

Abstract
Background and Purpose
Neuropathic pain is under‐treated, with a detrimental effect on quality of life, partly because of low treatment efficacy, but also because pathophysiological mechanisms are not fully elucidated. To clarify the pathobiology of neuropathic pain, we studied the contribution of neuroinflammation and oxidative stress in a model of peripheral neuropathy. We also assessed an innovative treatment for neuropathic pain by investigating the effects of histamine H4 receptor ligands in this model.
Experimental Approach
A peripheral mononeuropathy was induced in mice, by spared nerve injury (SNI). Neuroinflammation and oxidative stress parameters were evaluated by spectrophotometry. The mechanical (von Frey test) and thermal (plantar test) nociceptive thresholds were evaluated.
Key Results
SNI mice showed increased expression of the pro‐inflammatory cytokines IL‐1ß and TNF‐α, decreased antioxidant enzyme Mn‐containing SOD (MnSOD), increased levels of 8‐hydroxy‐2′‐deoxyguanosine (8‐OHdG), an indicator of oxidative DNA damage, and of PARP, nuclear enzyme activated upon DNA damage. Intrathecal administration of VUF 8430 (H4 receptor agonist) reversed the mechanical and thermal allodynia and was associated with decreased expression of IL‐1ß, TNF‐α, 8‐OHdG and PARP and with restoration of MnSOD activity in the spinal cord and sciatic nerve. These effects were prevented by JNJ 10191584 (H4 receptor antagonist).
Conclusion and Implications
In the SNI mouse model of neuropathic pain, neuronal H4 receptor stimulation counteracts hyperalgesia and reduces neuroinflammation and oxidative stress in the spinal cord and sciatic nerve. Targeting both oxidative stress and pro‐neuroinflammatory pathways through H4 receptor‐mediated mechanisms could have promising therapeutic potential for neuropathic pain management.
Abbreviations
- 8‐OHdG
8‐hydroxy‐2′‐deoxyguanosine
- CuZnSOD
CuZn superoxide dismutase
- MnSOD
Mn‐containing SOD
- SNI
spared nerve injury
Tables of Links
| TARGETS |
|---|
| GPCRs |
| Histamine H4 receptors |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Neuropathic pain is relatively common, occurring in about 7–8% of the population and will become increasingly common because of the ageing population, increasing incidence of diabetes and improved survival from cancer. Comorbidities such as poor sleep, depression and anxiety are common in neuropathic pain patients making it a syndrome that greatly impairs quality of life and with a high economic cost to society (Doth et al., 2010; Langley et al., 2013; Bennet, 2015).
Neuropathic pain is challenging to manage. Many patients have pain that is refractory to existing treatments despite the availability of numerous treatment options. Clinical trials that examined pharmacotherapy showed that not more than 50% of patients experience clinically meaningful pain relief, which is almost always partial but not complete. In addition, patients frequently experience troublesome adverse effects that often make the treatment difficult to be tolerated (Vranken, 2012; Finnerup et al., 2015). Inadequate response to drug treatments constitutes a substantial unmet need in patients with neuropathic pain. Therefore, there is a strong clinical need to develop novel and effective treatments for neuropathic pain.
Traditionally, neuropathic pain has been considered to originate from a purely neuronal response. However, extensive evidence indicates that damage to the peripheral nerves leads to a local inflammatory response, which contributes to the generation of behavioural hypersensitivity. In the periphery, after an event that causes direct nerve damage, peripheral immune cells produce mediators including cytokines (interleukins), protons and a variety of inflammatory mediators including histamine, 5‐HT, nerve growth factor and leukotrienes (Basbaum et al., 2009; Vranken, 2012; Dias et al., 2014). The inflammatory process that occurs in response to cellular injury contributes to neuropathic pain by activating nociceptive neurons (Basbaum et al., 2009). To assess an innovative therapeutic approach to neuropathic pain management, we focused on histamine because of its prominent role in inflammatory processes. Here we have concentrated on the histamine H4 receptors, the last discovered histamine receptor (Nakamura et al., 2000; Oda et al., 2000), which is expressed in several cell types of immune system, including mast cells, eosinophils, dendritic and T cells in peripheral tissues, such as spleen, thymus, colon, blood and bone marrow, and has been characterized as the immune system histamine receptor with a pro‐inflammatory role (Jutel et al., 2009; Leurs et al., 2009). However, H4 receptors are primarily, but not exclusively, distributed in immune cells, and the recently reported functional expression of H4 receptors on human and rodent neurons (Connelly et al., 2009; Strakhova et al., 2009) highlighted its possible involvement in neuronal processes. Recent studies suggested the participation of H4 receptors in the modulation of important neuronal functions including memory, anxiety and appetite (Galeotti et al., 2013). Histamine H4 receptors also modulate pain perception as they are involved in both acute (Connelly et al., 2009; Galeotti et al., 2013) and persistent inflammatory pain (Hsieh et al., 2010).
For these reasons, in search of an innovative treatment for neuropathic pain,we have evaluated the effects of modulation of neuronal H4 receptors, in an animal model. We also aimed to elucidate the role of H4 receptors on the alteration of neuronal processes activated by nerve damage. A better comprehension of the cellular processes modulated by neuronal H4 receptors might help clarify the physiopathological role of this receptor subtype in neuropathic pain and generate new approaches to better pain management.
Methods
Animals
All animal care and experimental protocols were in compliance with international laws and policies (Directive 2010/63/EU of the European parliament and of the council of 22 September 2010 on the protection of animals used for scientific purposes; Guide for the Care and Use of Laboratory Animals, US National Research Council, 2011) and were approved by the Animal Care and Research Ethics Committee of the University of Florence, Italy, under license from the Italian Department of Health (54/2014‐B). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). All effort was taken to minimize the number of animals used and their suffering.
Male CD1 mice (23‐40g; Harlan Laboratories, Bresso, Italy) were used. Mice were randomly assigned to standard cages, with four to five animals per cage. The cages were placed in the experimental room 24 h, for acclimatization before the behavioural test. The animals were fed a standard laboratory diet and tap water ad libitum and kept at 23 ± 1°C with a 12 h light/dark cycle, light on at 07:00 h. All tests were conducted during the light phase. Mice were killed by cervical dislocation for removal of spinal cord and sciatic nerve for in vitro analyses. The number of animals per experiment was based on a power analysis (Charan and Kantharia, 2013). For antinociceptive assays, all tested groups comprised 10 animals. For in vitro assays, data are mean of four individual experiments conducted in duplicate. A total of 180 mice were used to provide all the data.
