

Abstract
Background and Purpose
Chronic exposure to morphine increases spinal adrenomedullin (AM) bioactivity resulting in the development and maintenance of morphine tolerance. This study investigated the possible involvement of AM in morphine‐evoked alteration in μ‐opioid receptor‐coupled G proteins.
Experimental Approach
Agents were administered intrathecally (i.t.) in rats. Nociceptive behaviours and cumulative dose–response of morphine analgesia were assessed. Neurochemicals in the spinal dorsal horn were assayed by immunoprecipitation, Western blot analysis and ELISA.
Key Results
Intrathecal injection of AM (8 μg) for 9 days decreased and increased the levels of μ receptor‐coupled Gi and Gs proteins respectively. Morphine stimulation (5 μg) after chronic treatment with AM also induced an increase in cAMP production in the spinal dorsal horn. Co‐administration of the selective AM receptor antagonist AM22–52 inhibited chronic morphine‐evoked switch of G protein‐coupled μ receptor from Gi to Gs. Chronic exposure to AM increased the phosphorylation of cAMP‐responsive element‐binding protein (CREB) and ERK. Co‐administration of the PKA inhibitor H‐89 (5 μg) or MEK1 inhibitor PD98059 (1 μg) reversed the AM‐induced thermal/mechanical hypersensitivity, decline in morphine analgesic potency, switch of G protein‐coupled μ receptor and increase in cAMP.
Conclusions and Implications
The present study supports the hypothesis that an increase in AM activity in the spinal dorsal horn contributes to the switch of the μ receptor‐coupled G protein from Gi to Gs protein via the activation of cAMP/PKA/CREB and ERK signalling pathways in chronic morphine use.
Abbreviations
- AM
adrenomedullin
- IPP
immunoprecipitation
- MPE
maximum possible efffect
- MEK
MAP kinase kinase
- TFL
tail flick latency
- TRPV1
Transient receptor potential vanilloid 1
Tables of Links
| TARGETS | ||
|---|---|---|
| CGRPs a | Enzymes b | Other protein targets |
| μ receptor | ERK1 | CREB binding protein |
| AM1 receptor | ERK2 | |
| MEK1 | ||
| PKA |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
Introduction
Although many new analgesics have been developed, μ‐opioids typified by morphine remain the most effective analgesics for the treatment of severe pain (Arner et al., 1988). However, the therapeutic usefulness of opioids is greatly limited by the development of tolerance that usually occurs following repeated use. One of the mechanisms underlying morphine tolerance involves uncoupling of Gi protein from μ‐opioid receptors (Wong et al., 1992; Gintzler and Chakrabarti, 2000). μ receptors belong to the large superfamily of GPCRs and are predominantly coupled to the pertussis toxin‐sensitive heterotrimeric Gi/o proteins (Chalecka‐Franaszek et al., 2000). Upon μ receptor activation, Gi/o proteins interact with multiple cellular effector systems, inhibiting adenylyl cyclase (AC) activity (George et al., 2000; Mostany et al., 2008). As a result, nociceptive transmission is inhibited. Under normal conditions, only a small fraction of μ receptors are coupled to Gs protein (Shen and Crain, 1989; Fan et al., 1991). Repeated administration of morphine causes a switch of G protein coupled to μ receptors from Gi/o protein to Gs protein (Chakrabarti et al., 2005; Wang et al., 2005). Then, the activation of μ receptors produces opposite effects: activation of the AC/cAMP pathway and up‐regulation of PKA (Crain and Shen, 2000) and the cAMP response element‐binding protein (CREB) (Wang and Burns, 2009), leading to the decline of morphine‐induced analgesia efficacy (morphine tolerance) and paradoxical pain sensitization. However, the mechanisms underlying the switch from Gi/o protein to Gs protein coupled to μ receptors following chronic morphine exposure are basically unknown.
Adrenomedullin (AM) receptor activity may contribute to the switch of G protein coupled to μ receptors from Gi to Gs. AM is a newly demonstrated pronociceptive mediator (Ma et al., 2006) and a member of CGRP family (Poyner et al., 2002). The expression and release of this peptide are increased in the spinal dorsal horn and/or dorsal root ganglia (DRG) following the chronic administration of morphine (Hong et al., 2010), while chronic exposure to AM induces a decline of morphine analgesic potency, referred to as ‘naive morphine tolerance’ or naive tolerance and hyperalgesia (Wang et al., 2014b). The inhibition of AM receptor signalling abolishes morphine‐induced increase in CGRP (Hong et al., 2010) and neuronal NOS (nNOS) (Wang et al., 2014b) as well as the activation of spinal microglia and astrocytes (Zeng et al., 2014). These findings indicate that the mechanism underlying the contribution of AM to morphine tolerance involves the facilitation of nociceptive processing in the spinal cord. However, because the inhibition of AM receptor signalling prevents (Hong et al., 2010) and reverses (Wang et al., 2011) the development of morphine tolerance, it must affect the G proteins coupling by μ receptors, given that morphine analgesia depends on μ receptor–Gi/o protein coupling. The present study examined the hypothesis that the increased AM activity in the spinal dorsal horn contributes to the switch of G protein coupled to μ receptors from Gi to Gs protein in chronic morphine use. The mechanism was also addressed by determining the involvement of the cAMP/PKA–CREB and ERK signalling pathways.
Methods
Animals
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Male Sprague–Dawley rats (250–320 g) supplied by the Animal Centre of Fujian Medical University were housed at 22°C with 50% humidity under a 12‐h light/dark cycle (lights on at 07:00 h) and given free access to food and water. Care and treatment of animals were performed according to the guidelines for investigations of experimental pain in conscious animals (Zimmermann, 1983) and were approved by the Animal Care Committee of Fujian Normal University. Animals were divided randomly for various treatments in all studies. The samples for biochemical assays were identified with a code number and disclosed after data analysis. For assessment of nociceptive behaviour, the investigator was blind to the drug test conditions. All efforts were made to minimize animal suffering and the number of animals used in our experiments.
Intrathecal catheter implantation
Animals were implanted with chronic indwelling catheters (Pogatzki et al., 2000). Briefly, rats were injected with pentobarbital (50 mg·kg−1, i.p.) and shaved along the occiput and neck. The dura mater overlying the atlanto‐occipital junction was exposed by blunt dissection, and an incision was made in the dura. A polyethylene catheter (PE‐10; Stoelting, Wood Dale, IL, USA), with a loose knot at 8.0 cm from the end, was threaded caudally to position its tip at the L4–L5 segments of the spinal cord, and the knot was immobilized by suturing to the musculature. The rostral tip of the catheter was exteriorized at the back of the neck. The catheter was then flushed with 10 μL of saline and plugged. The rats were housed individually after surgery and allowed to recover for approximately 7 days before being used for behavioural testing. Only the animals with no evidence of neurological deficits after catheter placement were used for experiment.
The chronic morphine protocol consisted of intrathecal (i.t.) administration of morphine hydrochloride (20 μg) to rats once daily in the morning for 6 consecutive days. This protocol has been proven to generate tolerance to morphine antinociception (Johnston et al., 2004; Cai et al., 2007). Drug or vehicle (saline) was delivered in a volume of 10 μL via the i.t. catheter when the animals were conscious.
