

Abstract
Background
In-depth phylogeographic analysis can reveal migration patterns relevant for public health planning. Here, as a model, we focused on the provenance, in the current Italian HCV subtype 1a epidemic, of the NS3 resistance-associated variant (RAV) Q80K, known to interfere with the action of NS3/4A protease inhibitor simeprevir. HCV1a migration patterns were analysed using Bayesian phylodynamic tools, capitalising on newly generated and publicly available time and geo-referenced NS3 encoding virus genetic sequence data.
Results
Our results showed that both immigration and local circulation fuel the current Italian HCV1a epidemic. The United States and European continental lineages dominate import into Italy, with the latter taking the lead from the 1970s onwards. Since similar migration patterns were found for Q80K and other lineages, no clear differentiation of the risk for failing simeprevir can be made between patients based on their migration and travel history. Importantly, since HCV only occasionally recombines, these results are readily transferable to the genetic sequencing policy concerning NS5A RAVs.
Conclusions
The patient migration and travel history cannot be used to target only part of the HCV1a infected population for drug resistance testing before start of antiviral therapy. Consequently, it may be cost-effective to expand genotyping efforts to all HCV1a infected patients eligible for simeprevir-based therapies.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-017-0913-3) contains supplementary material, which is available to authorized users.
Keywords: HCV, HCV1a, Q80K, Italy, US, Europe, Phylogeography, Public health policy
Background
Hepatitis C virus (HCV) infected patients are nowadays treated with interferon-free regimens containing one or more antivirals that directly target virus proteins. These new direct-acting antivirals (DAAs) have a superb efficacy and, when combined, can clear the virus in more than 95% of the treated population, irrespective of the HCV genotype [1]. For most DAAs no clear association of particular naturally occurring resistance-associated variants (RAVs) with clinical outcome has been identified [2]. Because of this, baseline drug resistance testing is only recommended in well-defined cases such as the combination of NS3/4A protease inhibitor (PI) simeprevir with NS5B polymerase inhibitor sofosbuvir. This regimen is associated with reduced viral cure rates in HCV subtype 1a (HCV1a) cirrhotic patients who carry the naturally occurring, highly prevalent RAV Q80K [3–6]. The potential clinical relevance of the processes that shape the distribution of particular variants spurred an interest in the spatiotemporal aspects of the HCV1a evolutionary history in general, and in particular that of NS3 variant Q80K. Given the increased interest into the natural prevalence of several other RAVs, particularly located in NS5A [4, 7], insights into the patterns of HCV1a spread may be interesting for these variants as well.
Little over a decade after the discovery of the virus itself, it was revealed that there was a burst in the number of HCV1a transmissions starting from the second half of the 20th century, which is likely linked to the history of illicit drug use and iatrogenic intervention rates [8–11]. While in these earlier studies the emphasis was on the temporal and spatial aspects of HCV1a in general, the medical relevance of the Q80K polymorphism drew attention to the segregation of the global HCV1a variability into two distinct clades, clade I and clade II [12–15], with the first clade further structured into three subclades [16]. In particular, Q80K strains are concentrated in the so-called clade IA [16], and the Q80K prevalence is largely accounted for by a single substitution event dating back to 1940–1963 [17]. This ancestral lineage first arose in the United States (US) and has been introduced multiple times into Europe [13, 14, 17]. Importantly, in these studies the migration patterns of HCV1a were evaluated on the continental geographical level, while a more fine-grained resolution is required to provide actionable information for national or regional public health programs.
Detailed phylogeographic analyses are particularly pertinent for clinically relevant lineages. Due to the rapid evolution of HCV treatments that achieve high viral cure rates, the regimen simeprevir with sofosbuvir, largely used as first-line treatment so far in Europe and in Italy [18, 19], has recently been categorized as suboptimal for HCV1a infected patients [20]. Nonetheless, second-line NS5A inhibitor-based regimens can still be considered when the absence of relevant RAVs is confirmed [21]. Despite this shift from NS3-targeting DAAs to those that inhibit the NS5A protein, a thorough analysis of the patterns of virus flow based on the NS3 region remains valuable because HCV only rarely recombines [22], and migration patterns inferred from small genomic regions hence have a genome-wide representativeness. Moreover, because drug resistance testing before treatment initiation has always been recommended for HCV1a infected patients when simeprevir is part of the planned regimen, NS3-based phylogeographic reconstructions can capitalize on a comparatively large number of publicly available NS3 sequence data to obtain the most accurate insights as possible. However, the rapid evolution in preferred HCV treatment schemes makes that phylogeographically informed public health decisions are of temporal value, and need to be updated regularly.
Here, we reconstructed the historical spread of HCV1a using a within-country-level resolution, and focused on the potential public health implications of the recovered clustering patterns for Italy. To this end, new sequencing data of the NS3 gene from Italy were combined with a selection of globally sampled publicly available sequences to infer the plausible origins and frequencies of HCV1a migrations into and out of Italy, and to explore the within-country migration flows for Italy, with statistical phylogeographic tools. In doing so, we provide a blueprint of how similar studies with a focus on other genomic regions in the HCV genome like the NS5A protein, or even other viruses can analyze patterns of virus flow by integrating genetic, spatial and temporal information in a state-of-the-art Bayesian phylogenetic framework. Furthermore, the timing of the Q80K origins was refined and the historical context of their spread from the US to Europe and Italy was discussed.
Methods
Newly generated Italian sequences
This retrospective study includes 183 samples, collected between 2011–2015, from mostly DAA-naïve (N = 163, 89.1%) Italian patients infected with HCV1a. The Italian strains were collected in at least seven regions (Additional file 1: Table S1). The NS3 protease gene (181 amino acids) was sequenced as reported elsewhere [23] in the context of routine clinical care at University of Rome Tor Vergata, Italy (art. 6 and art. 9, leg. 211/2003 and 196/2003).
