

Abstract
GPCRs are the most successful pharmaceutical targets in history. Nevertheless, the pharmacology of many GPCRs remains inaccessible as their endogenous or exogenous modulators have not been discovered. Tools that explore the physiological functions and pharmacological potential of these ‘orphan’ GPCRs, whether they are endogenous and/or surrogate ligands, are therefore of paramount importance. Rates of receptor deorphanization determined by traditional reverse pharmacology methods have slowed, indicating a need for the development of more sophisticated and efficient ligand screening approaches. Here, we discuss the use of structure‐based ligand discovery approaches to identify small molecule modulators for exploring the function of orphan GPCRs. These studies have been buoyed by the growing number of GPCR crystal structures solved in the past decade, providing a broad range of template structures for homology modelling of orphans. This review discusses the methods used to establish the appropriate signalling assays to test orphan receptor activity and provides current examples of structure‐based methods used to identify ligands of orphan GPCRs.
Linked Articles
This article is part of a themed section on Molecular Pharmacology of G Protein‐Coupled Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v173.20/issuetoc
Abbreviations
- CRE
cAMP response element
- ECL
extracellular loop
- HTS
high‐throughput screening
- ICL
intracellular loops
- NFAT
nuclear factor of activated T cells
- RE
response element
- RMSD
root mean squared deviation
- SAR
structure activity relationship
- SBDD
structure‐based drug design
- SRE
serum response element
- SRF
serum response factor
- TM
transmembrane
- VLS
virtual ligand screening
Tables of Links
| LIGANDS |
|---|
| Acetate |
| ATP |
| cAMP |
| GW9508 |
| IP3 |
| Linoleic acid |
| Nateglinide |
| TAK‐875 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
GPCRs are membrane proteins that can be activated by a diverse range of extracellular stimuli, triggering intracellular signalling events that mediate numerous physiological responses. Consequently, GPCRs, especially those of the rhodopsin‐like family, are regularly exploited as drug targets (Garland, 2013; Kinch et al., 2015). However, the pairing of GPCRs with their respective endogenous agonists remains a challenge for pharmacologists. The sequencing and annotation of the human genome has revealed the full extent of the GPCR superfamily, identifying over 800 GPCR genes, of which 342 are non‐olfactory (Fredriksson et al., 2003). Related GPCRs were quickly recognized and sorted by phylogeny (Fredriksson et al., 2003; Bjarnadottir et al., 2006); however, more than 150 GPCRs remain orphans (Davenport et al., 2013), including 86 rhodopsin‐like receptors (Table 1). Furthermore, only a small subset of GPCRs (~10%) is actually targeted by existing drugs (Garland, 2013). Therapeutic candidates are currently under development for many more receptors whose pharmacology is well understood. However, orphan GPCRs may also prove to be valuable therapeutic targets and experiments with orphan receptor knockout mice may provide insights into the physiological roles and therapeutic potential of these receptors (Davenport et al., 2013). If the orphan GPCR in question is activated by a yet unknown endogenous ligand, the discovery of this ligand is of paramount importance; however, it is not clear that every orphan GPCR necessarily requires an endogenous ligand, as some of them instead rely upon constitutive activity or heteromerize to achieve activation (Civelli et al., 2013). Regardless of their endogenous means of stimulation, identification of surrogate (non‐natural) ligands that can modulate the activity of orphan GPCRs will be invaluable for unlocking both their pharmacology and function.
Table 1.
Rhodopsin‐like orphan GPCRs and the reach of crystal structure templates for homology modelling
| Rhodopsin‐like GPCRs a | Solved crystal structures (PDB b ) | Orphan GPCR | Best % TM identity within subclass (template) c | Best % TM identity overall (template) |
|---|---|---|---|---|
| α‐subclass 107 GPCRs | Bovine rhodopsin (1F88) | GPR3 | 28% (D3) | — |
| (26 orphans; 24%) | Human β2‐adrenoceptor (2RH1) | GPR6 | 26% (S1P1) | — |
| Turkey β1‐adrenoceptor (2VT4) | GPR12 | 28% (S1P1) | — | |
| Human adenosine A2A (3EML) | GPR21 | 28% (M3) | — | |
| Human dopamine D3 (3PBL) | GPR22 | 23% (H1) | — | |
| Human histamine H1 (3RZE) | GPR45 | 30% (5‐HT1B) | ‐ | |
| Human S1P1 (3V2Y) | GPR50 | 28% (D3) | — | |
| Human M2 muscarinic (3UON) | GPR52 | 28% (M3) | 29% (OX2) | |
| Rat M3 muscarinic (4DAJ) | GPR61 | 25% (D3) | — | |
| Human 5‐HT1B (4IAQ) | GPR62 | 20% (5‐HT1B) | 20% (NTS1) | |
| Human 5‐HT2B (4IB4) | GPR63 | 30% (β1‐adrenoceptor) | — | |
| Human LPA1 (4Z34) | GPR88 | 27% (D3) | — | |
| GPR119 | 29% (S1P1) | — | ||
| GPR135 | 29% (D3) | — | ||
| GPR152 | 21% (D3) | 23% (NTS1) | ||
| GPR153 | 15% (H1) | 19% (δ) | ||
| GPR160 | 15% (5‐HT2B) | 16% (P2Y12) | ||
| GPR162 | 17% (D3) | — | ||
| OPN3 | 35% (Rhodopsin) | — | ||
| OPN4 | 30% (β1‐adrenoceptor) | — | ||
| OPN5 | 28% (Rhodopsin) | — | ||
| TAAR2 | 33% (β2‐adrenoceptor) | — | ||
| TAAR5 | 39% (β1‐adrenoceptor) | — | ||
| TAAR6 | 33% (5‐HT1B) | — | ||
| TAAR8 | 32% (5‐HT1B) | — | ||
| TAAR9 | 35% (β2‐adrenoceptor) | — | ||
| β‐subclass 41 GPCRs | Rat NTS1 (4GRV) | BB3 receptor | 32% (OX2) | — |
| (7 orphans; 17%) | Human OX2 (4RNB) | GPR37 | 21% (OX2) | 24% (CCR5) |
| GPR37L1 | 23% (NTS1) | — | ||
| GPR39 | 38% (NTS1) | — | ||
| GPR75 | 20% (NTS1) | 21% (CXCR4) | ||
| GPR83 | 35% (OX2) | — | ||
| GPR150 | 18% (NTS1) | 19% (δ) | ||
| γ‐subclass 64 GPCRs | Human CXCR4 (3ODU) | GPR1 | 34% (AT1) | — |
| (7 orphans; 11%) | Human μ opioid receptor (4DKL) | GPR15 | 35% (AT1) | — |
| Human κ opioid receptor (4DJH) | GPR20 | 32% (κ) | — | |
| Human NOP receptor (4EA3) | GPR25 | 35% (AT1) | — | |
| Mouse δ opioid receptor (4EJ4) | GPR32 | 31% (δ) | — | |
| Human CCR5 (4MBS) | GPR151 | 22% (NOP) | — | |
| Human AT1 (4YAY) | GPR182 | 28% (NOP) | — | |
| δ‐subclass 74 GPCRs | Human PAR1 (3VW7) | GPR4 | 31% (P2Y1) | — |
| (39 orphans; 53%) | Human FFA1 (4PHU) | GPR17 | 33% (PAR1) | — |
| Human P2Y12 (4NTJ) | GPR18 | 28% (P2Y1) | 28% (AT1) | |
| Human P2Y1 (4XNV) | GPR19 | 20% (P2Y1) | 29% (D3) | |
| GPR26 | 19% (PAR1) | 24% (D3) | ||
| GPR27 | 18% (FFA1) | 28% (5‐HT2B) | ||
| GPR31 | 30% (P2Y1) | — | ||
| GPR34 | 32% (P2Y12) | — | ||
| GPR35 | 27% (P2Y1) | 30% (κ) | ||
| GPR55 | 24% (PAR1) | 29% (CCR5) | ||
| GPR65 | 32% (P2Y1) | — | ||
| GPR68 | 27% (PAR1) | 28% (δ) | ||
| GPR78 | 23% (FFA1) | — | ||
| GPR82 | 22% (P2Y1) | 23% (δ) | ||
| GPR84 | 24% (P2Y12) | 25% (D3) | ||
| GPR85 | 18% (FFA1) | 25% (5‐HT2B) | ||
| GPR87 | 47% (P2Y1) | — | ||
| GPR101 | 14% (PAR1) | 20% (β2‐adrenoceptor) | ||
| GPR132 | 31% (P2Y1) | — | ||
| GPR161 | 22% (P2Y1) | 27% (5‐HT1B) | ||
| GPR171 | 37% (P2Y12) | — | ||
| GPR173 | 16% (PAR1) | 24% (5‐HT2B) | ||
| GPR174 | 33% (P2Y1) | — | ||
| GPR183 | 29% (PAR1) | — | ||
| LGR4 | 19% (P2Y12) | 22% (AT1) | ||
| LGR5 | 16% (P2Y12) | 19% (S1P1) | ||
| LGR6 | 17% (P2Y12) | 20% (CCR5) | ||
| MAS1 | 20% (PAR1) | — | ||
| MAS1L | 17% (FFA1) | 19% (M2) | ||
| MRGPRD | 19% (PAR1) | 20% (κ) | ||
| MRGPRE | 19% (FFA1) | — | ||
| MRGPRF | 17% (FFA1) | 18% (M2) | ||
| MRGPRG | 20% (FFA1) | — | ||
| MRGPRX1 | 19% (P2Y1) | 23% (κ) | ||
| MRGPRX2 | 19% (P2Y1) | 24% (AT1) | ||
| MRGPRX3 | 18% (P2Y12) | 22% (μ) | ||
| MRGPRX4 | 20% (P2Y1) | 24% (μ) | ||
| P2RY8 | 38% (PAR1) | — | ||
| P2RY10 | 32% (P2Y1) | — | ||
| Unclassified orphans | GPR139 | — | 21% (δ) | |
| GPR141 | — | 19% (P2Y12) | ||
| GPR142 | — | 22% (NTS1) | ||
| GPR146 | — | 19% (CXCR4) | ||
| GPR148 | — | 16% (κ) | ||
| GPR149 | — | 14% (FFA1) | ||
| GPR176 | — | 23% (NOP) |
Receptor numbers are according to the IUPHAR/BPS Guide to PHARMACOLOGY (Alexander et al., 2015a); probable pseudogenes were excluded.
The PDB ID of the first instance of the unique GPCR structure is given.
Sequence identity was determined based on the Needleman and Wunch algorithm (Needleman and Wunsch, 1970), following alignment of the orphan receptor sequence to the TM regions of all crystallized GPCRs.