Assessment of nociceptive behaviour
Nociceptive threshold was determined by the tail flick assay using a Plantar Test and Tail Flick Meter (IITC Life Science Inc., Woodland Hills, CA, USA). Tail flick latency (TFL) was measured with 0.01 s precision. Radiant heat intensity was adjusted to produce on average a baseline of 2–3 s (high intensity for morphine response) or 7.0–9.0 s (low intensity for detection of hyperalgesia) in naive rats. The cut‐off latency was established at 10 and 20 s, respectively, to avoid tissue damage. TFL at any tested time point was measured three times at 2 min intervals, and the mean values of these measurements were taken.
Mechanical threshold was measured in the hindpaw using an automated von Frey‐type system (Dynamic Plantar Anesthesiometer 37400, Ugo Basile, Varese, Italy). Animals were acclimatized to the testing apparatus for 30 min each day for 3 days and briefly habituated to the test environment for 20 min on the day of the test to minimize intra‐individual and inter‐individual variability of behavioural measurements. Rats were placed on a metal mesh surface under a plastic enclosure. The stimulator unit was placed beneath the selected hindpaw with the filament below the plantar surface of the rat. A paw withdrawal response was elicited by applying an increase in force (measured in g) using a stainless‐steel filament (0.5 mm diameter). To start, the electrodynamic actuator unit lifted the filament and exerted a force. The force was increased at a rate of 2.5 g·s−1 until the rat moved its paw. A force of 50 g for 30 s was used as a cut‐off point to preclude possible injury to the paw. The force was measured three times at 2 min intervals to generate mean values. The investigator was blind to the drug test conditions. n = 6–7 in each group for this behavioural study.
For the assessment of the potency of morphine analgesia, a cumulative morphine dose–response curve was constructed. These dose–response experiments were conducted 24 h after the final injection of AM or saline given as a part of the chronic administration. Animals were given increasing doses of i.t. morphine every 30 min until a maximal level of antinociception was reached. TFL was examined 20 min after morphine administration, the time at which the peak effect of morphine is achieved (Jiang et al., 2006; Cai et al., 2007). The ED50 value was determined from the morphine dose–response curve. TFL in the behavioural test was converted to a percentage of maximum possible effect (MPE) using the following formula:
%MPE = ([post ‐ drug latency − baseline latency]/[cut ‐ off time − baseline latency]) × 100%
Spinal cord membrane fraction preparation
Animals were killed by decapitation 30 min following i.t. administration of morphine. The dorsal half of the lumbar spinal cord was quickly harvested. Tissue samples were homogenized in lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, 50 mM NaF, 1 mM NaVO3, 1% Igepal CA‐630 and 0.1% SDS) supplemented with protease inhibitors (5 mM PMSF, 10 μg·mL−1 leupeptin and 10 μg·mL−1 pepstatin); all chemicals were purchased from Sigma‐Aldrich (Saint Louis, MO, USA). A membrane fraction of the spinal dorsal horn samples was obtained using a membrane protein extraction kit (Beyotime Institute of Biotechnology, Shanghai, China) as recommended by the manufacturer and the fractions stored at −80°C until assay.
Immunoprecipitation
Membranes were solubilized in HEPES buffer containing 150 mM NaCl, 1% Nonidet P‐40, 0.5% Na‐deoxycholate, 0.1% SDS and 10% glycerol, agitated overnight at 4°C. The samples were centrifuged (14 000× g for 20 min at 4°C). An anti‐μ receptor antibody (1:50; Chemicon‐Millipore, Beijing, China) was covalently cross‐linked to protein G agarose from a protein G immunoprecipitation kit (Sigma, Shanghai, China), according to the manufacturer's instructions. Spinal dorsal horn lysates were incubated with anti‐μ receptor antibody or normal rabbit IgG (negative control) at 4°C overnight. Prewashed protein G agarose beads were added and mixed at 4°C overnight. After centrifugation at 10 000× g for 30 s and washing with lysis buffer, the immunoprecipitated complexes or the total proteins (positive control) were assayed by Western blot to detect G proteins or μ receptors. The specificity of the μ receptor antibody has been reported previously (Kasai et al., 2011); n = 4–5 in each group (Kawasaki et al., 2008; Ferrini et al., 2013).
Western blot
Animals were killed by decapitation and the dorsal half of the lumbar spinal cord was harvested. Tissue samples were homogenized and sonicated in lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, 50 mM NaF, 1 mM NaVO3, 1% Igepal CA‐630 and 0.1% SDS) supplemented with protease inhibitors (5 mM PMSF, 10 μg·mL−1 leupeptin and 10 μg·mL−1 pepstatin, all from Sigma). Homogenates were shaken at 4°C followed by centrifugation at 15 000× g for 30 min each. The supernatant was collected, aliquoted and then stored at −80°C. The BCA protein assay kit (Pierce Chemical, Rockford, IL, USA) was used to quantify proteins in the samples, and 20 μg of protein in SDS loading buffer was resolved on 7.5% SDS polyacrylamide gels. After protein transfer, the polyvinylidene difluoride membrane was blocked in 5% skimmed milk in Tween‐20/PBS for 1 h at room temperature. The membrane was then blotted with rabbit anti‐Gi (1:1000; Abcam, Cambridge, UK), Gs or Gq protein (1:300; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), phosphorylated CREB (pCREB) or phosphorylated ERK (pERK) (1:700; Santa Cruz Biotechnology Inc.) overnight at 4°C in 5% skimmed milk. Membranes were then incubated with horseradish peroxidase‐conjugated goat anti‐rabbit antibody (1:1000; Zhongshan Co., Beijing, China), and bands were detected using enhanced chemiluminescence detection (Amersham Biosciences UK, Ltd., Buckinghamshire, Little Chalfont, UK). The membrane was then blotted with a rabbit μ receptor antibody (1:300; Santa Cruz Biotechnology Inc.) or mouse polyclonal β‐actin antibody (1:2000; Santa Cruz Biotechnology Inc.) for 2 h at room temperature in 5% skimmed milk. Membranes were incubated with horseradish peroxidase‐conjugated goat anti‐rabbit or mouse antibody (1:5000) (Zhongshan Co.). Densitometry was performed using the Image J program and density of Gi, Gs or Gq protein, pCREB or pERK band was normalized to the μ receptor or β‐actin loading control. Results are expressed as relative density compared to the saline‐treated control. The specificity of the μ receptor antibody was as reported previously (Zagon et al., 2011); n = 5 in each group.
Enzyme immunoassay (ELISA)
Spinal dorsal horn samples were processed in a similar manner as those used for Western blot. A quantitative assay for cAMP was performed using a commercially available EIA kit (Rand D Systems Inc., Minneapolis, MN, USA). The ELISA was carried out according to the manufacturer's instructions. Microplates were read using a microplate reader (Molecular Devices Corporation, Sunnyvale, CA, USA). The average value from three to four rats was taken as the levels of cAMP in each group. The protein concentration of the tissue samples was determined in the crude supernatants using the bicinchoninic acid (BCA) protein assay kit (Bio‐Rad, Hercules, CA, USA). Levels of cAMP in tissues are expressed as fmol·mg−1 of tissue protein; n = 3–4 in each group (Kawasaki et al., 2008; Ferrini et al., 2013). A pseudo‐replication in triplicate was used to test the reliability of single values.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are expressed as mean ± SEM. Statistical significance for individual groups was examined using two‐way (groups × day) or one‐way (groups) anova followed by a post hoc Tukey's test when F achieved P < 0.05 and there was no significant variance in homogeneity. A P‐value less than 0.05 was considered statistically significant.