Sequence dataset compilation
The Italian sequence data were complemented with all publicly available HCV1a NS3 sequences from Genbank (N = 3032, http://www.ncbi.nlm.nih.gov/nucleotide) that fulfilled the following quality criteria. Only sequences that were unambiguously subtyped as HCV1a with subtyping tools Oxford HCV version 2 [24], COMET [25], and by phylogenetic analysis including a broad panel of reference sequences, were retained. The latter phylogenies were estimated on 1000 bootstrapped alignments with RAxML under a GTR + Γ substitution model, with subtypes assigned to strains clustering with ≥70% bootstrap support. Clonal sequences and strains from non-human hosts, duplicate sequences and strains covering <80% of the NS3 gene were removed. Remaining strains were aligned using an in house pairwise codon aware alignment tool (http://regatools.med.kuleuven.be/sequencetool/sequencetool.wt), followed by the removal of sequences with stop codons, leaving 930 taxa. Of these, 855 sequences with known sampling time and location were used for the phylogeographic reconstructions. A preliminary analysis revealed a large clade with only US-derived sequences that did not contain NS3 RAVs and that received 100% bootstrap support. As the within-US circulation was not the focus of this study, sequences from this clade were discarded to reduce computational burden in subsequent Bayesian analyses, resulting in a final set of 610 strains (Table 1).
Table 1.
Overview of the complete dataset used in this study
| Geographic origin | HCV1a dataset (n, % dataset) | Q80K (n, % country) | Sample time span |
|---|---|---|---|
| Italya | 280 (45.9) | 52 (18.6) | 2004-2015 |
| USb | 145 (23.8) | 58 (24.6)c | 1989-2008 |
| Germany | 67 (11.0) | 19 (28.4) | 2003-2013 |
| Switzerland | 46 (4.5) | 6 (13.0) | 2002-2006 |
| Belgium | 25 (4.1) | 8 (32.0) | 2007-2013 |
| Thailand | 19 (3.1) | 3 (15.8) | 2007-2010 |
| France | 13 (2.1) | / | 2007-2011 |
| Spain | 5 (0.8) | / | 2001-2002 |
| UK | 4 (0.7) | / | 2008-2010 |
| Brazil | 2 (0.3) | / | 2001-2003 |
| Australia | 2 (0.3) | / | 2007 |
| China | 1 (0.2) | / | 2009 |
| Japan | 1 (0.2) | / | 2009 |
| Total | 610 | 146 (23.9) | 1989-2015 |
The absolute and relative contribution of each country, the number of Q80K variants and their share by country, and the time span of the samples included per country, are detailed. a183 (63%) of the Italian sequences have not been published before. bTo assess the proportion of Q80K strains, the 91 taxa from the removed cluster were taken into account (see Methods). cSince the Q80K prevalence in the US was lower than expected, it was recalculated taking strains without sampling time information into account, resulting into a prevalence of 57.5%
To assess the robustness of the phylogeographic reconstructions against sampling biases, three random down-samples with a maximum of 30 sequences per location were created, resulting in three sets with a more balanced number of samples by location. Additionally, a diverse subset of 83 sequences was selected from clade I using the same procedure as in [26] to independently estimate the evolutionary rate in the Q80K clade. Briefly, from each sampling year the five most divergent sequences were selected with the Phylogenetic Diversity Algorithm [27], which selects the subtree of n taxa connected by the largest sum of branch lengths.
Identification of Italian transmission networks
Transmission clusters that represent the Italian transmission dynamics were identified using a posterior root node support cutoff of ≥90% and were required to consist at least for 90% of member taxa isolated in Italy.
Bayesian estimation of time-calibrated trees
All phylogenies were estimated using the Bayesian Evolutionary Analysis by Sampling Trees software (BEAST, v.1.8.2.) [28] with BEAGLE [29] to improve the computational performance. The skygrid or skyride model was used as a flexible non-parametric prior for the coalescent process in all analyses. A check for molecular clock signal [30] revealed that there was insufficient accumulation of divergence over the sampling time span to reliably estimate evolutionary rates. A first attempt to remedy this was specifying an informative prior distribution on the mean clock rate parameter of the relaxed clock model [31] that is based on a previously reported HCV1a dataset with strong temporal signal [32]. Exploratory runs with this model setup, however, indicated this still represented an overparameterization, which is why the mean clock rate was fixed to the mean of the NS3 rate estimate from the Gray et al. (2011) data, but while still estimating the variance of the evolutionary rate. A codon position partitioned HKY model was fitted, allowing for Γ-distributed among site rate heterogeneity at each position. Convergence and mixing properties of the Markov chain Monte Carlo (MCMC) chains were assessed with Tracer v1.6. Results of several MCMC chains were combined after removal of the burn-in. Maximum clade credibility (MCC) trees were summarized using TreeAnnotator included in the BEAST package. Trees were visualized with FigTree v 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Phylogeographic reconstructions
Source-sink relations were evaluated with a discrete diffusion model that allows for different rates depending on the direction of movement [33]. The migration pathways that are most relevant in the history of spread were identified with a Bayesian stochastic search variable selection (BSSVS) approach [34]. Bayes factor (BF) support (with BF ≥3 assumed to be relevant [35]) for all possible types of location exchanges was calculated using SpreaD3 [36]. Posterior expectations of the number of transition events between all possible pairs of locations (Markov jumps) were estimated with efficient stochastic mapping techniques [37].
Results
Sequence dataset
The final alignment included 610 sequences and covered 181 amino acids of the NS3 gene. Italy is the best-represented country in this final dataset, followed by the US (Table 1). Of the Western European strains, 36.4% was isolated in countries other than Italy. The Q80K variant was detected in 23.9% of all strains, and a somewhat lower prevalence of 19.3% was observed for Western Europe. The proportion of taxa carrying Q80K varied from 13.0% to 32.0% by country (Table 1).
Multiple independent introductions of HCV1a and Q80K in Italy
Bayesian phylogeographic methods were used to determine when and from where HCV1a strains were introduced into Italy. As only limited sequence data was available for most European countries, all non-Italian isolates from Europe were grouped as ‘Europe (not Italy)’. Similarly, isolates sampled outside the US and Europe were grouped as ‘others’. To mitigate the potentially biasing effect of sample sizes on ancestral reconstructions of the location states, three subsampled datasets with more equal sample numbers by location state were analyzed (US, Italy, Europe (not Italy) and others).
