, Rockford, IL, USA). Equal amounts (20 μg) of protein homogenate were subjected to SDS‐PAGE (10% for RXFP1 and 7.5% for NOS) and Western blot analysis with mouse/rabbit primary antibodies (all 1:1000, overnight, 4°C) against RXFP1 (gift from Prof. David Sherwood), inducible NOS (iNOS), phospho‐Ser1177 eNOS (all BD Transduction Laboratories, Lexington, KY, USA) and eNOS (Cell Signalling, Danvers, MA, USA). The rabbit RXFP1 antibody has been previously validated to be selective only for RXFP1 but not for RXFP2 (Jelinic et al., 2014). To normalize for the amount of protein, membranes were reprobed with a loading control antibody (actin). All proteins were detected by enhanced chemiluminescence (Amersham, GE Healthcare Life Sciences, Sydney, NSW, Australia) after incubation with anti‐mouse/rabbit secondary antibody (Millipore, Billerica, MA, USA) for 1 h at room temperature (1:2000). All protein bands were quantified by densitometry (Biorad Chemidoc, Sydney, NSW, Australia) and expressed as a ratio of the loading control.
RNA extraction and quantitative PCR
Frozen blood vessels were pulverized as described (Leo et al., 2014a). Pulverized tissues were resuspended in 1 mL TriReagent (Ambion Inc., Scoresby, Vic., Australia), and total RNA was then extracted according to the manufacturer's instructions. RNA pellets were resuspended in 15–20 μL RNA Secure™ (Ambion). Quality and quantity of RNA were analysed using the NanoDrop ND1000 Spectrophotometer (Thermo Fisher Scientific Australia Pty Ltd., Scoresby, Vic., Australia) with A260 : A280 ratios >1.8 indicating sufficient quality for quantitative PCR (qPCR) analysis. First strand cDNA synthesis used 1 μg of total RNA in a 20 μL reaction containing random hexamers (50 ng·μL−1) and 200 units of Superscript™ III (Invitrogen, Mulgrave, Vic., Australia). The comparative cycle threshold (2−ΔCt) method of quantitative real‐time PCR (qPCR) was used to analyse COX1 (Ptgs1) and COX2 (Ptgs2) and PGI2S (Ptgis) gene expression in serelaxin (48 and 72 h) and placebo‐treated rats. Rat‐specific forward/reverse primers and 6‐carboxyl fluorescein‐labelled TaqMan probes were designed and purchased from Biosearch Technologies (Novato, CA, USA). Primers were designed to span intron/exon boundaries. qPCR was performed on the Applied Biosystems ViiA7 PCR machine (Life Technologies, Mulgrave, Vic., Australia) using 96‐well plates with 10 μL volume reactions in triplicate containing SensiMix (Bioline) and 10 μmol·L−1 of primers and 6‐carboxyl fluorescein‐labelled probe. Ribosomal 18S (Rn18s) was used as the reference gene. Negative template controls substituting cDNA with water or RT negative controls substituting the reverse transcriptase in the cDNA synthesis were included on each plate. For each sample, the mean Rn18s CT triplicate value was subtracted from the mean gene of interest triplicate CT value and normalized to the reference gene. These normalized data (ΔCt) were then presented as a relative value (mean ± SEM).
6‐Keto PGF1α assay
In order to measure the release of 6‐keto PGF1α (metabolite of PGI2), segments of mesenteric arteries (~5 branches) were placed in 0.5 mL of Krebs‐HEPES buffer at 37°C. The equilibration time was 1 h during which the buffer was changed every 15 min. At the end of the incubation period, BK (1 μmol·L−1) was applied for 10 min. The mesenteric segments were removed, and the buffer was collected and snap‐frozen in liquid nitrogen and stored at −80°C for further analysis. The arteries were dried overnight, and dry tissue weight was measured. 6‐keto PGF1α was measured using EIA kits from Cayman Chemical (Ann Arbor, MI, USA) as described in the manufacturer's procedure booklet. The buffer was diluted 1:50 and used for the measurement of 6‐keto PGF1α. The absorbance counts were normalized to dry tissue weight.
Data analyses
All results are expressed as the mean ± SEM; n represents the number of animals per group or the number of assays when tissues from animals were pooled. Concentration–response curves were computer fitted to a sigmoidal curve using nonlinear regression (Prism version 5.0, GraphPad Software, San Diego, CA, USA) to calculate the sensitivity of each agonist (pEC50). Maximum relaxation (R max) to vasodilators was measured as a percentage of pre‐contraction to phenylephrine. Group pEC50 and R max values were compared via one‐way anova with post hoc analysis using Dunnett's test, Tukey's test or Student's unpaired t‐test as appropriate. Post hoc analysis was only performed when the F‐value was significant and there was no variance in homogeneity. P < 0.05 was considered statistically significant.
Materials
All drugs were purchased from Sigma‐Aldrich (St. Louis, MO, USA), except for BK (Auspep, Tullamarine, Vic., Australia), U51605, SC560 and NS398 (Cayman Chemical). All drugs were dissolved in distilled water, with the exception of indomethacin, which was dissolved in sodium carbonate (0.1 mol·L−1), and SC560 and NS398, which were dissolved in 100% DMSO. U51605 was dissolved in 100% EtOH.
Results
Effects of 48 and 72 h continuous i.v. serelaxin treatment on endothelial function in the mesenteric artery
Serelaxin was detected in the plasma of treated rats. Plasma serelaxin concentrations did not differ significantly between 48 h (mean: 69.86 ± 6.96 ng·mL−1) and 72 h (mean: 53.91 ± 9.75 ng·mL−1) of continuous i.v. infusion. Continuous i.v. serelaxin treatment for 48 h (P < 0.01) and 72 h (P < 0.001) significantly increased the sensitivity but not R max to BK when compared with responses from placebo‐treated rats (Figure 1A and E). There was no difference in the responses to BK between the 48 and 72 h treatment groups. In contrast, neither 48 nor 72 h continuous i.v. infusion of serelaxin had a significant effect on the sensitivity and R max to SNP (Figure 1F).
Figure 1.