Drug administration
Mice were randomly assigned to each treatment group by an individual other than the operator. To evaluate the role of H4 receptors within the CNS, the H4 receptor agonist VUF 8430 (10–40 μg per mouse) and histamine (40 μg per mouse) (Sigma, Milan, Italy) were dissolved in saline (0.9% NaCl) solution and administered intrathecally (i.t.), 15 min before the beginning of the tests. The H4 receptor antagonist JNJ 10191584 (3–6 mg·kg−1) (Sigma, Milan, Italy) was dispersed in 1% sodium carboxymethylcellulose (1% CMC) and administered by gavage 30 min before VUF 8430. H4 receptor antagonists (indolecarboxamide derivatives) readily cross the blood–brain barrier after peripheral administration allowing the study of effects due to interactions with receptors in the CNS. Pregabalin (30 mg·kg−1 i.p.) was administered 3 h before testing; WIN55252–2 (5 mg·kg−1 i.p.), MK‐801 (0.1 μg per mouse i.t.), α‐tocopherol (100 mg·kg−1 p.o.) and amitriptyline (10 mg·kg−1 i.p.) (Sigma, Milan, Italy) were administered 30 min before tests. All reference drugs were dissolved in saline solution, except α‐tocopherol which was dispersed in 1% CMC.
Drug concentrations were prepared so that the necessary dose could be administered in a volume of 10 mL·kg−1 by gavage (p.o.) or i.p. administration, or in a volume of 5 μL per mouse by intrathecal (i.t.) injection. These injections were performed as previously described (Sanna et al., 2015). Briefly, the animals were placed in a transparent plastic box and anaesthetized with a mixture of 4% isoflurane in O2/N2O (30:70 v/v). The lower half of the animal's back was shaved and scrubbed with povidone‐iodine. The animals were then placed in a prone position on a styrofoam board (25 × 13 × 6 cm), with the mask opening fixed 12 cm from the end of the board. Then the isoflurane concentration was lowered to 1.5–2% for the remainder of the procedure. The mouse forelimbs were extended towards the front and fixed to the board with tape, taking care not to force the neck. The hind limbs were left to hang off the board, lying on the table. In that way, the animal's vertebral column was flexed around the L3–L5 level, widening these intervertebral spaces. A lumbar puncture needle was introduced perpendicular to the surface through the widest intervertebral space and lowered until it came into contact with the vertebral body. Occasionally, a short flicking of the tail or of a limb was observed. Animals then allowed to recover in their home cage before testing.
Behavioural tests on treated animals were performed on postoperative day 7. For in vitro assays, samples were collected on day 7 post‐injury, 30 min after VUF 8430 administration. Control groups received saline i.t. or 1% CMC p.o.; treatment groups received VUF 8430 or JNJ 10191584 or VUF 8430 + JNJ 10191584. Vehicle‐treated groups have been used to control for drug effects.
Spared nerve injury (SNI)
Behavioural testing was performed before surgery to establish a baseline for comparison with post‐surgical values. The animal model of mono‐neuropathy has been in use for several years and was induced according to the method of Bourquin et al. (2006). Under sodium pentobarbital (60 mg·kg−1 i.p.) anaesthesia, the skin on the lateral surface of the thigh was incised, and a section made directly through the biceps femoris muscle exposing the sciatic nerve and its three terminal branches: the sural, common peroneal and tibial nerves. Both tibial and common peroneal nerves were ligated and transected together. A microsurgical forceps with curved tips was delicately placed below the tibial and common peroneal nerves to slide the thread (5.0 silk, Ethicon; Johnson & Johnson Intl, Brussels, Belgium) around the nerves. A tight ligation of both nerves was performed. Great care was taken to avoid any contact with or stretching of the intact sural nerve. Muscle and skin were closed in two distinct layers with silk 5.0 suture. At the end of the surgical procedure, xylocaine gel was applied to the wound and saline given subcutaneously.
Intense, reproducible and long‐lasting thermal hyperalgesia and mechanical allodynia‐like behaviours are measurable in the non‐injured sural nerve skin territory (Bourquin et al., 2006). The SNI model offers the advantage of a distinct anatomical distribution with an absence of co‐mingling of injured and uninjured nerve fibres distal to the lesion such that the injured and uninjured nerves and territories can be readily identified and manipulated for further analysis (i.e. behavioural assessment). The sham procedure consisted of the same surgery without ligation and transection of the nerves.
Nociceptive behaviour
Animals were habituated to the testing environment daily for at least 2 days before baseline testing. Nociceptive responses to a mechanical or thermal stimulus were measured every 30 min for 3 h before and after nerve surgery. Neuropathic mice were monitored up to postoperative day 25. All testing was performed by observers blinded to the treatment of the animals.
Mechanical allodynia
Mechanical allodynia was measured with the dynamic plantar aesthesiometer (von Frey instrument) (Ugo Basile, Comerio, Italy) before surgery and on postoperative days 3, 4, 5, 6, 7, 8, 11, 13, 14 and 21, as described (Sanna et al., 2014). The mice were placed in individual Plexiglas cubicles (8.5 × 3.4 × 3.4 cm) on a wire mesh platform. After approximately 1 h accommodation period, during which exploratory and grooming activity ended, the mechanical paw withdrawal threshold (PWT) was measured as the hind paw withdrawal responded to von Frey hair stimulation. The mechanical stimulus was delivered to the plantar surface of the hind paw of the mouse from below the floor of the test chamber by an automated testing device. A steel rod (2 mm) was pushed with electronic ascending force (0–5 g in 35 s). When the animal withdrew its hind paw, the mechanical stimulus was automatically withdrawn, and the force recorded to the nearest 0.1 g. Nociceptive response for mechanical sensitivity was expressed as PWT in grams, and both ipsilateral (injured) and contralateral (uninjured) hind paws were tested.
The mean PWT was calculated from six consecutive trials (each performed every 30 min) and averaged for each group of mice.
Hargreaves' plantar test
Thermal nociceptive threshold was measured using Hargreaves' device, as described (Hargreaves et al., 1988). Paw withdrawal latency in response to radiant heat (infrared) was assessed using the plantar test apparatus (Ugo Basile, Comerio, Italy) before surgery and on postoperative days 3, 4, 5, 6, 7, 8, 10, 12, 13, 14, 15 and 25. Each mouse was placed under a transparent Plexiglas box (7.0 × 12.5 × 17.0 cm) on a 0.6‐cm‐thick glass plate and allowed to acclimatize for 1–2 h before recording. The radiant heat source consisted of an infrared bulb (Osram halogen‐bellaphot bulb; 8 V, 50 W) that was positioned 0.5 cm under the glass plate directly beneath the hind paw. Paw withdrawal latency after switching on the infrared radiant heat stimulus was measured automatically. The infrared light intensity was chosen to give baseline latencies of 10 s in control mice. A cut‐off of 20 s was used to prevent tissue damage. Each hind paw was tested two to three times, alternating between paws with an interval of at least 1 min between tests, and averaged for each animal. The interval between two trials on the same paw was of at least 5 min. Nociceptive response for thermal sensitivity was expressed as thermal paw withdrawal latency in seconds.