Drugs and chemicals
Sodium pentobarbital was obtained from Shenwgong Co. (Shanghai, China). Morphine hydrochloride was purchased from Northeast (the first pharmaceutical industry of Shenyang, China). N‐[2‐(p‐bromocinnamylamino) ethyl]‐5‐isoquinolinesulfonamide dihydrochloride (H‐89), 2‐(2‐diamino‐3‐methoxyphenyl‐4H‐1‐benzopyran‐4‐one (PD98059) and D‐Phe‐Cys‐Tyr‐D‐Trp‐Arg‐Thr‐Pen‐Thr‐NH2 (CTAP) were purchased from Sigma‐Aldrich (Shanghai, China). Rat AM1–50 and AM22–52 were obtained from Huadatianyuan Biological Co. (Shanghai, China). PD98059 was dissolved with 10% DMSO and isotonic saline (0.1 μg·l−1 of PD98059; (Suzuki et al., 2013). All other agents were dissolved in 0.9% sterile physiological saline.
Results
Intrathecal administration of AM changes the expression of μ receptor‐coupled Gi and Gs, but not Gq, proteins in the spinal dorsal horn
Saline or AM (8 μg) was administered i.t. once daily for 9 days. The dorsal half of the lumbar spinal cord was harvested on day 10. Gi, Gs and Gq proteins coupled to μ receptors in the cell membrane were assayed by the IPP technique. To examine the specificity of antibody for the immunoprecipitation assays, total protein was used to identify μ receptors, Gi or Gs protein as a positive control. Spinal dorsal horn lysates were immunoprecipitated with an anti‐ μ receptor antibody or normal rabbit IgG (negative control). Then the total protein or immunoprecipitated complexes were subjected to Western blot analysis. The results showed that a unique band of μ receptors, Gi or Gs protein was detected in the total protein (positive control) and spinal cord lysates captured by the anti‐ μ receptor. However, IgG immunoprecipitation displayed no band (Figure 1).
Figure 1.
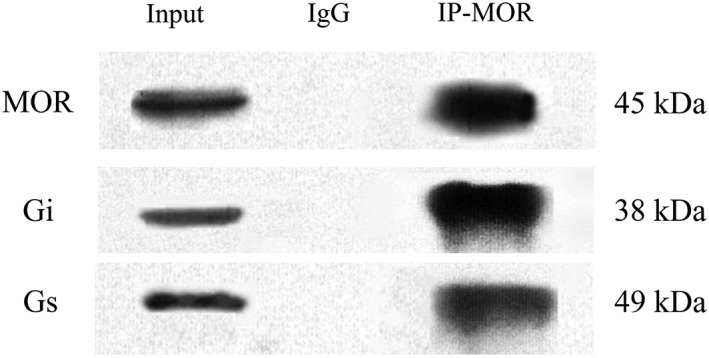
Immunoprecipitation of μ receptors (MOR). Total proteins (50 μg) were used as the ‘input’ positive control. Spinal dorsal horn lysates were immunoprecipitated with anti‐μ receptor antibody (IP) or normal rabbit IgG (IgG, negative control). Then the total protein or immunoprecipitated complexes were assayed to detect μ receptors, Gi or Gs protein using the Western blot technique.
As illustrated in Figure 2, chronic administration of AM induced a decrease and an increase in μ receptor‐coupled Gi (75 ± 3% of control, A) and Gs (135 ± 4%, B) proteins respectively. These alterations were significantly different from the corresponding saline group (P < 0.05). However, the AM treatment did not change the level of μ receptor‐coupled Gq protein (107 ± 8% of control, P > 0.05 vs. saline group, C).
Figure 2.
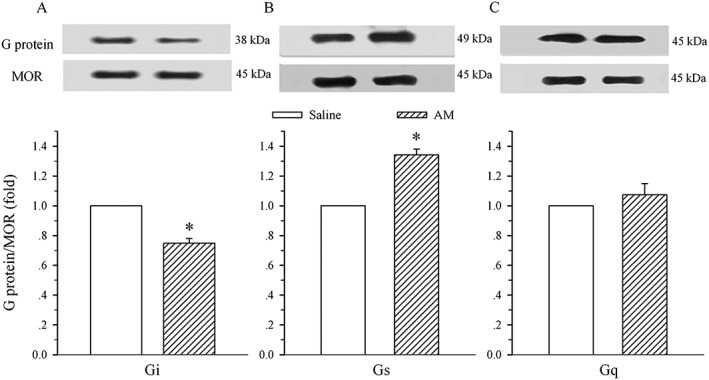
Effects of i.t. administration of AM on the expression of μ receptor (MOR)‐coupled G proteins in the spinal dorsal horn. Saline or AM (8 μg) was administered i.t. once daily for 9 days. The dorsal half of the lumbar spinal cord was harvested on day 10. Immunoprecipitation with immobilized μ receptors and Western blot analysis of the coupling of μ receptors to Gi (A), Gs (B) and Gq (C) proteins were performed on membrane extracts of the spinal dorsal horn. The μ receptor is the loading control. Density of Gi, Gs or Gq protein band was normalized to μ receptor loading control. Data are presented as mean ± SEM. * P < 0.05 compared with the saline group; n = 5 for each group.
Chronic exposure to morphine in the presence of AM22–52 does not change the expression of μ receptor‐coupled Gi and Gs proteins in the spinal dorsal horn
To determine if AM receptor signalling was involved in the morphine‐induced alteration of μ receptor‐coupled G proteins, saline, morphine (20 μg) or morphine plus AM22–52 (36 μg) was administered i.t. once daily for 6 days, and the dorsal half of the lumbar spinal cord was harvested on day 7. The 6 day treatment with morphine induced a decrease and an increase in μ receptor‐coupled Gi (76 ± 8%, Figure 3A) and Gs (120 ± 2%, Figure 3B) proteins, respectively, in the cell membrane of the spinal dorsal horn. These alterations were significantly different from the corresponding saline group (P < 0.05). However, the co‐administration of AM22–52 completely reversed the morphine‐induced alteration in μ receptor ‐coupled Gi (103 ± 7% of control) and Gs (100 ± 6%) proteins (P < 0.05 vs. morphine group).
Figure 3.
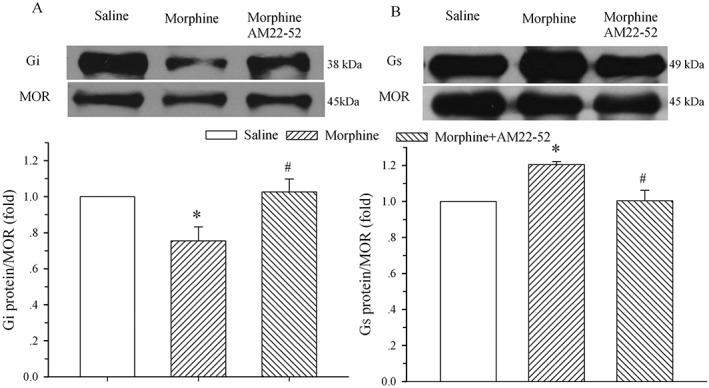
Effect of i.t. administration of AM22–52 on morphine‐evoked alteration of μ receptor (MOR)‐coupled G proteins in the spinal dorsal horn. Saline, morphine (20 μg) or morphine plus AM22–52 (36 μg) was administered i.t. once daily for 6 days, and the lumbar spinal dorsal horn was harvested on day 7. Immunoprecipitation with immobilized μ receptors and Western blot analysis of the coupling of μ receptors to Gi (A) and Gs (B) proteins were performed on membrane extracts of the spinal dorsal horn. The μ receptor is the loading control. The density of the Gi and Gs protein band was normalized to the μ receptor loading control. * P < 0.05 compared with the saline group. # P < 0.05 compared with the morphine group; n = 5 for each group.