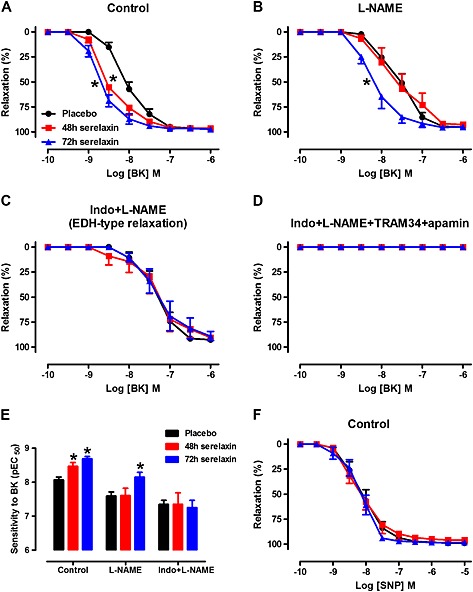
Concentration–response curves to BK in the absence (A, control) or presence of L‐NAME (B, NOS inhibitor), Indo + L‐NAME (C, inhibitors of COX and NOS) and Indo + L‐NAME + TRAM34 + apamin (D, inhibitors of COX, NOS, KCa3.1 and KCa2.x channels) in endothelium‐intact mesenteric arteries of rats treated with placebo, 48 h or 72 h of continuous i.v. infusion of serelaxin. Mesenteric artery sensitivity (E, pEC50) to BK in placebo, 48 or 72 h after continuous i.v. infusion of serelaxin. Concentration–response curves to SNP (F) in endothelium‐intact mesenteric arteries of rats treated with placebo, 48 h or 72 h of continuous i.v. infusion of serelaxin. n = 7–11 per group. * P < 0.05; pEC50 significantly different from placebo; one‐way anova, Dunnett's post hoc test.
To explore which endothelial factors were associated with the enhanced BK‐induced relaxation after serelaxin treatment, vascular reactivity to BK was evaluated in the presence of L‐NAME alone or in combination with Indo. In the presence of L‐NAME, the sensitivity to BK was still significantly (P < 0.05) increased in arteries at 72 h but not at 48 h of serelaxin infusion compared with arteries from placebo‐treated rats (Figure 1B and E). In the presence of Indo and L‐NAME, the sensitivity to BK was not significantly different between serelaxin and placebo‐treated rats (Figure 1C and E), indicating that serelaxin had no effect on BK‐dependent EDH‐type relaxation. The relaxation evoked by BK was abolished in both placebo‐treated and serelaxin‐treated mesenteric arteries with the blockade of COX, NOS and EDH (Figure 1D).
In order to investigate NO‐mediated relaxation, BK‐induced relaxation was evaluated after the inhibition of COX and EDH. The sensitivity to BK was significantly increased in the mesenteric arteries from 48 h (pEC50: 7.86 ± 0.17, n = 7, P < 0.05) but not 72 h (pEC50: 7.67 ± 0.15, n = 7, P = 0.09) serelaxin‐infused rats compared with placebo‐treated rats (pEC50: 7.34 ± 0.11, n = 9) (Figure 2A). Furthermore, the R max to BK was significantly increased in the mesenteric arteries from 48 h (R max: 78 ± 4%, n = 7, P < 0.05) to 72 h (R max: 82 ± 4%, n = 7) serelaxin‐infused rats compared with placebo‐treated rats (R max: 57 ± 7%, n = 9) (Figure 2A). There was no difference in the responses to BK between the 48 and 72 h treatment groups (Figure 2A). L‐NAME‐induced contraction was significantly greater in arteries from 48 h continuous i.v. serelaxin‐infused rats compared with those of placebo‐treated rats (Figure 2B), indicating that basal NOS activity was increased. This effect was absent in arteries from 72 h serelaxin‐treated rats compared with those of 48 h serelaxin‐treated rats (Figure 2B). Consistent with basal NOS activity, with 48 h i.v. serelaxin infusion, there was significant up‐regulation of total eNOS expression in the mesenteric arteries compared with those from placebo‐treated rats (Figure 2C). Phosphorylation of eNOSser1177 (Figure 2D) was also increased in arteries from 48 h serelaxin‐treated rats, but it did not reach significance (P = 0.06). Furthermore, total eNOS expression (Figure 2C) and phosphorylation of eNOSser1177 (Figure 2D) were significantly decreased in arteries from 72 h serelaxin‐treated rats compared to 48 h serelaxin‐treated rats and were not significantly different to placebo rats. Serelaxin treatment at either time point had no significant effect on iNOS protein expression (Figure 2E). RXFP1 protein expression in the mesenteric arteries was comparable between all treatment groups (Figure 2F).
Figure 2.
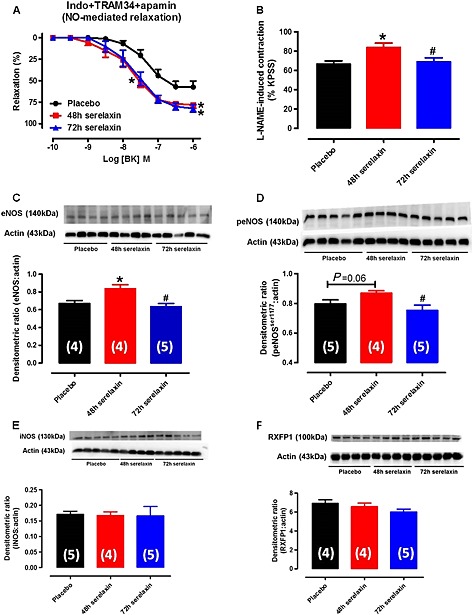
Concentration–response curves to BK in the presence of Indo + TRAM34 + apamin (A, inhibitors of COX, KCa3.1 and KCa2.x channels) in endothelium‐intact mesenteric arteries of rats treated with placebo, 48 h or 72 h of continuous i.v. infusion of serelaxin. n = 7–10 per group. (B) Basal NOS activity in endothelium‐intact mesenteric arteries of rats treated with placebo, 48 or 72 h of continuous i.v. infusion of serelaxin. In each group of experiments, mesenteric arteries were pre‐contracted with phenylephrine to a similar level (~20% of KPSS contraction) before the addition of L‐NAME (200 μmol·L−1), n = 8 per group. Western blot analysis of total eNOS (C), phospho‐eNOSser1177 (D), iNOS (E) and RXFP1 (F) protein expression in mesenteric arteries of rats treated with placebo, 48 h or 72 h of continuous i.v. infusion of serelaxin. Representative blots are shown above their respective panels. n are shown in parentheses. * P < 0.05; significantly different from placebo: # P < 0.05; significantly different from 48 h serelaxin treatment: one‐way anova, Tukey's post hoc test.