In vitro assays
SNI and control mice were killed on day 7 post‐injury, and spinal cord and sciatic nerve were removed. To obtain ipsilateral and contralateral parts of the spinal cord, samples were divided in the middle of the ventral and dorsal surfaces of the spinal cord in which are visible, the anterior median fissure and the posterior median sulcus that subdivide the cord in two equal and symmetrical halves (right and left).
A set of animals was used for the extraction of proteins and another one for DNA extraction (each set of animals for different determinations was composed of three animals, at least).
Determination of 8‐hydroxy‐2′‐deoxyguanosine (8‐OHdG)
Frozen spinal cord and sciatic nerve samples were thawed at room temperature, and DNA isolation was performed as previously described (Lodovici et al., 2000) with some modifications.
Samples were homogenized in 1 mL of 10 mM PBS, pH 7.4, sonicated on ice for 1 min. Then 1 mL of 10 mmol·L−1 Tris–HCl buffer, pH 8, containing 10 mmol·L−1 EDTA, 10 mmol·L−1 NaCl and 0.5% SDS was added, and the mixture incubated for 1 h at 37°C with 20 μg·mL−1 RNase 1 (Sigma‐Aldrich) and overnight at 37°C under argon in the presence of 100 μg·mL−1 proteinase K (Sigma‐Aldrich). The mixture was extracted with chloroform/isoamyl alcohol (10/2 v/v). DNA was precipitated from the aqueous phase with 0.2 volumes of 10 mmol·L−1 ammonium acetate, solubilized in 200 μL of 20 mmol·L−1 acetate buffer, pH 5.3, and denatured at 90°C for 3 min. The extract was then supplemented with 10 IU of P1 nuclease (Sigma‐Aldrich) in 10 μL and incubated for 1 h at 37°C with 5 IU of alkaline phosphatase (Sigma‐Aldrich) in 0.4 mol·L−1 phosphate buffer, pH 8.8. All of the procedures were performed in the dark under argon. The mixture was filtered by an Amicon Micropure‐EZ filter (Millipore Corporation, Billerica, MA), and 50 μL of each sample was used for 8‐hydroxy‐2‐deoxyguanosine (8‐OHdG) determination using an elisa kit (JalCA, Shizuoka, Japan), following the instructions provided by the manufacturer. The absorbance of the chromogenic product was measured at 450 nm and expressed as ng·mg−1 of DNA. The results were calculated from a standard curve based on known 8‐OHdG solutions.
Determination of TNF‐α and IL‐1β
The levels of IL‐1 β and TNF‐α, pro‐inflammatory cytokines, were measured on aliquots (100 μL) of spinal cord and sciatic nerve homogenate supernatants by using the mouse TNF‐α and the mouse IL‐1ß elisa ready‐SET‐Go!® assay (eBioscience, San Diego, USA), following the protocol provided by the manufacturer. Samples were homogenized in 1× Cell Lysis Buffer (#9803 Cell Signalling Technologies, Denver, MA, USA) according to manufacturer's instructions. Briefly, frozen spinal cord and sciatic nerve samples were homogenized with Ultraturrax in 200 μL of lysis buffer containing 20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton‐X, 2.5 mM sodium pyrophosphate (NaPP), 1 mM ß‐glycerophosphate, 1 mM Na3VO4, 1 μg·mL−1 leupeptin, 1 mM PMSF, followed by a 30 min sonication step. The supernatant was transferred in a clean vial and used for further determinations of IL‐1 β and TNF‐α. Values are expressed as pg·μg−1 of total proteins determined over an albumin standard curve.
Assay of PARP activity
PARP activity assay was performed as previously reported (Suzuki et al., 2004). Tissues were homogenized in 50 mM Tris HCI, pH 8, 4°C, containing 0.1% NP‐40, 200 mM KCI, 2 mM MgCl2, 50 μM ZnCl2, 2 mM dithiothreitol and protease inhibitors (1 mM PMSF, 5 μL·mL−1 leupeptin and aprotinin). Samples were then centrifuged, and 10 μL of each supernatant was incubated for 5 min at 25°C with 2 μL of [3H]NAD+(specific activity 25 Ci·nmol−1) in 50 mM Tris HCI, pH 8, containing 20 mM MgCl2, 1 mM dithiothreitol and 20 μM NAD+, in the absence or presence of activated calf thymus DNA, in a final volume of 100 μL. The reaction was stopped by the addition of 5% trichloroacetic acid. Samples were filtered, and radioactivity in the acid‐insoluble fraction was counted by a Beckman LS1801 liquid scintillation spectrometer. PARP activity estimated without activated DNA in the mixture was referred to as endogenous activity. Activity estimated in the presence of activated DNA in the assay mixture was referred to as total activity of PARP. The ratio between endogenous and total activities was considered as the measure of PARP activity in the tissues.
Measurement of MnSOD and CuZnSOD activity
Mn‐containing SOD (MnSOD) and CuZn SOD (CuZnSOD) activity was assayed as previously reported (Masini et al., 2005). Spinal cord and sciatic nerve frozen samples were homogenized with 10 mM PBS, pH 7.4, in a Polytron homogenizer and then sonicated on ice for 1 min and centrifuged at 1100 g for 10 min. Supernatants were used for SOD measurement. The assay is based on SOD‐induced inhibition of the conversion of nitroblue tetrazolium (NBT) into a blue tetrazolium salt mediated by superoxide radicals generated by xanthine oxidase. The reaction was performed in sodium carbonate buffer, 50 mM, pH 10.1, containing 0.1 mM EDTA, 25 μM NBT (Sigma), 0.1 mM xanthine and 2 nM xanthine‐oxidase (Boehringer). The rate of reduction of NBT was monitored with a Perkin Elmer Lambda 5 spectrophotometer (Milan, Italy) set at 560 nm. The amount of protein required to inhibit the rate of NBT reduction by 50% was defined as one unit (U) of SOD activity. CuZnSOD activity was determined by performing the assay in the presence of 2 mM NaCN, preincubated for 30 min. Values are expressed as U per mg of total proteins.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data were analysed using one‐way or two‐way ANOVA. Tukey's test was used for post hoc analysis following a significant one‐way ANOVA. Multiple comparisons following two‐way ANOVA were conducted with Bonferroni post hoc comparison. A P‐value of <0.05 was considered statistically significant. The computer program GraphPad Prism version 5.0 (GraphPad Software Inc., San Diego, CA, USA) was used in all statistical analyses. For in vitro studies, technical replicates were used to ensure the reliability of single values. All experimental results are given as the mean ± SEM.