Traditionally, reverse pharmacology has been used to identify endogenous and/or surrogate ligands for orphan GPCRs. This involves the expression of the orphan GPCR in a cellular system and testing large libraries of ligands, in a high‐throughput manner, for activity in a functional assay. Reverse pharmacology has proved successful for matching many orphan GPCRs with their cognate ligands (reviewed by Wise et al., 2004; Oh et al., 2006; Civelli et al., 2013), but newer strategies are needed as the success rate of this deorphanization approach has waned in the last few years. This is evident by the deorphanization of only 15 rhodopsin‐like GPCRs since the initial publication of IUPHAR's receptor list (Foord et al., 2005; Davenport et al., 2013; Southern et al., 2013) and the lack of independent validation of ligand‐receptor pairings (http://www.guidetopharmacology.org/latestPairings.jsp).
The past decade has seen the elucidation of an increasing number of GPCR X‐ray crystal structures, largely due to improved stabilization methods through protein engineering, the identification of tightly bound ligands and to the development of improved crystallization protocols [e.g. the development of lipidic cubic phase crystallization (Caffrey and Cherezov, 2009)] and crystallographic procedures [e.g. the development of femtosecond crystallography (Liu et al., 2014)]. Structures have typically been solved as fusion proteins with the T4‐lysozyme or thermostabilized apocytochrome bRIL substituted for the highly flexible third intracellular loop or N‐terminus; more recently, stabilizing mutations along with nanobodies have been used [discussed in Chun et al. (2012); Mujic‐Delic et al. (2014); Ghosh et al. (2015); Jazayeri et al. (2015)]. This increasing availability of GPCR structural information has allowed a shift from traditional high‐throughput screening (HTS) methods to cheaper and efficient virtual ligand screening (VLS) approaches for the identification of novel ligands at GPCRs.
This review will consider two aspects of these newer screening approaches: (i) establishing suitable screening assays for measuring orphan GPCR activation and (ii) the current status of structure‐based drug design (SBDD) in the context of orphan GPCRs. We have have also hypothesized what we expect could become a universal pipeline for orphan GPCR drug discovery, outlined in Figure 1.
Figure 1.
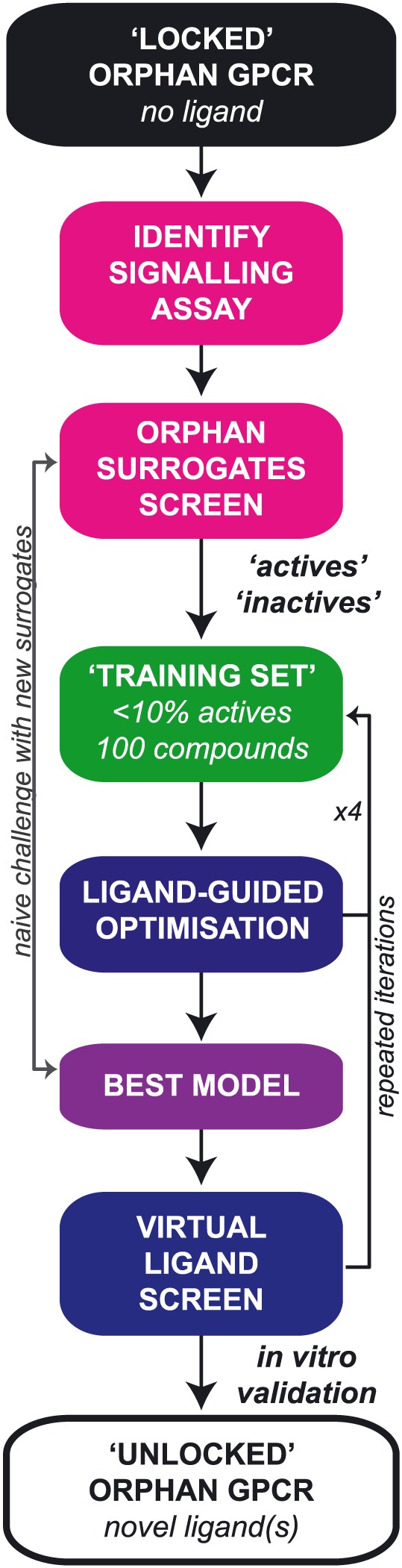
A putative pipeline for unlocking the pharmacology of orphan GPCRs by combining targeted HTS and SBDD. Combining both in vitro screening and hit validation with VLS at ligand‐optimized homology models is one rational approach to unlocking orphan GPCRs. First, an appropriate signalling assay must be identified, and then, a small library is screened for low affinity hits. This surrogate screen informs the composition of a ligand training set that can be used for the iterative ligand‐guided optimization of a homology model. Ideally, a subset of active and inactive ligands is set aside for naïve challenge of the best model to validate performance. VLS is then performed and hits validated in vitro by at least one assay readout. Additional site‐directed mutagenesis data and structure–activity relationships can further feed into repeated iterations of homology model optimization and VLS.
Experimental strategies for detection of orphan GPCR activation
Before attempting to deorphanize a GPCR, a number of issues need to be considered. Broadly speaking, it is important to establish whether there is sufficient preliminary evidence to suggest a significant physiological function of the orphan GPCR of interest, such as knockout mouse models, that would justify it being a potentially useful clinical target. Tissue expression profiles will also indicate the function of a particular orphan GPCR (Petryszak et al., 2015). Once an orphan target is identified, it is imperative that a robust, facile and relatively high‐throughput assay is established, either for screening of chemical libraries or for the validation of potential in silico ‘hits’. GPCRs mediate various physiological responses; however, as their name indicates, almost all of them signal via coupling to a heterotrimeric G protein (made up of α, β and γ subunits), particularly by interacting with the Gα subunit. Moreover, upon activation, most GPCRs recruit the internalization and scaffolding proteins β‐arrestins 1 and 2 (Luttrell and Lefkowitz, 2002). Several screening modalities based on both of these interactions have been developed and have their advantages and disadvantages. Other approaches could include, for example, the use of pathway‐specific inhibitors or siRNA screens.
If G protein‐coupling assays are used for detection of receptor activation, elucidation of the Gα subtype involved in receptor coupling is critical. A frequently employed strategy for identifying the coupling capacity of a GPCR is the use of receptor constitutive activity, a phenomenon whereby a GPCR (or indeed any receptor) signals in the absence of its activation by a ligand. Constitutive activity is now widely accepted as an intrinsic feature of GPCRs, particularly when they are overexpressed in heterologous in vitro expression systems, and it is also observed in in vivo studies (Damian et al., 2012; Corder et al., 2013). Constitutive activity upon overexpression of a GPCR can be used to identify the G protein involved in signalling by a particular orphan GPCR. However, given the lack of pharmacological controls when studying orphan GPCRs, it is imperative that any findings from such studies are validated using multiple second messenger assays (described in depth by Zhang and Xie, 2012a), particularly in mammalian cells. This is particularly important in overexpression studies as promiscuous coupling may result from high receptor abundance.
One of the simplest and least expensive assays for measuring multiple G protein pathways is that developed by Simon Dowell and Andrew Brown at GlaxoSmithKline, in which the yeast pheromone response pathway has been manipulated for the study of mammalian GPCRs (Brown et al., 2000; Dowell and Brown, 2009; Ngo et al., 2015). Different yeast strains have been engineered to express yeast Gpa1p/human Gα subunit chimeras, allowing the study of GPCR activation by the four Gα subunit families in a single assay. GPCR constitutive coupling is revealed by both growth and β‐galactosidase activity (Figure 2A and B). The first time this assay was used to identify orphan GPCR coupling through constitutive activity was with GPR43 (now known as FFA2), where it was found to be Gαi and Gαq coupled (Brown et al., 2003). The yeast strain expressing FFA2 was then screened with a small ligand library, and acetate was found to increase β‐galactosidase activity above constitutive levels. Importantly, acetate activation of FFA2 via Gαi or Gαq was then confirmed by [35S]‐GTPγS binding and increases in [Ca2 +] levels, respectively (Brown et al., 2003). Despite the versatility of the yeast system, which allows different G protein coupling pairs to be tested, it has not necessarily been the primary platform for initial G protein coupling screens of orphan GPCRs. In fact, the yeast assay was used as a secondary validation of Gαs coupling of GPR26 and Gα13 coupling of GPR35 (Jones et al., 2007; Jenkins et al., 2010).
Figure 2.
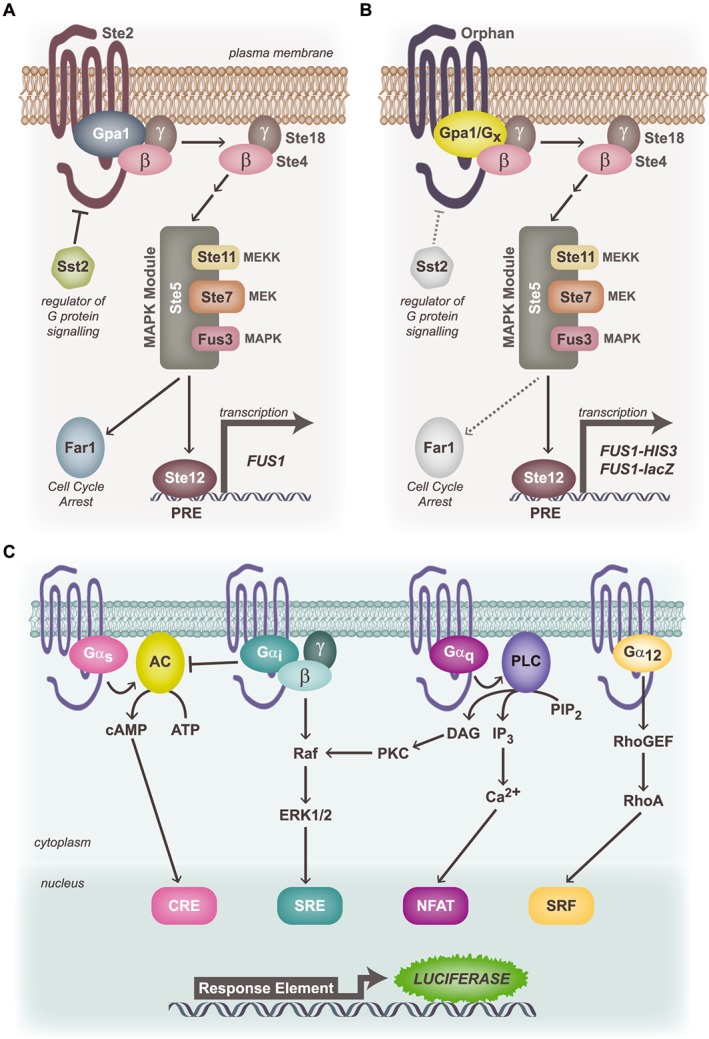
Assays for the identification of G protein signalling pathways using the constitutive activity of an orphan GPCR. (A) Yeast GPCR signalling pathway. The yeast pheromone receptor, Ste2, couples to the endogenous yeast heterotrimeric G proteins (Gα = Gpa1) to drive Gβγ‐mediated (MAPK) pathway activation and Ste12‐mediated transcription via the pheromone‐response element (PRE). MEK: MAPK kinase; MEKK: MEK kinase. (B) Modified yeast G protein signalling assay: chimeric versions of Gpa1 bearing the final five amino acids of each of the human Gα proteins have been introduced into yeast strains lacking each of the yeast GPCR, Ste2, the regulator of G protein signalling, Sst2, and the cell cycle arrest protein, Far1. Upon transformation with the orphan GPCR of interest, constitutive coupling to a specific G protein chimera drives the MAPK module to stimulate transcription of HIS3 (facilitates selection and expansion in HIS3‐deficient media) and the lacZ reporter via FUS1. (C) Mammalian reporter assays for transcription factors downstream of each of the major G protein families. Constitutive coupling via Gαs leads to stimulation of AC and generation of cAMP, which stimulates transcription of luciferase that lies downstream of a cAMP response element (CRE). Likewise, Gαi activity is reported via the serum response element (SRE) upon Gβγ signalling to ERK1/2 MAPK; Gαq via nuclear factor of activated T‐cells (NFAT) and Gα12 via serum response factor (SRF) response element.
