

Abstract
Classically, receptor‐mediated signalling was conceived as a linear process involving one agonist, a variety of potential targets within a receptor family (e.g. α‐ and β‐adrenoceptors) and a second messenger (e.g. cAMP)‐triggered response. If distinct responses were stimulated by the same receptor in different tissues (e.g. lipolysis in adipocytes vs. increased beating rate in the heart caused by adrenaline), the differences were attributed to different second messenger targets in the different tissues. It is now realized that an individual receptor can couple to multiple effectors (different G proteins and different β‐arrestins), even in the same cell, to drive very distinct responses. Furthermore, tailored agonists can mould the receptor conformation to activate one signal pathway versus another by a process termed ‘biased signalling’. Complicating issues further, we now know that activating one receptor can rapidly trigger the local release of agonists for a second receptor via a process termed ‘transactivation’. Thus, the end response can represent a cooperative signalling process involving two or more receptors linked by transactivation. This overview, with a focus on the GPCR, protease‐activated receptor‐1, integrates both of these processes to predict the complex array of responses that can arise when biased receptor signalling also involves the receptor transactivation process. The therapeutic implications of this signalling matrix are also briefly discussed.
Linked Articles
This article is part of a themed section on Molecular Pharmacology of G Protein‐Coupled Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v173.20/issuetoc
Abbreviations
- HB‐EGF
heparin‐binding EGF
- PAR
proteinase‐activated receptor
- TGFBR
TGF‐β receptor
- TRPV
transient receptor potential channel
- VSMC
vascular smooth muscle cell
Tables of Links
| TARGETS | ||
|---|---|---|
| GPCRs a | Voltage‐gated ion channels b | Enzymes d |
| EP receptors | TRPV1 | Cathepsin S |
| ET receptors | TRPV4 | COX |
| PAR1 | Catalytic receptors c | ERK |
| PAR2 | EGF receptor | MMP1 |
| PAR3 | TGFBR1 | Neutrophil elastase |
| PAR4 | Smad | |
| Src |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,dAlexander et al., 2015c, 2015a, 2015b, 2015d).
Introduction
GPCRs are integral to all physiology and much pathophysiology. The study of GPCRs and their signalling pathways has arguably proved to be the most successful route for generating highly efficacious therapeutic agents. Since the mid‐1960s when the first GPCR ligand‐binding studies were made using radiolabelled atropine (Paton and Rang, 1965), great strides have been made in understanding the molecular pharmacology of GPCR signalling ranging from detailed receptor structure‐activity studies to an understanding of the crystal structure of the β‐adrenoceptor interacting with its Gs protein effector (Rasmussen et al., 2011a, 2011b). For reference, guidance and clarity are greatly simplified; classical or historical views of GPCR signalling are provided in Figure 1.
Figure 1.
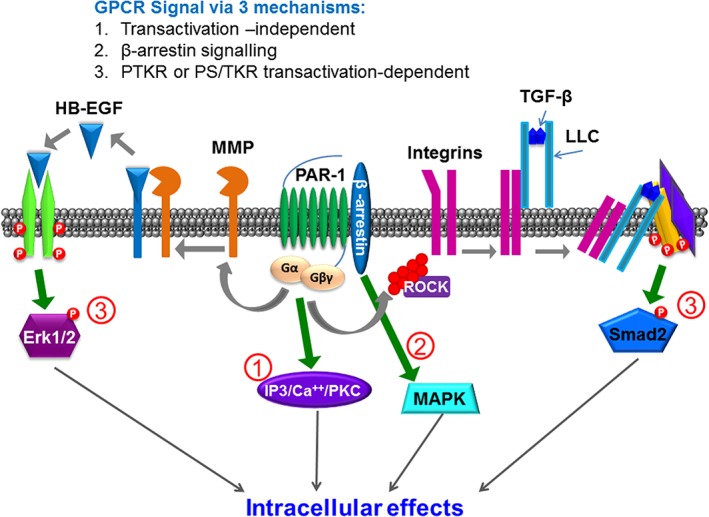
Classification of GPCR signalling developed from the perspective of the emerging importance of transactivation‐dependent signalling to the GPCR signalling paradigm.
The concept of the plasticity of the receptor has also advanced substantially, in terms of predicting either ligand‐stabilized (Changeux–Wyman–Monod model) or ligand‐induced (Koshland model) conformational changes that drive receptor–effector interactions (Christopoulos et al., 2014). In keeping with the mobile or floating receptor models of receptor function proposed some time ago (de Haen, 1976; Jacobs and Cuatrecasas, 1976), receptor plasticity permits different ligands to drive interactions of an individual GPCR in different ways with their G‐protein effectors leading to different cellular effects or hormone‐induced phenotypic outcomes. This ligand‐dependent differential interaction of a receptor with its effectors that results in distinct end responses has been termed ‘functional selectivity’ or ‘biased’ signalling (Kenakin and Miller, 2010; Kenakin, 2013; Kenakin and Christopoulos, 2013; Christopoulos, 2014) (Figure 2). This signalling bias for GPCRs applies not only to GPCR‐mediated signalling but also to Gα‐independent, β‐arrestin‐dependent scaffold‐mediated signalling by so‐called GPCRs (Defea, 2008). This conceptual framework for distinct signalling pathways triggered by an individual agonist–receptor interaction presumes that all of the receptor‐driven downstream signal pathways result directly from the individually but differentially activated receptor.