Results
Antihyperalgesic activity of the H4 receptor agonist VUF 8430
The involvement of histamine H4 receptors in the modulation of the neuropathic pain induced in mice by SNI was evaluated using a mechanical and a thermal noxious stimulus. SNI mice developed a persistent mechanical allodynia in the ipsilateral side that appeared 3 days after surgery and remained unmodified up to 21 days (Figure 1A). On the basis of this time course study, experiments to evaluate the role of H4 receptor modulation in neuropathic pain were performed on postoperative day 7.
Figure 1.
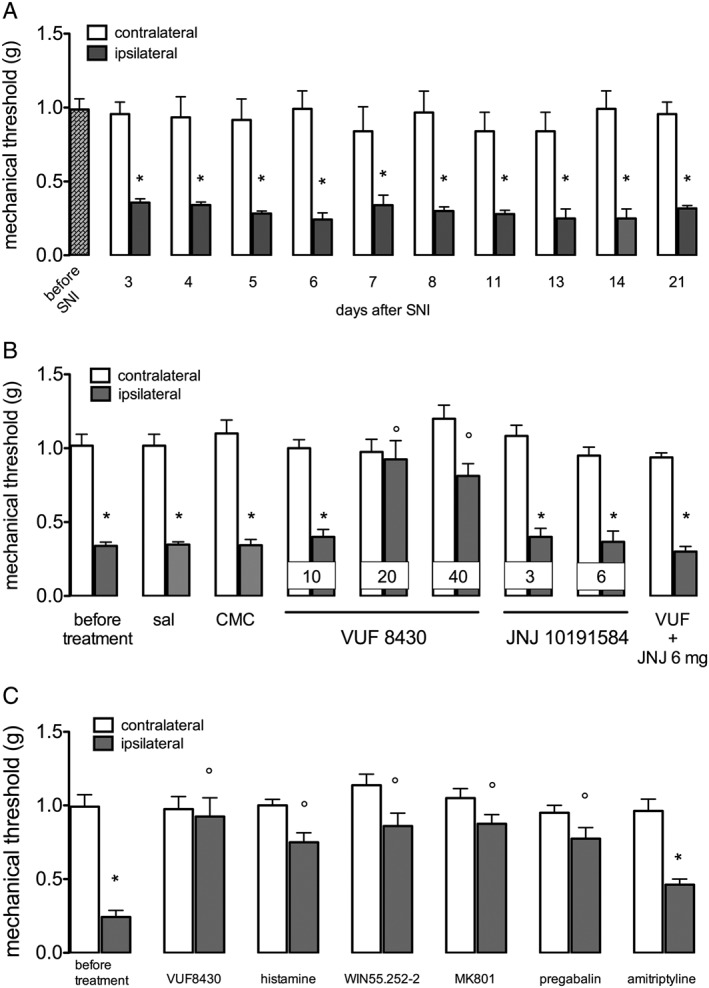
Prevention of mechanical allodynia by the H4 receptor agonist VUF 8430 in SNI neuropathic mice. (A) SNI produced a long‐lasting mechanical allodynia up to 21 days after surgery. (B) The H4 receptor agonist VUF 8430 (10–40 μg per mouse i.t.) dose‐dependently reversed mechanical allodynia. The antihyperalgesic effect of VUF 8430 (20 μg) was prevented by the H4 receptor antagonist JNJ 10191584 (6 mg·kg−1 p.o.). Doses administered are shown in each column. (C) The effect of VUF 8430 was of intensity comparable with that produced by histamine and by the reference drugs WIN‐55252‐2, MK‐801 and pregabalin. Amitriptyline was used as negative control. Data for the H4 receptor agonist were recorded 30 min after the beginning of the test. Data for reference drugs were recorded 30 min after the beginning of the test, except for pregabalin that was recorded after 3 h. Data points represent means ± SEM from experiments conducted on 10 mice. *P < 0.05 versus contralateral; # P < 0.05 versus ipsilateral before treatment.
The H4 receptor agonist VUF 8430 (20–40 μg per mouse; given i.t.) reduced mechanical allodynia without any significant effect on the contralateral side. The lower dose of 10 μg per mouse was ineffective (Figure 1B). The antihyperalgesic effect of VUF 8430 was prevented by the H4 receptor antagonist JNJ 10191584 (6 mg·kg−1 p.o.). No effect was produced on the contralateral side (Figure 1A). The intensity of the VUF 8430 antihyperalgesic effect was comparable with that of the reference drugs histamine, WIN55252.2, MK‐801 and pregabalin. Amitriptyline, used as a negative reference drug, was ineffective (Figure 1C). JNJ 10191584 (3–6 mg·kg−1 p.o.), when administered alone, did not modify the mechanical allodynia in SNI mice on the ispilateral side or induce analgesia in the contralateral side (Figure 1B), excluding the possibility of an antinociceptive effect due to blockade of peripheral pro‐inflammatory H4 receptors.
SNI mice developed a persistent thermal allodynia in the ipsilateral side that appeared 3 days after surgery and remained unmodified up to 25 days (Figure 2A), showing a time course comparable with the mechanical allodynia. VUF 8430 (20–40 μg) reversed thermal hypernociception. At the dose of 20 μg, the antiallodynic effect appeared 30 min after administration and persisted up to 60 min, with no effect on the contralateral side. At 40 μg, the nociceptive threshold was increased from 15 min and persisted up to 60 min, and antinociception on the contralateral side was induced 15 min after administration. The dose of 10 μg was devoid of any effect (Figure 2B). Pretreatment with JNJ 10151984 antagonized the H4 receptor agonist‐induced antihyperalgesic effect (Figure 2C). Thermal antinociception produced by VUF 8430 was comparable with that of WIN55252.2, MK‐801, α‐tocopherol and pregabalin. Amitriptyline, used as the negative reference drug, was ineffective (Figure 2D).
Figure 2.