Another approach for initially screening constitutive activity is to use a transcription factor response element (RE) reporter gene assay, a highly sensitive but not necessarily specific platform for ligand screening. In brief, a reporter gene construct, commonly luciferase, is cloned downstream of REs that are targeted by distinct second messengers (cAMP, ERK1/2, Ca2 +, RhoA; Figure 2C) (Cheng et al., 2010). For example, luciferase gene transcription is induced by cAMP RE (CRE) following cAMP activation (Gαs), or serum RE (SRE) activation downstream of Gαi/o mediated ERK1/2 phosphorylation (Cheng et al., 2010). This approach has been used to reveal the coupling mechanism of GPR133, a member of the adhesion GPCR family. Consistent with GPR133 being a Gαs‐coupled receptor, the assay revealed that of the three transcription factors, CRE, SRE and nuclear factor of activated T‐cells (NFAT), GPR133 signalled only via CRE to increase luciferase activity (Bohnekamp and Schoneberg, 2011). This observation was validated using chimeric Gαq(s), where the last five amino acids of Gαq were replaced by the corresponding Gαs amino acids, thus leading to increased inositol phosphate accumulation as a result of GPR133's constitutive signalling (Bohnekamp and Schoneberg, 2011).
Although the coupling mechanisms of a few orphan GPCRs have been proposed using similar approaches (Ge et al., 2008; Muller et al., 2013), they have not been validated with a second assay. The ambiguity and difficulty with working with orphan GPCRs is best highlighted by the case of GPR139. Here, multiple independent groups have used HTS to identify surrogate ligands for GPR139, but the signalling assays used to identify the cognate G protein involved produced contradictory results. GPR139 was proposed to be coupled to Gαs (Hu et al., 2009), Gαi/o (Susens et al., 2006) and/or Gαq (Susens et al., 2006; Shi et al., 2011; Isberg et al., 2014). Overall, the evidence favours GPR139 being a Gαq‐coupled receptor; however retesting the identified ligands in a second assay would provide better validation. This further highlights the necessity for careful validation of the proposed G protein interaction involved in orphan GPCR signalling, before moving forward.
The screening assays described above detect classical G protein coupled signalling, but it is clear that the recruitment of β‐arrestin by GPCRs results in signalling via several G protein‐independent pathways (Lefkowitz and Shenoy, 2005). Indeed, β‐arrestin screens should not be contingent on the G protein coupling repertoire of the orphan GPCR. However, ligand screening using various β‐arrestin recruitment approaches has met with variable success. For example, HTS of a panel of orphan GPCRs using a diverse library made up of natural and synthetic compounds (10 500 compounds) revealed novel surrogate ligands for the orphans MRGPRX2, GPR88 and GPR97, although the majority of orphans failed to register hits through the PathHunter β‐arrestin recruitment assay (Southern et al., 2013). Moreover, a screen of GPR35 with the Prestwick Chemical Library using BRET‐based β‐arrestin 2 recruitment both confirmed existing hits and identified a number of novel ligands at both the human and rat orthologues of the receptor (Jenkins et al., 2010). β‐arrestin recruitment assays have also been used to confirm proposed endogenous ligand pairings of orphan GPCRs, including CXCR7, GPR84, GPR92 and GPR99 (Yin et al., 2009; Southern et al., 2013). Finally, a very recent study by Roth and colleagues used a modified β‐arrestin Tango assay to identify and confirm that the KATP‐channel blocker, nateglinide, is a surrogate ligand for MRGPRX4 (Kroeze et al., 2015). Importantly, their open‐source platform proposed many other surrogate ligands at 91 orphan and poorly characterized GPCRs, providing an invaluable starting point for the scientific community in their quest to study orphan GPCRs (Kroeze et al., 2015). It should be noted however that β‐arrestin screens, in principle, should only capture agonists because antagonists or inverse agonists do not efficiently activate this pathway. This is a crucial point given that there are known endogenous inverse agonists of GPCRs (Milligan, 2003; Breit et al., 2006). It is also important to note that, particularly in the age of biased agonism where ligands can display distinct second messenger signals (Reiter et al., 2012; Kenakin, 2013), no single assay can be applied to all orphan GPCRs without the possibility of missing crucial hits. Indeed, it is possible that future screening campaigns will need to incorporate multiple endpoints and modalities [including allosterism (Wootten et al., 2013)].
Using GPCR crystal structures for VLS
Once the signalling pathway of the orphan GPCR of interest has been confirmed, the search for endogenous or surrogate ligands begins. Ideally, this would be performed directly via HTS; however, this is often not financially feasible, and there is no commercial or academic library of biological metabolites (in contrast to synthetic chemical compounds) including peptides and small proteins. Therefore, we need to look for alternative approaches such as structure‐based VLS for identifying the ligand in a direct screen of metabolites or indirectly for identifying chemical modulators as a first step towards the deorphanization.
Structure‐based VLS, or docking‐based VLS, has long been the mainstay of SBDD for the identification of novel ligands at various protein targets. It involves docking large and chemically diverse libraries of small molecules into a protein crystal structure or homology model of interest. The selection of small molecules for biological testing is generally based on docking score, chemical diversity, predicted interactions with key residues and other criteria. Small molecules that cause a biological response are termed hits and act as novel chemical scaffolds for hit‐to‐lead development. The first demonstration of the theoretical applicability of VLS of potential metabolites against a GPCR for deorphanization was shown in 2003 (Cavasotto et al., 2003). Thus, in the absence of crystal structures at that time, the challenge moved from the screen itself to the construction of a credible three‐dimensional model of the binding pocket.
As of November 2015, structures have been solved for 30 unique GPCRs, including two secretin‐like (Hollenstein et al., 2013; Siu et al., 2013), two glutamate‐like (Dore et al., 2014; Wu et al., 2014) and one frizzled‐like (Wang et al., 2013) families of GPCRs. Structure‐based VLS using the GPCR crystal structures themselves has been very successful in identifying new ligands, with particularly high hit rates (Table 2) [for more comprehensive examples, see de Graaf and Rognan (2009); Kooistra et al. (2013); Andrews et al. (2014)]. This shows that crystal structures are an accurate representation of a native GPCR and that structural accuracy of the docking target is important for VLS. However, one must consider the crystallized receptor conformation and the binding pocket space when docking and conducting VLS, as both depend on the nature of the co‐crystallized ligand, that is, whether it is an agonist or antagonist, a peptide or small molecule (Qin et al., 2015), and whether the structure reflects an inactive, active‐like or fully active receptor state (Lebon et al., 2011; Rasmussen et al., 2011a, 2011b). Studies conducted using an antagonist‐bound crystal structure identified only antagonists, with the exception of the κ‐opioid receptor and muscarinic M3 receptor screens, where one agonist and one partial agonist were identified respectively (Kruse et al., 2013; Negri et al., 2013). This contrasts with studies using crystal structures of GPCRs in an active‐like or fully activated state. Using the fully activated β2‐adrenoceptor crystal structure, Weiss and colleagues only retrieved novel agonists (27% hit rate; 6/22) (Weiss et al., 2013). However, in a separate study with the active crystal structure of the 5‐HT1B receptor, only three agonists were revealed out of five ligands tested (from 11 hits) (Rodriguez et al., 2014), while screening at a dopamine D2 receptor homology model based on the fully activated β2‐adrenoceptor was unsuccessful (Weiss et al., 2013). Intriguingly, VLS using the active‐like adenosine A2A receptor crystal structure only yielded novel antagonists (Rodriguez et al., 2015). The authors not only provide evidence of chemical bias towards A2A antagonists within the screening database, but also towards β2‐adrenoceptor and 5‐HT1B receptor agonists, thus explaining the discrepancies (Rodriguez et al., 2015). Despite this, GPCR agonists have been identified using homology models built on inactive structures (de Graaf and Rognan, 2009). Aside from chemical bias in screening databases, this highlights the importance of an accurate conformation of the binding pocket that can favourably accommodate agonist ligands, that is, optimization of the binding pocket is critical (discussed further below). It also reveals the need to further understand the intricate mechanism of receptor activation at the atomic level.
Table 2.
Examples of the types of hits identified from structure‐based virtual screening using published crystal structures
| Crystal structure (PDB ID) | Receptor conformation | Hit rate (actives/ligands tested) | Reference a |
|---|---|---|---|
| Adenosine A2A receptor (3EML) | Inactive | 41% (23/56) | (Katritch et al., 2010a) |
| β2‐adrenoceptor (2RH1) | Inactive | 24% (6/25) | (Kolb et al., 2009) |
| CXCR4 chemokine receptor (3ODU) | Inactive | 17% (4/23) | (Mysinger et al., 2012b) |
| Dopamine D3 receptor (3PBL) | Inactive | 20% (5/20) | (Carlsson et al., 2011) |
| Histamine H1 receptor (3RZE) | Inactive | 73% (19/26)b | (de Graaf et al., 2011) |
| κ‐opioid receptor (4DJH) | Inactive | 18% (4/22) (one agonist identified) | (Negri et al., 2013) |
| M3 muscarinic ACh receptor (4DAJ) | Inactive | 50% (8/16) (one partial agonist identified) | (Kruse et al., 2013) |
| Adenosine A2A receptor (2YDO and 2YDV) | Active‐like | 45% (9/20) (No agonists identified) | (Rodriguez et al., 2015) |
| 5‐HT1B receptor (4IAQ) | Active‐like | 50% (11/22) [three agonists identified (five tested)] | (Rodriguez et al., 2014) |
| β2‐adrenoceptor (2RH1) | Full active | 27% (6/22) (only agonists identified) | (Weiss et al., 2013) |
For a more comprehensive table of SBDD using published crystal structures, see Andrews et al. (2014).
Fragments (<22 heavy atoms) were screened.