Figure 2.
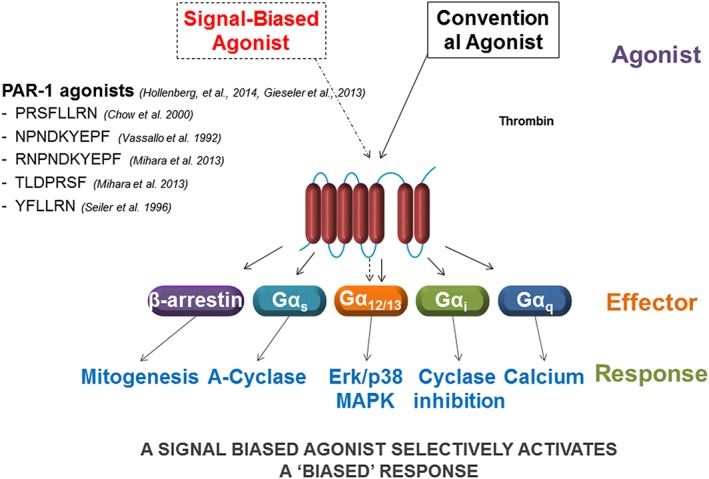
Schema encompassing the phenomena referred to as ‘biased signalling’.
What the above paradigms have not taken into account is the ability of a signalling process triggered by one receptor to cause a rapid sequential activation of a second cell surface receptor that is ultimately responsible for major downstream cellular signalling and cellular effects or hormone‐induced phenotypic outcomes. This sequential signalling, caused by the second receptor is most often qualitatively and quantitatively different from the canonical signalling pathways stimulated by the GPCR that was first activated. Indeed, sequential signalling can involve not only plasma membrane signalling (e.g. via growth factor receptors and ion channels; see below) but also rapid modulation of downstream signalling via steroid hormone receptors, like the receptors for the hormone, oestrogen.
For the purposes of our article, the transactivation of steroid hormone receptors will not be dealt with but cannot be discounted (Bunone et al., 1996; Kato et al., 1998; Busch et al., 2015). This type of sequential signalling, involving two or possibly more different receptors has been termed ‘receptor transactivation’. This transactivation process, as most commonly understood, involves the rapid generation by the first‐activated receptor of a ligand that in turn activates a second receptor. For instance, activation of the protease‐activated receptor‐1 (PAR1) for thrombin in gastric smooth muscle quickly (within a minute) releases COX‐generated prostanoids that in turn drive EP receptor‐driven contractile responses (Zheng et al., 1998). This receptor transactivation via a rapidly released ligand has, for GPCRs, also been found to activate the EGF receptor via the release of an EGF receptor‐activating ligand like heparin‐binding EGF (HB‐EGF). This ligand‐release process has been found to be proteinase dependent, in terms of the GPCR‐triggered transactivation of the EGF receptor. Thus, a membrane‐localized MMP triggered by GPCR stimulation releases an EGF‐activating ligand (HB‐EGF or, alternatively, amphiregulin). The released HB‐EGF in turn activates the EGF receptor to stimulate downstream Epidermal Growth Factor Receptor (EGFR) kinase signalling pathways (Daub et al., 1996; Prenzel et al., 1999). To date, most attention for this transactivation process has been focused on the sequential activation of the EGF receptor caused by a variety of GPCRs, including those for thrombin (PAR1), endothelin‐1 and lysophosphatidic acid (Daub et al., 1996; Prenzel et al., 1999). The transactivation of the EGF receptor, in parallel with GPCR activation, can also take place via an MMP‐independent process that involves a TK (Src)‐mediated cross‐phosphorylation/activation of the EGF‐receptor kinase. Thus, until recently, transactivation‐dependent signalling was thought to involve either the generation of locally produced prostanoids (a process unfortunately overlooked in many recent discussions of transactivation) or the release of an EGF receptor ligand that causes activation of the EGF‐receptor TK. Thus, downstream stimulation of growth factor‐associated pathways like MAPK/ERK caused by GPCR activation can be due either to the direct GPCR‐mediated regulation of G protein or β‐arrestin scaffold‐mediated signalling or, in parallel, to the transactivation of the EGF receptor. More recently, however, this paradigm has been extended from solely TK receptors to include the GPCR‐mediated transactivation of a receptor‐serine/threonine kinase, specifically the TGF‐β receptor (TGFBR1), leading to the formation of extreme carboxy‐terminal‐phosphorylated Smad2 (phosphoSmad2C). This process can be blocked by antagonising the GPCR PAR1 or the TGFBR1 (Burch et al., 2010; Burch et al., 2013). The mechanism of the GPCR transactivation of protein TK receptors has been the subject of almost 200 publications and is fairly well understood at least at a rudimentary level. Interestingly, and somewhat surprisingly, all of the data thus far reported for the GPCR transactivation of protein serine/threonine kinase receptors indicate that this mechanism is completely distinct from that for transactivation of protein TK receptors. There is a major caveat that the role of G proteins has been little explored, for reasons we have previously discussed (Bernard et al., 2014; Kamato et al., 2015b), and the potential that G proteins represent a point of commonality in GPCR to protein tyrosine, and serine/threonine kinase receptor transactivation signalling represents a very important piece of the puzzle in this area. This transactivation process can be even more complex in the setting of a cell culture or in vivo model where an independent rapid transcriptional event, for example, cell stretching, results in the generation of new GPCR agonists in the system (e.g. cytokines), which in turn regulate their own receptor systems.