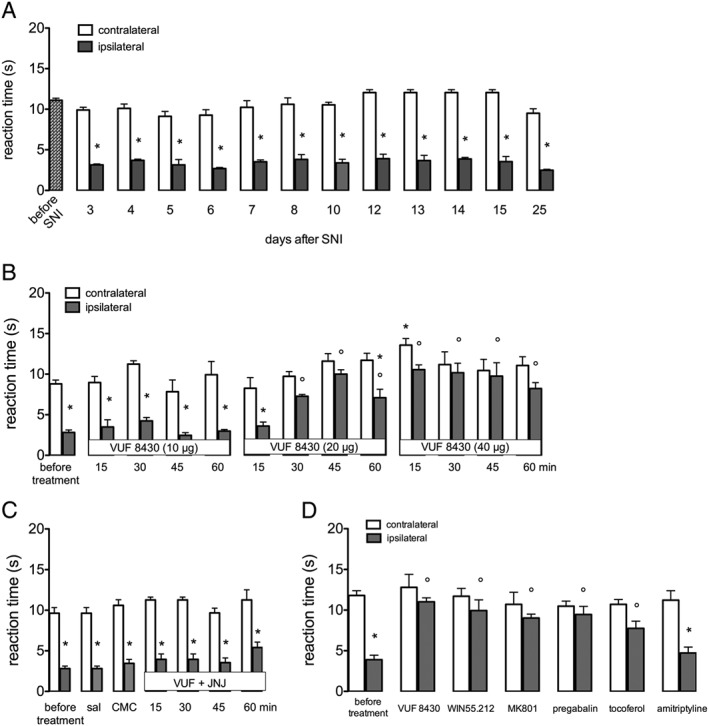
Prevention of thermal allodynia by the H4 receptor agonist VUF 8430 in SNI neuropathic mice. (A) SNI produced a long‐lasting thermal allodynia up to 25 days after surgery. (B) The H4 receptor agonist VUF 8430 (10–40 μg per mouse i.t.) dose‐dependently reversed thermal allodynia in the ipsilateral side and increased pain threshold in the contralateral side, at 40 μg. (C) The antihyperalgesic effect of VUF 8430 (20 μg i.t.) was prevented by the H4 receptor antagonist JNJ 10191584 (6 mg·kg−1 p.o.) at any time point. (D) The antihyperalgesic effect of VUF 8430 was of intensity comparable with that produced by the reference drugs WIN55252‐2, MK‐801, α‐tocopherol and pregabalin. Amitriptyline was used as negative control. Data for reference drugs were recorded 30 min after the beginning of the test, except for pregabalin that was recorded after 3 h. Data points represent means ± SEM from experiments conducted on 10 mice. *P < 0.05 versus contralateral; #P < 0.05 versus ipsilateral before treatment; §P < 0.05 versus contralateral before treatment.
Vehicle treatments (saline, 1% CMC) did not modify pain threshold of SNI mice (Figure 1B; Figure 2C). Sham operated mice, used as control mice, did not show any changes in mechanical and thermal nociceptive responses.
Effect of H4 receptor stimulation on neuroinflammation
The pro‐inflammatory cytokines IL‐1ß and TNF‐α were expressed in spinal cord and sciatic nerve. The SNI mice showed an increased spinal expression of IL‐1ß in both the ipsilateral and contralateral side that was reduced by VUF 8430 (20 μg). This effect was prevented by JNJ 10191584 (6 mg·kg−1) a dose that was ineffective when administered alone (Figure 3A). There was a greater increase of IL‐1ß in the sciatic nerve from SNI mice, in both the ipsilateral and contralateral side and and these effects were prevented by VUF 8430. Treatment with JNJ 10191584 reversed the H4 receptor agonist‐induced effect (Figure 3B).
Figure 3.
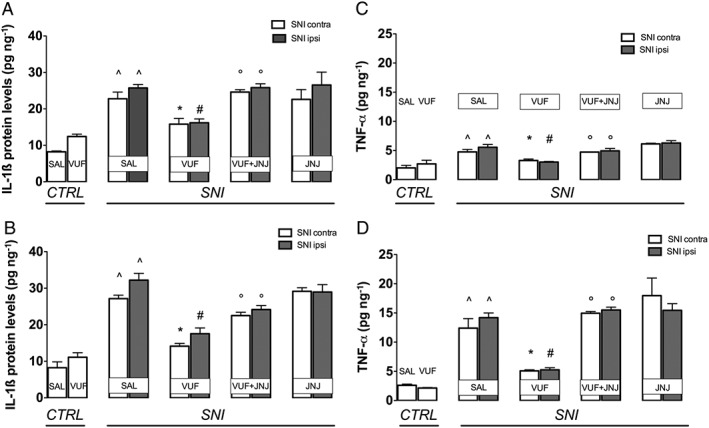
Prevention of the IL‐1ß and TNF‐α increase by H4 receptor stimulation in SNI mice. SNI mice showed increased expression of IL‐1ß in the spinal cord (A) and in the sciatic nerve (B) samples. The IL‐1ß increase was prevented by treatment with the H4 receptor agonist VUF 8430 (20 μg). Administration of the H4 receptor antagonist JNJ 10191584 (6 mg·kg−1 p.o.) reversed the effect of VUF 8430. (C) TNF‐α expression increased in SNI mice within spinal cord. (D) A three times higher increase was detected in the sciatic nerve. The TNF‐α increase was prevented by treatment with the H4 receptor agonist VUF 8430 (20 μg). This effect was reversed by administration of the H4 receptor antagonist JNJ 10191584 (6 mg·kg−1 p.o.). No influence on the IL‐1ß and TNF‐α content was produced by JNJ 10191584 when administered alone. ^P < 0.05 versus sham group; #P < 0.05 versus ipsilateral side of saline‐treated SNI mice; *P < 0.05 versus contralateral side of saline‐treated SNI mice; †P < 0.05 versus VUF 8430‐treated SNI mice.
TNF‐α was increased in the spinal samples on both the ipsilateral and contralateral side of SNI mice, an effect prevented by VUF 8430. A more marked increase of TNF‐α expression was found in sciatic nerve and this effect was also prevented by administration of the H4 receptor agonist (Figure 3D). JNJ 10191584, at a dose devoid of any influence on TNF‐α levels whne given alone, counteracted the effects of the H4 receptor agonist (Figure 3C–D).
Effect of H4 receptor stimulation on oxidative stress parameters
Oxidative stress has been proposed as a key process inducing nerve damage in models of diabetic and chemotherapy‐induced neuropathy (Naziroglu et al., 2012; Areti et al., 2014), and after injury (Guedes et al., 2008). This evidence encouraged us to investigate the presence of oxidative stress also in SNI mice and to elucidate the role of H4 receptors on the modulation of these processes.