GPCR homology models
Given that no bona fide orphan receptor has been crystallized to date, application of SBDD for orphan GPCRs must rely on the use of homology models. A homology model is an atomic‐level approximation of the structure of a protein, generated by comparative modelling of the target based upon the closest experimentally‐determined crystal structure (called a template). Template selection remains a critical step in building reliable homology models, particularly for GPCRs, given their sequence and/or ligand diversity. Considerations other than overall or local homology between a GPCR of interest and the template sequence, as discussed above, also include the receptor conformational state and the shape of the binding pocket of the crystal structure template. These issues are continually being addressed with the increasing number of crystal structures solved, leading to wider template selection for homology modelling of GPCRs.
It has been estimated that a receptor must have at least 30–35% sequence similarity to its template for accurate homology modelling in the absence of any additional refinement (which would usually involve ligand‐guided optimization or knowledge gained from site‐directed mutagenesis) (Kufareva et al., 2011; Beuming and Sherman, 2012; Katritch et al., 2012). Presently, all subclasses of the rhodopsin‐like family of GPCRs are represented by at least one crystal structure. Table 1 and Figure 3 highlight the transmembrane (TM) sequence homology of the remaining rhodopsin‐like orphan GPCRs in each of the subclasses, demonstrating not only the range of current crystal structures for orphan GPCR homology modelling but also the need for more crystal structures to continually expand this range (only 10 of 86 orphans have ≥35% identity to a GPCR with a solved structure; Figure 3). It is important to note, however, that some orphan GPCRs do not have ≥35% identity to any GPCR, crystallized or not. In these cases, further optimization of homology models will be necessary. Meanwhile, the ideal homology model template for a given orphan may not always reside in the same GPCR subclass, as different subclasses of GPCRs may share similar binding modes. For example, both β‐subclasses and γ‐subclasses bind to endogenous peptides and GPCR homology models built using distantly related templates can be successful (Kufareva et al., 2011; Rataj et al., 2014). Indeed, 31 of the 86 rhodopsin‐like orphan GPCRs have higher TM sequence homology with a template from another receptor subclass (Table 1).
Figure 3.
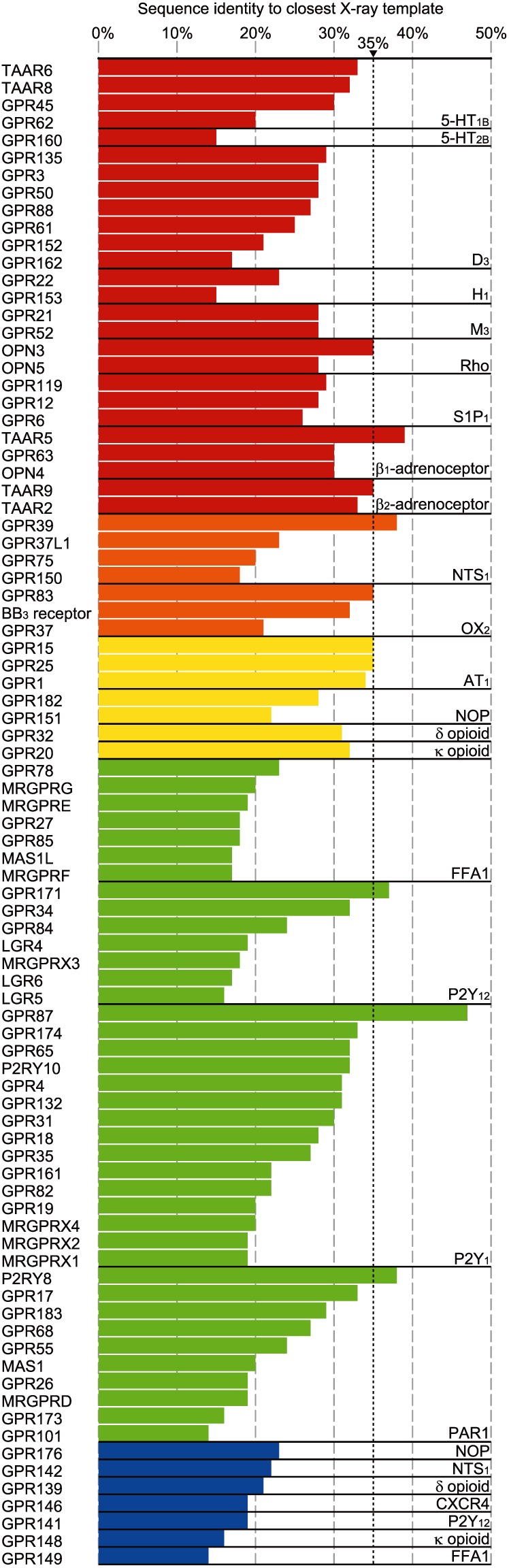
Orphan GPCRs ranked according to their closest crystal structure template. Each orphan GPCR (left) was ranked according to the crystal structure with which it shares the highest TM sequence identity, independent to GPCR subgroup (expressed as a percentage; listed in Table 1). The minimum identity required for VLS at a homology model in the absence of further refinement is 35% (dotted line). The homologous crystal structure is shown in the bottom right‐hand corner of each box. Red: α subgroup of rhodopsin‐like GPCRs; orange: β‐subgroup; yellow: γ‐subgroup; green: δ‐subgroup; blue: unclassified.
GPCR Dock is a community‐wide evaluation of the progress of molecular modelling and ligand docking methods for GPCRs (Michino et al., 2009; Kufareva et al., 2011; Kufareva et al., 2014). For a given competition round, a recently solved GPCR crystal structure(s) is withheld from publication, and research groups are called upon to submit their blinded prediction of the solved ligand–receptor complex, with particular focus on the ligand interactions within the receptor‐binding pocket. At the time of the CXCR4 GPCR Dock in 2010, no crystal structures of γ‐subgroup GPCRs had been solved. Thus, homology models could only rely on structures from the α‐subgroup. Despite distant homology (and ~25% sequence identity), the best performing CXCR4 model, built using the β2‐adrenoceptor crystal structure, achieved a TM backbone root mean squared deviation (RMSD) of 2.21 Å and captured 36% of ligand–receptor atomic contacts correctly when compared with the reference CXCR4 crystal structure. Interestingly, this was the only model that performed ‘well’ with respect to identifying ligand–receptor contacts.
Another approach to building homology models that has been gaining popularity is the use of multiple structural templates, which enable modelling of regions of the orphan GPCR based on higher local sequence identity, thereby creating a ‘chimeric’ template. Such an approach was applied to the orphan GPR17, where separate templates for the TM domains, and extracellular (ECL) and intracellular loops (ICL) were chosen with respect to sequence identity: the final model comprised TM regions based on bovine rhodopsin, ECL1 and ICL2 based on the human β2‐adrenoceptor, ECL2 on the turkey β1‐adrenoceptor, and ECL3 based on the A2A receptor (Eberini et al., 2011). Critically, five novel ligands were identified by screening against this mixed‐template homology model of GPR17 (100% hit rate in validation assays) (Eberini et al., 2011). The use of chimeric templates is not unprecedented; it has been shown previously that matching TMs to individual templates improved the RMSD of the binding site area of models to their reference crystal structure (Worth et al., 2011; Latek et al., 2013). However, other evaluation metrics were not considered, such as ligand–receptor contact‐based methods (Kufareva and Abagyan, 2012). Furthermore, homology models developed with multiple templates remain to be thoroughly tested in VLS studies.
Outside of community‐wide assessments like GPCR Dock, we do not have the luxury of a direct comparison between our homology model(s) and a reference crystal structure to provide feedback on how well we have portrayed the best representation of the binding pocket. Thus, other evaluations of the accuracy of a homology model are needed. In the context of this review, the most appropriate method to assess homology model(s) is by their performance in VLS, or more specifically, the ability of VLS to identify novel ligands that can be experimentally validated using the signalling assays discussed earlier (Figure 2). Furthermore, while building homology models with low sequence identity to the template structure can be daunting, the orientation of the binding pocket side chains is important (as mentioned above) – a process that can be optimized.
Optimization of orphan GPCR homology models and their use in VLS
Traditionally, homology models have been refined around a single ligand, with selection of the appropriate docking pose between ligand and receptor guided by existing site‐directed mutagenesis data. Explicit flexibility of the known interacting side chains is generally used to allow a degree of ligand‐induced fit of the binding pocket. However, invariably, this introduces some bias towards recognition of the original docked ligand. Similarly, a crystal structure is a static image of an otherwise dynamic entity that is capable of adopting multiple conformations. This is best exemplified by a study where a ligand‐guided optimized dopamine D3 receptor structure identified novel D3 ligands with a higher hit rate (56%) than the non‐refined crystal structure (20%) (Carlsson et al., 2011; Lane et al., 2013). For these reasons, it is important for VLS to optimize the modelled binding pocket residues to accommodate a diverse range of ligands.
Recently, multi‐step ligand‐guided optimization approaches have been developed to overcome this problem (Figure 1) (Carlsson et al., 2011; Mysinger et al., 2012b; Rueda et al., 2012; Kolaczkowski et al., 2013). Firstly, multiple conformations of the homology model are generated with specific importance given to the binding pocket, generally by building multiple models based on different templates or by elastic network models. The different conformations are then subjected to docking of a ligand training set made up of chemically diverse known ‘active’ ligands and ‘inactive’ decoy ligands. The screening performance of the models is then assessed by receiver operating characteristic curves, plotting the rate of true positives against false positives in the hit list ordered by decreasing predicted binding score, where the AUC can be calculated. Other measures have been introduced to improve the assessment of the early development of active ligands in VLS (Truchon and Bayly, 2007; Katritch et al., 2010b). Model conformations that can best distinguish between active and inactive ligands are selected, and this process is repeated iteratively until there is no further improvement in the model's performance. At this point, the fact that the model or ensemble of models can reliably retrieve known active ligands is taken as a sign that the same model will be able to identify novel hits from a large chemical database. The significance of considering VLS performance when optimizing models is evident in a comparison of the two best submitted models of the A2A receptor from GPCR Dock 2008 (Katritch et al., 2010b). Even though the traditionally optimized model by Costanzi was ranked first by the competition's criteria, the model's VLS performance was characterized by poor early enrichment of active ligands when compared with a ligand‐guided optimized model by Katritch/Abagyan (Katritch et al., 2010b). This highlights the importance of considering VLS performance when assessing homology models, particularly in cases where the motivation for constructing the homology model is to discover novel ligands.