Chronic treatment with AM increases the levels of cAMP, pCREB and pERK in the spinal dorsal horn
Because the treatment with chronic AM induced a μ receptor‐coupled Gi‐to‐Gs switch, cAMP production, a functional effect at the level of the receptor, was determined. As AM can activate cAMP/PKA signalling pathways via its own receptor (Xu and Krukoff, 2007; Ho et al., 2008; Wang et al., 2013), we first examined whether activation of μ receptors by morphine following the chronic AM pretreatment could increase cAMP production. Saline or AM (8 μg) was administered i.t. once daily for 9 days. Saline, morphine (5 μg) or morphine plus CTAP (10 nmol) were given i.t. on day 10, and the dorsal half of the lumbar spinal cord was harvested 30 min later. Figure 4A shows that chronic treatment with AM increased cAMP content to 12.7 ± 0.5 fmol·mg−1 from control level of 8.2 ± 1.1 fmol·mg−1 in the saline group (P < 0.05). The additional challenge with morphine further increased cAMP to 24.9 ± 0.5 fmol·mg−1, which was significantly higher than that in the AM group (P < 0.05). However, the effect of morphine on the cAMP level after chronic treatment with AM was reduced to 16.1 ± 0.5 fmol·mg−1 in the presence of CTAP. This value was significantly lower than that in the AM/morphine group (P < 0.05).
Figure 4.
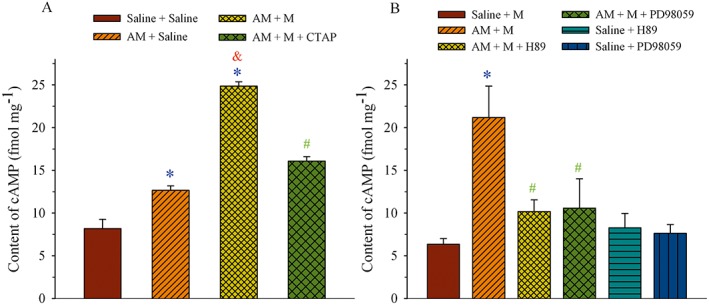
Effect of AM on μ receptor (MOR)‐associated cAMP production in the spinal dorsal horn. (A) Saline or AM (8 μg) was administered i.t. once daily for 9 days. Saline, morphine (M, 5 μg) or morphine plus CTAP (10 nmol) was given i.t. on day 10, and the dorsal half of the lumbar spinal cord was harvested 30 min later. (B) Saline, AM (8 μg), AM plus H‐89 (5 μg), H‐89, AM plus PD98059 (1 μg) or PD98059 was administered i.t. once daily for 9 days. Morphine (M, 5 μg) was given i.t. on day 10, and the dorsal half of the lumbar spinal cord was harvested 30 min later. The cAMP content was assayed by ELISA. * P < 0.05 compared with the saline group. & P < 0.05 compared with the saline + AM group. # P < 0.05 compared with the AM + M group; n = 3–4 for each group.
Saline, AM (8 μg), AM plus H‐89 (5 μg), H‐89, AM plus PD98059 (1 μg) or PD98059 was administered i.t. once daily for 9 days. Morphine (5 μg) was given i.t. on day 10 to stimulate the μ receptors and the dorsal half of the lumbar spinal cord was harvested 30 min later. As illustrated in Figure 4B, administration of morphine increased cAMP content to 21.2 ± 3.7 fmol·mg−1 following the AM treatment (AM + M group) and this value was significantly higher than that in the saline group (6.3 ± 0.7 fmol·mg−1, P < 0.05). The treatment with H‐89 (PKA inhibitor) or PD98059 (MEK1 inhibitor) blunted the effect of AM on morphine‐evoked cAMP production (P < 0.05 vs. AM + M group). When given alone, H‐89 or PD98059 did not change cAMP levels compared to the saline group (P > 0.05).
To further investigate the signalling transduction pathways that mediated the enhanced AM activity, pCREB and pERK protein levels were assayed. Immunoblot analysis showed the expression of pCREB (Figure 5A) and pERK (Figure 5B) proteins in animals that received chronic saline or AM. A nine‐day treatment with AM increased the levels of pCREB and pERK proteins to 147 ± 8 (P < 0.05) and 178 ± 7% of control (P < 0.05) respectively.
Figure 5.
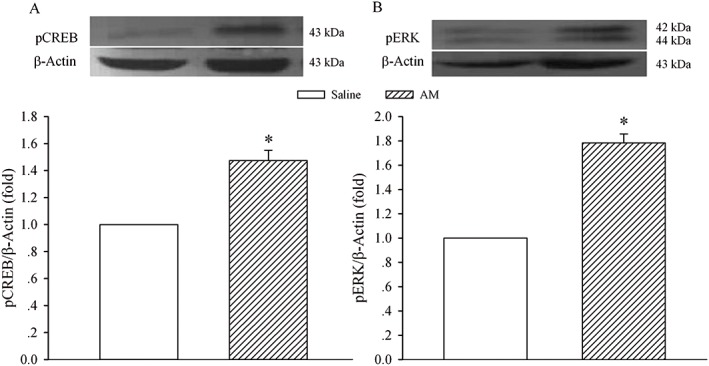
Effect of enhanced AM activity on the expression of pCREB and pERK in the spinal dorsal horn. Saline or AM (8 μg) was given i.t. once daily for 9 days. The dorsal half of the lumbar spinal cord was harvested on day 10 and processed for Western blot analysis for pCREB (A) and pERK (B). The density of the pCREB or pERK band was normalized to the β‐actin loading control. * P < 0.05 compared with the saline group; n = 5 for each group.
Co‐administration of H‐89 or PD98059 abolishes AM‐induced alteration in μ receptor‐coupled Gi and Gs proteins
To confirm the signalling transduction pathways that underlie the AM‐induced alteration in μ receptor‐coupled G proteins, saline, AM (8 μg), AM plus H‐89 (5 μg), H‐89, AM plus PD98059 (1 μg) or PD98059 was administered i.t. once daily for 9 days. The dorsal half of the lumbar spinal cord was harvested on day 10, and Gi and Gs proteins coupled to μ receptors in the cell membrane were assayed by the IPP technique. As illustrated in Figure 6A and 6B, the co‐administration of AM with H‐89 and PD98059 blocked the decrease in Gi protein and the increase in Gs protein induced by AM. These changes were significantly different from those in the AM alone groups (P < 0.05). Both H‐89 and PD98059 administration without AM did not affect the expression of Gi and Gs proteins compared with the saline group (Figure 6C and D, P > 0.05).
Figure 6.