Receptor transactivation in the context we will discuss here will be limited to the very rapid process, defined as the mechanism by which ‘the agonist occupancy of its cognate GPCR’ leads in a relatively short time (seconds to minutes) and in the absence of de novo protein synthesis to the activation of a second cell surface receptor (Kamato et al., 2015a). As mentioned above, it is necessary to take into account the rapid temporal characteristics of the response, because longer‐term receptor signalling can, via transcriptional or post‐translational mechanisms, lead to the generation of a multitude of secondary and tertiary signalling processes. For our overview, these longer‐term effects will be excluded from the definition of transactivation. For instance, this delayed effect may be due to the GPCR‐mediated up‐regulation of a cytokine, which in turn activates its own cytokine receptor in the cell microenvironment. This review deals with the challenge of integrating these paradigms of biased signalling or functional selectivity into a common paradigm that can include the receptor transactivation processes. The following sections deal with this challenge.
GPCR biased signalling and receptor transactivation
Classically, receptor‐mediated signalling was conceived as a linear process involving one agonist, a variety of potential targets within a receptor family (e.g. α‐ and β‐adrenoceptors) and a second messenger (e.g. cAMP)‐triggered response. If distinct responses were stimulated by the same receptor in different tissues (e.g. lipolysis in adipocytes vs. increased beating rate in the heart caused by adrenaline), the differences were attributed to different second messenger effectors in the different tissues. As already mentioned above, it is now appreciated and understood that an individual receptor can couple to multiple effectors (different G proteins and different β‐arrestins), even in the same cell, to drive very distinct responses. Furthermore, tailored agonists can mould the receptor conformation to activate one signal pathway versus another by a process termed biased signalling or functional selectivity (Kenakin and Miller, 2010; Kenakin, 2013; Kenakin and Christopoulos, 2013; Christopoulos, 2014). The plasticity of a receptor‐ligand interaction leads to signalling consequences for both agonists and antagonists in terms of their potential sites of interaction with the receptor [e.g. at the orthosteric ligand‐binding site or remotely at an allosteric‐binding site that can affect ligand signalling in a positive (positive allosteric modulation) or negative (negative allosteric modulation) way]. This process of modulating agonist action would also have its impact on the process of receptor transactivation, as will be dealt with briefly in the following sections.
To place our overview in a practical context, we will use as a prototype, signalling by PAR1, which is a major target for the serine protease, thrombin. We choose this example because thrombin has for some time been recognized as a rapid regulator of numerous cell responses, including platelet secretion, vascular contractility, tumour cell invasion and anabolic tissue responses involving transcriptional and translational mechanisms. Thus, this GPCR (PAR1) drives multiple and diverse responses that match the complexity of responses to growth factors like insulin, EGF and TGF‐β and to do so, PAR1 uses all of the tools for signalling, including functional selectivity and kinase receptor transactivation. Indeed, PAR1 has it all in terms of exploiting ligand‐driven receptor plasticity, and it does so all on its own in a unique manner.