Independent of the sampling, similar migration patterns were observed in the two major HCV1a clades as well as among the clade 1 subclades, including the clade that comprises virtually all Q80K lineages, to which we from now on refer to as the Q80K clade (Fig. 1a-d). The most clear re-emerging pattern in the complete and in the subsampled datasets is the seeding, but not receiving, role of the US in the global spread of HCV1a. Specifically, the migration rates involving the US as origin location always receive high support for being non-zero, while rates of migration towards the US are indistinguishable from zero (Table 2). The remaining three well-supported migration links in the complete dataset received substantial Bayes factor (BF) support in two out of three subsampled datasets. Two of these links involve Italy, and lend support for Italy as a source and a sink for European continental virus movements. An interactive web-based visualization and demo video of the phylogeographic history of HCV1a in general and in particular of the Q80K polymorphism is available at http://demo.luciad.com/rega/?hepa&2d (legend, see Additional file 2).
Fig. 1.
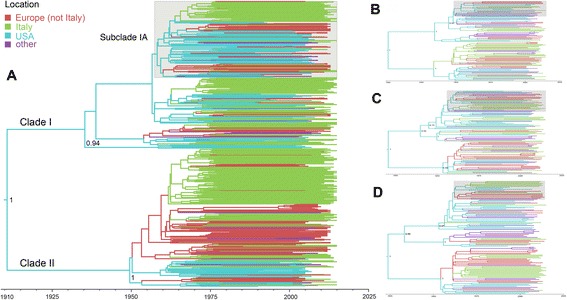
The United States as the immediate origin of HCV1a spread to Europe and Italy. The branches in the HCV1a maximum clade credibility (MCC) tree constructed from the complete dataset are colored according to sampling country with strains grouped into four locations (see legend). All but five Q80K strains are found in the same clade, which is highlighted in grey. We refer to Fig. 3 for further details on the Q80K history. Mixing of strains among the four locations can be observed in the entire tree. HCV entered Italy via the US and continental migration in the two major clades of HCV1a variability (Clade I and II) as well as in the Q80K clade. Posterior root node support is visualized in a selection of deeper nodes. a: entire dataset. b: MCC tree estimated from subsample 1 (see Methods). c: MCC tree estimated from subsample 2. d: MCC tree estimated from subsample 3
Table 2.
Posterior probabilities for all possible types of migrations
| Migration from | Migration to | Complete dataset | subsample 1 | subsample 2 | subsample 3 |
|---|---|---|---|---|---|
| Europe (not Italy) |
Italy | 29.12 | 2.88 | 8.48 | 19.43 |
| US | 0.01 | 0.08 | 0.09 | 0.08 | |
| Other | 7.66 | 0.81 | 5.88 | 11.07 | |
| Italy | Europe (not Italy) |
10.28 | 18.23 | 4.37 | 0.52 |
| US | 0 | 0.06 | 0.07 | 0.05 | |
| Other | 0.07 | 8.64 | 0.47 | 1.15 | |
| US | Europe (not Italy) |
21.68 | 19.08 | 23.34 | 21.42 |
| Italy | 22.37 | 22.88 | 24.37 | 12.93 | |
| Other | 8.74 | 25.61 | 31.01 | 29.45 | |
| Other (not Europe –not US) |
Europe (not Italy) |
0.03 | 1.3 | 0.37 | 3.61 |
| Italy | 0.03 | 0.33 | 1.43 | 0.23 | |
| US | 0 | 0.11 | 0.11 | 0.08 |
Posterior probabilities for migration rates supported by a BF ≥3 are marked in bold, indicated for the complete dataset as well as for the three subsamples created as described in methods
Additional file 2: Visualization. An interactive web-based visualization of the phylogeographic history of HCV1a. Migration patterns of HCV1a in general and in particular of the Q80K polymorphism are visualized in an interactive web application, available at http://demo.luciad.com/rega/?hepa&2d. A demo-movie with voice-over is accessible through a button implemented in the application, explaining the possibilities of the tool. More specifically, the world map is visualized in the upper panel, with the countries sampled in this study highlighted in a color and labeled by their name. The map shows how viral lineages are introduced/exported in/to different countries throughout time, represented by jumping lines on the upper panel. Viral lineages that circulate locally are represented by circles on the world map, with their size proportional to the number of branches. The visualization is simulated over time, using a fixed window of time as a filter. The length of a jump on the map, is inversely proportionate to the length of the branch in the phylogenetic tree. That is, longer geographic jumps on the map indicate branches that quickly traveled over large distances, while short jumps indicate branches that slowly traveled over relatively small distances. The timing of these jumps is indicated in the lower panel, which represents a timeline that plots new viral lineages over time. The colors in this timeline match the region of the same color on the geographic map. On the map, a darker color, indicates a larger viral lineage amount. Using the tool, we can apply a color filter on the branches of the tree, visualizing not only the phylogeographic history of HCV1a in general, but also of variant Q80K. Migration jumps will be colored by amino acid polymorphisms present at position 80 of the NS3 protein region, with amino acid Q indicated in red, and variant K in yellow. All other polymorphisms (being not Q or K) are colored blue, as indicated in the legend. (MP4 162 mb)
In addition to the significance of migration rates, the expected proportion of all possible migration events in the HCV1a history (Markov jumps) was also inferred (Table 2). Little over half of all expected migrations are virus movements from the US, and a majority of these are directed towards Western Europe. The virus flows involving Italy account for 83.4% of the remaining expected number of jumps, and Italy mostly acts as the destination location in this European continental spread (73.9%). Using the entire dataset, the first introduction of HCV1a into Italy was timed around 1957.7 (95% HPD: 1949.0-1964.4) (Fig. 1), and the first appearance of a Q80K lineage in Italy around 1961.5 (95% HPD: 1957.6-1965.7).
