

Abstract
Background and Purpose
Increasing evidence has suggested cadmium (Cd), as an inducer of ROS, is a potential pathogenic factor in human neurodegenerative diseases. Thus, it is important to find effective interventions for Cd‐induced oxidative stress in the CNS. Here, we have studied the effects of celastrol, a plant‐derived triterpene, on ROS production and cell death in neuronal cells, induced by Cd.
Experimental Approach
PC12, SH‐SY5Y cells and primary murine neurons were used to study celastrol neuroprotection against Cd‐poisoning. Cd‐induced changes in cell viability, apoptosis, ROS and AMP‐activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) pathway in the cells were analysed by Trypan blue exclusion, DAPI and TUNEL staining, ROS imaging, immunofluorescence staining and Western blotting. Pharmacological and genetic approaches were employed to investigate the mechanisms underlying Cd neurotoxicity.
Results
Celastrol attenuated Cd‐induced apoptosis by suppressing Cd activation of mTOR, which was attributed to preventing Cd inactivation of AMPK. Inhibition of AMPK with compound C or expression of dominant negative AMPKα prevented celastrol from hindering Cd‐induced dephosphorylation of AMPKα, activation of mTOR and apoptosis. Inhibition of mTOR with rapamycin or knockdown of mTOR potentiated prevention by celastrol, of Cd‐induced phosphorylation of p70 S6 kinase 1/eukaryotic initiation factor 4E binding protein 1 and apoptosis. Celastrol attenuated Cd‐induced cell death by suppressing induction of mitochondrial ROS.
Conclusions and Implications
Celastrol prevented the inactivation of AMPK by mitochondrial ROS, thus attenuating Cd‐induced mTOR activation and neuronal apoptosis. Celastrol may be a promising agent for prevention of Cd‐induced oxidative stress and neurodegenerative diseases.
Abbreviations
- 4E‐BP1
eukaryotic initiation factor 4E binding protein 1
- ACC
acetyl‐CoA carboxylase
- AD
Alzheimer's disease
- AICAR
5‐amino‐4‐imidazolecarboxamide ribose
- AMPK
AMP‐activated protein kinase
- CM‐H2DCFDA
5‐(and‐6)‐chloromethyl‐2′,7′‐dichlorodihydrofluorescein diacetate
- HD
Huntington's disease
- mTOR
mammalian target of rapamycin
- NAC
N‐acetyl‐L‐cysteine
- PD
Parkinson's disease
- PDL
poly‐D‐lysine
- S6K1
p70 S6 kinase 1
- TTFA
thenoyltrifluoroacetone
- TUNEL
the terminal deoxynucleotidyl transferase (TdT)‐mediated deoxyuridine triphosphate (dUTP) nick‐end labelling
Tables of Links
| TARGETS |
|---|
| Enzymes |
| Acetyl CoA carboxylase |
| AMPK, AMP kinase |
| Caspase‐3 |
| mTOR |
| PARP |
| LIGANDS |
|---|
| Compound C |
| Cadmium |
| NADPH |
| Rapamycin |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Cadmium (Cd), a well‐known heavy metal pollutant, easily traverses the blood–brain barrier and accumulates in the brain (Wang and Du, 2013). A number of studies have shown that Cd exposure results in elevated levels of ROS, thus causing apoptotic cell death in distinct brain regions of the CNS (Figueiredo‐Pereira et al. 1998; Bertin and Averbeck, 2006; Genovese and Cuzzocrea, 2008; Goncalves et al., 2010; Wang and Du, 2013; Chen et al., 2014b). Cd induction of ROS has been documented in the mitochondria of PC12 cells, anterior pituitary cells, cortical neurons and brain (Wang et al., 2004; Lopez et al., 2006; Xu et al., 2016). Excessive ROS induced by Cd can directly disrupt the structure of cell proteins, carbohydrates, nucleic acids and lipids alter their functions, and also activate or inhibit related signalling pathways, thereby leading to neuronal cell dysfunction/apoptosis or neurodegeneration (Figueiredo‐Pereira et al. 1998; Bertin and Averbeck, 2006; Genovese and Cuzzocrea, 2008; Wang and Du, 2013). Therefore, Cd induction of ROS is considered as a possible pathogenic factor in the development of neuronal cell death and, consequently, in neurodegenerative diseases, including Parkinson's desease (PD), Alzheimer's disease (AD) and Huntington's disease (HD) (Monroe and Halvorsen, 2006; Goncalves et al., 2010; Jomova and Valko, 2011; Wang and Du, 2013).
The mammalian target of rapamycin (mTOR), a serine/threonine (Ser/Thr) kinase, stands at a central position and the crossroad of various signal pathways (Laplante and Sabatini, 2012; Cornu et al., 2013). Through phosphorylating its downstream ribosomal p70 S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4E‐BP1), mTOR acts as a master kinase controlling cell growth/proliferation and survival (Laplante and Sabatini, 2012; Cornu et al., 2013). For neurons, mTOR could be activated or inhibited depending on the pathological status of neurons or CNS, for example brain tumours, tuberous sclerosis, cortical displasia and neurodegenerative disorders such as PD, AD and HD (Swiech et al., 2008; Laplante and Sabatini, 2012). The AMP‐activated protein kinase (AMPK), a sensor of cellular energy status, is activated under conditions of energy stress (Hardie et al., 2016). It is well known that AMPK is a negative regulator of mTOR pathway (Inoki et al., 2003; Gwinn et al., 2008). Some studies have revealed that AMPK activation due to oxidative stress is involved in neurodegenerative diseases (Vingtdeux et al., 2011; Jiang et al., 2013). Consistent with this idea, our group has demonstrated that hydrogen peroxide (H2O2), a major component of ROS, inhibits mTOR signalling by activation of AMPK, leading to apoptosis of neuronal cells (Chen et al., 2010). However, we have also noticed that Cd‐induced ROS can activate mTOR signalling, contributing to neuronal cell death (Xu et al., 2011; Chen et al., 2011b; Chen et al., 2011c; Chen et al., 2014b). Our results indicate that Cd activates the mTOR pathway by inhibiting AMPK, due to induction of ROS (Chen et al., 2011b). Hence, we proposed that a compound that could prevent Cd‐induced ROS from inactivating AMPK or activating the mTOR pathway might be useful to prevent the neurotoxicity of Cd.
Celastrol, a pentacyclic triterpene extracted from the roots of Tripterygium wilfordi [thunder god vine (TGV)] plant, is known to possess a wide variety of biological effects, including antioxidant, anti‐apoptotic, anti‐inflammatory, anti‐carcinogenic and anti‐obesity properties (Salminen et al., 2010; Kannaiyan et al., 2011a; Li et al., 2015; Liu et al., 2015). Recent studies have shown that celastrol induces AMPK activation and suppresses mTOR activation leading to apoptosis in gastric cancer (Lee et al., 2014). Celastrol inhibits growth and potentiates apoptosis in a variety of tumour cells by inducing ROS or ROS‐activated AMPK, and/or inhibiting the mTOR signalling pathway (Pang et al., 2010; Chen et al., 2011a; Kannaiyan et al., 2011b; Kim et al., 2013; Ma et al., 2014; Li et al., 2015; Sha et al., 2015). Of importance, celastrol has been identified as a neuroprotective agent in several different models of neurodegenerative diseases that are characterized by an overproduction of ROS, such as PD, AD and amyotrophic lateral sclerosis (ALS) (Allison et al., 2001; Cleren et al., 2005; Kiaei et al., 2005; Choi et al., 2014; Zhao et al., 2014). However, the underlying protective mechanism of celastrol remains to be elucidated. Recently, we have found that pretreatment with celastrol inhibits activation of the mTOR pathway by Cd and protects neuronal cells from Cd‐poisoning (Chen et al., 2014a). In the present study, we have further shown that this protection could be attributed to celastrol preventing mitochondrial ROS from inactivating AMPK. The results enhance our understanding of the molecular mechanism by which celastrol protects against Cd‐induced oxidative stress and neurodegenerative diseases.
Methods
Cell culture
All animal care and experimental procedures in this study were in compliance with the guidelines set by the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Rat pheochromocytoma (PC12) and human neuroblastoma SH‐SY5Y cell lines were from American Type Culture Collection (ATCC) (Manassas, VA, USA). Because of the replicative nature and cost‐effectiveness, these two cell lines are widely used as neuronal cell models, so they were employed in this study.
For culture, PC12 and SH‐SY5Y cells were seeded in a 6‐well plate (5 × 105 cells per well) or 96‐well plate (1 × 104 cells per well) pre‐coated with (for PC12) or without (for SH‐SY5Y) PDL (0.2 μg·mL−1). PC12 cells were grown in antibiotic‐free DMEM supplemented with 10% horse serum and 5% FBS, whereas SH‐SY5Y cells were grown in antibiotic‐free DMEM supplemented with 10% FBS. Cells were maintained in a humidified incubator of 5% CO2 at 37°C. To confirm the data obtained from PC12 or SH‐SY5Y cells, primary neurons were also used in this study. For this, primary murine neurons were isolated from fetal mouse cerebral cortexes of 16–18 days of gestation in pregnant female ICR mice, as described by Chen et al. (2010), and seeded in a 6‐well plate (5 × 105 cells per well) or 96‐well plate (1 × 104 cells per well) coated with 10 μg·mL−1 PDL for experiments after 6 days of culture.
Recombinant adenoviral constructs and infection of cells
The recombinant adenoviruses expressing myc‐tagged constitutively active mutant of rat AMPKα1 (T172D) (Ad‐AMPKα‐ca) (Zang et al., 2004) (a gift from Dr. Kenneth Walsh, Boston University School of Medicine, Boston, MA, USA), hemagglutinin (HA)‐tagged dominant‐negative mutant of human AMPKα1 (D159A) (Ad‐dn‐AMPKα) (Lu et al., 2008) (a gift from Dr. Nicholas J.G. Webster, University of California, San Diego, CA, USA), and the control virus expressing the GFP (Ad‐GFP) or β‐galactosidase (Ad‐LacZ) were described previously (Chen et al., 2010; Liu et al., 2010). For experiments, PC12 cells were grown in the growth medium and infected with the individual adenovirus for 24 h at 5 of multiplicity of infection (MOI = 5). Subsequently, cells were used for experiments. Ad‐GFP or Ad‐LacZ served as a control. Expression of myc‐tagged AMPKα‐ca and HA‐tagged AMPKα‐dn was determined by Western blot analysis with antibodies to myc and HA respectively.
Lentiviral shRNA cloning, production and infection
Lentiviral shRNAs to mTOR and GFP (for control) were generated as described (Liu et al., 2006; Liu et al., 2008). For use, monolayer PC12 cells, when grown to about 70% confluence, were infected with medium containing these lentiviruses, in the presence of 8 μg·mL−1 polybrene for 12 h twice at an interval of 6 h. Uninfected cells were eliminated by exposure to 2 μg·mL−1 puromycin for 48 h before use. After 5 days of culture, cells were used for experiments.