Optimized homology models have themselves still produced mixed hit rates (Table 3) [extensive examples are given in de Graaf and Rognan (2009); Kooistra et al. (2013)], relative to the crystal structures (Table 2), but the fact that hits have been found for orphan GPCRs is promising. Undoubtedly, the major limitation of orphan GPCR homology models for structure‐based VLS is the lack of ligands for model optimization and validation. Smaller screening campaigns [10–50k samples (Valler and Green, 2000)] can play a complementary role in SBDD in this instance, as any surrogate ligands identified can then be used to populate a VLS training set. This strategy was employed for the immune‐related orphan GPR34 (Liebscher et al., 2011), where a screening campaign of 17 565 compounds identified six novel ligands that were validated using two measures of cAMP (Diaz et al., 2013). Next, a crude GPR34 homology model was refined with iterative changes to rotamers of key side chains found in the binding cavity, until the model was optimized for the six identified ligands from the initial screen. Biological testing of 2954 compounds (from an in‐house database of 1.25 million small molecules), led to the identification of five hits (of which two hits were the same ligands identified in the initial screen; 0.17% hit rate, 5/2954) (Diaz et al., 2013). These results should be encouraging to orphan GPCR researchers, despite the dismal success rate, as they demonstrate that it is possible to optimize homology models and achieve novel hits from VLS by using ligands from a smaller biological screening campaign (Figure 1). Furthermore, there are a number of simple and modifiable methodological explanations for why the hit rate was low; for example, the composition of the training set was suboptimal with a ratio of active to inactive ligands at 0.03%, whereas the popular inactive or decoy generating database, DUD‐e (recommends 2% and up to 10% of active ligands) has been used in training sets (Carlsson et al., 2011; Katritch et al., 2012; Mysinger et al., 2012a, 2012b; Rueda et al., 2012).
Table 3.
Examples of structure‐based VLS using GPCR homology models
| Homology model | Model development strategy | Hit rate | Reference |
|---|---|---|---|
| Adenosine A2A receptor | Energy minimization; mapping of site‐directed mutagenesis data onto model | 4% (10/230) | (Langmead et al., 2012) |
| CXCR4 chemokine receptor | Ligand‐guided optimization | 4% (1/24) | (Mysinger et al., 2012b) |
| Dopamine D3 receptor | Ligand‐guided optimization | 23% (6/26) | (Carlsson et al., 2011) |
| Histamine H4 receptor | Manual adjustment of key residues; validated with known ligands and decoys | 6% (16/255) | (Kiss et al., 2008) |
| Orphan GPCRs | |||
| GPR17 | Multiple templates; energy minimization | 100% (5/5) | (Eberini et al., 2011) |
| GPR34 | Similar to ligand‐guided optimization; iterative changes to side chain rotamers until enriched for active ligands | 0.17% (5/2954) | (Diaz et al., 2013) |
| GPR40 (FFA1) | Monte Carlo torsional sampling of binding pocket residues | 10% (1/10) | (Tikhonova et al., 2008) |
A reasonable question concerning orphan GPCRs is whether a crude homology model may actually be sufficient to retrieve hits, without ligand optimization and validation of the model. Certainly, this was successful for the GPR17 example cited earlier, where the multi‐template‐based GPR17 homology model was only subjected to energy minimization (Eberini et al., 2011); in the absence of a training set, top ranking docked compounds were subjected to a further round of molecular dynamics‐based energy minimization steps before selection (Eberini et al., 2011). While a crude model cannot be validated prior to performing VLS, we can at least hypothesize which residues are necessary for ligand interactions by a binding pocket analysis: are charged residues compensated for? What types of residues are positioned at conserved interaction sites for GPCRs? Building upon these observations, the residue side chains can then be checked and made accessible for small molecule docking in the binding pocket, whether by manual adjustment or by fumigation methods (Nilmeier and Jacobson, 2008; Abagyan and Kufareva, 2009). One might suggest allowing some explicit flexibility within the hypothesized key binding pocket residues while conducting VLS; however, this remains computationally challenging when docking millions of compounds, leaving full conformational searches impractical. Some inherent flexibility in the binding pocket may be accounted for by screening using multiple binding pocket conformations (Totrov and Abagyan, 2008; Vilar and Costanzi, 2013). It must be stressed, however, that a robust signalling assay for the orphan GPCR should be established before conducting a blind VLS, as the hits are unlikely to have particularly high affinity.
Although there are few examples of successful VLS using orphan GPCR homology models, these models have been used to provide insights into the binding mode of proposed ligands. For example, postulated interactions based upon docking to orphan homology models were confirmed by site‐directed mutagenesis for FFA1/GPR40, FFA2/GPR43, FFA3/GPR41, GPR55, GPR183 and the bombesin BB3 receptor (Gonzalez et al., 2008; Kotsikorou et al., 2011; Schmidt et al., 2011; Zhang et al., 2012b), amongst many other examples. Such studies defining and validating the binding pocket of these orphans will be valuable for future VLS as they can inform docking selection criteria. Although these models were each optimized around a single ligand and thus have the potential to generate a biased homology model, this may not be limiting. Furthermore, the key interacting residues can also facilitate structure‐based pharmacophore development.
Application of pharmacophores for orphan GPCR ligand discovery
Pharmacophores are an alternative way to view the contacts made between ligand and receptor, where a pharmacophore defines the spatial arrangement of chemical features that characterize the interactions necessary for eliciting a desired biological effect at a protein target of interest. Structure‐based pharmacophore features can be selected from key residues that have been identified by site‐directed mutagenesis or those based on hypothesized ligand–protein interactions derived from grid‐based interaction energies of small chemical probes (Barillari et al., 2008; Sanders et al., 2012; Drwal and Griffith, 2013). These three‐dimensional features are then used to filter out and identify small molecules that map well onto the pharmacophore, taking into account the shape of the binding pocket. This is why optimizing the binding pocket of orphan GPCR homology models is important. Currently, the use of structure‐based pharmacophores to identify novel ligands for GPCRs has only been described for the β2‐adrenoceptor and GPR40 (now known as FFA1) (Tikhonova et al., 2007; Barillari et al., 2008). The application of structure‐based pharmacophores to orphan GPCRs is probably limited in cases where the binding pocket has been modelled without additional refinement, although mapping ligands of lower stringency can be considered.
Intuitively, ligand‐based pharmacophores must complement the structure‐based pharmacophores. Thus, instead of using known or hypothesized binding site residues as features, a ligand set is used to build the pharmacophore. Based upon the assumption that the ligands share a similar binding mode, compounds with a range of activity profiles for the target are compiled and common features likely to favour ligand binding inform pharmacophore composition. Ligand‐based pharmacophores offer an alternative way to identify novel ligands at GPCRs with little structural information but where a range of known ligands have been identified, for example, at the α1A‐adrenoceptor (Ngo et al., 2013). However, transferring this approach to orphan GPCRs is again hindered by a lack of ligands, although ligand‐based pharmacophores were used to identify aromatic l‐α‐amino acids as the putative endogenous agonists for GPR139 (Isberg et al., 2014). Here, a ligand scaffold was initially identified by HTS and then subjected to structure–activity relationship (SAR) studies, with a series of derivatives then used to build the ligand‐based pharmacophore (Shi et al., 2011; Isberg et al., 2014). The authors noted that the central linker of the identified ligands resembled peptide backbones and screened a dipeptide database against the pharmacophore. This resulted in the identification of aromatic l‐α‐amino acids as the putative endogenous agonists for GPR139 (Isberg et al., 2014), setting a precedent for inferring endogenous agonists of orphan GPCRs through identification of surrogate ligands (GPR139 is still considered an orphan GPCR according to the IUPHAR/BPS Concise Guide to PHARMACOLOGY; www.guidetopharmacology.org). It also further highlights the major complementary role that HTS can play in in silico identification of orphan GPCR ligands.
A case study of FFA1: from orphan to drug target
Drug discovery at the free fatty acid 1 receptor (FFA1; previously known as GPR40) is the prime example of an orphan receptor to drug target pipeline, combining both HTS and SBDD. FFA1 is a target for metabolic disorders, including type 2 diabetes, as stimulation of pancreatic beta cell expressing FFA1 receptors leads to an increase in glucose‐stimulated insulin secretion (reviewed by Mancini and Poitout, 2013). The GPR40 gene was discovered as part of a family of four genes (FFA1‐3 and GPR42, a presumed pseudogene) (Sawzdargo et al., 1997; Stoddart et al., 2008). The FFA1 receptor was deorphanized 6 years later via two independent screens of approximately 1000–1500 ligands, which demonstrated that long‐chain free fatty acids (>C6) activated FFA1 receptors in [Ca2 +] flux assays (Briscoe et al., 2003; Itoh et al., 2003). Although the FFA1 receptor was thus deorphanized, fatty acids are poor agonists and are not amenable to SAR studies [highlighted by a failure of one study to improve upon the ligand efficiency of short‐chain fatty acids at FFA2 and FFA3 receptors (Schmidt et al., 2011)], thereby preventing hit‐to‐lead development. Further efforts were needed to identify small molecules activating FFA1 receptors; a novel small molecule was discovered via HTS and, following SAR, led to the highly potent FFA1 agonist GW9508 (Figure 4) (Briscoe et al., 2006; Garrido et al., 2006).
Figure 4.
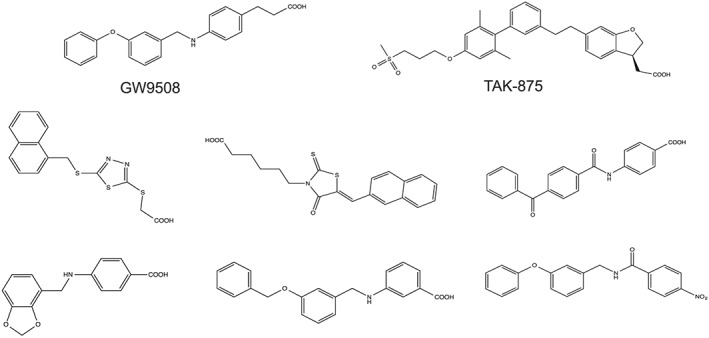
Structures of GW9508, TAK‐975 and novel FFA1 agonists identified by virtual screening.
Tikhonova and colleagues have used a computational approach to ligand discovery at FFA1 receptors, as detailed in a series of elegant papers combining molecular modelling with site‐directed mutagenesis (Sum et al., 2007; Tikhonova et al., 2007, 2008). Using bovine rhodopsin as the structural template, a FFA1 homology model was built, and Monte Carlo torsional sampling was applied to binding pocket residues. Both GW9508 and the endogenous agonist, linoleic acid, were docked into the model, and predicted interacting residues were validated experimentally by site‐directed mutagenesis. By building upon this knowledge (Sum et al., 2007; Tikhonova et al., 2007), the authors were able to subject their validated homology model to VLS studies that combined both traditional docking and structure‐based pharmacophore approaches, ultimately identifying six novel chemical scaffolds for the FFA1 receptor (Figure 4) (6/52; 11.5% hit rate) (Tikhonova et al., 2008). This provides further evidence of the strength of VLS, driving the discovery of multiple surrogate ligands from an initial HTS hit. Interestingly, comparison of hits with patented scaffolds of FFA1 agonists reveals that VLS was able to retrieve unique chemical motifs (Figure 4, Table 4), indicating both the utility of VLS and that opportunities remain for the development of second‐generation FFA1 agonists (Tikhonova et al., 2008).