Measurement of the antioxidant enzymes SOD revealed that MnSOD activity in the spinal cord was significantly decreased by SNI, compared with control mice. This decrease in MnSOD activity was particularly evident in the ipsilateral side, but it was present to a lesser extent also in the contralateral side. The decrease in MnSOD activity was prevented by treatment with the H4 receptor agonist VUF 8430 (20 μg), and this effect was reversed by co‐administration with the H4 receptor antagonist JNJ 10191584 (6 mg·kg−1) (Figure 4A). Similar results were obtained in the sciatic nerve (Figure 4B). There were no changes in CuZnSOD activity in either spinal cord (Figure 4C) or sciatic nerve (Figure 4D), following SNI and the administration of the H4 receptor agonist did not produce any effect (Figure 4B). The H4 receptor antagonist JNJ 10191584 (6 mg·kg−1), when administered alone, was devoid of any influence on both MnSOD and CuZnSOD activity (Figure 4A–D).
Figure 4.
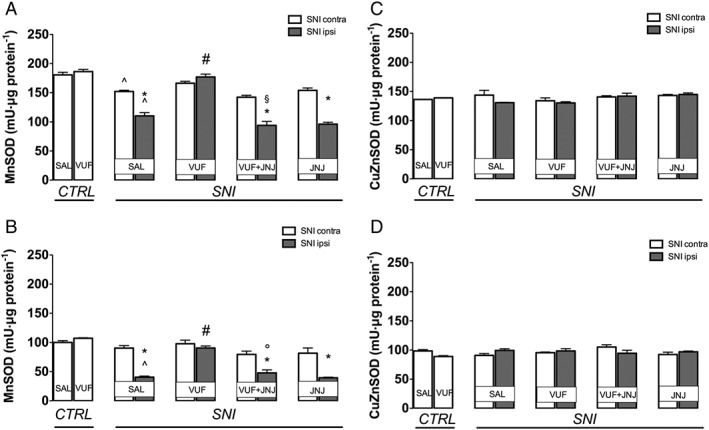
Effect of H4 receptor activation on SOD activity in SNI mice. A decreased activity of the antioxidant enzyme MnSOD was detected in the spinal cord (A) and in the sciatic nerve (B) of SNI neuropathic mice. Treatment with the H4 receptor agonist VUF 8430 (20 μg) restored MnSOD activity. This effect was reversed by administration of the H4 receptor antagonist JNJ 10191584 (6 mg·kg−1 p.o.). No changes in CuZnSOD were observed at spinal (C) and peripheral nerve levels (D). ^P < 0.05 in comparison with sham group; *P < 0.05 versus contralateral side of saline‐treated SNI mice; #P < 0.05 versus ipsilateral side of saline‐treated SNI mice; †P < 0.05 versus ipsilateral side of VUF 8430‐treated SNI mice.
Measurement of 8‐OHdG, an indicator of oxidative DNA damage, showed a marked increase in the spinal cord of SNI mice, but only on the ipsilateral side (Figure 5A). Similarly, in sciatic nerve samples, 8‐OHdG was increased but to a lesser extent than that in spinal cord samples. VUF 8430 restored basal levels of 8‐OHdG, and this effect was prevented by JNJ 10191584 (6 mg·kg−1) (Figure 5 A–B). The H4 receptor antagonist, when administered alone, did not modify levels of 8‐OHdG (Figure 5A–B).
Figure 5.
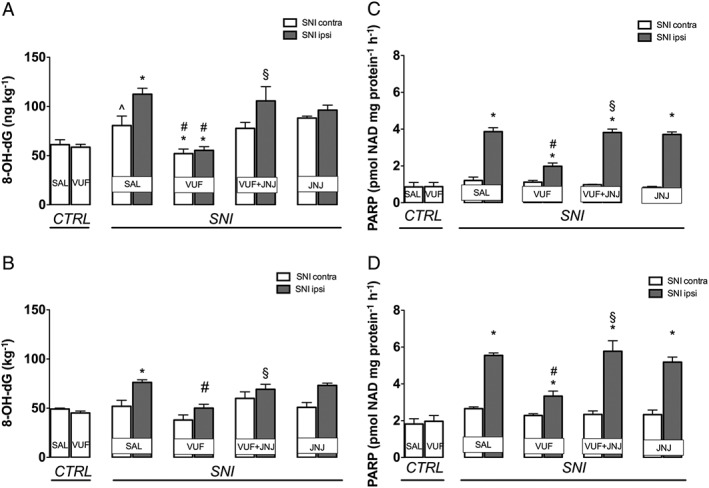
Prevention of the 8‐OHdG and PARP increase by H4 receptor agonist in SNI mice. SNI mice showed increased levels of 8‐OHdG within spinal cord (A) and sciatic nerve (B). An overexpression of PARP was also detected within spinal cord (C) and sciatic nerve (D). The increase of 8‐OHdG and PARP was prevented by treatment with the H4 receptor agonist VUF 8430 (20 μg). This effect was reversed by administration of the H4 receptor antagonist JNJ 10191584 (6 mg·kg−1 p.o.). No changes in the 8‐OHdG content were produced by JNJ 10191584 when administered alone. ^P < 0.05 versus saline‐treated control group; *P < 0.05 versus contralateral side of saline‐treated SNI mice; #P < 0.05 versus ipsilateral side of saline‐treated SNI mice; †P < 0.05 versus ipsilateral side of VUF 8430‐treated SNI mice.
Activity of PARP1/2, another marker of DNA damage, was markedly increased in spinal cord samples from SNI mice, only on the ipsilateral side (Figure 5C). Similarly, in sciatic nerve samples, PARP was increased following SNI (Figure 5D) and this increase was blocked by VUF 8430 (20 μg). Co‐administration with the H4 receptor antagonist JNJ 10191584 (6 mg·kg−1) completely prevented the VUF 8430‐induced effect (Figure 5C–D). JNJ 10191584, when administered alone, was ineffective (Figure 5C–D).