Assays for live cell number and caspase‐3/7 activity
As treatment with 10–20 μM Cd for 24 h is able to induce significant apoptosis in neuronal cells (PC12, SH‐SY5Y and primary neurons) (Chen et al., 2008a; Gerspacher et al., 2009; Jiang et al., 2014), treatment with 0.1–1 μM of celastrol for 24 h is not toxic to SH‐SY5Y and PC12 cells (Deng et al., 2013; Chen et al., 2014a), and at 1 μM, celastrol exhibits a best protection against Cd‐reduced viability in the cells (Chen et al., 2014a), Cd (10 and 20 μM) and celastrol (1 μM) were the concentrations chosen for this research. PC12 cells, SH‐SY5Y cells and/or primary neurons were seeded in a PDL‐uncoated or ‐coated 6‐well plate (5 × 105 cells per well) or 96‐well plate (1 × 104 cells per well). The next day, the cells were exposed to Cd (10 and 20 μM) for 24 h following pre‐incubation with/without celastrol (1 μM) for 1 h, which with five replicates of each treatment. Subsequently, live cells were monitored by counting viable cells, using Trypan blue exclusion. Caspase‐3/7 activity was determined using Caspase‐Glo® 3/7 Assay Kit (Promega, Madison, WI, USA), following the instructions of the supplier.
DAPI and TUNEL staining
PC12 cells, SH‐SY5Y cells and primary neurons, or PC12 cells infected with lentiviral shRNA to mTOR or GFP, or PC12 cells infected with Ad‐AMPKα‐ca or Ad‐GFP, respectively, were seeded at a density of 5 × 105 cells per well in a 6‐well plate containing a PDL‐uncoated or ‐coated glass coverslip per well. The next day, cells were treated with/without Cd (10 and/or 20 μM) for 24 h following pre‐incubation with/without celastrol (1 μM) for 1 h with five replicates of each treatment. In some cases, cells were pretreated with/without an AMPK inhibitor compound C (20 μM) (Emerling et al., 2007), an AMPK activator AICAR (2 mM) (Chen et al., 2011b), an antioxidant and ROS scavenger N‐acetyl cysteine (NAC; 5 mM) (Chen et al., 2008a; Chen et al., 2011b), or a mitochondria‐targeted antioxidant Mito‐TEMPO (10 μM) (Yeh et al., 2014) for 1 h, or a specific mTOR inhibitor rapamycin (200 ng/mL) for 48 h (Chen et al., 2008b; Chen et al., 2011b), and then with/without celastrol (1 μM) for 1 h, followed by exposure to Cd (10 μM) for 24 h. The concentrations of these compounds used and the time of treatment were based on the previous publications, as cited above. Afterwards, the cells with fragmented and condensed nuclei were determined using DAPI staining as described (Chen et al., 2008b). For the cells pretreated with/without 1 μM of celastrol for 1 h and then exposed to 10 and 20 μM of Cd for 24 h, following DAPI staining, TUNEL staining was performed according to the manufacturer's protocols of In Situ Cell Death Detection Kit® (Roche, Mannheim, Germany). Finally, photographs were taken under a fluorescence microscope (Leica DMi8, Wetzlar, Germany) equipped with a digital camera. For quantitative analysis of the fluorescence intensity using TUNEL staining, the integral OD (IOD) was measured by Image‐Pro Plus 6.0 software (Media Cybernetics Inc., Newburyport, MA, USA).
Immunofluorescence and imaging
PC12 cells, SH‐SY5Y cells and primary neurons were seeded at a density of 5 × 105 cells/well in a 6‐well plate containing a PDL‐uncoated or ‐coated glass coverslip per well. The next day, the cells were pre‐incubated with/without celastrol (1 μM) for 1 h, and then exposed to Cd (10 and 20 μM) for 24 h. Then, the cells on the cover‐slips were fixed with 4% paraformaldehyde and incubated with 3% normal goat serum to block non‐specific binding. Next, the cells were incubated with rabbit anti‐phospho‐AMPKα (Thr172) antibody (Cell Signaling, Danvers, MA, USA, 1:50, diluted in PBS containing 1% BSA) overnight at 4°C, washed three times (5 min per time) with PBS, and further incubated with FITC‐conjugated goat anti‐rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA, 1:500, diluted in PBS containing 1% BSA) for 1 h at room temperature. The cells were then washed three times (5 min per time) with PBS. Finally, slides were mounted in glycerol/PBS (1:1, v/v) containing 2.5% 1,4‐diazabiclo‐(2,2,2)octane. Cell images were captured under a fluorescence microscope (Leica DMi8, Wetzlar, Germany) equipped with a digital camera. IOD for fluorescence intensity was quantitatively analysed by Image‐Pro Plus 6.0 software as described above.
Cell ROS imaging
Intracellular ROS level was evaluated using CM‐H2DCFDA (MP Biomedicals), which is a stable nonfluorescent molecule and can passively diffuse into cells, where it is oxidized by ROS, and becomes green fluorescent, according to the supplier's information. In brief, PC12 cells, SH‐SY5Y cells and/or primary neurons, or PC12 cells infected with Ad‐dn‐AMPKα, Ad‐AMPKα‐ca and Ad‐LacZ, respectively, or PC12 cells infected with lentiviral shRNAs to mTOR and GFP, respectively, were seeded in a 6‐well plate (5 × 105 cells per well) containing a glass coverslip per well. The next day, cells were pretreated with/without celastrol (1 μM) or Mito‐TEMPO (10 μM) for 1 h, followed by exposure to Cd (10 and/or 20 μM) for 24 h, or to Cd (10 μM) in the presence or absence of mitochondrial complex I inhibitor rotenone (0.5 μM) (Zhou et al., 2015), mitochondrial complex II ubiquinone site inhibitor TTFA (10 μM) (Moreno‐Sanchez et al., 2013) or mitochondrial complex III inhibitor antimycin A (50 μM) (Lanju et al., 2014) for 24 h, with five replicates of each treatment. In some cases, cells were pretreated with/without NAC (5 mM) or Mito‐TEMPO (10 μM) for 1 h and then with/without celastrol (1 μM) for 1 h, followed by exposure to Cd (10 μM) for 24 h. In other cases, cells were pretreated with/without compound C (20 μM) or 5‐amino‐4‐imidazolecarboxamide ribose (AICAR; 2 mM) for 1 h or rapamycin (200 ng·mL−1) for 48 h, and then celastrol (1 μM) or Mito‐TEMPO (10 μM) for 1 h, followed by exposure to Cd (10 μM) for 24 h. The concentrations of these compounds and the time of treatment used were based on the previous publications, as cited above. The cells were then loaded with CM‐H2DCFDA (10 μM) for 1 h. To image intracellular ROS, all stained specimens were rinsed three times with PBS, followed by photographing under a fluorescence microscope, and quantitatively analysing IOD of the fluorescence intensity by Image‐Pro Plus 6.0 software as described above.
Western blot analysis
The indicated cells, after treatments, were washed with cold PBS, and then on ice, lysed in the radioimmunoprecipitation assay buffer [50 mM Tris, pH 7.2; 150 mM NaCl; 1% sodium deoxycholate; 0.1% sodium dodecyl sulfate (SDS); 1% Triton X‐100; 10 mM NaF; 1 mM Na3VO4; protease inhibitor cocktail (1:1000)]. Lysates were sonicated for 10 s and centrifuged at 16 000 × g for 2 min at 4°C. The supernatants were collected, and then Western blotting was performed as described previously (Chen et al., 2010). In brief, lysates containing equivalent amounts of protein were separated on 7–12% SDS–polyacrylamide gel and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). Membranes were incubated with PBS containing 0.05% Tween 20 and 5% non‐fat dry milk to block non‐specific binding, and then with primary antibodies against phosphorylated AMPKα (p‐AMPKα) (Thr172), p‐acetyl‐CoA carboxylase (p‐ACC) (Ser79), ACC, p‐mTOR (Ser2448), p‐S6K1 (Thr389), p‐4E‐BP1 (Thr70), 4E‐BP1, cleaved‐caspase‐3, PARP, myc epitope (Cell Signaling Technology, Danvers, MA, USA), AMPKα, S6K1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), mTOR, β‐tubulin (Sigma) overnight at 4°C, respectively, followed by incubating with appropriate secondary antibodies including horseradish peroxidase‐coupled goat anti‐rabbit IgG, goat anti‐mouse IgG or rabbit anti‐goat IgG (Pierce, Rockford, IL, USA) overnight at 4°C. Immunoreactive bands were visualized by using enhanced chemiluminescence solution (Pierce). The blots for detected proteins were semi‐quantified using NIH Image J software (National Institutes of Health, Bethesda, MD, USA).
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data were expressed as mean values ± SEM. Student's t‐test for non‐paired replicates was used to identify statistically significant differences between treatment means. Group variability and interaction were compared using either one‐way or two‐way ANOVA followed by Bonferroni's post‐tests to compare replicate means. Significance was accepted at P < 0.05.
Materials
Cadmium chloride, celastrol, DAPI, poly‐D‐lysine (PDL), rotenone (mitochondrial complex I inhibitor), antimycin A (mitochondrial complex III inhibitor), thenoyltrifluoroacetone (TTFA) (mitochondrial complex II ubiquinone site inhibitor), N‐acetyl‐L‐cysteine (NAC) and protease inhibitor cocktail were purchased from Sigma (St Louis, MO, USA), whereas compound C (AMPK inhibitor) was provided by Calbiochem (San Diego, CA, USA). 5‐(and‐6)‐chloromethyl‐2′,7′‐dichlorodihydrofluorescein diacetate (CM‐H2DCFDA) was purchased from MP Biomedicals (Solon, OH, USA). Mito‐TEMPO (mitochondria‐targeted antioxidant) and rapamycin (mTOR inhibitor) were acquired from ALEXIS Biochemicals Corporation (San Diego, CA, USA). 5‐amino‐4‐imidazolecarboxamide ribose (AICAR) (AMPK activator) was from Enzo Life Sciences (Farmingdale, NY, USA). Enhanced chemiluminescence solution was from Millipore (Billerica, MA, USA). DMEM, 0.05% Trypsin–EDTA, NEUROBASALTM Media and B27 Supplement were purchased from Invitrogen (Grand Island, NY, USA). Horse serum and FBS were supplied by Hyclone (Logan, UT, USA). Other chemicals were purchased from local commercial sources and were of analytical grade.
Results
Celastrol prevents Cd‐induced inactivation of AMPKα and apoptosis in neuronal cells
Our recent study has shown that treatment with 0.1–1 μM of celastrol for 24 h is not toxic to PC12 cells, and at 1 μM, celastrol exhibits the most effective protection against Cd‐reduced viability in the cells (Chen et al., 2014a). Here, we also observed that pretreatment with celastrol (1 μM) for 1 h ameliorated the cell death induced by 24‐h exposure to Cd (10 and 20 μM), as determined by Trypan blue exclusion in PC12 and SH‐SY5Y cells (Figure 1A). In addition, we investigated the cells with nuclear fragmentation and condensation, a hallmark of apoptosis (Hao et al., 2013), using DAPI staining, and concurrently analysed DNA strand breaks in the cells by TUNEL staining (Figure 1B). Imaged and quantified results revealed that celastrol significantly decreased the percentage of the cells with nuclear fragmentation and condensation (arrows) and the number of TUNEL‐positive cells with fragmented DNA (in green) in PC12 cells, SH‐SY5Y cells and primary neurons induced by Cd exposure, compared with the control (Figure 1B–D).
Figure 1.