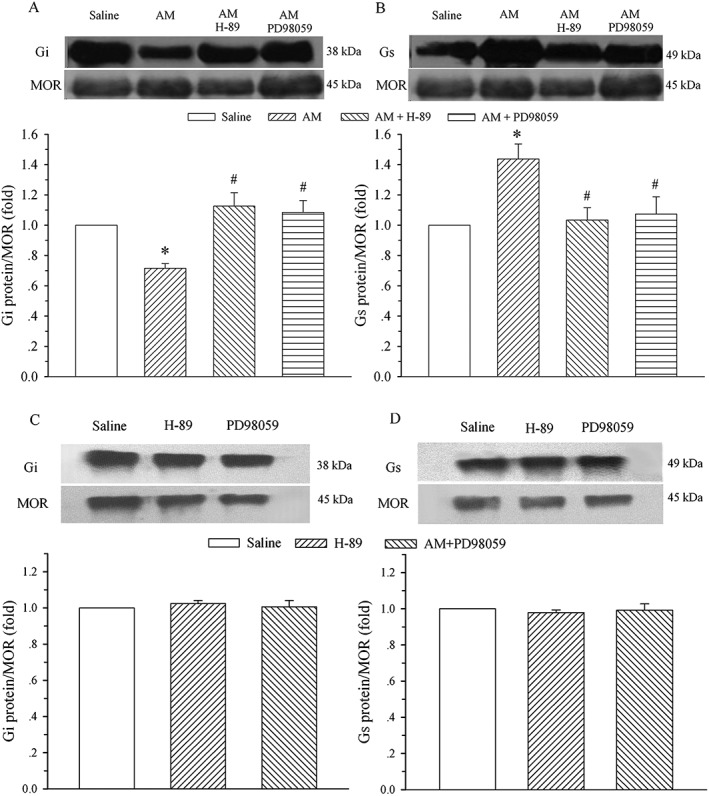
Effect of inhibition of PKA or ERK on AM‐induced alteration in μ receptor (MOR)‐coupled G proteins in the spinal dorsal horn. Saline, AM (8 μg) or AM plus H‐89 (5 μg) or plus PD98059 (1 μg), H‐89 or PD98059 was administered i.t. once daily for 9 days. The lumbar spinal dorsal horn was harvested on day 10, and μ receptors associated with Gi (A and C) and Gs (B and D) proteins were assayed on membrane extracts of the spinal dorsal horn using IPP. The μ receptor is the loading control. * P < 0.05 compared with the saline group. # P < 0.05 compared with the AM group; n = 4 for each group.
Co‐administration of H‐89 or PD98059 inhibits AM‐induced decrease in thermal and mechanical thresholds and naive morphine tolerance
To further confirm that PKA and ERK signalling pathways mediated the AM‐induced alteration in μ receptor‐coupled G proteins, saline, AM (8 μg), AM plus H‐89 (5 μg), AM plus PD98059 (1 μg), H‐89 or PD98059 was administered i.t. once daily for 9 days. The baseline tail flick latency or mechanical threshold before drug treatment was measured daily or every other day, and cumulative dose–response of morphine analgesia was assessed on day 10.
Chronic administration of AM decreased baseline thermal threshold. The TFL was significantly lower on day 9 than the pretreatment baseline level (P < 0.05, Figure 7A). When co‐administered with H‐89 or PD98059, the chronic treatment with AM did not change TFL compared with the baseline level on day 1. As illustrated in Figure 7B, AM administration significantly reduced the mechanical threshold starting on day 6 (P < 0.05). However, the treatment with AM plus H‐89 or PD98059 as well as H‐89 or PD98059 alone failed to change the mechanical threshold compared with the baseline levels on day 1, similar to the saline group (P > 0.05).
Figure 7.
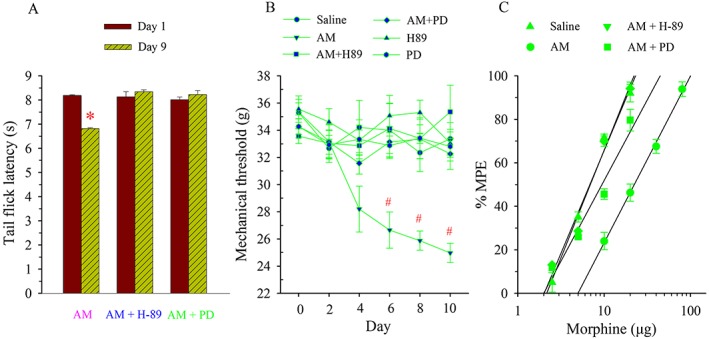
Effects of inhibition of PKA or ERK on AM‐induced alteration in tail flick latency, mechanical threshold and naive morphine tolerance. Saline, AM (8 μg), AM plus H‐89 (5 μg), AM plus PD (PD98059, 1 μg), H‐89 (5 μg) or PD (PD98059, 1 μg) was administered i.t. once daily for 9 days. Tail flick latency (A) or mechanical threshold (B) was measured on days 1 and 9 or from days 2 to 10. * P < 0.05 compared with that on day 1 in the AM group. # P < 0.05 compared with that on day 0. The antinociceptive effect of cumulative doses of morphine was generated on day 10 (C); n = 6–7 for each group.
Compared with the saline group, the cumulative dose–response curve of morphine analgesia following the AM administration was shifted to the right (Figure 7C), and the ED50 was increased by 3.4‐fold (Table 1). After the chronic treatment with AM plus H‐89 or PD98059, morphine was markedly potent as an analgesic with an ED50 of 6.8 ± 0.1 and 9.1 ± 0.3 μg respectively. These values were significantly lower than that in AM alone group (P < 0.05).
Table 1.
Potency of morphine analgesia
| Treatment | ED50 (μg) | 95% CI (μg) |
|---|---|---|
| Mean ± SEM | ||
| Saline | 6.9 ± 0.4 | 5.6 ‐ 8.4 |
| AM | 23.5 ± 1.7 * | 18.7 ‐ 27.4 |
| AM + H‐89 | 6.8 ± 0.1 # | 5.7–8.1 |
| AM + PD98059 | 9.1 ± 0.3 # | 6.8–12.4 |
Saline, AM (8 μg), AM + H‐89 (5 μg) or AM + PD98059 (1 μg) were administered i.t. once per day for 9 days. The cumulative dose–response of morphine analgesia was generated on day 10. The antinociceptive effect of morphine was assessed 20 min after its administration.
CI, confidence interval.
P < 0.001 compared with saline group.
P < 0.001 compared with AM group.
Discussion
The present study demonstrated that chronic i.t. administration of the AM receptor agonist AM1–50 reduced the expression of μ receptor‐coupled Gi protein and increased the level of μ receptor‐coupled Gs protein in the cell membrane of the spinal dorsal horn without changing the level of μ receptor‐coupled Gq protein. Blockade of the AM receptors reversed this morphine‐induced alteration in μ receptor ‐coupled Gi and Gs proteins. Furthermore, the chronic treatment with AM produced an increase in cAMP production and pCREB and pERK levels in the spinal dorsal horn. Inhibition of PKA and ERK abolished the AM‐induced reduction of TFL and mechanical threshold, decline in morphine analgesic efficacy, alterations in μ receptor ‐coupled Gi/Gs proteins and increase in cAMP production. These results suggest that the enhanced AM bioactivity in the spinal cord evoked by chronic morphine turns the switch of G protein coupled to μ receptors from Gi to Gs in a cAMP/PKA/CREB‐ and ERK‐dependent manner, leading to the development of morphine tolerance.