The sensitivity to BK was not affected by the presence of Indo in mesenteric arteries from placebo‐treated rats (Figure 3A) and 48 h serelaxin‐treated rats (Figure 3B), indicating no involvement of COX‐derived prostanoids. The involvement of COX‐derived PGI2 as the vasodilator prostanoid responsible for the improvement of BK‐mediated relaxation at 72 h after serelaxin infusion was elucidated using the PGI2S inhibitor, U51605. The sensitivity to BK was significantly (P < 0.001) reduced in the presence of Indo or U51605 in mesenteric arteries from 72 h serelaxin‐treated rats (Figure 3C and D). In the presence of the selective COX2 inhibitor NS398, but not the selective COX1 inhibitor SC560, the sensitivity to BK was also significantly (P < 0.01) reduced at this time point (Figure 3C and D). Furthermore, upon stimulation with BK, the release of 6‐keto PGF1α (metabolite of PGI2) was significantly (t 10 = 3.27, P = 0.009) higher in the incubation buffer of mesenteric arteries from 72 h, but not 48 h serelaxin‐treated rats as compared with placebo‐treated rats (Figure 3E and F). Similarly, expression of Pgts2 was significantly increased in the mesenteric arteries from 72 h serelaxin‐treated rats compared with placebo‐treated or 48 h serelaxin‐treated rats (Figure 4B). There was no effect of serelaxin on Pgts1 or Ptgis (Figure 4A and C) expression in mesenteric arteries at either time points.
Figure 3.
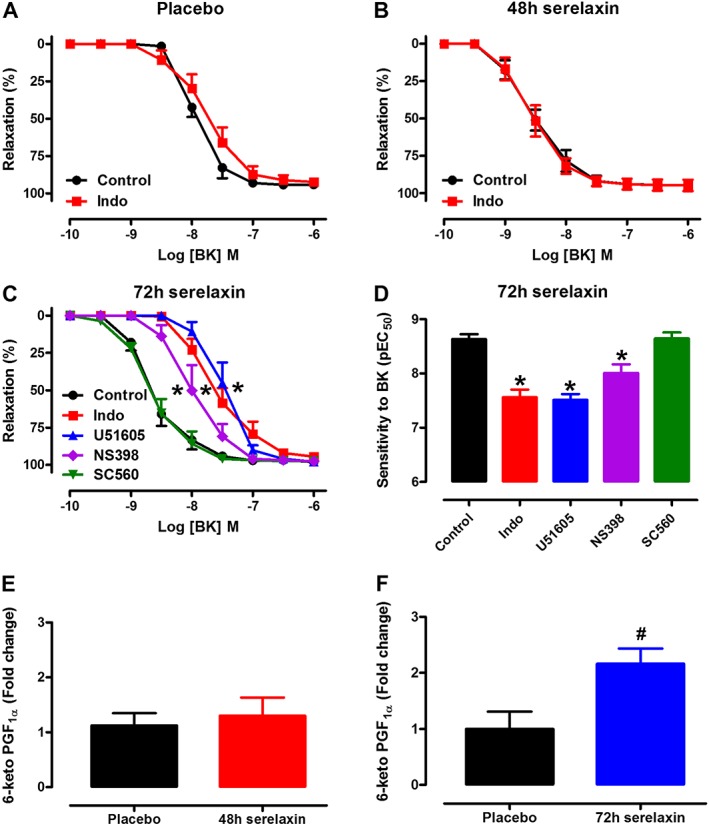
Concentration–response curves to BK in endothelium‐intact mesenteric arteries of rats treated with placebo (A), 48 h (B) or 72 h (C) of continuous i.v. infusion of serelaxin. Responses to BK (A–C) were evaluated either in the absence (control) or in the presence of Indo (1 μmol·L−1, non‐selective COX inhibitor), U51605 (1 μmol·L−1, PGI2S inhibitor), SC560 (1 μmol·L−1, COX1 inhibitor) or NS398 (1 μmol·L−1, COX2 inhibitor). Mesenteric artery sensitivity (D, pEC50) to BK following 72 h of serelaxin treatment. Levels of BK (1 μmol·L−1)‐induced 6‐keto PGF1α in endothelium‐intact mesenteric arteries of rats treated with placebo, 48 h (E) or 72 h (F) of continuous i.v. infusion of serelaxin. n = 5–9 per group. * P < 0.05; pEC50 significantly different from control; one‐way anova, Dunnett's post hoc test. # P < 0.05; significantly different from placebo; unpaired Student's t‐test.
Figure 4.
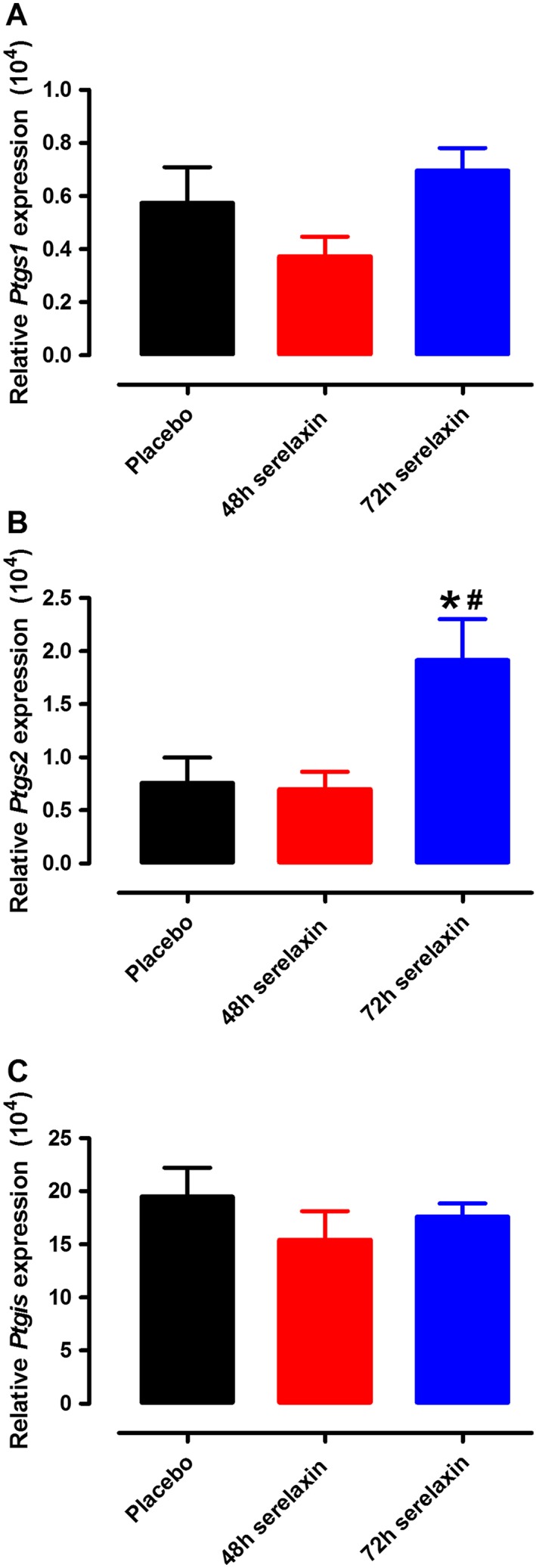
Quantitative analysis of Ptgs1 (A), Ptgs2 (B) and Ptgis (C) mRNA expression in mesenteric arteries of rats treated with placebo, 48 or 72 h of continuous i.v. infusion of serelaxin. n = 8–9 per group. Values are 2−ΔCt (means ± SEM). * P < 0.05; significantly different from placebo: # P < 0.05; significantly different from 48 h serelaxin treatment; one‐way anova, Tukey's post hoc test.