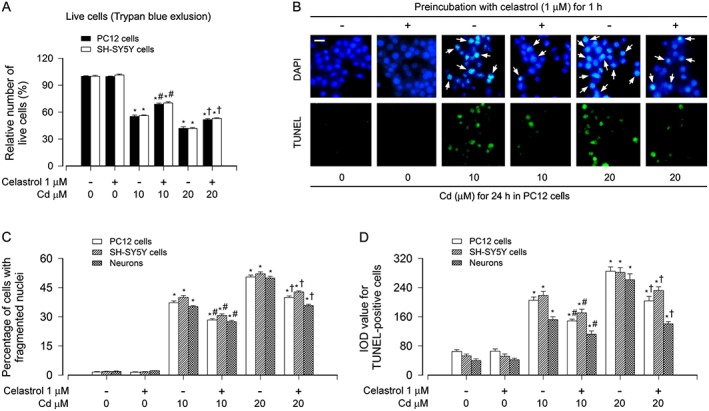
Celastrol attenuated Cd‐induced apoptosis in neuronal cells. PC12 cells, SH‐SY5Y cells and primary neurons were exposed to Cd (10 or 20 μM) for 24 h post pretreatment with celastrol (1 μM) for 1 h. A Live cells were detected by counting viable cells using Trypan blue exclusion. B Apoptotic cells were evaluated by nuclear fragmentation and condensation (arrows) using DAPI staining (upper panel) and concurrently by in situ detection of fragmented DNA (in green) using TUNEL staining (lower panel). Scale bar: 20 μm. C and D The percentages of cells with fragmented nuclei and the number of TUNEL‐positive cells were quantified. For A, C and D, all data were expressed as mean ± SE (n = 5). Using one‐way ANOVA, * P < 0.05, significantly different from control group; # P < 0.05, significantly different from 10 μM Cd group; † P < 0.05, significantly different from 20 μM Cd group.
Furthermore, our Western blot analysis also showed that celastrol significantly decreased Cd‐elicited robust cleavage of caspase‐3 in PC12, SH‐SY5Y cells and primary neurons (Figure 2A and B). Interestingly, we found that celastrol markedly prevented Cd‐reduced phosphorylation of AMPKα (Thr172) and its substrate ACC (Ser79) (Figure 2A and B), indicating that celastrol inhibits Cd‐inactivation of AMPKα, which may prevent Cd‐induced apoptotic cell death. To confirm this, we further conducted phospho‐AMPKα (p‐AMPKα) (Thr172) immunofluorescence staining and caspase3/7 activity assay in PC12, SH‐SY5Y cells and primary neurons respectively. Treatment with Cd (10 and 20 μM) for 24 h resulted in a drastic decrease in phosphorylation of AMPKα (Thr172) (in green), which was significantly attenuated by celastrol pretreatment (Figure 2C and D). In line with the inhibitory effect of celastrol on Cd‐increased cleaved‐caspase‐3, celastrol profoundly decreased Cd‐induced activation of caspases 3/7 in the cells (Figure 2E). These results demonstrate that celastrol prevents Cd‐induced inactivation of AMPKα and apoptotic cell death in neuronal cells.
Figure 2.
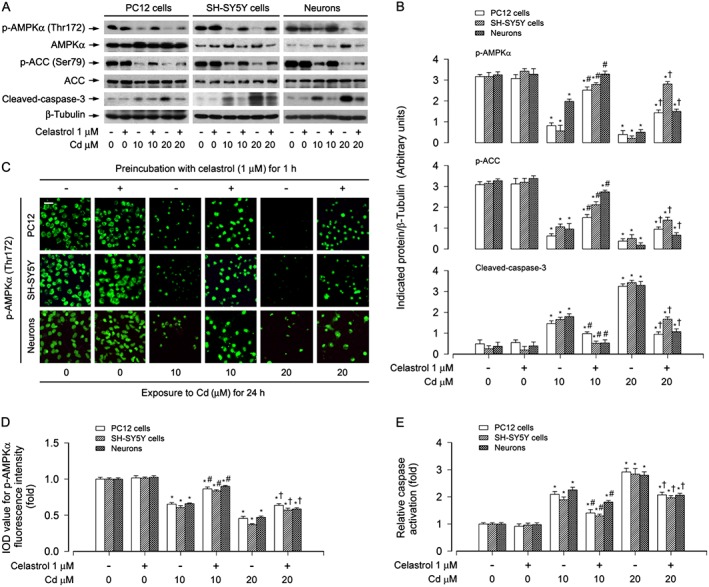
Celastrol attenuated Cd‐induced inactivation of AMPK in neuronal cells. PC12 cells, SH‐SY5Y cells and primary neurons were pretreated with celastrol (1 μM) for 1 h, and then exposed to Cd (10 and 20 μM) for 4 h (for Western blotting) or 24 h (for immunofluorescence staining and caspase‐3/7 activity assay). A Total cell lysates were performed for Western blotting using indicated antibodies. The blots were probed for β‐tubulin as a loading control. Similar results were observed in at least three independent experiments. B The blots for p‐AMPKα, p‐ACC and cleaved‐caspase‐3 were semi‐quantified. C and D Expression of p‐AMPKα (Thr172) was stained and imaged using immunofluorescence, showing that treatment of the cells with Cd for 24 h caused lower p‐AMPKα expression (in green), which was markedly increased by celastrol. Scale bar: 20 μm. E Caspase‐3/7 activities were detected using Caspase‐3/7 Assay Kit, showing that celastrol substantially inhibited Cd activation of caspases 3/7 in the cells. All data were expressed as mean ± SE (n = 3–5). Using one‐way ANOVA, *P < 0.05, significantly different from control group; # P < 0.05, significantly different from 10 μM Cd group; † P < 0.05, significantly different from 20 μM Cd group.
Celastrol attenuates Cd activation of mTOR by preventing Cd inactivation of AMPKα
To test whether celastrol inhibition of Cd‐induced apoptosis is attributable to its prevention of mTOR activation, PC12 cells and primary neurons were pre‐incubated with/without rapamycin (a specific mTOR inhibitor) alone, or in combination with celastrol. We found that rapamycin (200 ng·mL−1) or celastrol (1 μM) alone clearly suppressed the phosphorylation of mTOR (Ser2448), S6K1 (Thr389) and 4E‐BP1 (Thr70), as well as the cleavage of caspase‐3 in the cells induced by Cd exposure (Figure 3A and B). Especially, co‐treatment with celastrol/rapamycin exhibited a stronger inhibitory effect on Cd‐induced p‐mTOR, p‐S6K1, p‐4E‐BP1 and cleaved‐caspase‐3 (Figure 3A and B). Consistent with this finding, the combination of celastrol with rapamycin also exhibited more potent inhibition of Cd‐elicited apoptosis than celastrol or rapamycin alone, as shown by the proportion of cells with fragmented nuclei (Figure 3C).
Figure 3.
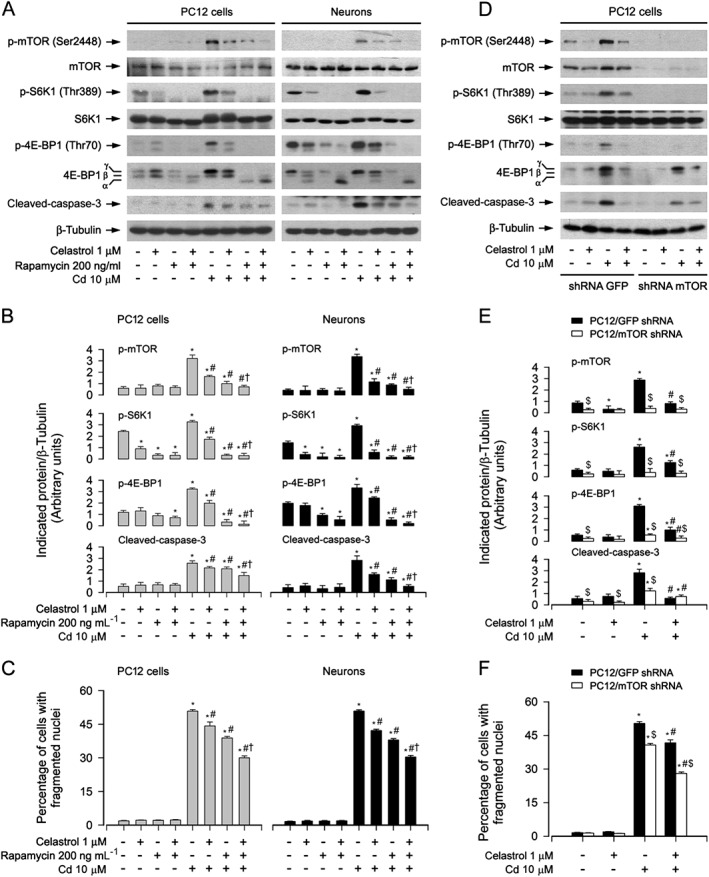
Inhibition of mTOR potentiated celastrol prevention of Cd‐induced apoptosis. PC12 cells and primary neurons, or PC12 cells infected with lentiviral shRNA to mTOR or GFP (as control), respectively, were pretreated with/without rapamycin (200 ng·mL−1) for 48 h and then celastrol (1 μM) for 1 h, or pretreated with/without celastrol for 1 h, followed by exposure to Cd (10 μM) for 4 h (for Western blotting) or 24 h (for cell apoptosis analysis). A and D Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β‐tubulin as a loading control. Similar results were observed in at least three independent experiments. (B and E) The blots for p‐mTOR, p‐S6K1, p‐4E‐BP1 and cleaved‐caspase‐3 were semi‐quantified. C and F Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. All data were expressed as mean ± SE (n = 3–5). Using one‐way or two‐way ANOVA or Student's t‐test, *P < 0.05, significantly different from control group; # P < 0.05, significantly different from 10 μM Cd group; † P < 0.05, significantly different from Cd+Celastrol group or Cd+Rapamycin group; $ P < 0.05, mTOR shRNA group significantly different from GFP shRNA group.
In agreement with the above findings, down‐regulation of mTOR using lentiviral shRNA to mTOR clearly prevented Cd‐induced p‐S6K1, p‐4E‐BP1, cleavage of caspase‐3 and cell apoptosis, and potentiated the inhibitory effect of celastrol in PC12 cells (Figure 3D–F). Taken together, our results support the concept that celastrol decreases Cd activation of mTOR‐dependent neuronal apoptosis.
Next, we determined whether celastrol inhibits Cd activation of mTOR‐dependent neuronal apoptosis by preventing Cd inactivation of AMPKα. For this, PC12 cells and primary neurons were pretreated with/without the AMPK inhibitor compound C (20 μM) or the AMPK activator AICAR (2 mM) for 1 h and then with/without celastrol (1 μM) for 1 h, followed by exposure to Cd (10 μM) for 4 h or 24 h. As demonstrated in Figure 4A and B, Cd markedly induced the decline of p‐AMPKα and p‐ACC, and the elevation of p‐mTOR, p‐S6K1, p‐4E‐BP1 and cleaved‐caspase‐3 in the absence or presence of compound C. The effects appeared more potent in the cells co‐treated with Cd and compound C than in those treated with Cd or compound C alone (Figure 4A and B). Compound C alone profoundly affected the basal levels of p‐AMPKα, p‐ACC, p‐mTOR, p‐S6K1, p‐4E‐BP1 and cleaved‐caspase‐3 in the absence of celastrol and particularly hindered the inhibitory effect of celastrol on Cd‐triggered events (Figure 4A and B). We also observed that compound C alone strengthened the basal or Cd‐induced apoptosis (Figure 4C). Of note, compound C conferred high resistance to celastrol's inhibition of Cd‐evoked apoptosis in the cells (Figure 4C). On the contrary, AICAR alone caused partial restoration of decreased p‐AMPKα and p‐ACC with a concomitant repression of elevated p‐mTOR, p‐S6K1 and p‐4E‐BP1 in the cells in response to Cd (Figure 4D and E). Interestingly, AICAR powerfully reinforced the inhibitory effect of celastrol on Cd‐elicited events (Figure 4D and E). Also, co‐treatment with celastrol and AICAR protected cells from Cd‐induced cleaved‐caspase‐3 and apoptosis more potently than celastrol or AICAR alone (Figure 4D–F). These results support the idea that celastrol inhibits Cd‐induced mTOR activation and apoptosis in part by activating AMPK in neuronal cells.