Table 4.
Patented chemical scaffolds as FFA1 agonists
| Pharmaceutical company | Patent application number | Patented scaffold (and derivatives thereof) |
|---|---|---|
| Amgen | US2011190330 |
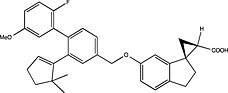
|
| Astellas Pharma | US2012035196 |
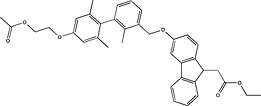
|
| Boehringer Ingelheim | WO2012072691 |
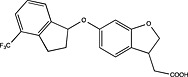
|
| Connexios Life Sciences | WO2012011125 |
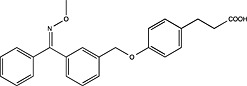
|
| Mochida Pharmaceutical | US2012157459 |
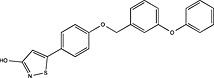
|
| Sanofi | WO2012004269 |
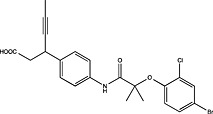
|
| Schering Corporation | US2011312995 |
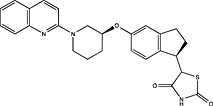
|
| Takeda | US8088821 |
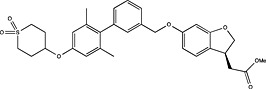
|
Adapted from (Harrison, 2012).
Pharmaceutical companies have been actively chasing FFA1 as a therapeutic target, demonstrated by the number of patented compounds in Table 4. Although identified by traditional HTS (Sasaki et al., 2011), Takeda's TAK‐875 was the first FFA1 allosteric agonist to complete clinical development and significantly improved glycaemic control of type 2 diabetes in two independent phase‐II and one phase‐III trials (Burant et al., 2012; Kaku et al., 2013, 2015). The therapeutic development of TAK‐875 appears to have been abandoned due to liver toxicity (Takeda, 2015); however, it proved useful in receptor crystallization (Srivastava et al., 2014). The crystal structure provided visual validation of the FFA1 binding pocket, with the carboxylic acid moiety of TAK‐875 interacting with R183(5.39) and R258(7.35) as predicted with GW9508 and linoleic acid from homology modelling (Sum et al., 2007; Tikhonova et al., 2007; Srivastava et al., 2014) [Ballesteros and Weinstein numbers in parentheses (Ballesteros and Weinstein, 1995)]. The FFA1 crystal structure also confirmed the allosteric nature of TAK‐875 and suggested other potential binding pockets that can be targeted, which, in turn, may prove useful in future SBDD efforts aimed at identifying potent agonists with better safety profiles.
Concluding remarks
Orphan GPCRs represent an experimental black box – we rarely have information about their activation, signal transduction or (patho)physiological functions. However, we have reasons to believe that they will become easy to use once we start to fill in the gaps, because GPCRs have historically been good therapeutic targets. Clearly, the process of GPCR deorphanization and in vivo phenotyping would be greatly hastened by the identification of surrogate ligands, either through traditional HTS or via VLS. Currently, the use of SBDD for the identification of surrogate ligands for orphan GPCRs is somewhat limited and will probably still require some kind of small screening campaign for scaffold identification (Figure 1). Importantly, our experimental toolkit is constantly expanding, with a steady supply of new GPCR crystal structures and methods for homology model optimization, although approaches for assessing the performance of homology models is an area needing further development. Furthermore, it is still unclear just how ‘accurate’ a homology model needs to be to reliably predict hits for orphan GPCRs, although visual inspection of the positioning of key conserved residues such as that in transmembrane 3 (3.32) (Venkatakrishnan et al., 2013), and generation of multiple homology models based upon multiple templates, will increase confidence in their use. Finally, it is important to consider ligand‐independent roles of orphan GPCRs, including modulation of signalling through heteromerization (Smith and Milligan, 2010), for example, GPR50 and the MT1 melatonin receptor (Levoye et al., 2006), GPR88 and the D1 dopamine receptor (Marley et al., 2013) and even GPR3 complexes with β‐arrestin and amyloid precursor protein (Thathiah et al., 2009; Nelson and Sheng, 2013). Although it is likely that more than the abovementioned GPCRs signal through such non‐canonical mechanisms (i.e. without an endogenous ligand), this does not preclude the development of surrogate ligands. Much remains to be discovered about orphan GPCR pharmacology; surrogate ligands from VLS can only augment the rate of translation of these interesting receptors to genuine drug targets.
Note added during revision
During the revision of our manuscript, a seminal paper was published by Bryan Roth and Brian Shoichet in which they used a very similar combined experimental and computational approach to that discussed herein to identify surrogate ligands for the proton receptors, GPR68 and GPR65 (Huang et al., 2015).
Conflict of interest
R. A. is a founder of Molsoft, LLC. Other authors declare no conflicts of interest.
Acknowledgements
This work was supported in part by NHMRC Program Grants 573732 and 1074386 (RMG), an NHMRC & NHF CJ Martin Fellowship (NJS), Australian Postgraduate Awards (TN & JLJC), an Endeavour Research Fellowship to TN and a Simon and Michal Wilkenfeld Scholarship to JLJC. IK and R. A. are supported by NIH Grants R01 GM071872, U01 GM094612, U54 GM094618, and R01 AI118985. The authors would like to thank Dr Fiona McRobb for helpful discussions.
Ngo, T. , Kufareva, I. , Coleman, J. LJ. , Graham, R. M. , Abagyan, R. , and Smith, N. J. (2016) Identifying ligands at orphan GPCRs: current status using structure‐based approaches. British Journal of Pharmacology, 173: 2934–2951. doi: 10.1111/bph.13452.
References
- Abagyan R, Kufareva I (2009). The flexible pocketome engine for structural chemogenomics In: Jacoby E. (ed). Chemogenomics: Methods and Applications, Methods in Molecular Biology, Vol. 575 Human Press: New York, pp. 249–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SP, Brown GA, Christopher JA (2014). Structure‐based and fragment‐based GPCR drug discovery. ChemMedChem 9: 256–275. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H (1995). Integrated methods for the construction of three‐dimensional models and computational probing of structure–function relations in G protein‐coupled receptors In: Sealfon SC. (ed). Receptor Molecular Biology, Methods in Neuroscience, Vol. 25 Academic Press: New York, pp. 366–428. [Google Scholar]
- Barillari C, Marcou G, Rognan D (2008). Hot‐spots‐guided receptor‐based pharmacophores (HS‐Pharm): a knowledge‐based approach to identify ligand‐anchoring atoms in protein cavities and prioritize structure‐based pharmacophores. J Chem Inf Model 48: 1396–1410. [DOI] [PubMed] [Google Scholar]
- Beuming T, Sherman W (2012). Current assessment of docking into GPCR crystal structures and homology models: successes, challenges, and guidelines. J Chem Inf Model 52: 3263–3277. [DOI] [PubMed] [Google Scholar]
- Bjarnadottir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schioth HB (2006). Comprehensive repertoire and phylogenetic analysis of the G protein‐coupled receptors in human and mouse. Genomics 88: 263–273. [DOI] [PubMed] [Google Scholar]
- Bohnekamp J, Schoneberg T (2011). Cell adhesion receptor GPR133 couples to Gs protein. J Biol Chem 286: 41912–41916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breit A, Wolff K, Kalwa H, Jarry H, Buch T, Gudermann T (2006). The natural inverse agonist agouti‐related protein induces arrestin‐mediated endocytosis of melanocortin‐3 and ‐4 receptors. J Biol Chem 281: 37447–37456. [DOI] [PubMed] [Google Scholar]
- Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, et al. (2003). The orphan G protein‐coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem 278: 11303–11311. [DOI] [PubMed] [Google Scholar]
- Briscoe CP, Peat AJ, McKeown SC, Corbett DF, Goetz AS, Littleton TR, et al. (2006). Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol 148: 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AJ, Dyos SL, Whiteway MS, White JH, Watson MA, Marzioch M, et al. (2000). Functional coupling of mammalian receptors to the yeast mating pathway using novel yeast/mammalian G protein alpha‐subunit chimeras. Yeast 16: 11–22. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. (2003). The orphan G protein‐coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 278: 11312–11319. [DOI] [PubMed] [Google Scholar]
- Burant CF, Viswanathan P, Marcinak J, Cao C, Vakilynejad M, Xie B, et al. (2012). TAK‐875 versus placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomised, double‐blind, placebo‐controlled trial. Lancet 379: 1403–1411. [DOI] [PubMed] [Google Scholar]
- Caffrey M, Cherezov V (2009). Crystallizing membrane proteins using lipidic mesophases. Nat Protoc 4: 706–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, et al. (2011). Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat Chem Biol 7: 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavasotto CN, Orry AJ, Abagyan R (2003). Structure‐based identification of binding sites, native ligands and potential inhibitors for G‐protein coupled receptors. Proteins 51: 423–433. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Garvin D, Paguio A, Stecha P, Wood K, Fan F (2010). Luciferase reporter assay system for deciphering GPCR pathways. Curr Chem Genomics 4: 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, Katritch V, et al. (2012). Fusion partner toolchest for the stabilization and crystallization of G protein‐coupled receptors. Structure 20: 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelli O, Reinscheid RK, Zhang Y, Wang Z, Fredriksson R, Schioth HB (2013). G protein‐coupled receptor deorphanizations. Annu Rev Pharmacol Toxicol 53: 127–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, He Y, et al. (2013). Constitutive mu‐opioid receptor activity leads to long‐term endogenous analgesia and dependence. Science 341: 1394–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damian M, Marie J, Leyris JP, Fehrentz JA, Verdie P, Martinez J, et al. (2012). High constitutive activity is an intrinsic feature of ghrelin receptor protein: a study with a functional monomeric GHS‐R1a receptor reconstituted in lipid discs. J Biol Chem 287: 3630–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport AP, Alexander SP, Sharman JL, Pawson AJ, Benson HE, Monaghan AE, et al. (2013). International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein‐coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol Rev 65: 967–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf C, Rognan D (2009). Customizing G Protein‐coupled receptor models for structure‐based virtual screening. Curr Pharm Des 15: 4026–4048. [DOI] [PubMed] [Google Scholar]
- de Graaf C, Kooistra AJ, Vischer HF, Katritch V, Kuijer M, Shiroishi M, et al. (2011). Crystal structure‐based virtual screening for fragment‐like ligands of the human histamine H(1) receptor. J Med Chem 54: 8195–8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz C, Labit‐Le Bouteiller C, Yvon S, Cambon‐Kernëis A, Roasio A, Jamme M‐F, et al. (2013). a strategy combining differential low‐throughput screening and virtual screening (dls‐vs) accelerating the discovery of new modulators for the orphan GPR34 receptor. Mol Inform 32: 213–229. [DOI] [PubMed] [Google Scholar]