Discussion
Neuropathic pain involves damage to the peripheral or central nervous system of different aetiology, but, across the different neuropathic pain syndromes, the clinical manifestation of the pain is similar. Changes in the sensory nervous system alter pain sensitivity producing positive and negative sensory symptoms. Pain hypersensitivity is the dominating positive symptom which is mainly manifested as cold and heat hyperalgesia and mechanical allodynia (Baron et al., 2010). To investigate the effects of modulation of H4 receptors in neuropathic pain, we assessed thermal and mechanical allodynia. In agreement with clinical symptoms, animals subjected to SNI showed a persistent and long‐lasting mechanical and thermal allodynia that was reversed by the H4 receptor agonist VUF 8430. Because H4 receptors are not restricted to the CNS, VUF 8430 was given i.t. to avoid any interference from actions at peripheral H4 receptors. The positive effect on neuropathic pain induced by neuronal H4 receptor stimulation indicates different roles for the neuronal and peripheral H4 receptors: pro‐inflammatory and pronociceptive for the peripheral receptors due to their regulatory role in immune and inflammatory responses, as widely reported; antinociceptive for the neuronal receptors. Previous studies with VUF 8430 describing its analgesic activity in acute thermal nociception (Galeotti et al., 2013) and in animal models of neuropathic pain (Smith et al., 2007; Sanna et al., 2015, b) further support this hypothesis. An anti‐allodynic effect, in models of neuropathic pain, induced by systemic administration of the H4 receptor antagonist JNJ‐7777120 has also been reported (Hsieh et al., 2010), but this anti‐neuropathic effect was weaker than the anti‐inflammatory effect, suggesting that the beneficial effect on nociception might originate from a reduced inflammatory response at the site of injury, due to antagonism of the H4 receptors on immune system cells. However, great caution must be exerted when interpreting in vivo effects of JNJ‐7777120 as evidence for H4 receptor antagonism. Although JNJ‐7777120 had been considered a standard H4 receptor antagonist, recent findings suggest that it can act as both antagonist/partial inverse agonist and agonist of H4 receptors (Seifert et al., 2011). As JNJ‐7777120 readily crosses the blood–brain barrier, we cannot exclude the possibility of stimulation of neuronal H4 receptors rather than antagonism of peripheral receptors might be involved in the anti‐allodynic effect at the H4 receptors.
Neuropathic pain is undertreated, partly because of low treatment efficacy, but also because the molecular mechanisms are poorly understood. The elucidation of the pathophysiological events involved would greatly improve the therapy. In recent years, many investigations have focused on the role of neuroinflammation in the pathogenesis of neuropathic pain (Austin and Moalem Taylor, 2010; Calvo et al., 2012). The inflammatory response consists of a pro‐inflammatory phase followed by a resolution phase. T‐helper cells, dependent on their class, release cytokines that are classified on the basis of their biological activity as pro‐inflammatory cytokines (IL‐1ß, TNF‐α, IL‐6), or anti‐inflammatory cytokines (IL‐4, IL‐10) (Dinarello, 2007). Under physiological conditions, spinal expression of pro‐inflammatory cytokines is minimal, with rapid up‐regulation following peripheral nerve injury (Ji et al., 2013; Sacerdote et al., 2013). In our experiments, SNI was accompanied by a significant increase in the spinal cord levels of TNF‐α and IL‐1β. An even greater increase in the pro‐inflammatory cytokines was detected in the sciatic nerve. Damage to a peripheral nerve induces the activation of a descending pathway which could produce a retrograde response from the spinal cord. It is possible that the effect on cytokine expression detected in the sciatic nerve following acute i.t. VUF 8430 might be related to a retrograde modulation of the nerve activity. This increase was more evident in the ipsilateral side but was significant also in the contralateral side 7 days after injury, indicating the presence of a widespread and long‐lasting neuroinflammatory process. VUF 8430 treatment counteracted the increase of IL‐1β and TNF‐α, in both the spinal cord and sciatic nerve of SNI mice. Because agents that suppress inflammatory cytokine elevation have been advocated to be useful for the treatment of neuropathic pain (Okoro et al., 2010; Kandhare et al., 2012), this anti‐inflammatory effect mediated by neuronal H4 receptors might, at least in part, explain the antinociceptive activity of VUF 8430.
Although IL‐1β is largely produced and secreted by glial cells (Liao et al., 2011; Ji et al., 2013), neuronal expression is also observed following models of peripheral neuropathy (DeLeo et al., 1997). In humans, patients with a range of painful peripheral neuropathies exhibit enhanced IL‐1β levels in their cerebrospinal fluid compared with normal controls (Alexander et al., 2005; Backonja et al., 2008). Similarly, TNF‐α is expressed by both glial cells and neurons (Ohtori et al., 2004). A selective neuronal expression of H4 receptors within spinal cord and dorsal root ganglia have been described (Sanna et al., 2015, b). It is plausible that the H4 receptor‐mediated anti‐inflammatory mechanism occurs within neurons, further differentiating the cellular and physiological functions of neuronal H4 receptors from the pro‐inflammatory activity of peripheral H4 receptors.
The defences against the ROS, such as superoxide anion, hydroxyl radical and hydrogen peroxide, are crucial in inflammatory responses, and it is generally believed that oxidative stress is the key pathological process inducing nerve damage in models of neuropathies such as diabetic neuropathy and Charcot–Marie neuropathy (Saifi et al., 2003; Nazıroğlu et al., 2012). Oxidative stress has also recently been invoked as a contributing factor to painful symmetrical sensorimotor polyneuropathy (Nazıroğlu et al., 2012). Mammalian nerves are known to be more susceptible to oxidative stress because of their high content of phospholipids, mitochondria rich axoplasm and also due to weak cellular antioxidant defences (Low et al., 1997). These observations laid the foundation for investigating a possible involvement of oxidative stress in neuropathic SNI mice. Oxidative damage to peripheral neurons can cause damage to the myelin sheath, mitochondrial proteins and antioxidant enzymes and promote apoptosis and DNA damage. Hence, monitoring levels of 8‐OHdG, an indicator of oxidative stress damage, can be helpful in following the course of peripheral neuropathy and response of neuropathy to the treatment. The PARPs are nuclear enzymes activated upon DNA damage, and inhibition of these enzymes provides a resistance mechanism against DNA damage (Hong et al., 2004). However, when overactivated by massive DNA damage, PARP initiates an energy consuming cycle by catalysing the transfer of ADP‐ribose units from NAD+ to nuclear proteins. This leads to exhaustion of the cellular NAD+ and ATP pools, affecting mitochondrial respiration and ultimately resulting in cellular dysfunction and death. PARP has been targeted as a treatment for cancer (Hong et al., 2004), but it is localized to sensory neurons of the peripheral and central nervous systems (Drel et al., 2007), and PARP inhibitors attenuate pain‐like behaviours in experimental diabetic (Negi et al., 2010) and chemotherapy‐induced neuropathy (Brederson et al., 2012).
A major mechanism for the control of ROS and of consequent damage is provided by the antioxidant enzymes and a crucial component of these is the SOD family, composed of mitochondrial MnSOD and cytoplasmatic CuZnSOD (Zelko et al., 2002). MnSOD and CuZnSOD rapidly scavenge and dismutate superoxide, acting as a first line of defence against electron transport chain‐derived oxidative stress (Zelko et al., 2002; Candas and Li, 2014). MnSOD is localized within the mitochondrial matrix, a site of significant oxygen‐free radical production during energy metabolism and also supposed to be linked to the aetiology of neuropathic pain (Sui et al., 2013). CuZnSOD is located in the cytosol, in greater concentration than MnSOD, and functions to detoxify superoxide anion generated from many sources. Following peripheral nerve injury, the production of NO and superoxide is increased. Superoxide reacts with NO to form peroxynitrite, which is involved in cellular injury by continuously generating superoxide and inactivating intrinsic MnSOD, the enzyme that normally keeps superoxide under tight control, through nitration of Tyr34 in a Mn‐catalysed process (Salvemini et al., 2011). Thus, knowledge of the levels of PARP and 8‐OHdG, in addition to SOD activity, should provide important evidence for the presence of an oxidative stress condition in trauma‐induced neuropathy.