Discovery of thrombin action via PAR1 and biased PAR signalling
Proteinase‐activated receptors (PARs), discovered as a result of the search for the target of thrombin action (Rasmussen et al., 1991; Vu et al., 1991; Coughlin, 2000; Adams et al., 2011; Alexander et al.,2015a) belong to the group A rhodopsin‐like GPCR subfamily comprising four members (PAR1, 2, 3 and 4) (Alexander et al., 2015a). PARs have the unusual mechanism of activation that involves the proteolytic unmasking of a cryptic receptor‐activating peptide sequence that remains attached to the receptor N‐terminus and functions as a tethered ligand (Vu et al., 1991). When first cloned, it was not appreciated that the thrombin receptor belongs to a GPCR family with four members and that the multiple cellular actions of thrombin involve the activation of two different receptors, namely, PAR1 and 4. Further, the second family member to be cloned, PAR2, which is preferentially activated by trypsin but not thrombin, was found to be co‐expressed with PAR1 in the vasculature and was therefore able to work in a coordinated way with PAR1 to regulate vasoconstriction. Thus, it took some time to sort out the multiple signalling effects of the PARs in terms of which PAR can trigger which response in a target tissue. Issues of biased signalling and receptor transactivation involving the PARs were therefore difficult to evaluate until about the late 2000s, when the actions of PAR‐selective peptide agonists and PAR‐selective antagonists became available. Furthermore, although it was realized that multiple serine proteinases could mimic the actions of thrombin and trypsin to activate PAR1, 2 and 4 by cleaving at a common arginine to unmask a tethered canonical ligand in each PAR, it was not appreciated that some enzymes like MMP1 and neutrophil elastase can activate PAR signalling by cleaving the N‐terminal sequence at sites distinct from those targeted by thrombin and trypsin (Boire et al., 2005; Trivedi et al., 2009; Ramachandran et al., 2011; Mihara et al., 2013; Hollenberg et al., 2014). In a similar manner, the coagulation factor, activated protein‐C (APC) for PAR1 (Mosnier et al., 2012; Schuepbach et al., 2012), and cathepsin‐S for PAR2 (Zhao et al., 2014a) are also able to cleave PARs at distinct N‐terminal sites to result in signalling that is distinct from that for thrombin (for PAR1) and trypsin (for PAR2). These distinct non‐canonical tethered ligand sequences unmasked by MMP1, elastase, APC and cathepsin S drive biased PAR signalling, so as to result in quite distinct cellular responses (Zhao et al., 2014a; Zhao et al., 2014b). As for the canonical PAR‐activating tethered ligand sequences unmasked by thrombin (PAR1 and 4) and trypsin (PAR2), synthetic peptides derived from the non‐canonical tethered ligands revealed in the PAR1 N‐terminus by MMP1, APC and neutrophil elastase can, in the absence of receptor proteolysis, trigger biased signalling (Boire et al., 2005; Mosnier et al., 2012; Schuepbach et al., 2012; Mihara et al., 2013; Zhao et al., 2014a). As reviewed more extensively elsewhere, GPCRs in general can be stimulated to activate a selective signalling pathway (Kenakin and Christopoulos, 2013; Christopoulos, 2014), and many of these GPCRs are also able to mediate the transactivation of other receptors including the one for EGF.
PAR1 signalling and receptor transactivation
As already mentioned, in addition to biased PAR signalling, it was appreciated early on in studies of thrombin action that PAR1 can signal via the transactivation of a prostanoid EP receptor, due to the rapid generation of prostaglandins in the course of tissue activation (Zheng et al., 1998). Further, PAR1 joined other GPCRs as a receptor able to transactivate the EGF receptor via the MMP‐catalysed release of amphiregulin or HB‐EGF (Daub et al., 1996; Daub et al., 1997). What is not yet clear is whether or not the processes of biased signalling and PAR‐driven signal transactivation does or does not involve the participation of PAR homo or heterodimers (Gieseler et al., 2013). This issue is of a complexity beyond the scope of our overview but will need to be taken into account in the future. Thus, it is essential to distinguish biased PAR1 signalling (either as a receptor monomer or as a potential homo or heterodimer with another PAR) from a cell response that results from PAR1‐mediated transactivation of a second receptor. In principle, depending on the host cell in which PAR1 is activated, the transactivation process can be driven by a biased PAR1 activation process, where a unique signal pathway is triggered (Figure 2).
For PAR1‐mediated signalling via transactivation of other receptors, attention has been focused primarily on targeting the receptor for EGF (Gschwind et al., 2001). This process is now known to involve not only participation of a membrane‐localized MMP that liberates an EGF receptor agonist but also via an MMP‐independent process that involves Src (Kawao et al., 2005; Caruso et al., 2006; van der Merwe et al., 2008) and a number of other recently identified factors, including a Rho‐GTP‐exchange factor (TRIO), an apoptosis‐targeted TK (BMX) and choline‐kinase‐α (George et al., 2013). These complex signal pathways lead to the rapid transphosphorylation/activation of the EGF receptor to drive MAPK activation and thereby stimulate a number of complex downstream signalling pathways that are targets of EGF action. In a similar way, the PDGF receptor can be transactivated by an Src‐dependent direct phosphorylation of the receptor when PAR2 is activated by the Tissue Factor/Factor VIIa complex (Siegbahn et al., 2008).There is no reason to doubt that the Src‐mediated process will also coordinately regulate other receptor targets in addition to the EGF and PDGF receptors.
An example of the complexity of this transactivation process can be seen in the regulation of primary cultures of human saphenous vein‐derived vascular smooth muscle cells (VSMCs) via PAR1. Thus, when VSMCs are treated with thrombin, there is a rapid increase in ERK/MAPK phosphorylation activation. There are several possible mechanisms downstream of PAR1 that can lead to activation of this pathway including not only the kinase suppressor of Raf scaffolding protein (KSR; Dhanasekaran et al., 2007) but also pathways relying on the transactivation of the EGF receptor. This EGFR transactivation, via its GRB2‐Ras activation mechanism would lead to ERK/MAPK signals. In VSMCs, almost all of the increased ERK/MAPK activation in response to thrombin treatment can be blocked by the EGF receptor kinase inhibitor, AG1478. Thus, the EGF receptor‐kinase is integrally involved, although its precise mechanism of transactivaton in the smooth muscle cells (i.e. MMP mediated or not) was not determined (Burch et al., 2013).