AM is a member of the CGRP family and shares the cardiovascular (Brain and Grant, 2004) and pain modulation properties (Ma et al., 2006; Hong et al., 2009) of CGRP. Similar to CGRP (Menard et al., 1996), AM is involved in the development of morphine tolerance as its expression is up‐regulated in the spinal dorsal horn and DRG and the inhibition of AM receptor signalling prevents (Hong et al., 2010) and reverses (Wang et al., 2011) morphine tolerance. AM is actually released when morphine is given acutely at even a moderate dose, and this release results in the attenuation of morphine analgesia (Wang et al., 2014a). Therefore, it appears that with the repeated μ receptor stimulation, accumulated AM could eventually abolish morphine analgesia. As the development of morphine tolerance is a chronic process, we administered AM i.t. for 9 days at a dose that did not change pain threshold after acute use. This treatment has been shown to induce naive morphine tolerance and thermal hyperalgesia (Wang et al., 2014b) and mechanical allodynia (present study). These results demonstrate that an enhanced AM bioactivity underlies the decline in morphine analgesic potency regardless of whether morphine is used acutely or repeatedly and plays a pivotal role in morphine tolerance. It has been documented that morphine analgesia depends on μ receptor–Gi/o protein coupling (Corbett et al., 2006) and a switch of G protein coupled to μ receptors from Gi to Gs is believed to contribute to the development of morphine tolerance (Chakrabarti et al., 2005; Wang et al., 2005). Because blockade of AM receptors reverses morphine tolerance (Wang et al., 2011), we reasoned that the enhanced AM receptor signalling would be involved in the morphine‐induced switch from Gi to Gs protein coupled to μ receptors. The present study showed that chronic treatment with AM decreased and increased the levels of μ receptor‐coupled Gi and Gs proteins, respectively, in the spinal dorsal horn. Moreover, the cAMP content, a functional effect of Gs protein activation at the level of the receptors (Neves et al., 2002), was also increased. This was demonstrated by the results showing that morphine treatment following the chronic AM further increased cAMP production and this additional increase in cAMP level was abolished by the co‐administration of CTAP, the selective μ receptor antagonist (Pelton et al., 1985). An increase in cAMP following morphine treatment was reported in previous studies (Wang et al., 2005; Wang and Burns, 2009). As μ receptor‐coupled Gs protein is more effective at signalling relative to the more abundant μ receptor‐coupled Gi/o proteins (Crain and Shen, 1998), an additive or synergistic effect of a loss of AC inhibition by Gi and a stimulation of AC by Gs can have a striking effect on overall signalling, leading to morphine tolerance. Therefore, these results showed that enhanced AM receptor signalling can mimic chronic morphine to induce the changes in Gi and Gs proteins/ μ receptor coupling.
Previous studies have shown that the mechanisms underlying AM's contribution to morphine tolerance involve a nociception‐facilitating property (Hong et al., 2009) and the recruitment of CGRP and nNOS (Hong et al., 2010; Wang et al., 2013; Wang et al., 2014b), as well as the activation of spinal microglia and astrocytes (Zeng et al., 2014). We now examined if AM activity was involved in the chronic morphine‐induced alteration in Gi and Gs proteins/ μ receptor coupling. In accord with results from previous studies (Chakrabarti et al., 2005; Wang et al., 2005), chronic morphine exposure resulted in a decrease in μ receptor‐coupled Gi protein and an increase in μ receptor‐coupled Gs protein in the spinal dorsal horn. Importantly, inhibition of the AM receptor signalling by co‐administration of the AM receptor antagonist AM22–52 (Hay et al., 2003) completely abolished the morphine‐induced alteration in μ receptor‐coupled Gi and Gs proteins. These data were comparable with the behavioural observations showing that the blockade of AM receptors inhibits morphine tolerance (Hong et al., 2010; Wang et al., 2011). Given that the chronic AM exposure mimics morphine‐induced analgesic tolerance and hyperalgesia (Wang et al., 2014a) and alterations in μ receptor‐coupled Gi and Gs proteins (present study), we suggest that the switch from Gi protein to Gs protein coupled to μ receptors underlies the contribution of AM receptor signalling to the development of morphine tolerance during its chronic use.
The AM receptor complex is known to be coupled with Gs protein (Mittra and Bourreau, 2006). We have demonstrated that treatment with AM dose‐dependently increases PKA activity and cAMP in cultured DRG neurons (Wang et al., 2013), suggesting that cAMP/PKA is a downstream signalling pathway of AM bioactivity. However, the cAMP/PKA signalling pathway has been demonstrated to play an important role in the pathogenesis of morphine tolerance, as PKA activity in the spinal cord is increased following chronic morphine (Dalton et al., 2005) and inhibition of spinal cAMP/PKA signalling abolishes morphine‐induced antinociceptive tolerance and hyperalgesia (Tumati et al., 2011). Therefore, we examined the role of PKA in AM‐induced naive tolerance. The results showed that following the co‐administration of the PKA inhibitor H‐89, chronic AM failed to induce thermal hyperalgesia, mechanical allodynia and naive tolerance. Moreover, the chronic treatment with AM induced an increase in pCREB, the downstream nuclear target of PKA. This was in agreement with the findings that the activation of PKA results in a phosphorylation of CREB (Montminy, 1997; Daniel et al., 1998) and the expression of pCREB is up‐regulated in the spinal dorsal horn after chronic morphine (Lim et al., 2005; Ko et al., 2006). The present study also determined the involvement of ERK activity in AM's bioactivity, as the ERK signal transduction pathway can mediate AM receptor signalling (Chen et al., 2012; Lausson and Cressent, 2011; Jin et al., 2008; Iemura‐Inaba et al., 2008; Uzan et al., 2008) and phosphorylation of ERK is increased following chronic morphine and contributes to the development of morphine tolerance (Chen et al., 2008; Wang et al., 2009; Berta et al., 2013). In particul, pERK is expressed in spinal astrocytes following morphine exposure (Wang et al., 2009; Berta et al., 2013), while chronic i.t. AM activates spinal astrocytes, and the blockade of AM receptors inhibits morphine‐induced astrocyte activation (Zeng et al., 2014). The results from the present study showed that chronic AM increases the expression of pERK and the inhibition of ERK by the MEK1 inhibitor PD98059 abolished AM‐induced thermal/mechanical hypersensitivity and naïve tolerance. Furthermore, the co‐administration of H‐89 or PD98059 inhibited the AM‐induced alteration in μ receptor‐coupled Gi and Gs proteins cAMP production and abnormal behaviours (thermal hyperalgesia, mechanical allodynia and morphine analgesic potency). Taken together, the results of the present study suggest that the increase in AM receptor signalling induced a switch from Gi protein to Gs protein coupled to μ receptors via the activation of the cAMP/PKA/CREB and ERK transduction pathways and these mechanisms underlie its involvement in the development of morphine tolerance.
The development of morphine tolerance is attributed to the up‐regulation of pronociceptive mediators in the spinal cord, such as glutamate (Zeng et al., 2006), CGRP (Menard et al., 1996), substance P (Powell et al., 2000), NO (Kielstein et al., 2007), TRPV1 (Chen et al., 2008) and AM (Hong et al., 2010). It is generally believed that the mechanism underlying the contribution of these mediators to morphine tolerance is the prolonged excitation of dorsal horn neurons (King et al., 2005). The fact that inhibiting the action of these mediators by i.t. administration of a corresponding antagonist or inhibitor attenuates morphine tolerance implies their involvement in morphine‐induced alterations of μ receptor‐coupled G proteins. To the best of our knowledge, this is the first study showing that the enhanced bioactivity of a pronociceptive mediator (AM) can induce the switch from Gi protein to Gs protein coupled to μ receptors, which represents a mechanism of morphine tolerance.
Author contributions
D. W., J. Z. and Q. L. performed the behavioural study, Western blot, IPP and ELISA techniques. D. W., J. Z., Q. L. and J. H. analysed the data. Y. H. contributed to the experimental design and interpretation of the findings. R. C., D. W. and J. H. contributed to the discussion of the findings. All authors approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This study was funded by grants from the Natural Science Foundation of China (31171072) and Natural Science Foundation of Fujian Province (2014J05039, 2012J01124).