Effects of 48 and 72 h continuous i.v. serelaxin treatment on endothelial function in the abdominal aorta
Continuous i.v. serelaxin treatment for 48 h (P < 0.05) but not 72 h significantly increased the sensitivity but not R max to ACh (Figure 5A and B). The relaxation to ACh was almost abolished by the presence of L‐NAME in all groups, and these responses did not significantly differ between placebo and serelaxin treatments (Figure 5C). This suggests that continuous i.v. serelaxin treatment for 48 h did not augment endothelial function via a NOS‐independent pathway. Neither 48 nor 72 h continuous i.v. infusion of serelaxin had a significant effect on the sensitivity and R max to SNP (Figure 5D). L‐NAME‐induced contraction was significantly (P < 0.05) increased in aortae from 48 h, but not 72 h serelaxin‐treated rats compared with those of placebo‐treated rats (Figure 5E), indicating that basal NOS activity was increased at 48 h of serelaxin treatment.
Figure 5.
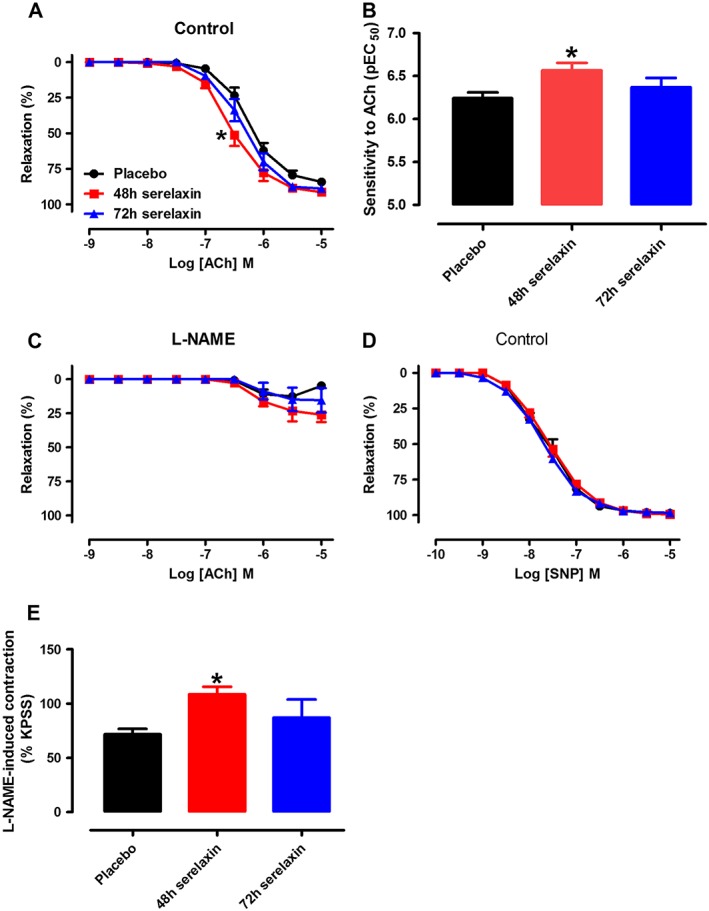
Concentration–response curves to ACh in the absence (A, control) or presence of L‐NAME (C, NOS inhibitor) and aortic sensitivity to ACh (B). Concentration–response curves to SNP (D) in endothelium‐intact aorta of rats treated with placebo, 48 h or 72 h of continuous i.v. infusion of serelaxin. (E) Basal NOS activity in endothelium‐intact aorta of rats treated with placebo, 48 or 72 h of continuous i.v. infusion of serelaxin. In each group of experiments, aortae were pre‐contracted with phenylephrine to a similar level (~20% of KPSS contraction) before the addition of L‐NAME (200 μmol·L−1). n = 5–7 per group. * P < 0.05; significantly different from placebo; one‐way anova, Tukey's post hoc test.
In the aorta, phosphorylation of eNOS‐Ser1177 (Figure 6A) appeared to have increased (P = 0.07, n = 7–8) in 48 h serelaxin‐treated rats, and to a lesser extent in 72 h serelaxin‐treated rats; however, this failed to reach statistical significance. Total eNOS expression (Figure 6B) was unaffected by 48 or 72 h of serelaxin treatment. Similarly, serelaxin treatment had no effect on iNOS expression (Figure 6C) or RXFP1 expression (Figure 6D) in the aorta.
Figure 6.