To add to the depth of this transactivation process, stimulation of the VSMCs by thrombin, along with affecting EGF‐kinase activity, simultaneously transactivates the serine/threonine kinase TGFBR1, which directly phosphorylates Smad2 and/or Smad3 in their extreme carboxy‐terminal domains in what is known as canonical TGFBR1 signalling (Derynck and Zhang, 2003; Massague, 2012). The carboxy‐terminal phosphoSmad2 or 3 can translocate to the nucleus and is thus fully functional in terms of TGF‐β signalling. Smad2 or 3 phosphorylation is followed by Smad4 heterodimerization with the phosphoSmads and ultimately the regulation of TGF‐β‐stimulated gene expression (Massague, 2012; Burch et al., 2013; Macias et al., 2015). Notably, concomitant PAR1 stimulation and transactivation of both the EGF receptor and TGF‐β receptors mediates the action of thrombin to stimulate hyperelongation of the glycosaminoglycan chains on the proteoglycan, biglycan, in a mechanism under investigation as the initiating event in the development of atherosclerosis (Little et al., 2008; Ballinger et al., 2010; Getachew et al., 2010; Burch et al., 2013). Thus, understanding the signal mechanisms that can ultimately lead to transactivation of both the EGF and TGF‐β receptors is of pathophysiological importance.
We have also shown that as well as thrombin acting at PAR1, endothelin‐1 acting through its cognate receptor, ET receptor, can transactivate the TGFBR1 in cultured human VSMCs with both models producing a time‐dependent increase in phosphoSmad2 and downstream regulation of glycosaminoglycan synthesis on biglycan (Little et al., 2010). Cheng et al. have recently reported nearly identical results for the transactivation of TGFBR1 by the PAR2 agonist 2f‐LI, in primary human proximal tubular epithelial cells (Chung et al., 2013). There was only one substantial difference in the mechanism of GPCR (PAR1 and PAR2) to serine/threonine kinase receptor TGFBR1 transactivation in these two reports in that Chung et al. (2013 reported that the response was dependent upon MMPs, implying a role for the release of a TGFBR1‐activating ligand analogous to the role of HB‐EGF in angiotensin II to EGF receptor transactivation, whereas we had earlier reported that the response was not blocked by the broad spectrum MMP inhibitor, GM6001, and was therefore independent on MMPs (Burch et al., 2013). This important mechanistic point requires experimental clarification. Our preliminary data indicate that the phenomenon of PAR1 transactivation of the TGFBR1 is cell‐type specific. We have found that the response is not present in human cardiac fibroblasts or in bovine aortic endothelial cells but the transactivation appears to be present in HaCaT keratinocytes, which show an increase in phosphoSmad2C in response to several GPCR agonists (unpublished results); it is notable that immortalized human keratinocyte HaCaT cells were also used in the early work demonstrating the GPCR‐mediated transactivation of the EGF receptor (Daub et al., 1997).
PAR‐mediated transregulation of transient receptor potential channels
In addition to regulation of receptor function, transactivation processes can also affect ion channel function to stimulate signal transduction pathways. Thus, activation of PAR2, in addition to triggering the transactivation of the EGF receptor can also enhance the function of transient receptor potential channels TRPV1 and 4. The amplification of TRPV1 function by PAR2 activation involves phosphorylation of PKC‐ε as well as PKA (Amadesi et al., 2006). Although not evaluated, it is possible that a direct TRPV1 phosphorylation due to PAR2 action in a sensory nerve leads to increased calcium influx and thus, calcium‐mediated signalling events due to channel transregulation. In a comparable way, PAR2 stimulation can augment the function of TRPV4. In this instance, the transregulation of the channel is due to the phosphorylation of tyrosine residue 110 in TRPV4 (Poole et al., 2013). No doubt other GPCRs will be found to affect TRPV4 channel function, as we have found that TRPV4 signalling is augmented in endothelial cells stimulated by angiotensin II as well as by a PAR agonist (Saifeddine et al., 2015). In this situation, the transregulation of TRPV4 is a result of transactivation of the EGF receptor. Thus, two quite distinct signal events, one involving transactivation of the EGF receptor and a second due to transregulation of TRPV4 would have an immediate effect on cell function. This example illustrates the complexity of the transactivation process. Whether or not biased PAR or angiotensin II signalling can differentially affect this dual transactivation process remains to be evaluated.