Figure 4.
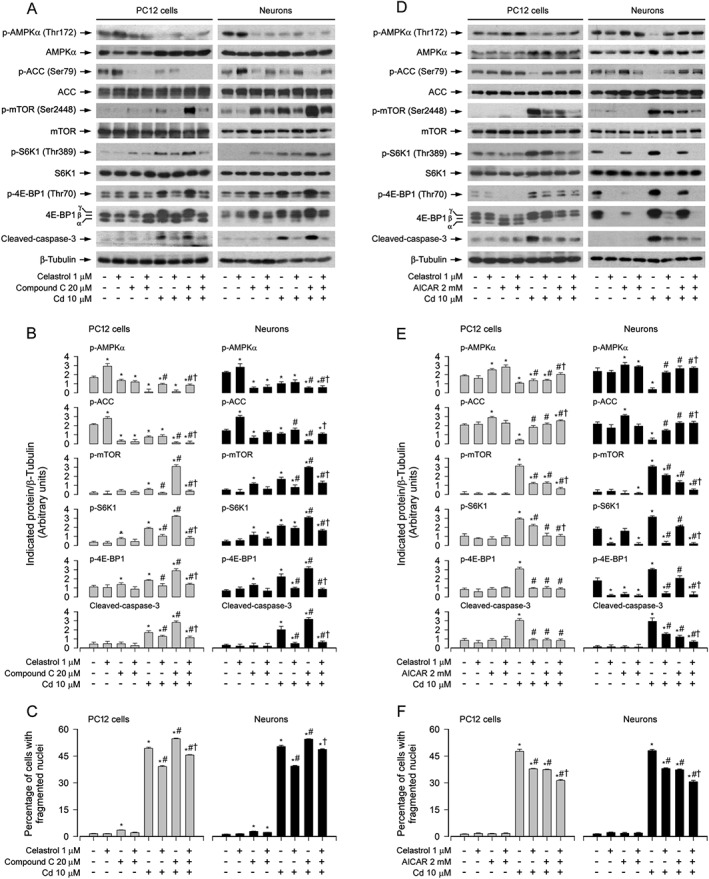
Celastrol prevented Cd activation of mTOR pathway and apoptosis by suppressing Cd inactivation of AMPK. PC12 cells and primary neurons were pretreated with/without compound C (20 μM) or AICAR (2 mM) for 1 h and then celastrol (1 μM) for 1 h, followed by exposure to Cd (10 μM) for 4 h (for Western blotting) or 24 h (for cell apoptosis analysis). A and D Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β‐tubulin as a loading control. Similar results were observed in at least three independent experiments. B and E The blots for p‐AMPKα, p‐ACC, p‐mTOR, p‐S6K1, p‐4E‐BP1 and cleaved‐caspase‐3 were semi‐quantified. C and F Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. All data were expressed as mean ± SE (n = 3–5). Using one‐way or two‐way ANOVA, * P < 0.05, significantly different from control group; # P < 0.05, significantly different from 10 μM Cd group; † P < 0.05, † Cd+Celastrol group, Cd+Compound C group or Cd+AICAR group.
To confirm the above findings, PC12 cells, infected with recombinant adenoviruses expressing HA‐tagged dominant negative AMPKα1 (Ad‐dn‐AMPKα), myc‐tagged continuous‐active AMPKα (Ad‐AMPKα‐ca) and control virus encoding GFP alone (Ad‐GFP), respectively, were exposed to Cd (10 μM) for 4 or 24 h following pretreatment with/without celastrol (1 μM) for 1 h. As expected, a high level of HA‐tagged dn‐AMPKα or myc‐tagged AMPKα‐ca was seen in Ad‐dn‐AMPKα‐ or Ad‐AMPKα‐ca‐infected cells, but not in Ad‐GFP‐infected cells (control) (Figure 5A and D). Ectopic expression of dn‐AMPKα or AMPKα‐ca strongly reduced or raised the AMPK activity, because the basal or Cd‐inhibited p‐AMPK and p‐ACC was substantially down‐regulated (Figure 5A and B) or up‐regulated (Figure 5D and E), as shown by Western blotting. Of importance, expression of dn‐AMPKα markedly enhanced the basal or Cd‐increased p‐mTOR, p‐S6K1, p‐4E‐BP1, cleaved‐caspase‐3 and cell apoptosis and displayed marked resistance to celastrol inhibition of Cd‐induced events (Figure 5A–C). However, in contrast, expression of AMPKα‐ca profoundly inhibited Cd‐induced p‐mTOR, p‐S6K1, p‐4E‐BP1, cleaved‐caspase‐3 and cell apoptosis, which was potentiated by celastrol (Figure 5D–E). Taken together, our data demonstrate the importance of celastrol's prevention of Cd‐inactivated AMPKα in suppressing Cd activation of mTOR and neuronal apoptosis.
Figure 5.
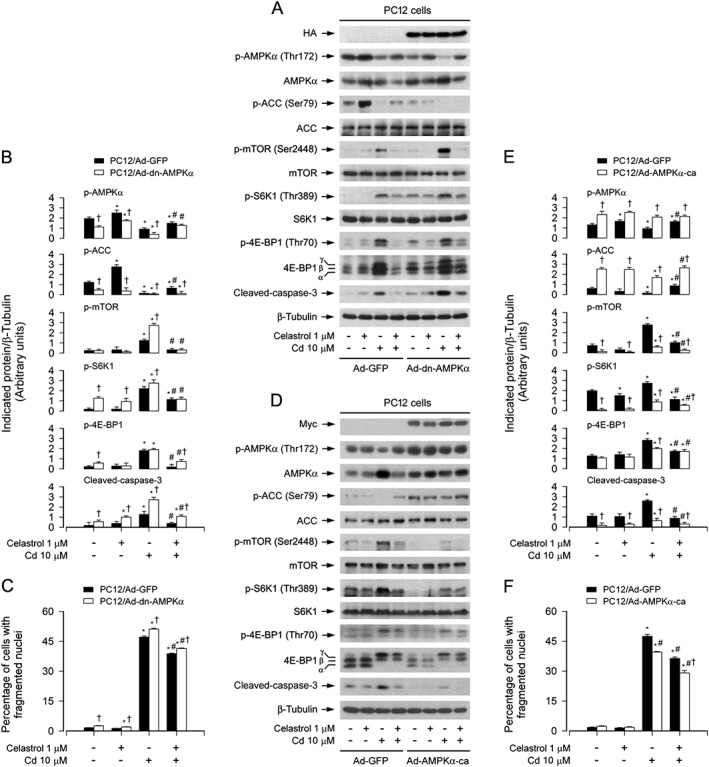
Ectopic expression of dominant negative AMPKα enhanced celastrol preventing Cd activation of mTOR pathway and apoptosis. PC12 cells, infected with Ad‐dn‐AMPKα, Ad‐AMPKα‐ca or Ad‐GFP (as control), respectively, were pretreated with/without celastrol (1 μM) for 1 h, followed by exposure to Cd (10 μM) for 4 h (for Western blotting) or 24 h (for cell apoptosis analysis). A and D Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β‐tubulin as a loading control. Similar results were observed in at least three independent experiments. (B and E) The blots for p‐AMPKα, p‐ACC, p‐mTOR, p‐S6K1, p‐4E‐BP1 and cleaved‐caspase‐3 were semi‐quantified. C and F Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. All data were expressed as mean ± SE (n = 3–5). Using one‐way ANOVA or Student's t‐test, * P < 0.05, significantly different from control group; # P < 0.05, significantly different from 10 μM Cd group; † P < 0.05, significantly different from Ad‐dn‐AMPKα group or Ad‐AMPKα‐ca group significantly different from Ad‐GFP group.
Celastrol attenuates Cd‐induced apoptosis by suppressing induction of mitochondrial ROS
To investigate whether celastrol prevents Cd induction of ROS‐mediated neuronal apoptosis, ROS assay was performed. We observed that pretreatment with celastrol (1 μM) for 1 h significantly attenuated the intracellular ROS production in PC12, SH‐SY5Y cells and primary neurons induced by 24 h exposure to Cd (Figure S1A and B), as measured by imaging and quantifying using an oxidant‐sensitive probe, CM‐H2DCFDA. This was consistent with the findings of decreased live cells and increased apoptosis observed by counting viable cells as well as by DAPI and TUNEL staining (Figure 1) respectively. Pretreatment with NAC, an antioxidant and ROS scavenger, clearly inhibited Cd‐induced ROS levels (Figure 6A) and markedly strengthened celastrol's prevention of Cd‐evoked ROS in PC12, SH‐SY5Y cells and primary neurons (Figure 6A). Of note, co‐treatment with celastrol and NAC reduced the percentage of cells with fragmented nuclei more potently than treatment with NAC or celastrol alone in the cells (Figure 6B). NAC also diminished cleavages of caspase‐3 and PARP in the cells in response to Cd, and substantially enhanced the inhibitory effect of celastrol (Figure 6C and D). These findings indicate that celastrol inhibits ROS‐dependent cell death triggered by Cd.
Figure 6.
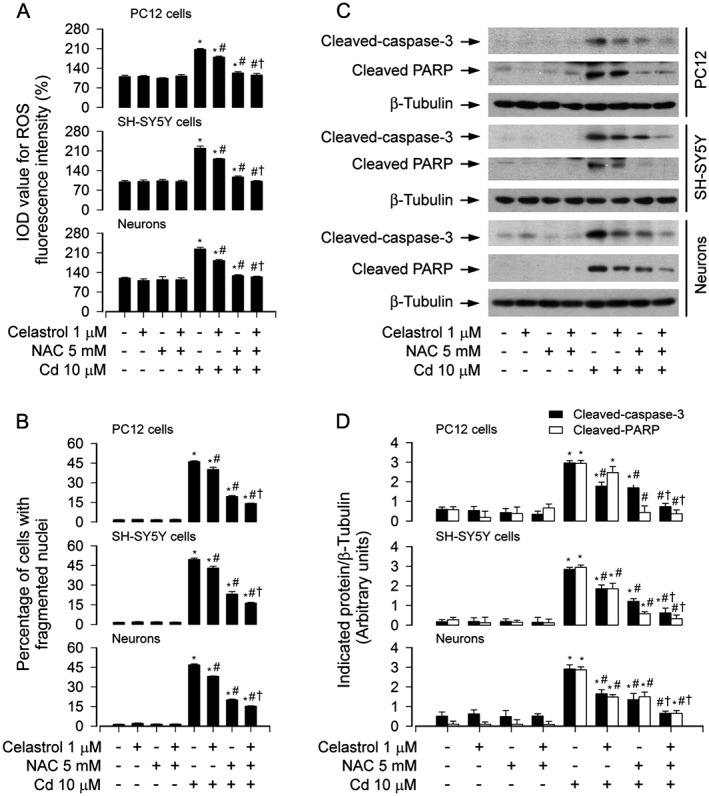
NAC potentiated celastrol's protection against Cd‐induced apoptosis. PC12 cells, SH‐SY5Y cells and primary neurons were pretreated with/without NAC (5 mM) for 1 h and then celastrol (1 μM) for 1 h, followed by exposure to Cd (10 μM) for 4 h (for Western blotting) or 24 h (for cell ROS imaging and apoptosis analysis). A Cell ROS was imaged and quantified using an oxidant‐sensitive probe CM‐H2DCFDA. B Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. C Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β‐tubulin as a loading control. Similar results were observed in at least three independent experiments. D The blots for cleaved‐caspase‐3 and cleaved‐PARP were semi‐quantified. All data were expressed as mean ± SE (n = 3–5). Using one‐way or two‐way ANOVA, * P < 0.05, significantly different from control group; # P < 0.05, significantly different from 10 μM Cd group; † P < 0.05, significantly different from Cd+Celastrol group or Cd+NAC group.