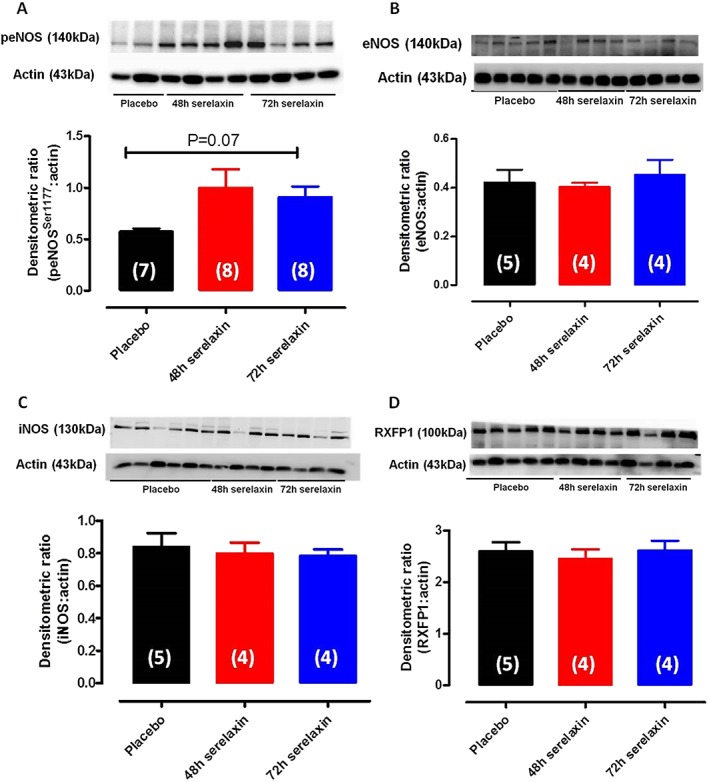
Western blot analysis of phospho‐eNOSser1177 (A), total eNOS (B), iNOS (C) and RXFP1 (D) protein expression in aorta of rats treated with placebo, 48 or 72 h of continuous i.v. infusion of serelaxin. Representative blots are shown above their respective panels. n are shown in parentheses.
Effects of 48 h continuous i.v. serelaxin treatment on endothelial function in mesenteric veins
Serelaxin treatment for 48 h had no effect on the sensitivity and R max to BK in the mesenteric veins (Figure 7A). Similarly, following inhibition of NOS alone (Figure 7B) or the combination of NOS and COX (Figure 7C), the responses to BK were comparable between mesenteric veins from placebo‐treated and serelaxin‐treated rats. The addition of L‐NAME to the mesenteric veins caused a spontaneous contraction, which is considered to reflect the basal level of NOS activity (Figure 7D). The L‐NAME‐induced contraction was comparable between placebo‐treated and serelaxin‐treated rat mesenteric veins, indicating that basal NOS activity was not affected by serelaxin treatment (Figure 7D).
Figure 7.
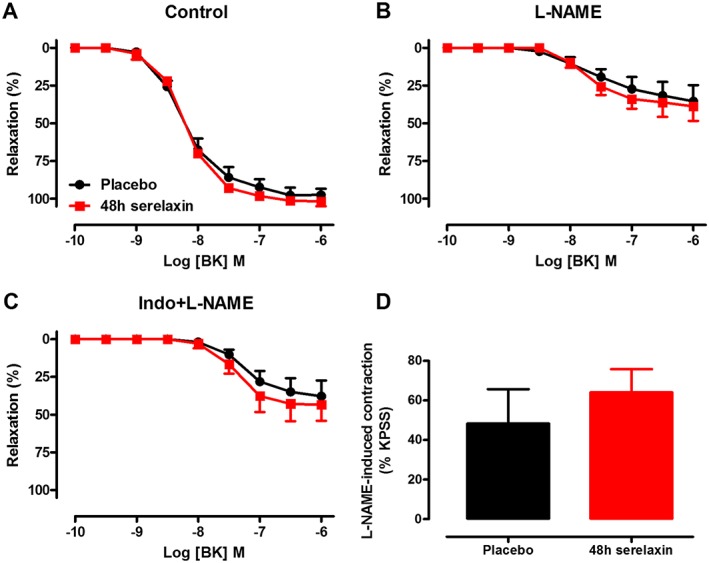
Concentration–response curves to BK in the absence (A, control) or presence of L‐NAME (B, NOS inhibitor) or Indo + L‐NAME (C, inhibitors of COX and NOS) in endothelium‐intact mesenteric veins of rats treated with placebo or 48 h of continuous i.v. infusion of serelaxin. (D) Basal NOS activity in endothelium‐intact mesenteric veins of rats treated with placebo or 48 h of continuous i.v. infusion of serelaxin. n = 5 per group. In each group of experiments, mesenteric veins spontaneously contracted with the addition of L‐NAME (200 μmol·L−1).
Discussion
This study demonstrates that continuous i.v. serelaxin infusion augmented endothelial vasodilator function in arteries (mesenteric and aorta) but not in the mesenteric veins of healthy rats. Although circulating levels of serelaxin did not differ between 48 and 72 h post‐infusion, the potentiated vasodilator effects of BK in mesenteric arteries were mediated by activation of different endothelium‐derived relaxing factor pathways. Specifically, 48 h i.v. serelaxin infusion increased basal NOS activity, and potentiated BK‐evoked NO‐mediated relaxation, which was associated with an up‐regulation of total eNOS expression. A 72 h i.v. serelaxin treatment also enhanced BK‐mediated relaxation that involved NO and COX2‐derived PGI2. Interestingly, after a prolonged period of i.v. serelaxin infusion (72 h), basal NOS activity, eNOS phosphorylation at Ser1177 and total eNOS protein expression in mesenteric arteries returned to levels comparable with those from placebo‐treated rats. In contrast to mesenteric arteries, continuous i.v. serelaxin infusion only augmented endothelial function at 48 h in the aorta, and this was mediated by NO.