Therapeutic implications of biased signalling and receptor transactivation
The substantial impact of biased signalling on the development of therapeutic agents has already been reviewed in depth (Kenakin, 2013, Kenakin and Christopoulos, 2013). However, the differential transactivation of multiple receptor targets in the same cellular environment has yet to be taken into account for the process of biased signalling, as outlined in Figure 3. Thus, the transactivation of multiple target receptors could in principle shower the cell with a large matrix of downstream signalling pathways as illustrated in Figure 2. However, as shown in Figure 3, a biased agonist could in principle transactivate only one of the potential feed‐forward receptors in the system. Thus, if a therapeutic agent targeted only one of the downstream receptors that can be transactivated (e.g. an EGF receptor‐selective kinase inhibitor like AG1478), the remainder of the signalling triggered by the upstream GPCR would be unaffected. Thus, one can predict that the distinct transcriptome readout for the biased GPCR agonist would be due not to the direct action of the GPCR and its effectors alone but due to the absence of signalling by the other transactivated receptors that are activated by the unbiased agonist number 1. Potentially, this kind of situation could lead to what might be interpreted as off‐target effects of the two distinct agonists. That interpretation would miss the mark made by the selective transactivation process. This issue merits consideration, because many of the paradigms for developing biased agonists or antagonists use reductionist signal readout systems, which are very often devoid of secondary receptor transactivation signals. To complement this reductionist approach, it will be of value in the future to assess the ability of biased agents to drive (or not) biased signalling in several cell and intact tissue systems.
Figure 3.

This schema illustrates the potential simultaneous transactivation of three different receptor systems resulting from the unbiased activation of a GPCR by agonist number 1 (black arrows). However, a biased agonist, number 2, is shown selectively to transactivate only receptor number 3.
Conclusion
The main message we wish to give is that GPCR signalling provides a rich source of stimuli, including not only the potential activation of multiple G proteins and a β‐arrestin‐scaffold‐stimulated signal pathway but also via the rapid coordinated transactivation of other independent receptors, which also signal to the cell originally activated by the first GPCR. The transactivation process can affect multiple membrane receptors with intrinsic tyrosine and serine/threonine kinase activity as well as activating prostanoid EP/TP receptors due to the rapid generation of COX products. Further, the transactivation process is able to signal not only via pharmacological receptors but also via ion channels like TRPV1 and TRPV4.
To sum up, established pathways of classical multiplex signalling can be termed transactivation‐independent. The multiple downstream signals generated by a single receptor can, for this process, be seen to be due either to the activation of multiple effectors (de Haen, 1976; Jacobs and Cuatrecasas, 1976) involving either distinct G proteins or a Gα‐protein‐independent signal pathway involving an arrestin scaffold. This arrestin‐mediated signalling can occur either with or without receptor internalizaton. Added to these multiple signals triggered by the GPCR itself, transactivation‐dependent signalling involves a rapid stimulation of a sequential receptor mechanism that can involve the immediate generation of released autacoids, like prostaglandins or growth factor‐activating agonists, or the direct cross‐activation of receptor kinases, which in turn phosphorylate either tyrosine or serine/threonine targets to generate signals (e.g. PDGF, EGF or TGFBRs respectively). These transactivation‐independent and transactivation‐dependent signal processes initiated by a GPCR will of necessity overlap with the biased signalling paradigm, in which an individual receptor like PAR1 can produce different responses when stimulated with different activating ligands, as outlined above. Thus, a future issue to sort out will be to determine if biased agonists in a bioassay system of relevance do or do not trigger the same transactivation signalling process as the non‐biased agonists. This information will be of therapeutic relevance as more biased receptor regulators are discovered for use in the clinic. One can therefore look forward to exciting developments in this area of GPCR signalling in the future.
Conflict of interest
The authors declare no conflicts of interest.
Little, P. , Hollenberg, M. , Kamato, D. , Thomas, W. , Chen, J. , Wang, T. , Zheng, W. , and Osman, N. (2016) Integrating the GPCR transactivation‐dependent and biased signalling paradigms in the context of PAR1 signalling. British Journal of Pharmacology, 173: 2992–3000. doi: 10.1111/bph.13398.