- Dore AS, Okrasa K, Patel JC, Serrano‐Vega M, Bennett K, Cooke RM, et al. (2014). Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511: 557–562. [DOI] [PubMed] [Google Scholar]
- Dowell SJ, Brown AJ (2009). Yeast assays for G protein‐coupled receptors In: Leifert WR. (ed). G Protein‐Coupled Receptors in Drug Discovery, Methods in Molecular Biology, Vol. 552 Humana Press: New York, pp. 213–229. [DOI] [PubMed] [Google Scholar]
- Drwal MN, Griffith R (2013). Combination of ligand‐ and structure‐based methods in virtual screening. Drug Discov Today Technol 10: e395–e401. [DOI] [PubMed] [Google Scholar]
- Eberini I, Daniele S, Parravicini C, Sensi C, Trincavelli ML, Martini C, et al. (2011). In silico identification of new ligands for GPR17: a promising therapeutic target for neurodegenerative diseases. J Comput Aided Mol Des 25: 743–752. [DOI] [PubMed] [Google Scholar]
- Foord SM, Bonner TI, Neubig RR, Rosser EM, Pin JP, Davenport AP, et al. (2005). International Union of Pharmacology. XLVI. G protein‐coupled receptor list. Pharmacol Rev 57: 279–288. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB (2003). The G‐protein‐coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63: 1256–1272. [DOI] [PubMed] [Google Scholar]
- Garland SL (2013). Are GPCRs still a source of new targets? J Biomol Screen 18: 947–966. [DOI] [PubMed] [Google Scholar]
- Garrido DM, Corbett DF, Dwornik KA, Goetz AS, Littleton TR, McKeown SC, et al. (2006). Synthesis and activity of small molecule GPR40 agonists. Bioorg Med Chem Lett 16: 1840–1845. [DOI] [PubMed] [Google Scholar]
- Ge H, Weiszmann J, Reagan JD, Gupte J, Baribault H, Gyuris T, et al. (2008). Elucidation of signaling and functional activities of an orphan GPCR, GPR81. J Lipid Res 49: 797–803. [DOI] [PubMed] [Google Scholar]
- Ghosh E, Kumari P, Jaiman D, Shukla AK (2015). Methodological advances: the unsung heroes of the GPCR structural revolution. Nat Rev Mol Cell Biol 16: 69–81. [DOI] [PubMed] [Google Scholar]
- Gonzalez N, Hocart SJ, Portal‐Nunez S, Mantey SA, Nakagawa T, Zudaire E, et al. (2008). Molecular basis for agonist selectivity and activation of the orphan bombesin receptor subtype 3 receptor. J Pharmacol Exp Ther 324: 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison C (2012). Patent watch. Nat Rev Drug Discov 11: 592–593. [DOI] [PubMed] [Google Scholar]
- Hollenstein K, Kean J, Bortolato A, Cheng RK, Dore AS, Jazayeri A, et al. (2013). Structure of class B GPCR corticotropin‐releasing factor receptor 1. Nature 499: 438–443. [DOI] [PubMed] [Google Scholar]
- Hu LA, Tang PM, Eslahi NK, Zhou T, Barbosa J, Liu Q (2009). Identification of surrogate agonists and antagonists for orphan G‐protein‐coupled receptor GPR139. J Biomol Screen 14: 789–797. [DOI] [PubMed] [Google Scholar]
- Huang XP, Karpiak J, Kroeze WK, Zhu H, Chen X, Moy SS, et al. (2015). Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 527: 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg V, Andersen KB, Bisig C, Dietz GP, Brauner‐Osborne H, Gloriam DE (2014). Computer‐aided discovery of aromatic l‐alpha‐amino acids as agonists of the orphan G protein‐coupled receptor GPR139. J Chem Inf Model 54: 1553–1557. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, et al. (2003). Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422: 173–176. [DOI] [PubMed] [Google Scholar]
- Jazayeri A, Dias JM, Marshall FH (2015). From G Protein‐coupled Receptor Structure Resolution to Rational Drug Design. J Biol Chem 290: 19489–19495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins L, Brea J, Smith NJ, Hudson BD, Reilly G, Bryant NJ, et al. (2010). Identification of novel species‐selective agonists of the G‐protein‐coupled receptor GPR35 that promote recruitment of beta‐arrestin‐2 and activate Galpha13. Biochem J 432: 451–459. [DOI] [PubMed] [Google Scholar]
- Jones PG, Nawoschik SP, Sreekumar K, Uveges AJ, Tseng E, Zhang L, et al. (2007). Tissue distribution and functional analyses of the constitutively active orphan G protein coupled receptors, GPR26 and GPR78. Biochim Biophys Acta 1770: 890–901. [DOI] [PubMed] [Google Scholar]
- Kaku K, Araki T, Yoshinaka R (2013). Randomized, double‐blind, dose‐ranging study of TAK‐875, a novel GPR40 agonist, in Japanese patients with inadequately controlled type 2 diabetes. Diabetes Care 36: 245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaku K, Enya K, Nakaya R, Ohira T, Matsuno R (2015). Efficacy and safety of fasiglifam (TAK‐875), a GPR40 agonist, in Japanese patients with type 2 diabetes inadequately controlled by diet and exercise: A randomized, double‐blind, placebo‐controlled, phase III trial. Diabetes Obes Metab 17: 675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Jaakola VP, Lane JR, Lin J, Ijzerman AP, Yeager M, et al. (2010a). Structure‐based discovery of novel chemotypes for adenosine A(2 A) receptor antagonists. J Med Chem 53: 1799–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Rueda M, Lam PC, Yeager M, Abagyan R (2010b). GPCR 3D homology models for ligand screening: lessons learned from blind predictions of adenosine A2a receptor complex. Proteins 78: 197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Rueda M, Abagyan R (2012). Ligand‐guided receptor optimization In: Orry AJW, Abagyan R. (eds). Homology Modelling: Methods and Protocols, Methods in Molecular Biology, Vol. 857 Humana Press: New York, pp. 189–205. [DOI] [PubMed] [Google Scholar]
- Kenakin T (2013). New concepts in pharmacological efficacy at 7TM receptors: IUPHAR review 2. Br J Pharmacol 168: 554–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinch MS, Hoyer D, Patridge E, Plummer M (2015). Target selection for FDA‐approved medicines. Drug Discov Today 20: 784–789. [DOI] [PubMed] [Google Scholar]
- Kiss R, Kiss B, Könczöl A, Szalai F, Jelinek I, László V, et al. (2008). Discovery of novel human histamine H4 receptor ligands by large‐scale structure‐based virtual screening. J Med Chem 51: 3145–3153. [DOI] [PubMed] [Google Scholar]
- Kolaczkowski M, Bucki A, Feder M, Pawlowski M (2013). Ligand‐optimized homology models of D1 and D2 dopamine receptors: application for virtual screening. J Chem Inf Model 53: 638–648. [DOI] [PubMed] [Google Scholar]
- Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK (2009). Structure‐based discovery of beta2‐adrenergic receptor ligands. Proc Natl Acad Sci U S A 106: 6843–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra AJ, Roumen L, Leurs R, de Esch IJ, de Graaf C (2013). From heptahelical bundle to hits from the Haystack: structure‐based virtual screening for GPCR ligands In: Conn PM. (ed). G Protein Coupled Receptors Modelling, Activation, Interactions and Virtual Screening, Methods in Enzymology, Vol. 522 Academic Press: New York, pp. 279–336. [DOI] [PubMed] [Google Scholar]
- Kotsikorou E, Madrigal KE, Hurst DP, Sharir H, Lynch DL, Heynen‐Genel S, et al. (2011). Identification of the GPR55 agonist binding site using a novel set of high‐potency GPR55 selective ligands. Biochemistry 50: 5633–5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze WK, Sassano MF, Huang XP, Lansu K, McCorvy JD, Giguere PM, et al. (2015). PRESTO‐Tango as an open‐source resource for interrogation of the druggable human GPCRome. Nat Struct Mol Biol 22: 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Weiss DR, Rossi M, Hu J, Hu K, Eitel K, et al. (2013). Muscarinic receptors as model targets and antitargets for structure‐based ligand discovery. Mol Pharmacol 84: 528–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I, Abagyan R (2012). Methods of protein structure comparison In: Orry AJW, Abagyan R. (eds). Homology Modelling: Methods and Protocols, Methods in Molecular Biology, Vol. 857 Humana Press: New York, pp. 231–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R, GPCR Dock Participants (2011). Status of GPCR modeling and docking as reflected by community‐wide GPCR Dock 2010 assessment. Structure 19: 1108–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I, Katritch V, Participants of GPCR Dock , Stevens RC, Abagyan R (2014). Advances in GPCR modeling evaluated by the GPCR Dock 2013 assessment: meeting new challenges. Structure 22: 1120–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane JR, Chubukov P, Liu W, Canals M, Cherezov V, Abagyan R, et al. (2013). Structure‐based ligand discovery targeting orthosteric and allosteric pockets of dopamine receptors. Mol Pharmacol 84: 794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead CJ, Andrews SP, Congreve M, Errey JC, Hurrell E, Marshall FH, et al. (2012). Identification of novel adenosine A(2 A) receptor antagonists by virtual screening. J Med Chem 55: 1904–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latek D, Pasznik P, Carlomagno T, Filipek S (2013). Towards improved quality of GPCR models by usage of multiple templates and profile‐profile comparison. PLoS One 8: e56742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, et al. (2011). Agonist‐bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474: 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK (2005). Transduction of receptor signals by beta‐arrestins. Science 308: 512–517. [DOI] [PubMed] [Google Scholar]
- Levoye A, Dam J, Ayoub MA, Guillaume JL, Couturier C, Delagrange P, et al. (2006). The orphan GPR50 receptor specifically inhibits MT1 melatonin receptor function through heterodimerization. EMBO J 25: 3012–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebscher I, Muller U, Teupser D, Engemaier E, Engel KM, Ritscher L, et al. (2011). Altered immune response in mice deficient for the G protein‐coupled receptor GPR34. J Biol Chem 286: 2101–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Wacker D, Wang C, Abola E, Cherezov V (2014). Femtosecond crystallography of membrane proteins in the lipidic cubic phase. Philos Trans R Soc Lond B Biol Sci 369: 20130314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM, Lefkowitz RJ (2002). The role of beta‐arrestins in the termination and transduction of G‐protein‐coupled receptor signals. J Cell Sci 115: 455–465. [DOI] [PubMed] [Google Scholar]
- Mancini AD, Poitout V (2013). The fatty acid receptor FFA1/GPR40 a decade later: how much do we know? Trends Endocrinol Metab 24: 398–407. [DOI] [PubMed] [Google Scholar]
- Marley A, Choy RW, von Zastrow M (2013). GPR88 reveals a discrete function of primary cilia as selective insulators of GPCR cross‐talk. PLoS One 8: e70857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michino M, Abola E, Participants of GPCR Dock , Brooks CL, III , Dixon JS, Moult J, et al (2009). Community‐wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat Rev Drug Discov 8: 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G (2003). Constitutive activity and inverse agonists of G protein‐coupled receptors: a current perspective. Mol Pharmacol 64: 1271–1276. [DOI] [PubMed] [Google Scholar]