Continuous i.v. serelaxin treatment for 48 and 72 h augmented endothelial function in the mesenteric artery: involvement of NO and PGI2
A number of previous studies report that the direct vasodilator action of serelaxin in resistance arteries is primarily attributed to endothelium‐derived NO (Jeyabalan et al., 2003, 2007; McGuane et al., 2011a, 2011b). Consistent with previous findings in resistance arteries (Jelinic et al., 2014; Leo et al., 2014b), we demonstrated that continuous i.v. infusion of serelaxin, both 48 and 72 h, selectively improved endothelial function in the mesenteric arteries. Specifically, serelaxin augmented BK‐mediated endothelium‐dependent relaxation, without affecting endothelium‐independent, smooth muscle relaxation. We further investigated the mechanisms underlying the augmentation of BK‐mediated endothelium‐dependent relaxation 48 and 72 h after continuous i.v. infusion of serelaxin. Consistent with earlier observations (Conrad, 2010; Jelinic et al., 2014; Leo et al., 2014b), the augmentation of BK‐mediated relaxation was normalized by the NOS inhibitor, L‐NAME at 48 h, but not 72 h post‐infusion of serelaxin. This suggests that the augmentation of BK‐mediated relaxation at 48 h serelaxin treatment is mediated entirely by NO. Indeed, we demonstrated that serelaxin treatment at 48 h increased basal NOS activity, which is associated with the up‐regulation of total eNOS expression/activity. Interestingly, basal NOS activity, phosphorylation of eNOS at Ser1177 and total eNOS expression/activity were down‐regulated at 72 h of serelaxin treatment to levels similar to placebo‐treated animals. It was well established that prolonged nitrovasodilator treatment increases superoxide production and arginase expression, contributing to eNOS uncoupling and endothelial dysfunction (Münzel et al., 2005, 2011; Khong et al., 2012). Thus, it is possible that after prolonged NO activation, there may be compensatory down‐regulation of NO production or bioavailability via multiple pathways including increased superoxide production and arginase expression, and eNOS uncoupling. The exact mechanisms underlying the down‐regulation of eNOS phosphorylation and expression remain to be investigated. Interestingly, endothelial function remained enhanced at 72 h, suggesting that there is possible involvement of an NOS‐independent factor at this time point, which serves to prolong the enhancement of endothelial function beyond 48 h. In contrast to other studies in which serelaxin was infused for 24 h or less (Collino et al., 2013; Leo et al., 2014b), iNOS expression was not up‐regulated in this study, suggesting that the activation of iNOS expression may be a rapid response (24 h or less) to synthesize NO.
Although we previously reported that acute i.v. serelaxin injection augments EDH‐type relaxation (Leo et al., 2014b), in this study, our data demonstrated that 72 h continuous i.v. infusion of serelaxin had no effect on EDH‐type relaxation in the mesenteric arteries. Thus, we proposed that the effect of serelaxin on the EDH component is a rapid effect and that the contribution of EDH is not apparent with the longer duration of serelaxin treatment. Therefore, in 72 h serelaxin‐treated mesenteric arteries, there was possible involvement of a vasodilator prostanoid, likely PGI2 that contributed to enhancement of BK‐mediated relaxation. The synthesis of PGI2 involves a cascade of enzymes such as COX1, COX2 and PGI2S. PGI2 stimulates smooth muscle PGI2 receptors leading to accumulation of cAMP and vasodilation (Moncada et al., 1976; Narumiya et al., 1999). Our data indicated that following 72 h, but not after 48 h, of serelaxin infusion, the enhanced response to BK was significantly reduced by blockade of COX and PGI2S, suggesting up‐regulation of COX‐derived PGI2 at 72 h of serelaxin treatment. Consistent with our functional data, 72 h but not 48 h of serelaxin treatment significantly increased the production of 6‐keto PGF1α (metabolite for PGI2) when mesenteric arteries were stimulated with BK, reflecting the increased production of PGI2. The contribution of COX1 and COX2 in the responses evoked by serelaxin treatment was elucidated using pharmacological inhibition. The selective COX1 inhibitor SC560 had no effect on BK response, whereas the selective COX2 inhibitor NS398 significantly reduced BK‐evoked relaxation at 72 h of serelaxin infusion. Furthermore, there was an up‐regulation of COX2 but not COX1 and PGI2S mRNA expression after 72 h of serelaxin infusion. These findings indicate that the up‐regulation of BK‐evoked PGI2‐mediated relaxation is dependent on COX2 but not on COX1 activity. Consistent with this, serelaxin treatment for 24 h has been shown to increase expression of COX2 in normal human endometrial stromal cells (Unemori et al., 2001). Taken together, we propose that the prolonged effect of serelaxin on endothelial function is underpinned by COX2‐derived PGI2 production and this occurs at a time when eNOS expression/activity has returned to baseline levels.
Continuous i.v. serelaxin treatment for 48 h but not 72 h augmented endothelial function in the abdominal aorta: involvement of NO
In contrast to the findings in mesenteric arteries, ACh‐mediated endothelium‐dependent relaxation in the rat aorta was enhanced at 48 h, but not 72 h of serelaxin infusion. There is relatively limited information on the effects of serelaxin treatment on endothelial function in conduit vessels such as the aorta. In rat aortic rings treated in vitro with TNF‐α, co‐incubation with serelaxin for 48 h improved ACh‐mediated relaxation (Dschietzig et al., 2012). This was associated with enhanced eNOS phosphorylation at Ser1177 and Ser633 and dephosphorylation at Thr495 without affecting total eNOS expression (Dschietzig et al., 2012). Furthermore, a recent study demonstrated reduced basal NOS activity in the aorta of relaxin‐deficient mice (Ng et al., 2015). In our study, we also demonstrated that 48 h of i.v. serelaxin infusion increased basal NOS activity and this was independent of changes in total eNOS and iNOS expression. Previous studies demonstrated that serelaxin treatment increased eNOS phosphorylation at Ser1177 (McGuane et al., 2011b; Dschietzig et al., 2012), and although our study is consistent with this, we found that the difference did not reach statistical significance. Thus, it is also possible that 48 h of continuous i.v. serelaxin infusion enhanced NOS activity and NO‐mediated relaxation in the aorta through eNOS phosphorylation at Ser633 and dephosphorylation at Thr495 (Dschietzig et al., 2012). Interestingly, activation of the NO pathway by serelaxin was not prolonged to 72 h in the aorta as we did not observe augmentation to ACh‐mediated relaxation and basal NOS activity. Our functional data also indicate that relaxation to the endothelium‐independent agonist, SNP that targets soluble guanylate cyclase within the vascular smooth muscle cells, was not affected by prolonged (72 h) serelaxin treatment. This rules out the possible down‐regulation of pathways within the vascular smooth muscle cells for the loss of serelaxin's vascular action after 72 h. Another possible explanation for this phenomenon is the down‐regulation of the receptors for serelaxin, RXFP1, after prolonged exposure to the agonist. However, our Western data indicate that RXFP1 expression was not altered after serelaxin infusion. Furthermore, molecular investigation of the RXFP1 signalling pathway also revealed that RXFP1 lacks of the capacity to recruit β‐arrestins to the cell surface, a key process for the internalization of GPCRs to terminate its receptor activation, which accounts for the prolonged activation of RXFP1 (Callander et al., 2009). Taken together, we propose that with prolonged infusion (>48 h) of serelaxin under physiological conditions, it is likely that there are compensatory mechanisms within endothelial cells to counter for the activation/overproduction of NO in the aorta.