The picture painted by the Markov jumps analysis above is a static one and does not inform us on changes over time of the relative importance of the migration flows to and from Italy. A plot of the expected number of migration events from Italy reveals a stable pattern of seeding almost uniquely towards other Western European countries throughout the history (Fig. 2, left panel). In contrast, a similar plot for the incoming migrations shows that the initial dominance of the US as origin location has decreased in favor of European continental HCV1a circulation, starting from 1970 (Fig. 2, right panel).
Fig. 2.

Immigration and emigration rates of HCV1a strains from and to Italy, over time period 1940–2015. Left panel: Over 99% of exportation events from Italy are directed towards other European countries (in blue). Note that the rate decline in the most recent years reflects the loss of branches in the sampling time period, rather than the start of an actual tendency. Right panel: The origin of virtually all strains entering Italy can be traced back to the United States (US) and other European countries (see Table 2). At the onset of the Italian HCV1a epidemic, immigration was most intense from the US (in blue). From 1970 onwards, introductions from the European continent started to dominate (in red). The introduction rate (y-axis) was defined as the median posterior estimate of the expected number of migration events from that particular location per time unit
Migration patterns within Italy
Twelve highly supported small clusters with only Italian isolates (size range: 2–7) and one pure Italian large cluster (size 34, cluster 13 in Table 3) were identified in the MCC summary tree of the time evolutionary histories (Additional file 3: Figure S1). Information on the sampling region (North, Central, South) was available only for a limited number of the members of the smaller clusters and for the majority of samples from the large cluster (Table 3). Both homogenous (n = 1, cluster 4) and mixed-origin (n = 1, cluster 2) small clusters were observed. Phylogeographic analysis of cluster 13 with the same (asymmetric) model as before painted a picture of migration by proximity where both North and South regions interact with the Central region (BF support >3) and vice versa (BF support >20), but not with each other (BF support for North-to-South and South-to-North migration rates <3). There is one larger Italian cluster in the Q80K clade (Fig. 1), but this has a posterior root node support of 81%, which is below the used cut-off. Nonetheless, the distribution of its lineages over at least five regions (Additional file 4: Table S2) also points to an interregional mixing of strains within Italy.
Table 3.
Highly supported Italian clusters identified in the MCC tree obtained from the complete dataset
| Sample size (number of strains) |
Tree root support (posterior probability) |
Presence of Q80K (N, %) | Time span (years) |
Regional information | |
|---|---|---|---|---|---|
| Cluster 1 | 2 | 1 | 0 (0) | 2.23 | Unknown: 2 |
| Cluster 2 | 2 | 1 | 0 (0) | 3.4 | North: 1 – South: 1 |
| Cluster 3 | 2 | 1 | 2 (100) | 8.4 | Unknown: 2 |
| Cluster 4 | 2 | 1 | 2 (100) | 15.0 | Central: 2 |
| Cluster 5 | 2 | 1 | 0 (0) | 17.6 | Unknown: 2 |
| Cluster 6 | 2 | 0.98 | 2 (100) | 22.8 | Unknown: 2 |
| Cluster 7 | 2 | 0.99 | 0 (0) | 32.3 | Unknown: 2 |
| Cluster 8 | 2 | 0.97 | 0 (0) | 32.8 | Central: 1 – Unknown: 1 |
| Cluster 9 | 2 | 0.92 | 0 (0) | 33.1 | Unknown: 2 |
| Cluster 10 | 5 | 0.99 | 0 (0) | 13.9 | Unknown: 5 |
| Cluster 11 | 6 | 1 | 0 (0) | 12.6 | Central: 1 – Unknown: 5 |
| Cluster 12 | 7 | 1 | 0 (0) | 7.8 | Central: 6 – Unknown: 1 |
| Cluster 13 | 34 | 0.98 | 3 (8.8) | 35.4 | North: 2 - Central: 21 - South: 7 – Unknown: 4 |
In total, 13 highly supported clusters were identified (posterior root node support ≥0.90), with 2 to 34 Italian member taxa. Sampling region information was unavailable for many taxa. For each cluster, the sample size, posterior root node support, presence of Q80K and the time spanned by the cluster, is listed. Clusters are ordered according to sample size and time span
Refining Q80K divergence times
As investigating the virus migration patterns entails estimating time-calibrated histories, we seized the opportunity to briefly re-evaluate the temporal aspects of the Q80K origins, but now taking among lineage rate variation explicitly into account. The Q80K variant has evolved independently on multiple occasions, and one lineage was particularly successful and gave rise to almost all currently circulating Q80K strains. This major Q80K clade had a low posterior support (58.6%), but upon further inspection it became clear that this was due to one or a few taxa that frequently clustered outside this dominant Q80K clade (see Fig. 3b-c). The genesis of the most recent common ancestor of the most successful Q80K clade was estimated around 1957 (95% highest posterior density (HPD): 1947.3-1958.6).
Fig. 3.
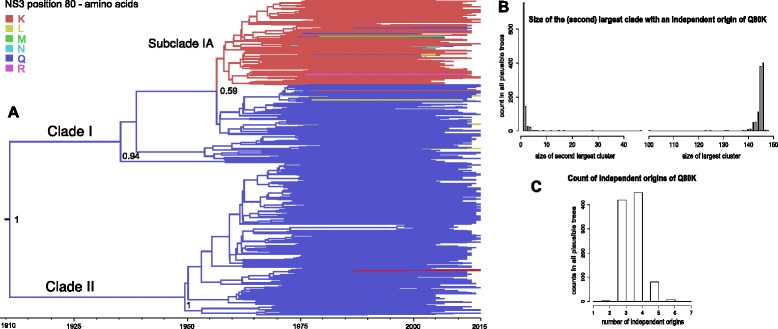
a: A founder effect dominates the history of the Q80K clade. The branches in the HCV1a MCC tree are colored according to the inferred amino acid at NS3 position 80 (see legend). All except five Q80K variants descend from a common ancestor that is inferred to have existed around 1957 (95% HPD: 1947–1959). Posterior root node support is visualized in a selection of deeper nodes. Amino acids other than Q or K were found in only 13 lineages. b: Histogram of the size of the (second) largest clade with an independent origin of Q80K among the plausible trees. The size of the largest clade remains more or less constant, and the limited size of the second largest clade shows that most contemporaneous Q80K lineages reside in the major Q80K clade. c: Histogram of the number of independent origins of a Q80K lineage in the plausible trees. This shows that in the most plausible trees three to four de novo appearances of the Q80K variant appear
In this analysis the mean clock rate was fixed to the mean of the NS3 rate estimate obtained from the Gray et al. (2011) HCV1a full-genome data [32]. To verify whether this did not bias our timing of the origin of the Q80K clade, a ‘Q80K clade rate’ was estimated independently using a subset of this clade’s taxa selected in the same way as in [26] (see Methods). Reassuringly, the NS3 mean rate estimates in both analyses were nearly identical to each other (1.02x10−3 substitutions/site/year for the Q80K clade, and 1.00x10−3 for the HCV1a NS3 gene using the dataset of [32]), showing that the latter was an appropriate external calibration.