- Mujic‐Delic A, de Wit RH, Verkaar F, Smit MJ (2014). GPCR‐targeting nanobodies: attractive research tools, diagnostics, and therapeutics. Trends Pharmacol Sci 35: 247–255. [DOI] [PubMed] [Google Scholar]
- Muller A, Kleinau G, Piechowski CL, Muller TD, Finan B, Pratzka J, et al. (2013). G‐protein coupled receptor 83 (GPR83) signaling determined by constitutive and zinc(II)‐induced activity. PLoS One 8: e53347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger MM, Carchia M, Irwin JJ, Shoichet BK (2012a). Directory of useful decoys, enhanced (DUD‐E): better ligands and decoys for better benchmarking. J Med Chem 55: 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger MM, Weiss DR, Ziarek JJ, Gravel S, Doak AK, Karpiak J, et al. (2012b). Structure‐based ligand discovery for the protein–protein interface of chemokine receptor CXCR4. Proc Natl Acad Sci U S A 109: 5517–5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD (1970). A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48: 443–453. [DOI] [PubMed] [Google Scholar]
- Negri A, Rives ML, Caspers MJ, Prisinzano TE, Javitch JA, Filizola M (2013). Discovery of a novel selective kappa‐opioid receptor agonist using crystal structure‐based virtual screening. J Chem Inf Model 53: 521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CD, Sheng M (2013). GPR3 Stimulates Abeta Production via Interactions with APP and beta‐Arrestin2. PLoS One 8: e74680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo T, Nicholas TJ, Chen J, Finch AM, Griffith R (2013). 5‐HT1A receptor pharmacophores to screen for off‐target activity of alpha1‐adrenoceptor antagonists. J Comput Aided Mol Des 27: 305–319. [DOI] [PubMed] [Google Scholar]
- Ngo T, Coleman JL, Smith NJ (2015). Using constitutive activity to define appropriate high‐throughput screening assays for orphan g protein‐coupled receptors In: Prazeres DMF, Martins SAM. (eds). G Protein‐Coupled Receptors Screening Assays, Methods in Molecular Biology, Vol. 1272 Humana Press: New York, pp. 91–106. [DOI] [PubMed] [Google Scholar]
- Nilmeier J, Jacobson M (2008). Multiscale Monte Carlo sampling of protein sidechains: application to binding pocket flexibility. J Chem Theory Comput 4: 835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Kim K, Kwon HB, Seong JY (2006). Cellular and molecular biology of orphan G protein‐coupled receptors. Int Rev Cytol 252: 163–218. [DOI] [PubMed]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al., NC‐IUPHAR(2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryszak R, Keays M, Tang YA, Fonseca NA, Barrera E, Burdett T, et al. (2015). Expression Atlas update‐an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res 44: D746–D752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, et al. (2015). Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347: 1117–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, et al. (2011a). Structure of a nanobody‐stabilized active state of the beta(2) adrenoceptor. Nature 469: 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. (2011b). Crystal structure of the beta2 adrenergic receptor‐Gs protein complex. Nature 477: 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rataj K, Witek J, Mordalski S, Kosciolek T, Bojarski AJ (2014). Impact of template choice on homology model efficiency in virtual screening. J Chem Inf Model 54: 1661–1668. [DOI] [PubMed] [Google Scholar]
- Reiter E, Ahn S, Shukla AK, Lefkowitz RJ (2012). Molecular mechanism of beta‐arrestin‐biased agonism at seven‐transmembrane receptors. Annu Rev Pharmacol Toxicol 52: 179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez D, Brea J, Loza MI, Carlsson J (2014). Structure‐based discovery of selective serotonin 5‐HT(1B) receptor ligands. Structure 22: 1140–1151. [DOI] [PubMed] [Google Scholar]
- Rodriguez D, Gao ZG, Moss SM, Jacobson KA, Carlsson J (2015). Molecular docking screening using agonist‐bound GPCR structures: probing the A2A adenosine receptor. J Chem Inf Model 55: 550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda M, Totrov M, Abagyan R (2012). ALiBERO: evolving a team of complementary pocket conformations rather than a single leader. J Chem Inf Model 52: 2705–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MPA, McGuire R, Roumen L, de Esch IJP, de Vlieg J, Klomp JPG, et al. (2012). From the protein's perspective: the benefits and challenges of protein structure‐based pharmacophore modeling. Med Chem Commum 3: 28–38. [Google Scholar]
- Sasaki S, Kitamura S, Negoro N, Suzuki M, Tsujihata Y, Suzuki N, et al. (2011). Design, synthesis, and biological activity of potent and orally available G protein‐coupled receptor 40 agonists. J Med Chem 54: 1365–1378. [DOI] [PubMed] [Google Scholar]
- Sawzdargo M, George SR, Nguyen T, Xu S, Kolakowski LF, O'Dowd BF (1997). A cluster of four novel human G protein‐coupled receptor genes occurring in close proximity to CD22 gene on chromosome 19q13.1. Biochem Biophys Res Commun 239: 543–547. [DOI] [PubMed] [Google Scholar]
- Schmidt J, Smith NJ, Christiansen E, Tikhonova IG, Grundmann M, Hudson BD, et al. (2011). Selective orthosteric free fatty acid receptor 2 (FFA2) agonists: identification of the structural and chemical requirements for selective activation of FFA2 versus FFA3. J Biol Chem 286: 10628–10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Shen JK, Chen D, Fog K, Thirstrup K, Hentzer M, et al. (2011). Discovery and SAR of a series of agonists at orphan G protein‐coupled receptor 139. ACS Med Chem Lett 2: 303–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu FY, He M, de Graaf C, Han GW, Yang D, Zhang Z, et al. (2013). Structure of the human glucagon class B G‐protein‐coupled receptor. Nature 499: 444–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NJ, Milligan G (2010). Allostery at G protein‐coupled receptor homo‐ and heteromers: uncharted pharmacological landscapes. Pharmacol Rev 62: 701–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southern C, Cook JM, Neetoo‐Isseljee Z, Taylor DL, Kettleborough CA, Merritt A, et al. (2013). Screening beta‐arrestin recruitment for the identification of natural ligands for orphan G‐protein‐coupled receptors. J Biomol Screen 18: 599–609. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Yano J, Hirozane Y, Kefala G, Gruswitz F, Snell G, et al. (2014). High‐resolution structure of the human GPR40 receptor bound to allosteric agonist TAK‐875. Nature 513: 124–127. [DOI] [PubMed] [Google Scholar]
- Stoddart LA, Smith NJ, Milligan G (2008). International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1, −2, and −3: pharmacology and pathophysiological functions. Pharmacol Rev 60: 405–417. [DOI] [PubMed] [Google Scholar]
- Sum CS, Tikhonova IG, Neumann S, Engel S, Raaka BM, Costanzi S, et al. (2007). Identification of residues important for agonist recognition and activation in GPR40. J Biol Chem 282: 29248–29255. [DOI] [PubMed] [Google Scholar]
- Susens U, Hermans‐Borgmeyer I, Urny J, Schaller HC (2006). Characterisation and differential expression of two very closely related G‐protein‐coupled receptors, GPR139 and GPR142, in mouse tissue and during mouse development. Neuropharmacology 50: 512–520. [DOI] [PubMed] [Google Scholar]
- Takeda (2015). Takeda Announces Termination of Fasiglifam (TAK‐875) Development. Available at: http://www.takeda.com/news/2013/20131227_6117.html (accessed 17 April 2015)
- Thathiah A, Spittaels K, Hoffmann M, Staes M, Cohen A, Horre K, et al. (2009). The orphan G protein‐coupled receptor 3 modulates amyloid‐beta peptide generation in neurons. Science 323: 946–951. [DOI] [PubMed] [Google Scholar]
- Tikhonova IG, Sum CS, Neumann S, Thomas CJ, Raaka BM, Costanzi S, et al. (2007). Bidirectional, iterative approach to the structural delineation of the functional "chemoprint" in GPR40 for agonist recognition. J Med Chem 50: 2981–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova IG, Sum CS, Neumann S, Engel S, Raaka BM, Costanzi S, et al. (2008). Discovery of novel agonists and antagonists of the free fatty acid receptor 1 (FFAR1) using virtual screening. J Med Chem 51: 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totrov M, Abagyan R (2008). Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr Opin Struct Biol 18: 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truchon JF, Bayly CI (2007). Evaluating virtual screening methods: good and bad metrics for the "early recognition" problem. J Chem Inf Model 47: 488–508. [DOI] [PubMed] [Google Scholar]
- Valler MJ, Green D (2000). Diversity screening versus focussed screening in drug discovery. Drug Discov Today 5: 286–293. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM (2013). Molecular signatures of G‐protein‐coupled receptors. Nature 494: 185–194. [DOI] [PubMed] [Google Scholar]
- Vilar S, Costanzi S (2013). Application of Monte Carlo‐based receptor ensemble docking to virtual screening for GPCR ligands. Methods Enzymol 522: 263–278. [DOI] [PubMed] [Google Scholar]
- Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, et al. (2013). Structure of the human smoothened receptor bound to an antitumour agent. Nature 497: 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss DR, Ahn S, Sassano MF, Kleist A, Zhu X, Strachan R, et al. (2013). Conformation guides molecular efficacy in docking screens of activated beta‐2 adrenergic G protein coupled receptor. ACS Chem Biol 8: 1018–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise A, Jupe SC, Rees S (2004). The identification of ligands at orphan G‐protein coupled receptors. Annu Rev Pharmacol Toxicol 44: 43–66. [DOI] [PubMed] [Google Scholar]
- Wootten D, Christopoulos A, Sexton PM (2013). Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov 12: 630–644. [DOI] [PubMed] [Google Scholar]
- Worth CL, Kreuchwig A, Kleinau G, Krause G (2011). GPCR‐SSFE: a comprehensive database of G‐protein‐coupled receptor template predictions and homology models. BMC Bioinf 12: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wang C, Gregory KJ, Han GW, Cho HP, Xia Y, et al. (2014). Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 344: 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Chu A, Li W, Wang B, Shelton F, Otero F, et al. (2009). Lipid G protein‐coupled receptor ligand identification using beta‐arrestin PathHunter assay. J Biol Chem 284: 12328–12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Xie X (2012a). Tools for GPCR drug discovery. Acta Pharmacol Sin 33: 372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Shih AY, Yang XV, Kuei C, Wu J, Deng X, et al. (2012b). Identification of structural motifs critical for epstein–barr virus‐induced molecule 2 function and homology modeling of the ligand docking site. Mol Pharmacol 82: 1094–1103. [DOI] [PubMed] [Google Scholar]