Continuous i.v. serelaxin treatment for 48 h had no effect on endothelial function in the mesenteric veins
RXFP1s are localized to endothelial cells and vascular smooth muscle cells of mesenteric veins (Jelinic et al., 2014). However, in the present study, 48 h serelaxin treatment had no effect on endothelial function in the mesenteric veins. Consistent with our data, previous studies also demonstrated that s.c. serelaxin treatment for 5 days had no effect on endothelial function (Jelinic et al., 2014) in mesenteric veins. Although we did not assess endothelial function in the mesenteric veins after 72 h of serelaxin treatment, we predict that endothelial function will not be altered. A recent study revealed that increasing concentrations of serelaxin caused cGMP and cAMP accumulation in human umbilical vein endothelial cells (Sarwar et al., 2015). However, serelaxin‐induced cGMP and cAMP accumulation were not concentration dependent because serelaxin had a bell‐shaped rather than a sigmoidal concentration–response curve (Sarwar et al., 2015). These findings suggest that the activation of signalling mechanisms by serelaxin in veins does not occur when the concentration of serelaxin is either too low or too high. In our study, the plasma concentration of serelaxin after 48 h of continuous i.v. infusion was reported in the range of high nanomolar concentration. Thus, it is possible that the concentrations of serelaxin achieved in vivo in our study and possibly others (Li et al., 2005; Jelinic et al., 2014) are too high so that activation of vasodilator signalling pathways in veins does not occur.
In conclusion, continuous i.v. infusion of serelaxin activated a number of endothelial vasodilator pathways that were differentially up‐regulated, depending on the duration of treatment (48 or 72 h) and the type of vasculature. Continuous i.v. infusion for 48 h, which mimics the treatment duration for the phase III RELAX‐AHF clinical trial (Teerlink et al., 2013), augmented endothelial function through a NO‐dependent pathway in both mesenteric arteries and aortae. When treatment duration was extended to 72 h, the NO pathway was down‐regulated, and there was an up‐regulation of COX2‐derived PGI2 production, which sustained augmentation of endothelial function in the mesenteric arteries. This suggests that serelaxin has prolonged vasodilator effects in mesenteric arteries, which may be, at least in part, a mechanism by which serelaxin regulates peripheral resistance in either physiological or (patho)physiological settings.
Authorship contribution
C. H. L., M. J. and H. H. N. performed the research. C. H. L., M. J., M. T. and L. J. P. designed the study. C. H. L. and H. H. N. analysed the data. C. H. L. wrote the paper. M. J., H. H. N., M. T. and L. J. P. revised the paper critically for important intellectual content. C. H. L., M. J., H. H. N., M. T. and L. J. P. approved the final submission of paper.
Conflict of interest
The authors disclose that this project was partially funded by Novartis Pharma AG, who also provided the serelaxin as a condition of an Australian Research Council Linkage Grant. L. J. P. was also a paid consultant for Novartis Pharma AG.
Acknowledgements
The authors thank Dr Dennis Stewart, the coordinator of the research partnership (Novartis Pharma AG, Basel, Switzerland), for his helpful advice with the experimental design and critical analysis of the data. We thank Prof. David Sherwood for the rat RXFP1 antibody. We also thank Ms Kelly O’ Sullivan for her technical assistance in this study. The research was funded by an Australian Research Council Linkage Grant (L. J. P. and M. T.). Part of this research was also funded by Early Career Researcher Grant and JN Peter's Research Fellowship (C. H. L.). M. J. received an Australian Postgraduate Award, and H. H. N. received a Melbourne International Fee Remission Scholarship and a Melbourne International Research Scholarship.
Leo, C. H. , Jelinic, M. , Ng, H. H. , Tare, M. , and Parry, L. J. (2016) Time‐dependent activation of prostacyclin and nitric oxide pathways during continuous i.v. infusion of serelaxin (recombinant human H2 relaxin). British Journal of Pharmacology, 173: 1005–1017. doi: 10.1111/bph.13404.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callander GE, Thomas WG, Bathgate RA (2009). Prolonged RXFP1 and RXFP2 signaling can be explained by poor internalization and a lack of beta‐arrestin recruitment. Am J Physiol Cell Physiol 296: C1058–C1066. [DOI] [PubMed] [Google Scholar]
- Collino M, Rogazzo M, Pini A, Benetti E, Rosa AC, Chiazza F, et al. (2013). Acute treatment with relaxin protects the kidney against ischaemia/reperfusion injury. J Cell Mol Med 17: 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KP (2010). Unveiling the vasodilatory actions and mechanisms of relaxin. Hypertension 56: 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dschietzig T, Brecht A, Bartsch C, Baumann G, Stangl K, Alexiou K (2012). Relaxin improves TNF‐α‐induced endothelial dysfunction: the role of glucocorticoid receptor and phosphatidylinositol 3‐kinase signalling. Cardiovasc Res 95: 97–107. [DOI] [PubMed] [Google Scholar]
- Jelinic M, Leo CH, Post Uiterweer ED, Sandow SL, Gooi JH, Wlodek ME, et al. (2014). Localization of relaxin receptors in arteries and veins, and region‐specific increases in compliance and bradykinin‐mediated relaxation after in vivo serelaxin treatment. FASEB J 28: 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyabalan A, Novak J, Danielson LA, Kerchner LJ, Opett SL, Conrad KP (2003). Essential role for vascular gelatinase activity in relaxin‐induced renal vasodilation, hyperfiltration, and reduced myogenic reactivity of small arteries. Circ Res 93: 1249–1257. [DOI] [PubMed] [Google Scholar]
- Jeyabalan A, Novak J, Doty KD, Matthews J, Fisher MC, Kerchner LJ, et al. (2007). Vascular matrix metalloproteinase‐9 mediates the inhibition of myogenic reactivity in small arteries isolated from rats after short‐term administration of relaxin. Endocrinology 148: 189–198. [DOI] [PubMed] [Google Scholar]