Discussion
In this work, we elucidated patterns in the HCV1a transmission dynamics that are relevant for the Italian HCV sequencing policy, with a focus on the NS3 variant Q80K, which can affect success of simeprevir containing drug regimens and serves as a model for other clinically relevant RAVs. To this end, newly obtained Italian NS3 sequences were combined with all publicly available sequences worldwide obtained from isolates with known sampling time and location. Sequence evolution was integrated with spatial diffusion at a country-level resolution to assess the relative importance of local spread versus virus importation into Italy. The within-Italy migration dynamics were tentatively explored and the opportunity was taken to concisely revisit the dating of the highly successful Q80K founder event.
The clustering patterns of Italian strains with globally sampled ones showed that HCV1a, including Q80K strains, entered Italy on multiple occasions. The origin of these lineages can be traced to both the US and Western Europe, and no significant link with other locations was found (Table 2). Few samples were available from other regions with a considerable HCV1a burden, such as the Andean Latin American and Caribbean regions (Table 1, [38]). While this may have affected the inferred role of the US as the origin (but see [8]), it does not affect our findings that HCV1a, including Q80K strains, has been introduced into Italy on multiple occasions.
The plot of the expected number of migration events per year from the dominant origin locations (e.g. the US and Western Europe) to Italy shows that in the hypothetical situation of a 100% effective screening and treatment of immigrants and short-term travelers of these regions, approximately five to six lineages can be prevented from initiating an ‘Italian’ transmission network each year. Note that, because the sampling density impacts the extent of clustering, this estimate is a lower bound. Temporally resolving the seeding intensity by origin region also revealed a switch in the relative importance between both locations, initially mainly from the US, with European continental spread dominating from the late 1960s, early 1970s onwards (Fig. 2a, left panel).
Importantly, the Italian lineages are dispersed among all clade 1 subclades and there are no clear differences in migration pathways between clade I, which encompasses all but one of the Q80K strains, and clade II (Fig. 1). This has several potential public health policy implications. Because both virus import and local spread shape the Italian HCV1a, and thus also the Italian Q80K epidemic, immigrants and short-term travelers or other (non-travelling) Italian citizens cannot be specifically targeted or disregarded in a sequencing program. Moreover, since the origins of imported lineages in the Q80K and other clades are shared, and the Italian epidemic is usually seeded from those locations that contribute most to the Q80K clade, also referred to as clade 1A [16], this too cannot be used to distinguish populations in their relative risk of failing with simeprevir.
The national patterns of virus movements were explored by coarsely dividing Italy in a North, Central and South region, not only because detailed geographical data are only available for a subset of the Italian samples (see Additional file 1: Table S1) but also because using a higher-level resolution for this small dataset precludes well-informed parameter estimations. Although it remains difficult to extract general migration patterns from a limited sampling, the extensive mixing of lineages among different regions identified for both wild-type and Q80K lineages shows that the Italian epidemic likely is quite uniform and does not evolve in well-segregated local sub-epidemics. Of note, because of this it is likely that many unsampled Italian HCV1a lineages are part of already detected Italian transmission networks and thus represent the same introduction events (and origins) as those of sampled lineages.
HCV is characterised by substantial amounts of among lineage rate variation [32]. This cannot only interfere with divergence time estimation [39, 40] but also with phylogenetic inference [41, 42], and is to the best of our knowledge for the first time explicitly modelled in our timing of the Q80K origins. Moreover, unlike in earlier work, the phylogeny and divergence times were co-estimated using an integrated Bayesian approach that appropriately takes the shared ancestry into account and also avoids the risk of error propagation typical for rate-smoothing procedures that rely on a pre-specified topology. Our dating places the origin of the most successful lineage in 1955 (95% HPD: 1947.3-1958.7). This is far more recent than the penalised likelihood based point estimate by McCloskey et al. [17] and demarcates a more narrow interval than their linear regression based estimate (1955, 95% CI: 1945–1963). This makes that the earliest Q80K origin no longer overlaps with the dates of World War II (WOII) and, in turn, renders the hypothesis that the Q80K lineage was introduced into Europe through large-scale movements of military troops unlikely. Rather, this successful Q80K lineage found itself at the right time at the right place to profit from the post-WOII increase in parenteral iatrogenic procedures and illicit drug use, both in the US and elsewhere, including Italy [43, 44]. The majority of Italian Q80K strains for which sampling region information was available (156/183) were isolated in Central to Southern and Insular areas, the regions from where most Italy-to-US emigrants originate [45]. As many of these emigrants eventually returned - estimates range from 11% to 73% for the first half of the 20th century [46, 47] - the introduction of HCV1a via remigration is an alternative hypothesis that deserves further exploration. Other Western European countries, in particular Germany and the UK, also have a history of intense travel, migration and trade with the US, but large discrepancies in the reported numbers of immigrants arriving and leaving the US hamper a comparison of the migration flow intensities (statistics US Homeland Security). In this respect it is noteworthy that the group of non-Italian European samples includes a reasonable 67 German samples but only four UK isolates (Table 1). The inclusion of additional data, in particular from the UK, can therefore be expected to ‘break’ some of the direct links between the US and Italy. Additionally, Latin America and South-East Asia, for which high HCV1a prevalence rates have been reported [38], were underrepresented in our dataset. This implies that a number of introductions into Western Europe (or Italy) from these locations likely were not detected. However, because Brockmann et al. [48] reported less intensive migration links between Europe and Latin America as compared to between Europe and North America, and virus dispersal is usually linked to the degree of human connectivity [49], we believe this did not heavily impact our results.