We next sought to validate whether celastrol prevention against Cd‐induced neuronal apoptosis is dependent on mitochondrial ROS induction. To answer this question, PC12 cells, SH‐SY5Y cells and primary neurons were pretreated with/without celastrol (1 μM) for 1 h, followed by exposure to Cd (10 μM) in the presence or absence of rotenone (0.5 μM), a mitochondrial complex I inhibitor, known to stimulate ROS production (Moreno‐Sanchez et al., 2013), TTFA (10 μM), a mitochondrial complex II ubiquinone site inhibitor with blockade of electron supply to ubiquinol and consequential limiting the formation of ubisemiquinone (Moreno‐Sanchez et al., 2013), or antimycin A (50 μM), a mitochondrial complex III inhibitor that increases the lifetime of ubisemiquinone (Lanju et al., 2014), for 24 h. We showed that co‐treatment with celastrol and TTFA relieved Cd‐induced ROS fluorescence more potently than treatment with celastrol or TTFA alone in the cells (Figure S2A). In contrast, treatment with rotenone or antimycin A alone increased levels of ROS and there was a further increase in ROS levels after exposure to Cd in the presence of rotenone or antimycin A, which was markedly inhibited by celastrol pretreatment (Figure S2B and C). Of importance, pretreatment with Mito‐TEMPO (10 μM), a mitochondria‐targeted antioxidant (Yeh et al., 2014), markedly repressed Cd‐induced ROS levels (Figure 7A) and cell apoptosis (Figure 7B), and dramatically strengthened the inhibitory effects of celastrol on Cd‐induced events in PC12 cells and primary neurons (Figure 7A and B). In addition, we noticed that Mito‐TEMPO reversed the phosphorylated status of AMPKα, ACC, mTOR, S6K1 and 4E‐BP1, as well as cleavage of caspase‐3 in the cells, in response to Cd (Figure 7C and D). Especially, co‐treatment with celastrol and Mito‐TEMPO exhibited a stronger intervention for Cd‐evoked events in the cells (Figure 7C and D). Taken together, these data confirmed that celastrol attenuated Cd‐induced mitochondrial ROS, thereby preventing inactivation of AMPKα and consequent activation of the mTOR signalling pathway, and, finally, decreasing apoptosis in neuronal cells.
Figure 7.
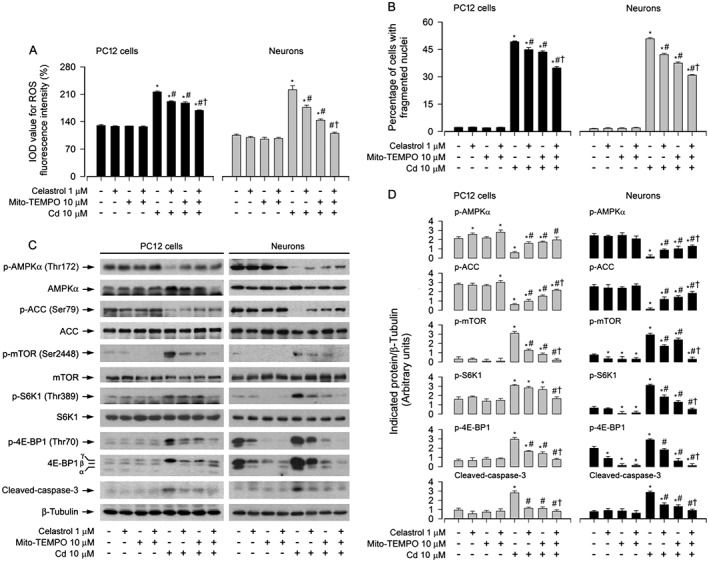
Celastrol prevented Cd‐induced mitochondrial ROS‐dependent AMPK inactivation, mTOR activation and apoptosis. PC12 cells and primary neurons were pretreated with/without Mito‐TEMPO (10 μM) for 1 h, celastrol (1 μM) for 1 h and then exposed to Cd (10 μM) for 4 h (for Western blotting) or 24 h (for cell ROS imaging and apoptosis analysis). A Cell ROS was imaged and quantified using an oxidant‐sensitive probe CM‐H2DCFDA. B Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. C Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β‐tubulin as a loading control. Similar results were observed in at least three independent experiments. D The blots for p‐AMPKα, p‐ACC, p‐mTOR, p‐S6K1, p‐4E‐BP1 and cleaved‐caspase‐3 were semi‐quantified. All data were expressed as mean ± SE (n = 3–5). Using one‐way or two‐way ANOVA, * P < 0.05, significantly different from control group; # P < 0.05, significantly different from 10 μM Cd group; † P < 0.05, significantly different from Cd+Celastrol group or Cd+Mito‐TEMPO group.
Modulation of AMPK/mTOR activity interferes with the inhibitory effect of celastrol on Cd‐induced mitochondrial ROS
To dissect whether AMPKα and mTOR positively mediates Cd‐induced mitochondrial ROS and the role of celastrol in preventing Cd‐induced mitochondrial ROS from inactivation of AMPKα and activation of mTOR, PC12 cells and primary neurons were pretreated with/without rapamycin (200 ng·mL−1) for 48 h, or compound C (20 μM) or AICAR (2 mM) for 1 h, and then with/without celastrol (1 μM) or Mito‐TEMPO (10 μM) for 1 h, followed by exposure to Cd (10 μM) for 24 h. We showed that treatment with compound C alone greatly potentiated Cd‐induced ROS generation (Figure 8A and B), whereas treatment with AICAR or rapamycin alone significantly decreased Cd‐induced ROS generation (Figure 8A and B), consistent with our previous findings (Chen et al., 2011b). Of importance, there were more inhibitory effects on Cd‐induced ROS in the cells co‐treated with celastrol and AICAR or celastrol and rapamycin, or with Mito‐TEMPO and AICAR or Mito‐TEMPO and rapamycin, than in the cells treated with celastrol, Mito‐TEMPO, AICAR or rapamycin alone (Figure 8A and B). However, pretreatment with compound C conferred resistance to celastrol's or Mito‐TEMPO's intervention in Cd‐induced ROS (Figure 8A and B).
Figure 8.
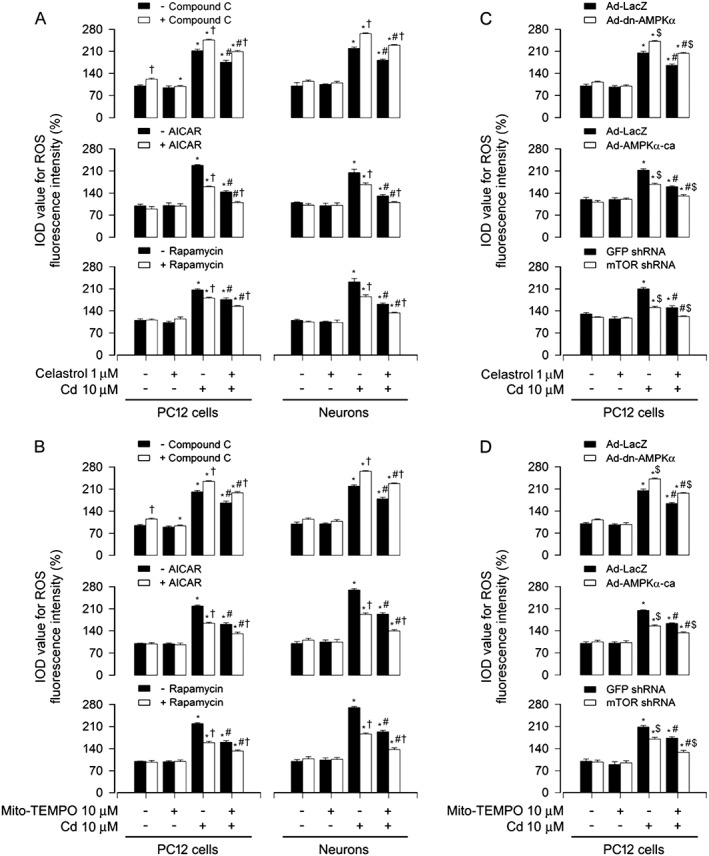
Modulation of AMPK/mTOR activity influenced celastrol's inhibition of Cd‐induced mitochondrial ROS. PC12 cells and primary neurons, or PC12 cells infected with Ad‐dn‐AMPKα, Ad‐AMPKα‐ca or Ad‐LacZ (as control) or with lentiviral shRNA to mTOR or GFP (as control), respectively, were pretreated with/without rapamycin (200 ng·mL−1) for 48 h or compound C (20 μM) or AICAR (2 mM) for 1 h, and then celastrol (1 μM) or Mito‐TEMPO (10 μM) for 1 h, or pretreated with/without celastrol (1 μM) or Mito‐TEMPO (10 μM) for 1 h, followed by exposure to Cd (10 μM) for 24 h. A‐D Cell ROS was imaged and quantified using an oxidant‐sensitive probe CM‐H2DCFDA. All data were expressed as mean ± SE (n = 5). Using one‐way ANOVA or Student's t‐test, * P < 0.05, significantly different from control group; # P < 0.05, significantly different from 10 μM Cd group; † P < 0.05, significant effect of Compound C, significant effect of AICAR or significant effect of rapamycin; $ P < 0.05, Ad‐dn‐AMPKα group or Ad‐AMPKα‐ca group significantly different from Ad‐LacZ group, or mTOR shRNA group significantly different from GFP shRNA group.
Next, PC12 cells were infected with Ad‐dn‐AMPKα, Ad‐AMPKα‐ca or Ad‐LacZ (as control), or with lentiviral shRNA to mTOR or GFP, and then exposed to Cd (10 μM) for 24 h post pre‐incubation with or without celastrol (1 μM) or Mito‐TEMPO (10 μM) for 1 h respectively. The results showed that expression of AMPKα‐ca or down‐regulation of mTOR partly prevented the cells from ROS overproduction caused by Cd, yet expression of dn‐AMPKα substantially enhanced ROS overproduction in the cells in response to Cd (Figure 8C and D). Notably, expression of AMPKα‐ca or silencing mTOR strengthened the inhibitory effects of celastrol or Mito‐TEMPO on Cd‐induced ROS in the cells, whereas expression of dn‐AMPKα inhibited the protection by celastrol or Mito‐TEMPO against Cd‐induced ROS (Figure 8C and D). On the basis of these data, we conclude that celastrol exerts a beneficial role in preventing Cd‐induced mitochondrial ROS from inactivation of AMPK and activation of mTOR, which, in turn, also positively mediates Cd‐induced mitochondrial ROS.