Wang, D. , Zeng, J. , Li, Q. , Huang, J. , Couture, R. , and Hong, Y. (2016) Contribution of adrenomedullin to the switch of G protein‐coupled μ‐opioid receptors from Gi to Gs in the spinal dorsal horn following chronic morphine exposure in rats. British Journal of Pharmacology, 173: 1196–1207. doi: 10.1111/bph.13419.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner S, Rawal N, Gustafsson LL (1988). Clinical experience of long‐term treatment with epidural and intrathecal opioids – a nationwide survey. Acta Anaesthesiol Scand 32: 253–259. [DOI] [PubMed] [Google Scholar]
- Berta T, Liu YC, Xu ZZ, Ji RR (2013). Tissue plasminogen activator contributes to morphine tolerance and induces mechanical allodynia via astrocytic IL‐1beta and ERK signaling in the spinal cord of mice. Neuroscience 247: 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brain SD, Grant AD (2004). Vascular actions of calcitonin gene‐related peptide and adrenomedullin. Physiol Rev 84: 903–934. [DOI] [PubMed] [Google Scholar]
- Cai Q, Jiang J, Chen T, Hong Y (2007). Sensory neuron‐specific receptor agonist BAM8‐22 inhibits the development and expression of tolerance to morphine in rats. Behav Brain Res 178: 154–159. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Regec A, Gintzler AR (2005). Biochemical demonstration of mu‐opioid receptor association with Gsalpha: enhancement following morphine exposure. Brain Res Mol Brain Res 135: 217–224. [DOI] [PubMed] [Google Scholar]
- Chalecka‐Franaszek E, Weems HB, Crowder AT, Cox BM, Cote TE (2000). Immunoprecipitation of high‐affinity, guanine nucleotide‐sensitive, solubilized mu‐opioid receptors from rat brain: coimmunoprecipitation of the G proteins G(alpha o), G(alpha i1), and G(alpha i3). J Neurochem 74: 1068–1078. [DOI] [PubMed] [Google Scholar]
- Chen P, Pang X, Zhang Y, He Y (2012). Effect of inhibition of the adrenomedullin gene on the growth and chemosensitivity of ovarian cancer cells. Oncol Rep 27: 1461–1466. [DOI] [PubMed] [Google Scholar]
- Chen Y, Geis C, Sommer C (2008). Activation of TRPV1 contributes to morphine tolerance: involvement of the mitogen‐activated protein kinase signaling pathway. J Neurosci 28: 5836–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett DA, Henderson G, McKnigh AT, Paterson SJ (2006). 75 years of opioid research: the exciting but vain quest for the Holy Grail. Br J Pharmacol 147: S153–S162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crain SM, Shen KF (1998). Modulation of opioid analgesia, tolerance and dependence by Gs‐coupled, GM1 ganglioside‐regulated opioid receptor functions. Trends Pharmacol Sci 19: 358–365. [DOI] [PubMed] [Google Scholar]
- Crain SM, Shen KF (2000). Antagonists of excitatory opioid receptor functions enhance morphine's analgesic potency and attenuate opioid tolerance/dependence liability. Pain 84: 121–131. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA, et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton GD, Smith FL, Smith PA, Dewey WL (2005). Protein Kinase A activity is increased in mouse lumbar spinal cord but not brain following morphine antinociceptive tolerance for 15 days. Pharmacol Res 52: 204–210. [DOI] [PubMed] [Google Scholar]
- Daniel PB, Walker WH, Habener JF (1998). Cyclic AMP signaling and gene regulation. Annu Rev Nutr 18: 353–383. [DOI] [PubMed] [Google Scholar]
- Fan SF, Shen KF, Crain SM (1991). Opioids at low concentration decrease openings of K+ channels in sensory ganglion neurons. Brain Res 558: 166–170. [DOI] [PubMed] [Google Scholar]
- Ferrini F, Trang T, Mattioli TA, Laffray S, Del'Guidice T, Lorenzo LE, et al. (2013). Morphine hyperalgesia gated through microglia‐mediated disruption of neuronal Cl(−) homeostasis. Nat Neurosci 16: 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George SR, Fan T, Xie Z, Tse R, Tam V, Varghese G, et al. (2000). Oligomerization of mu‐ and delta‐opioid receptors. Generation of novel functional properties J Biol Chem 275: 26128–26135. [DOI] [PubMed] [Google Scholar]
- Gintzler AR, Chakrabarti S (2000). Opioid tolerance and the emergence of new opioid receptor‐coupled signaling. Mol Neurobiol 21: 21–33. [DOI] [PubMed] [Google Scholar]
- Hay DL, Howitt SG, Conner AC, Schindle M, Smith DM, Poyner DR (2003). CL/RAMP2 and CL/RAMP3 produce pharmacologically distinct adrenomedullin receptors: a comparison of effects of adrenomedullin22‐52, CGRP8‐37 and BIBN4096BS. Br J Pharmacol 140: 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho LK, Chen K, Ho IC, Shen YC, Yen DH, Li FC, et al. (2008). Adrenomedullin enhances baroreceptor reflex response via cAMP/PKA signaling in nucleus tractus solitarii of rats. Neuropharmacology 55: 729–736. [DOI] [PubMed] [Google Scholar]
- Hong Y, Liu Y, Chabot JG, Fournier A, Quirion R (2009). Upregulation of adrenomedullin in the spinal cord and dorsal root ganglia in the early phase of CFA‐induced inflammation in rats. Pain 146: 105–113. [DOI] [PubMed] [Google Scholar]
- Hong Y, Wang D, Chabot JG, Ma W, Chen P, Quirion R (2010). A role for protein kinase C‐dependent upregulation of adrenomedullin in the development of morphine tolerance in male rats. J Neurosci 30: 12508–12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iemura‐Inaba C, Nishikimi T, Akimoto K, Yoshihara F, Minamino N, Matsuoka H (2008). Role of adrenomedullin system in lipid metabolism and its signaling mechanism in cultured adipocytes. Am J Physiol Regul Integr Comp Physiol 295: R1376–R1384. [DOI] [PubMed] [Google Scholar]
- Jiang J, Chen Y, Hong Y (2006). Differential reversal effect of intrathecal bovine adrenal medulla peptide 22 on morphine tolerance in rats. Sheng Li Xue Bao 58: 529–535. [PubMed] [Google Scholar]
- Jin D, Harada K, Ohnishi S, Yamahara K, Kangawa K, Nagaya N (2008). Adrenomedullin induces lymphangiogenesis and ameliorates secondary lymphoedema. Cardiovasc Res 80: 339–345. [DOI] [PubMed] [Google Scholar]
- Johnston IN, Milligan ED, Wieseler‐Frank J, Frank MG, Zapata V, Campisi J, et al. (2004). A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci 24: 7353–7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai S, Yamamoto H, Kamegaya E, Uhl GR, Sora I, Watanabe M, et al. (2011). Quantitative detection of micro opioid receptor: western blot analyses using micro opioid receptor knockout mice. Curr Neuropharmacol 9: 219–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, et al. (2008). Distinct roles of matrix metalloproteases in the early‐ and late‐phase development of neuropathic pain. Nat Med 14: 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielstein A, Tsikas D, Galloway GP, Mendelson JE (2007). Asymmetric dimethylarginine (ADMA) – a modulator of nociception in opiate tolerance and addiction? Nitric Oxide 17: 55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Ossipov MH, Vanderah TW, Porreca F, Lai J (2005). Is paradoxical pain induced by sustained opioid exposure an underlying mechanism of opioid antinociceptive tolerance? Neurosignals 14: 194–205. [DOI] [PubMed] [Google Scholar]