The rate at which new virus genetic data is generated is increasing as a result of upscaled sequencing efforts in the context of DAA combination therapies. These are frequently extensively annotated with socio-demographic and clinical metadata, which can be used for a fine-grained identification of processes that fuel an epidemic (see [50] for a recent example for HCV). Unfortunately, such metadata are usually not deposited along with the virus genetic data in public databases for reasons of privacy. Here, it has been highlighted how scrutinizing the population level transmission dynamics can still reveal information relevant for intervention strategies provided basic annotations such as sampling time and space are available.
Conclusions
In-depth phylogeographical analyses revealed that the Italian HCV1a epidemic is shaped by complex patterns of virus importation and sustained local and interregional transmission. Because this precludes a clear differentiation of Q80K presence/absence between sub-populations, it may be cost-effective to test all HCV1a patients eligible for therapy with simeprevir for the absence of this polymorphism. Furthermore, this study demonstrates the usefulness of a versatile analysis strategy that can readily be applied in different regions or countries, to other genetic regions in the HCV genome such as the NS5A protein and to other measurably evolving pathogens.
Acknowledgements
The authors wish to thank Philippe Lemey for his assistance and advise in the phylogeographic reconstructions.
Funding
Lize Cuypers was supported by a PhD grant of the FWO (Fonds Wetenschappelijk Onderzoek – Vlaanderen, Asp/12). Part of this research was sponsored by two FWO grants G.A029.11 N and G.0E84.16 N, and by the Italian Ministry of Instruction, University & Research (MIUR) (Accordi di Programma 2011: RBAP11YS7K_001, Bandiera InterOmics Protocollo PB05 1°) and by the Aviralia Foundation. The VIROGENESIS project receives funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No 634650. This work was made possible by funding of the Bijzonder Onderzoeksfonds KU Leuven (BOF) No. OT/14/115. Nadia Marascio was supported by a postdoctoral grant of QOR (Regional Operating Program) CALABRIA FSE (European Social Founding) 2007/2013. Lavinia Fabeni was supported by the Italian Ministry of Health (Ricerca corrente 2015, linea n.2, progetto n.3). Andrea-Clemencia Pineda- Peña was funded by the project Bio-Molecular and Epidemiological Surveillance of HIV Transmitted Drug Resistance, Hepatitis Co-Infections and Ongoing Transmission Patterns in Europe (BEST HOPE) (funded through HIVERA: Harmonizing Integrating Vitalizing European Research on HIV/AIDS, grant 249697); by L’Oréal Portugal Medals of Honor for Women in Science 2012 (financed through L’Oréal Portugal, Comissão Nacional da Unesco and Fundação para a Ciência e Tecnologia - FCT) and by funds to GHTM (UID/Multi/04413/2013). A part of the computational resources and services used in this work were provided by the Hercules Foundation and the Flemish Government – department EWI-FWO Krediet aan Navorsers (Theys, KAN2012 1.2.249.12).
Availability of data and materials
The datasets generated and analysed during the current study are available on the public database Genbank (https://www.ncbi.nlm.nih.gov/nucleotide).
Authors’ contributions
LC gathered the NS3 publicly available data, constructed the final dataset, performed phylogeographic analyses and wrote subsequent drafts of the manuscript. BV performed all Bayesian phylogeographic analyses and wrote subsequent drafts of the manuscript. LB participated in the compilation of the new Italian sequences, quality control of the dataset and assisted in writing the manuscript, supported by her supervisor CFP. NM participated in the study design and assisted in writing the manuscript, supported by her supervisors AF and CT. VC, VCDM and MA were responsible for the sequencing and collection of the new NS3 Italian sequence dataset. ACP participated in the study design and the construction of the final dataset, and assisted in writing the manuscript. YS, KVL and LC were responsible for the sequencing and processing of NS3 sequences from Belgian patients, once FN gave permission for their use. DB was responsible for the web-based interactive visualization of the obtained results. FCS and AMV supervised the design of the study, and assisted in writing the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no other competing interests.
Consent for publication
Not applicable
Ethics approval and consent to participate
In total, 183 samples from Italian patients infected with HCV1a were collected between 2011–2015. The NS3 protease gene was sequenced in the context of routine clinical care at University of Rome Tor Vergata in Italy. As only anonymized samples were used and research was not conducted within the context of a clinical trial, ethical approval was not required according to the Italian law (art. 6 and art. 9, leg. 211/2003 and 196/2003). The analysis of the combined dataset was approved by the Leuven ethical committee (ML9219, ML10770 and ML8635).