Discussion
Recent advances in redox biology have suggested Cd‐induced ROS as a potential pathogenic factor in human neurodegenerative diseases, such as PD, AD and HD (Monroe and Halvorsen, 2006; Goncalves et al., 2010; Jomova and Valko, 2011; Wang and Du, 2013). A series of studies from our group have shown that Cd induction of ROS activated the mTOR pathway contributing to neuronal cell death (Xu et al., 2011; Chen et al., 2011b; Chen et al., 2011c; Chen et al., 2014b), and that ROS‐mediated inhibition of AMPK was a critical step in this effect (Chen et al., 2011b). This prompted us to seek a compound, which could be effective in inhibiting Cd‐induced, ROS‐dependent, neuronal apoptosis. Celastrol, a potent anti‐inflammatory and antioxidant, has been reported to exert neuroprotective effects in different models of neurodegenerative diseases (such as PD, AD and ALS) (Allison et al., 2001; Cleren et al., 2005; Kiaei et al., 2005; Choi et al., 2014; Zhao et al., 2014). However, high concentrations of celastrol have detrimental effects on neuronal cells (Deng et al., 2013; Chen et al., 2014a). For example, treatment with 5 μM of celastrol reduced cell viability by about 50% in SH‐SY5Y cells (Deng et al., 2013). Celastrol, at high concentrations (>1.5 μM), reduced cell viability of PC12 cells in a dose‐dependent manner, with an IC50 of ~8 μM (Chen et al., 2014a). Therefore, much more attention should be paid to rational use of celastrol and its related preparations. Especially, detailed pharmacokinetics data of celastrol is in great demand to achieve the best clinical therapeutic efficacy and the least toxicity of celastrol.
Recently, our group has shown that treatment with 0.1–1 μM of celastrol for 24 h is not toxic to PC12 cells, and at 1 μM, celastrol exhibits the most effective protection against Cd‐reduced viability in the cells (Chen et al., 2014a). So, 1 μM of celastrol was used in the present study. We found that treatment with 1 μM of celastrol attenuated Cd‐induced apoptosis in PC12 cells, SH‐SY5Y cells and primary neurons, which was through preventing Cd inactivation of AMPK and thus inhibiting Cd activation of mTOR in the cells. Overproduction of ROS was also dramatically attenuated by celastrol treatment in Cd‐exposed neuronal cells. This is consistent with the finding that celastrol inhibits ROS generation and thus exerts its neuroprotective effect on rotenone‐exposed SH‐SY5Y cells (Deng et al., 2013). More interestingly, celastrol significantly attenuated Cd‐induced mitochondrial ROS, thereby preventing inactivation of AMPK, activation of mTOR, and apoptosis in neuronal cells. The results provide new evidence supporting a role of celastrol in combating the effects of Cd‐induced ROS associated with neurodegenerative disorders.
Given the potential therapeutic effects of celastrol, efforts have been made to study pharmacokinetic profiles of celastrol in animals and human, such as plasma celastrol manifestation in rats by oral administration or intravenous injection of celastrol (Zhang et al., 2012). Plasma levels of celastrol in male and female subjects after oral administration of celastrol tablets for more than 2 days have also been measured (Xu et al., 2007). Interestingly, when Sprague–Dawley rats were administered orally with 1000 μg·kg−1 of pure celastrol and TGV tablets (corresponding to 534 μg·kg−1 of celastrol), respectively, the bioavailability of celastrol increased significantly from 17% for pure celastrol to 94% for TGV tablets containing equivalent celastrol (Zhang et al., 2012). Following oral administration of TGV tablets, the maximum serum concentration (Cmax) of celastrol was 32 ± 8.4 μg·L−1 in female rats (Zhang et al., 2012). Celastrol's bioavailability in brain has not been reported, but the neuroprotective effects of celastrol on neurodegenerative diseases imply that celastrol could cross the blood brain barrier. Undoubtedly, more studies on the bioavailability and pharmacokinetics of celastrol are needed to facilitate the in vivo studies of celastrol for prevention of Cd neurotoxicity in animal models.
Our recent studies have revealed that celastrol inhibits Cd‐induced activation of mTOR pathway and neuronal apoptosis (Chen et al., 2014a). In this study, we observed that celastrol inhibited Cd‐induced decline of p‐AMPKα (Thr172) and cleavage of caspase‐3 in PC12 cells, SH‐SY5Y cells and primary neurons (Figure 2), implying that celastrol prevents Cd inactivation of AMPK, contributing to the protection of neuronal cells from apoptosis. We also confirmed that celastrol prevented Cd activation of mTOR‐dependent neuronal apoptosis, as inhibiting mTOR with rapamycin or silencing mTOR by RNA interference potentiated celastrol's inhibition of Cd‐induced phosphorylation of mTOR, S6K1 and 4E‐BP1 and apoptosis in neuronal cells (Figure 3). It is known that AMPK negatively regulates the mTOR pathway (Inoki et al., 2003; Gwinn et al., 2008), and Cd activates mTOR pathway by inhibiting AMPK (Chen et al., 2011b). Next, we asked whether celastrol inhibited Cd activation of mTOR‐dependent neuronal apoptosis by preventing Cd inactivation of AMPK. For this, pharmacological or genetic inhibition, or rescue experiments for AMPKα were carried out. We found that pharmacological inhibition of AMPKα with compound C or expression of dn‐AMPKα prevented celastrol from hindering Cd‐induced dephosphorylation of AMPK, phosphorylation of mTOR, S6K1 and 4E‐BP1, as well as cleaved‐caspase‐3 and cell apoptosis (Figure 4 and 5). In contrast, pharmacological activation of AMPKα with AICAR or ectopic expression of AMPKα‐ca significantly attenuated Cd‐induced p‐mTOR, p‐S6K1, p‐4E‐BP1, cleaved‐caspase‐3 and cell apoptosis, and strengthened the inhibitory effect of celastrol on Cd‐induced events in neuronal cells (Figure 4 and 5). Taken together, these observations support the proposition that celastrol counteracts Cd neurotoxicity by inhibiting Cd inactivation of AMPK, thereby preventing activation of mTOR pathway.
Abundant evidence has placed ROS at a critical position in the pathogenesis of various CNS injuries/disorders (Chen et al., 2010; Mates et al., 2010; Jomova and Valko, 2011). We have demonstrated that Cd triggers ROS‐dependent inactivation of AMPK leading to activation of mTOR pathway and apoptosis of neuronal cells (Chen et al., 2011b), whereas H2O2 inhibits mTOR signalling by activation of AMPK, leading to apoptosis of neuronal cells (Chen et al., 2010), suggesting that the main functional ingredient of ROS induced by Cd may not be H2O2 in neuronal cells. It is well known that ROS comprise oxygen radicals, including O2 ˉ•, hydroxyl (•OH), peroxyl (RO2•), alkoxyl (RO•) and certain non‐radicals that are oxidizing agents and/or are readily converted into radicals, such as hypochlorous acid (HOCl), ozone (O3), singlet oxygen (1O2) and H2O2 (Zhou et al., 2015). Currently, we do not know what kinds of ROS are induced by Cd. Further research is needed to address this issue.
Celastrol belongs to the triterpene family. The natural triterpenoids, such as quercetin, lupeol and naringenin, have a potentiality for attenuating Cd toxicity (Sunitha et al., 2001; Renugadevi and Prabu, 2010a; Renugadevi and Prabu, 2010b), and act as anti‐oxidative agents, increasing resistance to oxidative stress (Dumont et al., 2009). In the current study, we found that celastrol could also detoxify Cd in neuronal cells, which is consistent with the findings that celastrol possesses antioxidant property (Salminen et al., 2010; Jaquet et al., 2011). Therefore, in this study, we next investigated celastrol's effects on Cd‐induced ROS generation in neuronal cells. We found that when celastrol‐pretreated PC12 cells, SH‐SY5Y cells and primary neurons were exposed to Cd for 24 h, cellular ROS overproduction was significantly reduced compared to the vehicle‐pretreated cells (Figure S1). Of note, co‐treatment with celastrol and NAC resulted in more ROS reduction in Cd‐exposed cells (Figure 6A). NAC also substantially enhanced the inhibitory effect of celastrol on Cd‐induced cell death (Figure 6B–D). The findings highlight that celastrol may be exploited for the prevention of Cd‐induced ROS‐dependent neuronal apoptosis.
Maintaining the functional integrity of mitochondria is pivotal for neuronal cell survival and homeostasis (Karbowski and Neutzner, 2012). However, neuronal mitochondria are especially vulnerable to oxidative stress due to their role in energy supply and use, causing excess ROS to be formed in the mitochondria and reduction of mitochondrial biogenesis (Seo et al., 2010; Palacios et al., 2011; Lu et al., 2012), which, in turn, further promotes ROS induction and mitochondrial dysfunction (Palacios et al., 2011; Woo and Shadel, 2011; Karbowski and Neutzner, 2012; Chaturvedi and Flint Beal, 2013). Cd induces high ROS levels in the mitochondria of PC12 cells, anterior pituitary cells, cortical neurons and brain (Wang et al., 2004; Lopez et al., 2006; Xu et al., 2016). In this study, using TTFA, rotenone, antimycin A or Mito‐TEMPO, we demonstrated that celastrol decreased Cd‐induced mitochondrial ROS elevation in PC12 cells, SH‐SY5Y cells and primary neurons (Figure S2 and 7). Subsequently, we found that Cd‐induced inactivation of AMPK and consequential activation of mTOR and caspase‐3 were more effectively ameliorated in the cells co‐treated with celastrol/Mito‐TEMPO than in the ones treated with celastrol or Mito‐TEMPO alone (Figure 7). To our knowledge, this is the first report showing that celastrol prevents Cd‐induced apoptosis in neuronal cells by suppressing mitochondrial ROS‐dependent AMPK‐mTOR signalling pathway.
Our previous studies suggested that the following three events formed a loop: (1) treatment with Cd initially generates ROS, (2) the ROS subsequently inactivates AMPK, and (3) eventually activates the mTOR signalling pathway leading to increased translation of ROS‐generating NADPH oxidase 2 (NOX2) protein and its regulatory proteins (p22phox, p40phox, p47phox, p67phox and Rac1), which, in turn, produces more ROS and further inactivates AMPK and activates mTOR (Chen et al., 2011b). This is supported by the time course observations that treatment of PC12 cells with Cd (10 μM) increases ROS level significantly within 2 h, but does not elevate NOX2 protein expression until 4–6 h (Chen et al., 2011b). The results support the notion that Cd induces ROS generation initially coming from non‐NOX systems in the cells that secondarily up‐regulate the expression of the ROS generating enzyme NOX2 and its regulatory proteins. In the present study, we have shown that celastrol directly attenuated Cd‐induced ROS elevation, which caused inactivation of AMPK and subsequent activation of mTOR, suggesting that celastrol suppression of the initial stimulation by Cd of ROS production was the most critical action.