- Ko SW, Jia Y, Xu H, Yim SJ, Jang DH, Lee YS, et al. (2006). Evidence for a role of CaMKIV in the development of opioid analgesic tolerance. Eur J Neurosci 23: 2158–2168. [DOI] [PubMed] [Google Scholar]
- Lausson S, Cressent M (2011). Signal transduction pathways mediating the effect of adrenomedullin on osteoblast survival. J Cell Biochem 112: 3807–3815. [DOI] [PubMed] [Google Scholar]
- Lim G, Wang S, Zeng Q, Sung B, Yang L, Mao J (2005). Expression of spinal NMDA receptor and PKCgamma after chronic morphine is regulated by spinal glucocorticoid receptor. J Neurosci 25: 11145–11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Chabot JG, Quirion R (2006). A role for adrenomedullin as a pain‐related peptide in the rat. Proc Natl Acad Sci U S A 103: 16027–16032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard DP, van Rossum D, Kar S, St PS, Sutak M, Jhamandas K, et al. (1996). A calcitonin gene‐related peptide receptor antagonist prevents the development of tolerance to spinal morphine analgesia. J Neurosci 16: 2342–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittra S, Bourreau JP (2006). Gs and Gi coupling of adrenomedullin in adult rat ventricular myocytes. Am J Physiol Heart Circ Physiol 290: H1842–H1847. [DOI] [PubMed] [Google Scholar]
- Montminy M (1997). Transcriptional regulation by cyclic AMP. Annu Rev Biochem 66: 807–822. [DOI] [PubMed] [Google Scholar]
- Mostany R, Diaz A, Valdizan EM, Rodriguez‐Munoz M, Garzon J, Hurle MA (2008). Supersensitivity to mu‐opioid receptor‐mediated inhibition of the adenylyl cyclase pathway involves pertussis toxin‐resistant Galpha protein subunits. Neuropharmacology 54: 989–997. [DOI] [PubMed] [Google Scholar]
- Neves SR, Ram PT, Iyengar R (2002). G protein pathways. Science 296: 1636–1639. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al., NC‐IUPHAR (2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelton JT, Gulya K, Hruby VJ, Duckle SP, Yamamura HI (1985). Conformationally restricted analogs of somatostatin with high mu‐opiate receptor specificity. Proc Natl Acad Sci U S A 82: 236–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogatzki EM, Zahn PK, Brennan TJ (2000). Lumbar catheterization of the subarachnoid space with a 32‐gauge polyurethane catheter in the rat. Eur J Pain 4: 111–113. [DOI] [PubMed] [Google Scholar]
- Powell KJ, Ma W, Sutak M, Doods H, Quirion R, Jhamandas K (2000). Blockade and reversal of spinal morphine tolerance by peptide and non‐peptide calcitonin gene‐related peptide receptor antagonists. Br J Pharmacol 131: 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W, et al. (2002). International Union of Pharmacology. XXXII. The mammalian calcitonin gene‐related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev 54: 233–246. [DOI] [PubMed] [Google Scholar]
- Shen KF, Crain SM (1989). Dual opioid modulation of the action potential duration of mouse dorsal root ganglion neurons in culture. Brain Res 491: 227–242. [DOI] [PubMed] [Google Scholar]
- Suzuki I, Tsuboi Y, Shinoda M, Shibuta K, Honda K, Katagiri A, et al. (2013). Involvement of ERK phosphorylation of trigeminal spinal subnucleus caudalis neurons in thermal hypersensitivity in rats with infraorbital nerve injury. PLoS One 8: e57278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumati S, Roeske WR, Largent‐Milnes TM, Vanderah TW, Varga EV (2011). Intrathecal PKA‐selective siRNA treatment blocks sustained morphine‐mediated pain sensitization and antinociceptive tolerance in rats. J Neurosci Methods 199: 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzan B, Villemin A, Garel JM, Cressent M (2008). Adrenomedullin is anti‐apoptotic in osteoblasts through CGRP1 receptors and MEK–ERK pathway. J Cell Physiol 215: 122–128. [DOI] [PubMed] [Google Scholar]
- Wang D, Chen P, Li Q, Quirion R, Hong Y (2011). Blockade of adrenomedullin receptors reverses morphine tolerance and its neurochemical mechanisms. Behav Brain Res 221: 83–90. [DOI] [PubMed] [Google Scholar]
- Wang D, Huo Y, Quirion R, Hong Y (2014a). Involvement of adrenomedullin in the attenuation of acute morphine‐induced analgesia in rats. Peptides 54: 67–70. [DOI] [PubMed] [Google Scholar]
- Wang D, Li J, Chen P, Hong Y (2014b). Upregulation of pronociceptive mediators and downregulation of opioid peptide by adrenomedullin following chronic exposure to morphine in rats. Neuroscience 280: 31–39. [DOI] [PubMed] [Google Scholar]
- Wang D, Ruan L, Hong Y, Chabot JG, Quirion R (2013). Involvement of PKA‐dependent upregulation of nNOS‐CGRP in adrenomedullin‐initiated mechanistic pathway underlying CFA‐induced response in rats. Exp Neurol 239: 111–119. [DOI] [PubMed] [Google Scholar]
- Wang HY, Burns LH (2009). Naloxone's pentapeptide binding site on filamin A blocks Mu opioid receptor‐Gs coupling and CREB activation of acute morphine. PLoS One 4: e4282. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang HY, Friedman E, Olmstead MC, Burns LH (2005). Ultra‐low‐dose naloxone suppresses opioid tolerance, dependence and associated changes in mu opioid receptor‐G protein coupling and Gbetagamma signaling. Neuroscience 135: 247–261. [DOI] [PubMed] [Google Scholar]
- Wang Z, Ma W, Chabot JG, Quirion R (2009). Cell‐type specific activation of p38 and ERK mediates calcitonin gene‐related peptide involvement in tolerance to morphine‐induced analgesia. FASEB J 23: 2576–2586. [DOI] [PubMed] [Google Scholar]
- Wong CS, Su YF, Watkins WD, Chang KJ (1992). Continuous intrathecal opioid treatment abolishes the regulatory effects of magnesium and guanine nucleotides on mu opioid receptor binding in rat spinal membranes. J Pharmacol Exp Ther 262: 317–326. [PubMed] [Google Scholar]
- Xu Y, Krukoff TL (2007). Adrenomedullin stimulates nitric oxide production from primary rat hypothalamic neurons: roles of calcium and phosphatases. Mol Pharmacol 72: 112–120. [DOI] [PubMed] [Google Scholar]
- Zagon IS, Donahue RN, Bonneau RH, McLaughlin PJ (2011). T lymphocyte proliferation is suppressed by the opioid growth factor ([Met(5)]‐enkephalin)‐opioid growth factor receptor axis: implication for the treatment of autoimmune diseases. Immunobiology 216: 579–590. [DOI] [PubMed] [Google Scholar]
- Zeng J, Thomson LM, Aicher SA, Terman GW (2006). Primary afferent NMDA receptors increase dorsal horn excitation and mediate opiate tolerance in neonatal rats. J Neurosci 26: 12033–12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Lin MY, Wang D, Zhang Y, Hong Y (2014). Involvement of adrenomedullin in spinal glial activation following chronic administration of morphine in rats. Eur J Pain 18: 1323–1332. [DOI] [PubMed] [Google Scholar]
- Zimmermann M (1983). Ethical guidelines for invesstigations of experimental painin conscious animals. Pain 16: 109–110. [DOI] [PubMed] [Google Scholar]