References
- Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD et al. (2011). Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther 130: 248–282. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadesi S, Cottrell GS, Divino L, Chapman K, Grady EF, Bautista F et al. (2006). Protease‐activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon‐ and A‐dependent mechanisms in rats and mice. J Physiol 575: 555–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinger ML, Osman N, Hashimura K, de Hann J, Jandeleit‐Dahm K, Allen TJ et al. (2010). Imatinib inhibits vascular smooth muscle proteoglycan synthesis and reduces LDL binding in vitro and aortic lipid deposition in vivo. J Cell Mol Med 14: 1408–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard R, Thach L, Kamato D, Osman N, Little AMP (2014). Assessing the role of Gaq/11 in cellular responses: An analysis of investigative tools. Clin Exp Pharmacol 4: 164. [Google Scholar]
- Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A (2005). PAR1 is a matrix metalloprotease‐1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120: 303–313. [DOI] [PubMed] [Google Scholar]
- Bunone G, Briand PA, Miksicek RJ, Picard D (1996). Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J 15: 2174–2183. [PMC free article] [PubMed] [Google Scholar]
- Burch ML, Ballinger ML, Yang SN, Getachew R, Itman C, Loveland K et al. (2010). Thrombin stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by protease‐activated receptor‐1 transactivation of the transforming growth factor beta type I receptor. J Biol Chem 285: 26798–26805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch ML, Getachew R, Osman N, Febbraio MA, Little PJ (2013). Thrombin‐mediated proteoglycan synthesis utilizes both protein‐tyrosine kinase and serine/threonine kinase receptor transactivation in vascular smooth muscle cells. J Biol Chem 288: 7410–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch S, Sims AH, Stal O, Ferno M, Landberg G (2015). Loss of TGFbeta receptor type 2 expression impairs estrogen response and confers tamoxifen resistance. Cancer Res 75: 1457–1469. [DOI] [PubMed] [Google Scholar]
- Caruso R, Pallone F, Fina D, Gioia V, Peluso I, Caprioli F et al. (2006). Protease‐activated receptor‐2 activation in gastric cancer cells promotes epidermal growth factor receptor trans‐activation and proliferation. Am J Pathol 169: 268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A (2014). Advances in G protein‐coupled receptor allostery: from function to structure. Mol Pharmacol 86: 463–478. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Changeux JP, Catterall WA, Fabbro D, Burris TP, Cidlowski JA et al. (2014). International Union of Basic and Clinical Pharmacology. XC. Multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev 66: 918–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Ramachandran R, Hollenberg MD, Muruve DA (2013). Proteinase‐activated receptor‐2 transactivation of epidermal growth factor receptor and transforming growth factor‐beta receptor signaling pathways contributes to renal fibrosis. J Biol Chem 288: 37319–37331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SR (2000). Thrombin signalling and protease‐activated receptors. Nature 407: 258–264. [DOI] [PubMed] [Google Scholar]
- Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A (1997). Signal characteristics of G protein‐transactivated EGF receptor. Embo J 16: 7032–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub H, Weiss FU, Wallasch C, Ullrich A (1996). Role of transactivation of the EGF receptor in signalling by G‐protein‐coupled receptors. Nature 379: 557–560. [DOI] [PubMed] [Google Scholar]
- de Haen C (1976). The non‐stoichiometric floating receptor model for hormone sensitive adenylyl cyclase. J Theor Biol 58: 383–400. [DOI] [PubMed] [Google Scholar]
- Defea K (2008). Beta‐arrestins and heterotrimeric G‐proteins: collaborators and competitors in signal transduction. Br J Pharmacol 153 (Suppl 1): S298–S309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang YE (2003). Smad‐dependent and Smad‐independent pathways in TGF‐beta family signalling. Nature 425: 577–584. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran DN, Kashef K, Lee CM, Xu H, Reddy EP (2007). Scaffold proteins of MAP‐kinase modules. Oncogene 26: 3185–3202. [DOI] [PubMed] [Google Scholar]
- George AJ, Purdue BW, Gould CM, Thomas DW, Handoko Y, Qian H et al. (2013). A functional siRNA screen identifies genes modulating angiotensin II‐mediated EGFR transactivation. J Cell Sci 126: 5377–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getachew R, Ballinger ML, Burch ML, Reid JJ, Khachigian LM, Wight TN et al. (2010). Platelet‐derived growth factor {beta}‐receptor kinase activity and ERK1/2 mediate glycosaminoglycan elongation on biglycan and increases binding to low‐density lipoprotein. Endocrinology 151: 4356–4367. [DOI] [PubMed] [Google Scholar]
- Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R (2013). Proteinase‐activated receptors (PARs) – focus on receptor‐receptor‐interactions and their physiological and pathophysiological impact. Cell Commun Signal 11: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A (2001). Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene 20: 1594–1600. [DOI] [PubMed] [Google Scholar]
- Hollenberg MD, Mihara K, Polley D, Suen JY, Han A, Fairlie DP et al. (2014). Biased signalling and proteinase‐activated receptors (PARs): targeting inflammatory disease. Br J Pharmacol 171: 1180–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs S, Cuatrecasas P (1976). The mobile receptor hypothesis and "cooperativity" of hormone binding. Application to insulin. Biochim Biophys Acta 433: 482–495. [DOI] [PubMed] [Google Scholar]