Abbreviations
- BEAGLE
Broad-platform Evolutionary Analysis General Likelihood Evaluator
- BEAST
Bayesian Evolutionary Analysis Sampling Trees
- BF
Bayes Factor
- BSSVS
Bayesian stochastic search variable selection
- CI
Confidence interval
- DAA
Direct-acting antiviral
- GTR
Generalized time reversible model
- HCV
Hepatitis C virus
- HIV
Human immunodeficiency virus
- HKY
Hasegawa-Kishino-Yano
- HPD
Highest posterior density interval
- MCC
Maximum clade credibility
- MCMC
Markov Chain Monte Carlo
- n
Number
- PI
Protease inhibitor
- RAV
Resistance-associated variant
- RAxML
Randomized Axelerated Maximum-Likelihood
- US
United States
- WOII
World War II
Additional files
The country-wide distribution of Italian samples. Overall, 183 sequences were newly obtained from seven different regions. The publicly available data trace back to the work by de Luca et al. [14] (n = 67) and Paolluci et al. [51] (n = 30). The former samples have an unknown distribution among the Lazio, Lombardy, Tuscany and Apulia region. For the latter, it is only known that the patients visited the Fondazione IRCCS Policlinico San Matteo, Pavia, Lombardy. (DOC 31 kb)
The thirteen highly supported Italian clusters indicated in the maximum clade credibility summary tree. The highly supported Italian-only clusters are highlighted in green, and the cluster number corresponding to the numbering in Table 3 is also detailed. The red background marks the dominant Q80K clade. The evolutionary distance bar at the bottom indicates the percentage of nucleotide substitutions per site along each lineage. (PDF 50 kb)
Geographical distribution of Italian Q80K lineages. For each Italian strain within the large Q80K clade, more detailed regional information is listed. (DOC 29 kb)
References
- 1.Cuypers L, Ceccherini Silberstein F, Van Laethem K, et al. Impact of HCV genotype on treatment regimens and drug resistance: a snapshot in time. Rev Med Virol. 2016 doi: 10.1002/rmv.1895. [DOI] [PubMed] [Google Scholar]
- 2.Schneider MD, Sarrazin C. Antiviral therapy of hepatitis C in 2014: do we need resistance testing? Antiviral Res. 2014;105:64–71. doi: 10.1016/j.antiviral.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 3.EASL recommendations on treatment of hepatitis C, 2015. http://www.easl.eu/medias/cpg/HEPC-2015/Full-report.pdf
- 4.Cuypers L, Li G, Libin P, et al. Genetic diversity and selective pressure in hepatitis C virus genotypes 1–6: significance for direct-acting antiviral treatment and drug resistance. Viruses. 2015;7:5018–5039. doi: 10.3390/v7092857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawitz E, Matusow G, DeJesus E, et al. Simeprevir plus sofosbuvir in patients with chronic hepatitis C virus genotype 1 infection and cirrhosis: a phase 3 study (OPTIMIST-2) Hepatology. 2016;64:360–369. doi: 10.1002/hep.28422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarrazin C, Lathouwers E, Peeters M, et al. Prevalence of the hepatitis C virus NS3 polymorphism Q80K in genotype 1 patients in the European region. Antiviral Res. 2015;116:10–16. doi: 10.1016/j.antiviral.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Cuypers L, Li G, Neumann-Haefelin C, et al. Mapping the genomic diversity of HCV subtypes 1a and 1b: implications of structural and immunological constraints for vaccine and drug development. Virus Evolution. 2016;2. doi:10.1093/ve/vew024. [DOI] [PMC free article] [PubMed]
- 8.Magiorkinis G, Magiorkinis E, Paraskevis D, et al. The global spread of hepatitis C virus 1a and 1b: a phylodynamic and phylogeographic analysis. PLoS Med. 2009;6:e1000198. doi: 10.1371/journal.pmed.1000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakano T, Lu L, Liu P, Pybus OG. Viral gene sequences reveal the variable history of hepatitis C virus infection among countries. J Infect Dis. 2004;190:1098–1108. doi: 10.1086/422606. [DOI] [PubMed] [Google Scholar]
- 10.Pybus OG, Charleston MA, Gupta S, et al. The epidemic behavior of the hepatitis C virus. Science. 2001;292:2323–2325. doi: 10.1126/science.1058321. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka Y, Hanada K, Mizokami M, et al. A comparison of the molecular clock of hepatitis C virus in the United States and Japan predicts that hepatocellular carcinoma incidence in the United Status will increase over the next two decades. Proc Natl Acad Sci U S A. 2002;99:15584–15589. doi: 10.1073/pnas.242608099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bagaglio S, Uberti-Foppa C, Messina E, et al. Distribution of natural resistance to NS3 protease inhibitors in hepatitis C genotype 1a separated into clades 1 and 2 and in genotype 1b of HIV-infected patients. Clin Microbiol Infect. 2016;22:386. doi: 10.1016/j.cmi.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Beloukas A, King S, Childs K, et al. Detection of the NS3 Q80K polymorphism by Sanger and deep sequencing in hepatitis C virus genotype 1a strains in the UK. Clin Microbiol Infect. 2015;21:1033–1039. doi: 10.1016/j.cmi.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 14.De Luca A, Di Giambenedetto S, Lo Presti A, et al. Two distinct hepatitis C virus genotype 1a clades have different geographica distribution and association with natural resistance to NS3 protease inhibitors. Open Forum Infect Dis. 2015;2:ofv043. doi: 10.1093/ofid/ofv043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pickett BE, Striker R, Lefkowitz EJ. Evidence for separation of HCV subtype 1a into two distinct clades. J Viral Hepat. 2011;9:608–618. doi: 10.1111/j.1365-2893.2010.01342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santos AF, Bello G, Vidal LL, et al. In-depth phylogenetic analysis of hepatitis C virus subtype 1a and occurrence of 80 K and associated polymorphisms in the NS3 protease. Sci Rep. 2016;6:31780. doi: 10.1038/srep31780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCloskey RM, Liang RH, Joy JB, et al. Global origin and transmission of hepatitis C virus nonstructural protein 3 Q80K polymorphism. J Infect Dis. 2015;211:1288–1295. doi: 10.1093/infdis/jiu613. [DOI] [PubMed] [Google Scholar]
- 18.Aghemo AM, Cologni G, Maggiolo F, et al. Safety and efficacy of directly acting antivirals in 2432 HCV patients with advanced fibrosis: an interim analysis of the Lombardia regional network for viral hepatitis. Barcelona, Spain: 51st EASL; 2016. [Google Scholar]
- 19.Mauss S, Buggisch P, Böker KHW, et al. Treatment outcomes for hepatitis C genotype 1 infection with direct acting antivirals: data from the German hepatitis C-registry. Barcelona, Spain: 51st EASL; 2016. [Google Scholar]
- 20.EASL recommendations on treatment of hepatitis C, 2016. http://www.easl.eu/medias/cpg/HCV2016/English-report.pdf.