In this study, interestingly, pretreatment with AICAR or rapamycin, or over‐expression of AMPKα‐ca or down‐regulation of mTOR potentiated the inhibitory effects of celastrol or Mito‐TEMPO on Cd‐induced ROS (Figure 8), yet pretreatment with compound C or expression of dn‐AMPKα conferred resistance to the effects of celastrol or Mito‐TEMPO on Cd‐induced ROS in the cells (Figure 8), suggesting that activated AMPK or deactivated mTOR may feedback to up‐regulate Cd‐induced mitochondrial ROS. Taken together, we conclude that celastrol suppresses Cd‐induced mitochondrial ROS, which is essential for preventing Cd inactivation of AMPK and activation of the mTOR pathway and, ultimately, neuronal apoptosis. However, it is unclear whether celastrol modulates ROS homeostasis by hindering ROS‐generating and/or ‐eliminating systems in neuronal cells in response to Cd. Further study is also needed to uncover why pharmacological deactivation/activation of AMPKα or deactivation of mTOR, or genetic modulation to deactivate/activate AMPKα or to inhibit mTOR affects the inhibition by celastrol, of Cd‐induced mitochondrial ROS in neuronal cells.
In conclusion, we have shown that celastrol attenuated Cd activation of mTOR‐dependent apoptosis by preventing Cd inactivation of AMPK in neuronal cells. Further investigation revealed that celastrol relieved Cd‐evoked neuronal apoptosis by preventing mitochondrial ROS inactivation of AMPK, thus suppressing activation of the mTOR pathway (Figure 9). Our results suggest that celastrol may be a promising agent for prevention of Cd‐induced oxidative stress and neurodegenerative diseases.
Figure 9.
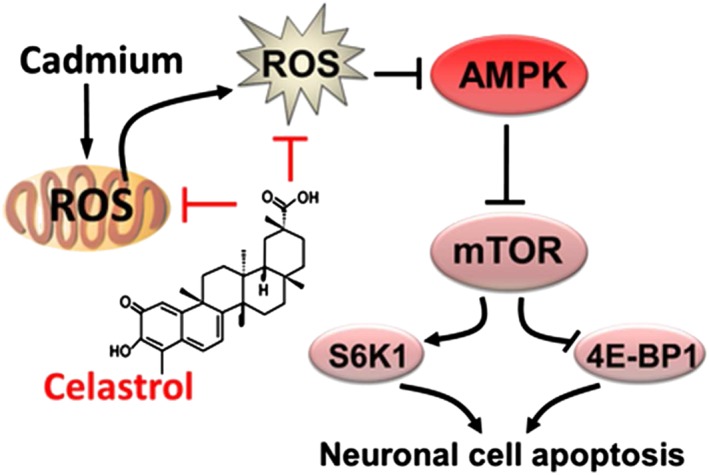
Diagram illustrating the neuroprotective effects of celastrol on Cd‐induced apoptosis in neuronal cells. Celastrol mitigates neuronal cell apoptosis induced by Cd, by preventing mitochondrial ROS inactivation of AMPK, thus suppressing activation of the mTOR pathway.
Author contributions
S.H. and L.C. conceived and designed the experiments; R.Z., N.Z., H.Z. and C.L. performed the experiments; R.Z., N.Z. and L.C. analysed the data; X.D., X.W., Y.Z., C.X., L.L. and S.Y. contributed reagents/materials/analysis tools; and R.Z., S.H. and L.C. wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Celastrol suppressed Cd induction of ROS in neuronal cells. PC12 cells, SH‐SY5Y cells and primary neurons were pretreated with/without celastrol for 1 h, followed by exposure to Cd (10 and 20 μM) 24 h. (A and B) Cell ROS was imaged and quantified using an oxidant‐sensitive probe 5‐(and‐6)‐chloromethyl‐2′,7′‐dichlorodihydrofluorescein diacetate (CM‐H2DCFDA), showing that Cd‐evoked strong ROS fluorescence (in green) was significantly inhibited by celastrol in the cells. All data were expressed as mean ± SE (n = 5). Using one‐way ANOVA, a P < 0.05, difference with control group; b P < 0.05, difference with 10 μM Cd group; c P < 0.05, difference with 20 μM Cd group.
Figure S2 Celastrol attenuated Cd‐induced mitochondrial ROS in neuronal cells. PC12 cells, SH‐SY5Y cells and primary neurons were pretreated with/without celastrol (1 μM) for 1 h and then exposed to Cd (10 μM) in the presence or absence of thenoyltrifluoroacetone (TTFA) (10 μM), rotenone (0.5 μM) or antimycin A (50 μM) for 24 h. (A‐C) Cell ROS was imaged and quantified using an oxidant‐sensitive probe CM‐H2DCFDA. All data were expressed as mean ± SE (n = 5). Using one‐way or two‐way ANOVA, a P < 0.05, difference with control group; b P < 0.05, difference with 10 μM Cd group; c P < 0.05, difference with Cd/Celastrol group, Cd/TTFA group, Cd/Rotenone or Cd/Antimycin A group.
Supporting info item
Supporting info item
Acknowledgements
This work was supported in part by the grants from National Natural Science Foundation of China (no. 30971486, 81271416; L.C.), NIH (CA115414; S.H.), Project for the Priority Academic Program Development of Jiangsu Higher Education Institutions of China (PAPD‐14KJB180010; L.C.), American Cancer Society (RSG‐08‐135‐01‐CNE; S.H.), Louisiana Board of Regents (NSF‐2009‐PFUND‐144; S.H.) and Innovative Research Program of Jiangsu College Graduate of China (KYLX16_1284; R.Z.).
Zhang, R. , Zhang, N. , Zhang, H. , Liu, C. , Dong, X. , Wang, X. , Zhu, Y. , Xu, C. , Liu, L. , Yang, S. , Huang, S. , and Chen, L. (2017) Celastrol prevents cadmium‐induced neuronal cell death by blocking reactive oxygen species‐mediated mammalian target of rapamycin pathway. British Journal of Pharmacology, 174: 82–100. doi: 10.1111/bph.13655.
Contributor Information
Shile Huang, Email: shuan1@lsuhsc.edu.
Long Chen, Email: lchen@njnu.edu.cn.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C (2001). Celastrol, a potent antioxidant and anti‐inflammatory drug, as a possible treatment for Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry 25: 1341–1357. [DOI] [PubMed] [Google Scholar]
- Bertin G, Averbeck D (2006). Cadmium: cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review). Biochimie 88: 1549–1559. [DOI] [PubMed] [Google Scholar]
- Chaturvedi RK, Flint Beal M (2013). Mitochondrial diseases of the brain. Free Radic Biol Med 63: 1–29. [DOI] [PubMed] [Google Scholar]
- Chen G, Zhang X, Zhao M, Wang Y, Cheng X, Wang D et al. (2011a). Celastrol targets mitochondrial respiratory chain complex I to induce reactive oxygen species‐dependent cytotoxicity in tumor cells. BMC Cancer 11: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu L, Huang S (2008a). Cadmium activates the mitogen‐activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic Biol Med 45: 1035–1044. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu L, Luo Y, Huang S (2008b). MAPK and mTOR pathways are involved in cadmium‐induced neuronal apoptosis. J Neurochem 105: 251–261. [DOI] [PubMed] [Google Scholar]
- Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H et al. (2010). Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab Invest 90: 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Xu B, Liu L, Luo Y, Zhou H, Chen W et al. (2011b). Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic Biol Med 50: 624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Gu C, Xu C, Zhang J, Xu Y, Ren Q et al. (2014a). Celastrol prevents cadmium‐induced neuronal cell death via targeting JNK and PTEN‐Akt/mTOR network. J Neurochem 128: 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Ren Q, Zhang J, Ye Y, Zhang Z, Xu Y et al. (2014b). N‐acetyl‐L‐cysteine protects against cadmium‐induced neuronal apoptosis by inhibiting ROS‐dependent activation of Akt/mTOR pathway in mouse brain. Neuropathol Appl Neurobiol 40: 759–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Xu Y, Xu B, Guo M, Zhang Z, Liu L et al. (2011c). CaMKII is involved in cadmium activation of MAPK and mTOR pathways leading to neuronal cell death. J Neurochem 119: 1108–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BS, Kim H, Lee HJ, Sapkota K, Park SE, Kim S et al. (2014). Celastrol from ‘Thunder God Vine’ protects SH‐SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson's disease. Neurochem Res 39: 84–96. [DOI] [PubMed] [Google Scholar]
- Cleren C, Calingasan NY, Chen J, Beal MF (2005). Celastrol protects against MPTP‐ and 3‐nitropropionic acid‐induced neurotoxicity. J Neurochem 94: 995–1004. [DOI] [PubMed] [Google Scholar]
- Cornu M, Albert V, Hall MN (2013). mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev 23: 53–62. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng YN, Shi J, Liu J, Qu QM (2013). Celastrol protects human neuroblastoma SH‐SY5Y cells from rotenone‐induced injury through induction of autophagy. Neurochem Int 63: 1–9. [DOI] [PubMed] [Google Scholar]
- Dumont M, Wille E, Calingasan NY, Tampellini D, Williams C, Gouras GK et al. (2009). Triterpenoid CDDO‐methylamide improves memory and decreases amyloid plaques in a transgenic mouse model of Alzheimer's disease. J Neurochem 109: 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerling BM, Viollet B, Tormos KV, Chandel NS (2007). Compound C inhibits hypoxic activation of HIF‐1 independent of AMPK. FEBS Lett 581: 5727–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerspacher C, Scheuber U, Schiera G, Proia P, Gygax D, Di Liegro I (2009). The effect of cadmium on brain cells in culture. Int J Mol Med 24: 311–318. [DOI] [PubMed] [Google Scholar]
- Figueiredo‐Pereira ME, Yakushin S, Cohen G (1998). Disruption of the intracellular sulfhydryl homeostasis by cadmium‐induced oxidative stress leads to protein thiolation and ubiquitination in neuronal cells. J Biol Chem 273: 12703–12709. [DOI] [PubMed] [Google Scholar]
- Genovese T, Cuzzocrea S (2008). Role of free radicals and poly(ADP‐ribose)polymerase‐1 in the development of spinal cord injury: new potential therapeutic targets. Curr Med Chem 15: 477–487. [DOI] [PubMed] [Google Scholar]
- Goncalves JF, Fiorenza AM, Spanevello RM, Mazzanti CM, Bochi GV, Antes FG et al. (2010). N‐acetylcysteine prevents memory deficits, the decrease in acetylcholinesterase activity and oxidative stress in rats exposed to cadmium. Chem Biol Interact 186: 53–60. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30: 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao B, Cheng S, Clancy CJ, Nguyen MH (2013). Caspofungin kills Candida albicans by causing both cellular apoptosis and necrosis. Antimicrob Agents Chemother 57: 326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Schaffer BE, Brunet A (2016). AMPK: an energy‐sensing pathway with multiple inputs and outputs. Trends Cell Biol 26: 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115: 577–590. [DOI] [PubMed] [Google Scholar]
- Jaquet V, Marcoux J, Forest E, Leidal KG, McCormick S, Westermaier Y et al. (2011). NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br J Pharmacol 164: 507–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Yuan Y, Hu F, Wang Q, Zhang K, Wang Y et al. (2014). Cadmium induces PC12 cells apoptosis via an extracellular signal‐regulated kinase and c‐Jun N‐terminal kinase‐mediated mitochondrial apoptotic pathway. Biol Trace Elem Res 158: 249–258. [DOI] [PubMed] [Google Scholar]