- Kamato D, Rostam MA, Bernard R, Piva TJ, Mantri N, Guidone D et al. (2015a). The expansion of GPCR transactivation‐dependent signalling to include serine/threonine kinase receptors represents a new cell signalling frontier. Cell Mol Life Sci 72: 799–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamato D, Thach L, Bernard R, Chan V, Zheng W, Kaur H et al. (2015b). Structure, function, pharmacology and therapeutic potential of the G protein, Gα/q,11. Front Cardiovasc Med 2: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Kitamoto T, Masuhiro Y, Yanagisawa J (1998). Molecular mechanism of a cross‐talk between estrogen and growth‐factor signaling pathways. Oncology 55 (Suppl 1): 5–10. [DOI] [PubMed] [Google Scholar]
- Kawao N, Nagataki M, Nagasawa K, Kubo S, Cushing K, Wada T et al. (2005). Signal transduction for proteinase‐activated receptor‐2‐triggered prostaglandin E2 formation in human lung epithelial cells. J Pharmacol Exp Ther 315: 576–589. [DOI] [PubMed] [Google Scholar]
- Kenakin T (2013). New concepts in pharmacological efficacy at 7TM receptors: IUPHAR review 2. Br J Pharmacol 168: 554–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A (2013). Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12: 205–216. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ (2010). Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev 62: 265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little PJ, Burch ML, Getachew R, Al‐Aryahi S, Osman N (2010). Endothelin‐1 stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by endothelin receptor transactivation of the transforming growth factor‐[beta] type I receptor. J Cardiovasc Pharmacol 56: 360–368. [DOI] [PubMed] [Google Scholar]
- Little PJ, Osman N, O'Brien KD (2008). Hyperelongated biglycan: the surreptitious initiator of atherosclerosis. Curr Opin Lipidol 19: 448–454. [DOI] [PubMed] [Google Scholar]
- Macias MJ, Martin‐Malpartida P, Massague J (2015). Structural determinants of Smad function in TGF‐beta signaling. Trends Biochem Sci 40: 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J (2012). TGFbeta signalling in context. Nat Rev Mol Cell Biol 13: 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara K, Ramachandran R, Renaux B, Saifeddine M, Hollenberg MD (2013). Neutrophil elastase and proteinase‐3 trigger G protein‐biased signaling through proteinase‐activated receptor‐1 (PAR1). J Biol Chem 288: 32979–32990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH (2012). Biased agonism of protease‐activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 120: 5237–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton WD, Rang HP (1965). The uptake of atropine and related drugs by intestinal smooth muscle of the guinea‐pig in relation to acetylcholine receptors. Proc R Soc Lond B Biol Sci 163: 1–44. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole DP, Amadesi S, Veldhuis NA, Abogadie FC, Lieu T, Darby W et al. (2013). Protease‐activated receptor 2 (PAR2) protein and transient receptor potential vanilloid 4 (TRPV4) protein coupling is required for sustained inflammatory signaling. J Biol Chem 288: 5790–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C et al. (1999). EGF receptor transactivation by G‐protein‐coupled receptors requires metalloproteinase cleavage of proHB‐EGF. Nature 402: 884–888. [DOI] [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA et al. (2011). Neutrophil elastase acts as a biased agonist for proteinase‐activated receptor‐2 (PAR2). J Biol Chem 286: 24638–24648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS et al. (2011a). Structure of a nanobody‐stabilized active state of the beta(2) adrenoceptor. Nature 469: 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Devree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS et al. (2011b). Crystal structure of the beta2 adrenergic receptor‐Gs protein complex. Nature 477: 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen UB, Vouret‐Craviari V, Jallat S, Schlesinger Y, Pages G, Pavirani A et al. (1991). cDNA cloning and expression of a hamster alpha‐thrombin receptor coupled to Ca2+ mobilization. FEBS Lett 288: 123–128. [DOI] [PubMed] [Google Scholar]
- Saifeddine M, El‐Daly M, Mihara K, Bunnett NW, Mcintyre P, Altier C et al. (2015). GPCR‐mediated EGF receptor transactivation regulates TRPV4 action in the vasculature. Br J Pharmacol 172: 2493–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuepbach RA, Madon J, Ender M, Galli P, Riewald M (2012). Protease‐activated receptor‐1 cleaved at R46 mediates cytoprotective effects. J Thromb Haemost 10: 1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegbahn A, Johnell M, Nordin A, Aberg M, Velling T (2008). TF/FVIIa transactivate PDGFRbeta to regulate PDGF‐BB‐induced chemotaxis in different cell types: involvement of Src and PLC. Arterioscler Thromb Vasc Biol 28: 135–141. [DOI] [PubMed] [Google Scholar]
- Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O'Callaghan K et al. (2009). Platelet matrix metalloprotease‐1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell 137: 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Merwe JQ, Hollenberg MD, Macnaughton WK (2008). EGF receptor transactivation and MAP kinase mediate proteinase‐activated receptor‐2‐induced chloride secretion in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 294: G441–G451. [DOI] [PubMed] [Google Scholar]
- Vu TK, Wheaton VI, Hung DT, Charo I, Coughlin SR (1991). Domains specifying thrombin‐receptor interaction. Nature 353: 674–677. [DOI] [PubMed] [Google Scholar]
- Zhao P, Lieu T, Barlow N, Metcalf M, Veldhuis NA, Jensen DD et al. (2014a). Cathepsin S causes inflammatory pain via biased agonism of PAR2 and TRPV4. J Biol Chem 289: 27215–27234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Metcalf M, Bunnett NW (2014b). Biased signaling of protease‐activated receptors. Front Endocrinol (Lausanne) 5: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XL, Renaux B, Hollenberg MD (1998). Parallel contractile signal transduction pathways activated by receptors for thrombin and epidermal growth factor‐urogastrone in guinea pig gastric smooth muscle: blockade by inhibitors of mitogen‐activated protein kinase‐kinase and phosphatidyl inositol 3'‐kinase. J Pharmacol Exp Ther 285: 325–334. [PubMed] [Google Scholar]