- 21.Craxì A, Perno CF, Viganò M, et al. From current status to optimization of HCV treatment: Recommendations from an expert panel. Dig Liver Dis. 2016;48:995–1005. doi: 10.1016/j.dld.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 22.González-Candelas F, López-Labrador FX, Bracho MA. Recombination in hepatitis C virus. Viruses. 2011;3:2006–2024. doi: 10.3390/v3102006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cento V, Mirabelli C, Salpini R, et al. HCV genotypes are differently prone to the development of resistance to linear and macrocyclic protease inhibitors. PLoS One. 2012;7:e39652. doi: 10.1371/journal.pone.0039652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Oliveira T, Deforche K, Cassol S, et al. An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics. 2005;21:3797–3800. doi: 10.1093/bioinformatics/bti607. [DOI] [PubMed] [Google Scholar]
- 25.Struck D, Lawyer G, Ternes AM, et al. COMET: adaptive context-based modeling for ultrafast HIV-1 subtype identification. Nucleic Acids Res. 2014;42:e144. doi: 10.1093/nar/gku739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vrancken B, Rambaut A, Suchard MA, et al. The genealogical population dynamics of HIV-1 in a large transmission chain: bridging within and among host evolutionary rates. PLoS Comput Biol. 2014;10:e1003505. doi: 10.1371/journal.pcbi.1003505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chernomor O, Minh BQ, Forest F, et al. Split diversity in constrained conservation prioritization using integer linear programming. Methods Ecol Evol. 2015;6:83–91. doi: 10.1111/2041-210X.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suchard MA, Rambaut A. Many-core algorithms for statistical phylogenies. Bioinformatics. 2009;25:1370–1376. doi: 10.1093/bioinformatics/btp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rambaut A, Lam TT, Carvalho M, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evolution. 2016;2. doi:10.1093/ve/vew007. [DOI] [PMC free article] [PubMed]
- 31.Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gray RR, Parker J, Lemey P, et al. The mode and tempo of hepatitis C virus evolution within and among hosts. BMC Evol Biol. 2011;11:131. doi: 10.1186/1471-2148-11-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edwards CJ, Suchard MA, Lemey P, et al. Ancient hybridization and an Irish origin for the modern polar bear matriline. Curr Biol. 2011;21:1251–1258. doi: 10.1016/j.cub.2011.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLoS Comput Biol. 2009;5:e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kass RE, Raftery AE. Bayes factors. J Am Stat Assoc. 1995;90:773–795. doi: 10.1080/01621459.1995.10476572. [DOI] [Google Scholar]
- 36.Bielejec F, Baele G, Vrancken B, et al. SpreaD3 : interactive visualization of spatiotemporal history and trait evolutionary processes. Mol Biol Evol. 2016. doi: 10.1093/molbev/msw082. [DOI] [PMC free article] [PubMed]
- 37.Minin VN, Suchard MA. Counting labeled transitions in continuous-time Markov models of evolution. J Math Biol. 2008;56:391–412. doi: 10.1007/s00285-007-0120-8. [DOI] [PubMed] [Google Scholar]
- 38.Gower E, Estes C, Blach S, et al. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol. 2014;61:S45–S57. doi: 10.1016/j.jhep.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 39.Yoder AD, Yang Z. Estimation of primate speciation dates using local molecular clocks. Mol Biol Evol. 2000;17:1081–1090. doi: 10.1093/oxfordjournals.molbev.a026389. [DOI] [PubMed] [Google Scholar]
- 40.Drummond AJ, Suchard MA. Bayesian random local clocks, or one rate to rule them all. BMC Biol. 2010;8:114. doi: 10.1186/1741-7007-8-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Felsenstein J. Cases in which parsimony or compatibility methods will be positively misleading. Syst Biol. 1978;27:401–410. doi: 10.1093/sysbio/27.4.401. [DOI] [Google Scholar]
- 42.Worobey M, Han GZ, Rambaut A. A synchronized global sweep of the internal genes of modern avian influenza virus. Nature. 2014;508:254–257. doi: 10.1038/nature13016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cochrane A, Searle B, Hardie A, et al. A genetic analysis of hepatitis C virus transmission between injection drug users. J Infect Dis. 2002;186:1212–1221. doi: 10.1086/344314. [DOI] [PubMed] [Google Scholar]
- 44.Silini E, Bono F, Cividini A, et al. Molecular epidemiology of hepatitis C virus infection among intravenous drug users. J Hepatol. 1995;22:691–695. doi: 10.1016/0168-8278(95)80225-8. [DOI] [PubMed] [Google Scholar]
- 45.Wepman D. Immigration: from the founding of Virginia to the closing of Ellis Island. New York: Facts on File Book; 2002. p. 430.
- 46.Monticelli GL. Italian emigration: basic characteristic and trends with special reference to the last twenty years. International Migration Review. 1967;1:10–24. doi: 10.2307/3002737. [DOI] [Google Scholar]
- 47.Cometti E. Trends in Italian Emigration. The Western Political Quarterly. 1958;11:820–834. doi: 10.2307/443655. [DOI] [Google Scholar]
- 48.Brockmann D, Hufnagel L, Geisel T. The scaling laws of human travel. Nature. 2005;439:462–465. doi: 10.1038/nature04292. [DOI] [PubMed] [Google Scholar]
- 49.Pybus OG, Tatem AJ, Lemey P. Virus evolution and transmission in an ever more connected world. Proc Biol Sci. 2015;282:20142878. doi: 10.1098/rspb.2014.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jacka B, Applegate T, Poon AF, et al. Transmission of hepatitis C virus infection among younger and older people who inject drugs in Vancouver. Canada J Hepatol. 2016;64:1247–1255. doi: 10.1016/j.jhep.2016.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paolucci S, Fiorina A, Piralla R, et al. Naturally occurring mutations to HCV protease inhibitors in treatment-naive patients. Virol J. 2012;9:245. doi: 10.1186/1743-422X-9-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and analysed during the current study are available on the public database Genbank (https://www.ncbi.nlm.nih.gov/nucleotide).