- Jiang P, Gan M, Ebrahim AS, Castanedes‐Casey M, Dickson DW, Yen SH (2013). Adenosine monophosphate‐activated protein kinase overactivation leads to accumulation of alpha‐synuclein oligomers and decrease of neurites. Neurobiol Aging 34: 1504–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomova K, Valko M (2011). Advances in metal‐induced oxidative stress and human disease. Toxicology 283: 65–87. [DOI] [PubMed] [Google Scholar]
- Kannaiyan R, Manu KA, Chen L, Li F, Rajendran P, Subramaniam A et al. (2011a). Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c‐Jun N‐terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis 16: 1028–1041. [DOI] [PubMed] [Google Scholar]
- Kannaiyan R, Shanmugam MK, Sethi G (2011b). Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett 303: 9–20. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Neutzner A (2012). Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol 123: 157–171. [DOI] [PubMed] [Google Scholar]
- Kiaei M, Kipiani K, Petri S, Chen J, Calingasan NY, Beal MF (2005). Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neurodegener Dis 2: 246–254. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lee JO, Lee SK, Kim N, You GY, Moon JW et al. (2013). Celastrol suppresses breast cancer MCF‐7 cell viability via the AMP‐activated protein kinase (AMPK)‐induced p53‐polo like kinase 2 (PLK‐2) pathway. Cell Signal 25: 805–813. [DOI] [PubMed] [Google Scholar]
- Lanju X, Jing X, Shichang L, Zhuo Y (2014). Induction of apoptosis by antimycin A in differentiated PC12 cell line. J Appl Toxicol 34: 651–657. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM (2012). mTOR signaling in growth control and disease. Cell 149: 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HW, Jang KS, Choi HJ, Jo A, Cheong JH, Chun KH (2014). Celastrol inhibits gastric cancer growth by induction of apoptosis and autophagy. BMB Rep 47: 697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HY, Zhang J, Sun LL, Li BH, Gao HL, Xie T et al. (2015). Celastrol induces apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells: an in vitro and in vivo study. Cell Death Dis 6: e1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, Ozcan U (2015). Treatment of obesity with celastrol. Cell 161: 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Chen L, Chung J, Huang S (2008). Rapamycin inhibits F‐actin reorganization and phosphorylation of focal adhesion proteins. Oncogene 27: 4998–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li F, Cardelli J, Martin K, Blenis J, Huang S (2006). Rapamycin inhibits cell motility by suppression of mTOR‐mediated S6 K1 and 4E‐BP1 pathways. Oncogene 25: 7029–7040. [DOI] [PubMed] [Google Scholar]
- Liu L, Luo Y, Chen L, Shen T, Xu B, Chen W et al. (2010). Rapamycin inhibits cytoskeleton reorganization and cell motility by suppressing RhoA expression and activity. J Biol Chem 285: 38362–38373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez E, Arce C, Oset‐Gasque MJ, Canadas S, Gonzalez MP (2006). Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radic Biol Med 40: 940–951. [DOI] [PubMed] [Google Scholar]
- Lu J, Wu DM, Zheng YL, Hu B, Cheng W, Zhang ZF (2012). Purple sweet potato color attenuates domoic acid‐induced cognitive deficits by promoting estrogen receptor‐alpha‐mediated mitochondrial biogenesis signaling in mice. Free Radic Biol Med 52: 646–659. [DOI] [PubMed] [Google Scholar]
- Lu M, Tang Q, Olefsky JM, Mellon PL, Webster NJ (2008). Adiponectin activates adenosine monophosphate‐activated protein kinase and decreases luteinizing hormone secretion in LbetaT2 gonadotropes. Mol Endocrinol 22: 760–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Han LZ, Liang H, Mi C, Shi H, Lee JJ et al. (2014). Celastrol inhibits the HIF‐1alpha pathway by inhibition of mTOR/p70S6K/eIF4E and ERK1/2 phosphorylation in human hepatoma cells. Oncol Rep 32: 235–242. [DOI] [PubMed] [Google Scholar]
- Mates JM, Segura JA, Alonso FJ, Marquez J (2010). Roles of dioxins and heavy metals in cancer and neurological diseases using ROS‐mediated mechanisms. Free Radic Biol Med 49: 1328–1341. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe RK, Halvorsen SW (2006). Cadmium blocks receptor‐mediated Jak/STAT signaling in neurons by oxidative stress. Free Radic Biol Med 41: 493–502. [DOI] [PubMed] [Google Scholar]
- Moreno‐Sanchez R, Hernandez‐Esquivel L, Rivero‐Segura NA, Marin‐Hernandez A, Neuzil J, Ralph SJ et al. (2013). Reactive oxygen species are generated by the respiratory complex II – evidence for lack of contribution of the reverse electron flow in complex I. FEBS J 280: 927–938. [DOI] [PubMed] [Google Scholar]
- Palacios HH, Yendluri BB, Parvathaneni K, Shadlinski VB, Obrenovich ME, Leszek J et al. (2011). Mitochondrion‐specific antioxidants as drug treatments for Alzheimer disease. CNS Neurol Disord Drug Targets 10: 149–162. [DOI] [PubMed] [Google Scholar]
- Pang X, Yi Z, Zhang J, Lu B, Sung B, Qu W et al. (2010). Celastrol suppresses angiogenesis‐mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res 70: 1951–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renugadevi J, Prabu SM (2010a). Cadmium‐induced hepatotoxicity in rats and the protective effect of naringenin. Exp Toxicol Pathol 62: 171–181. [DOI] [PubMed] [Google Scholar]
- Renugadevi J, Prabu SM (2010b). Quercetin protects against oxidative stress‐related renal dysfunction by cadmium in rats. Exp Toxicol Pathol 62: 471–481. [DOI] [PubMed] [Google Scholar]
- Salminen A, Lehtonen M, Paimela T, Kaarniranta K (2010). Celastrol: molecular targets of thunder god vine. Biochem Biophys Res Commun 394: 439–442. [DOI] [PubMed] [Google Scholar]
- Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C (2010). New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci 123: 2533–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha M, Ye J, Luan ZY, Guo T, Wang B, Huang JX (2015). Celastrol induces cell cycle arrest by MicroRNA‐21‐mTOR‐mediated inhibition p27 protein degradation in gastric cancer. Cancer Cell Int 15: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunitha S, Nagaraj M, Varalakshmi P (2001). Hepatoprotective effect of lupeol and lupeol linoleate on tissue antioxidant defence system in cadmium‐induced hepatotoxicity in rats. Fitoterapia 72: 516–523. [DOI] [PubMed] [Google Scholar]
- Swiech L, Perycz M, Malik A, Jaworski J (2008). Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys Acta 1784: 116–132. [DOI] [PubMed] [Google Scholar]
- Vingtdeux V, Chandakkar P, Zhao H, d'Abramo C, Davies P, Marambaud P (2011). Novel synthetic small‐molecule activators of AMPK as enhancers of autophagy and amyloid‐beta peptide degradation. FASEB J 25: 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Du Y (2013). Cadmium and its neurotoxic effects. Oxid Med Cell Longev 2013: 898034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Fang J, Leonard SS, Rao KM (2004). Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic Biol Med 36: 1434–1443. [DOI] [PubMed] [Google Scholar]
- Woo DK, Shadel GS (2011). Mitochondrial stress signals revise an old aging theory. Cell 144: 11–12. [DOI] [PubMed] [Google Scholar]
- Xu B, Chen S, Luo Y, Chen Z, Liu L, Zhou H et al. (2011). Calcium signaling is involved in cadmium‐induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS One 6: e19052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Wang X, Zhu Y, Dong X, Liu C, Zhang H et al. (2016). Rapamycin ameliorates cadmium‐induced activation of MAPK pathway and neuronal apoptosis by preventing mitochondrial ROS inactivation of PP2A. Neuropharmacology 105: 270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Huang M, Jin M, Ren Q (2007). LC‐APCI‐MS–MS for the determination of celastrol in human whole blood. Chromatographia 66: 735–739. [Google Scholar]
- Yeh YT, Yeh H, Su SH, Lin JS, Lee KJ, Shyu HW et al. (2014). Phenethyl isothiocyanate induces DNA damage‐associated G2/M arrest and subsequent apoptosis in oral cancer cells with varying p53 mutations. Free Radic Biol Med 74: 1–13. [DOI] [PubMed] [Google Scholar]
- Zang M, Zuccollo A, Hou X, Nagata D, Walsh K, Herscovitz H et al. (2004). AMP‐activated protein kinase is required for the lipid‐lowering effect of metformin in insulin‐resistant human HepG2 cells. J Biol Chem 279: 47898–47905. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li CY, Xu MJ, Wu T, Chu JH, Liu SJ et al. (2012). Oral bioavailability and gender‐related pharmacokinetics of celastrol following administration of pure celastrol and its related tablets in rats. J Ethnopharmacol 144: 195–200. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Zhao H, Lobo N, Guo X, Gentleman SM, Ma D (2014). Celastrol enhances cell viability and inhibits amyloid‐beta production induced by lipopolysaccharide in vitro. J Alzheimers Dis 41: 835–844. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Liu C, Liu W, Zhang H, Zhang R, Liu J et al. (2015). Rotenone induction of hydrogen peroxide inhibits mTOR‐mediated S6K1 and 4E‐BP1/eIF4E pathways, leading to neuronal apoptosis. Toxicol Sci 143: 81–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Celastrol suppressed Cd induction of ROS in neuronal cells. PC12 cells, SH‐SY5Y cells and primary neurons were pretreated with/without celastrol for 1 h, followed by exposure to Cd (10 and 20 μM) 24 h. (A and B) Cell ROS was imaged and quantified using an oxidant‐sensitive probe 5‐(and‐6)‐chloromethyl‐2′,7′‐dichlorodihydrofluorescein diacetate (CM‐H2DCFDA), showing that Cd‐evoked strong ROS fluorescence (in green) was significantly inhibited by celastrol in the cells. All data were expressed as mean ± SE (n = 5). Using one‐way ANOVA, a P < 0.05, difference with control group; b P < 0.05, difference with 10 μM Cd group; c P < 0.05, difference with 20 μM Cd group.
Figure S2 Celastrol attenuated Cd‐induced mitochondrial ROS in neuronal cells. PC12 cells, SH‐SY5Y cells and primary neurons were pretreated with/without celastrol (1 μM) for 1 h and then exposed to Cd (10 μM) in the presence or absence of thenoyltrifluoroacetone (TTFA) (10 μM), rotenone (0.5 μM) or antimycin A (50 μM) for 24 h. (A‐C) Cell ROS was imaged and quantified using an oxidant‐sensitive probe CM‐H2DCFDA. All data were expressed as mean ± SE (n = 5). Using one‐way or two‐way ANOVA, a P < 0.05, difference with control group; b P < 0.05, difference with 10 μM Cd group; c P < 0.05, difference with Cd/Celastrol group, Cd/TTFA group, Cd/Rotenone or Cd/Antimycin A group.
Supporting info item
Supporting info item