- Khong SM, Andrews KL, Huynh NN, Venardos K, Aprico A, Michell DL, et al. (2012). Arginase II inhibition prevents nitrate tolerance. Br J Pharmacol 166: 2015–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo CH, Hart JL, Woodman OL (2011a). Impairment of both nitric oxide‐mediated and EDHF‐type relaxation in small mesenteric arteries from rats with streptozotocin‐induced diabetes. Br J Pharmacol 162: 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo CH, Hart JL, Woodman OL (2011b). 3′,4′‐Dihydroxyflavonol reduces superoxide and improves nitric oxide function in diabetic rat mesenteric arteries. PLoS One 6: e20813‐e20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo CH, Hart JL, Woodman OL (2011c). 3′,4′‐Dihydroxyflavonol restores endothelium dependent relaxation in small mesenteric artery from rats with type 1 and type 2 diabetes. Eur J Pharmacol 659: 193–198. [DOI] [PubMed] [Google Scholar]
- Leo CH, Joshi A, Hart JL, Woodman OL (2012). Endothelium‐dependent nitroxyl‐mediated relaxation is resistant to superoxide anion scavenging and preserved in diabetic rat aorta. Pharmacol Res 66: 383–391. [DOI] [PubMed] [Google Scholar]
- Leo CH, Jelinic M, Gooi JH, Tare M, Parry LJ (2014a). A vasoactive role for endogenous relaxin in mesenteric arteries of male mice. PLoS One 9: e107382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo CH, Jelinic M, Parkington HC, Tare M, Parry LJ (2014b). Acute intravenous injection of serelaxin (recombinant human relaxin‐2) causes rapid and sustained bradykinin‐mediated vasorelaxation. J Am Heart Assoc 3: e000493–e000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Brookes ZL, Kaufman S (2005). Acute and chronic effects of relaxin on vasoactivity, myogenic reactivity and compliance of the rat mesenteric arterial and venous vasculature. Regul Pept 132: 41–46. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuane JT, Danielson LA, Debrah JE, Rubin JP, Novak J, Conrad KP (2011a). Angiogenic growth factors are new and essential players in the sustained relaxin vasodilatory pathway in rodents and humans. Hypertension 57: 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuane JT, Debrah JE, Sautina L, Jarajapu YP, Novak J, Rubin JP, et al. (2011b). Relaxin induces rapid dilation of rodent small renal and human subcutaneous arteries via PI3 kinase and nitric oxide. Endocrinology 152: 2786–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Gryglewski R, Bunting S, Vane JR (1976). An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibit platelet aggregation. Nature 263: 663–665. [DOI] [PubMed] [Google Scholar]
- Münzel T, Daiber A, Mülsch A (2005). Explaining the phenomenon of nitrate tolerance. Circ Res 30: 618–628. [DOI] [PubMed] [Google Scholar]
- Münzel T, Daiber A, Gori T (2011). Nitrate therapy: new aspects concerning molecular action and tolerance. Circulation 123: 2132–2144. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F (1999). Prostanoid receptors: structures, properties, and functions. Physiol Rev 79: 1193–1226. [DOI] [PubMed] [Google Scholar]
- Ng HH, Jelinic M, Parry LJ, Leo CH (2015). Increased superoxide production and altered nitric oxide‐mediated relaxation in the aorta of young but not old male relaxin‐deficient mice. Am J Physiol Heart Circ Physiol 309: H285–H296. [DOI] [PubMed] [Google Scholar]
- Novak J, Ramirez RJ, Gandley RE, Sherwood OD, Conrad KP (2002). Myogenic reactivity is reduced in small renal arteries isolated from relaxin‐treated rats. Am J Physiol Regul Integr Comp Physiol 283: R349–R355. [DOI] [PubMed] [Google Scholar]
- Novak J, Parry LJ, Matthews JE, Kerchner LJ, Indovina K, Hanley‐Yanez K, et al. (2006). Evidence for local relaxin ligand‐receptor expression and function in arteries. FASEB J 20: 2352–2362. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SPH, Buneman OP et al, NC‐IUPHAR (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponikowski P, Mitrovic V, Ruda M, Fernandez A, Voors AA, Vishnevsky A, et al. (2014). A randomized, double‐blind, placebo‐controlled, multicentre study to assess haemodynamic effects of serelaxin in patients with acute heart failure. Eur Heart J 35: 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarwar M, Samuel CS, Bathgate RA, Stewart DR, Summers RJ (2015). Serelaxin‐mediated signal transduction in human vascular cells: bell‐shaped concentration‐response curves reflect differential coupling to G proteins. Br J Pharmacol 172: 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, et al. (2013). Serelaxin, recombinant human relaxin‐2, for treatment of acute heart failure (RELAX‐AHF): a randomised, placebo‐controlled trial. Lancet 381: 29–39. [DOI] [PubMed] [Google Scholar]
- Unemori EN, Lewis M, Grove BH, Deshpande U (2001). Relaxin Induces Specific Alterations in Gene Expression in the Human Endometrium In: {0} (eds)Tregear GW, Ivell R, Bathgate RA, Wade JD. Kluwer Academic Publishers, The Netherlands Relaxin, Vol. 2000, pp. 65–72. [Google Scholar]
- van Drongelen J, Ploemen IH, Pertijs J, Gooi JH, Sweep FC, Lotgering FK, et al. (2011). Aging attenuates the vasodilator response to relaxin. Am J Physiol Heart Circ Physiol 300: H1609–H1615. [DOI] [PubMed] [Google Scholar]
- van Drongelen J, van Koppen A, Pertijs J, Gooi JH, Sweep FC, Lotgering FK, et al. (2013). Impaired effect of relaxin on vasoconstrictor reactivity in spontaneous hypertensive rats. Peptides 49: 41–48. [DOI] [PubMed] [Google Scholar]
- van Drongelen J, van Koppen A, Pertijs J, Gooi JH, Parry LJ, Sweep FC, et al. (2012). Impaired vascular responses to relaxin in diet‐induced overweight female rats. J Appl Physiol 112: 962–969. [DOI] [PubMed] [Google Scholar]
- Vodstrcil LA, Tare M, Novak J, Dragomir N, Ramirez RJ, Wlodek ME, et al. (2012). Relaxin mediates uterine artery compliance during pregnancy and increases uterine blood flow. FASEB J 26: 4035–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]