Our results indicate that the antioxidant enzyme MnSOD is greatly reduced in the ipsilateral side of the spinal cord and sciatic nerve following SNI, in agreement with results from other peripheral nerve injury models, such as sciatic nerve transection and chronic constriction injury (Guedes et al., 2006; Nie et al., 2013). Conversely, no modification in the CuZnSOD activity was detected. The different changes in the contents of MnSOD and CuZnSOD following nerve injury may reflect their respective subcellular localization and site of action. In contrast to the cytosolic localization of the CuZnSOD isoform, MnSOD has a mitochondrial localization (Zelko et al., 2002), and mitochondrial dysfunction has been linked to the aetiology of neuropathic pain (Sui et al., 2013). The SOD enzyme working at mitochondrial levels, even if less abundantly expressed, appears the most likely to play a prominent protective role on SNI mice. The finding that nerve injury‐induced oxidative stress was not associated with changes of CuZnSOD activity is consistent with previous studies (Yoneda et al., 1992; Chen et al., 2010). Simultaneously, a robust increase of 8‐OHdG and PARP was detected both in the spinal cord and in the sciatic nerve, confirming the presence of a condition of oxidative stress. Stimulation of H4 receptors restored the MnSOD activity, as well as 8‐OHdG and PARP levels in the absence of any effect induced by H4 receptor blockade. These data indicate that, in addition to the anti‐neuroinflammatory activity, activation of H4 receptors in the CNS exerts an antioxidant activity, which further supports the hypothesis of a specific antinociceptive and antineuropathic property for neuronal H4 receptors.
In conclusion, these results demonstrated that activation of neuronal H4 receptors counteracted mechanical and thermal allodynia in an animal model of peripheral neuropathy through reduction of neuroinflammation and oxidative stress. Despite their wide usage and clinical efficacy, the presently available antioxidants only provide mild to moderate pain relief in peripheral neuropathy (Schloss et al., 2013). Due to the poor clinical efficacy of antioxidant drugs, targeting both oxidative stress and pro‐neuroinflammatory pathways could have promising therapeutic potential for neuropathic pain management, with a wide clinical and economic benefit.
Author contributions
M.D.S. conducted the behavioural experiments, acquired and analysed the data. N.G designed and directed the protocol and drafted the article. L.L. and M.D. carried out the biochemical measurements. E.M. participated in the protocol design and critically revised the article. C.G. was involved in the study design and critically revised the article.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Acknowledgements
This work was supported by grants from Ministry of Instruction, University and Research (MIUR) and Ente Cassa di Risparmio di Firenze.
Sanna, M. D. , Lucarini, L. , Durante, M. , Ghelardini, C. , Masini, E. , and Galeotti, N. (2017) Histamine H4 receptor agonist‐induced relief from painful peripheral neuropathy is mediated by inhibition of spinal neuroinflammation and oxidative stress. British Journal of Pharmacology, 174: 28–40. doi: 10.1111/bph.13644.
References
- Alexander GM, van Rijn MA, van Hilten JJ, Perreault MJ, Schwartzman RJ (2005). Changes in cerebrospinal fluid levels of pro‐inflammatory cytokines in CRPS. Pain 116: 213–219. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Areti A, Yerra VG, Naidu VGM, Kumar A (2014). Oxidative stress and nerve damage: role in chemotherapy induced peripheral neuropathy. Redox Biol 2: 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin PJ, Moalem Taylor G (2010). The neuroimmune balance in neuropathic pain: involvement of inflammatory immune cell, immune‐like glia cells and cytokines. J Neuroimmunol 229: 26–50. [DOI] [PubMed] [Google Scholar]
- Backonja MM, Coe CL, Muller DA, Schell K (2008). Altered cytokine levels in the blood and cerebrospinal fluid of chronic pain patients. J Neuroimmunol 195: 157–163. [DOI] [PubMed] [Google Scholar]
- Baron R, Binder A, Wasner G (2010). Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol 9: 807–819. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D (2009). Cellular and molecular mechanisms of pain. Cell 139: 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet DLH (2015). Informed drug choices for neuropathic pain. Lancet Neurol 14: 129–130. [DOI] [PubMed] [Google Scholar]
- Bourquin AF, Süveges M, Pertin M, Gilliard N, Sardy S, Davison AC et al. (2006). Assessment and analysis of mechanical allodynia‐like behavior induced by spared nerve injury (SNI) in the mouse. Pain 122: 14.e1–14.e14. [DOI] [PubMed] [Google Scholar]
- Brederson JD, Joshi SK, Browman KE, Mikusa J, Zhong C, Gauvin D et al. (2012). PARP inhibitors attenuate chemotherapy‐induced painful neuropathy. J Peripher Nerv Syst 17: 324–330. [DOI] [PubMed] [Google Scholar]
- Calvo M, Dawes JM, Bennet DLH (2012). The role of the immune system in the generation of neuropathic pain. Lancet Neurol 11: 629–642. [DOI] [PubMed] [Google Scholar]
- Candas D, Li JJ (2014). MnSOD in oxidative stress response‐potential regulation via mitochondrial protein influx. Antioxid Redox Signal 20: 1599–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charan J, Kantharia ND (2013). How to calculate sample size in animal studies? J Pharm Pharmacol 4: 303–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Muscoli C, Doyle T, Bryant L, Cuzzocrea S, Mollace V et al. (2010). NMDA‐receptor activation and nitroxidative regulation of the glutamatergic pathway during nociceptive processing. Pain 149: 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly WM, Shenton FC, Lethbridge N, Leurs R, Waldvogel HJ, Faull RL et al. (2009). The histamine H4 receptor is functionally expressed on neurons in the mammalian CNS. Br J Pharmacol 157: 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeo JA, Colbur RW, Rickman AJ (1997). Cytokine and growth factor immunohistochemical spinal profiles in two animal models of mononeuropathy. Brain Res 759: 50–57. [DOI] [PubMed] [Google Scholar]
- Dias JM, de Brito TV, de Aguiar Magalhães D, da Silva Santos PW, Batista JA, do Nascimento Dias EG et al. (2014). Gabapentin, a synthetic analogue of gamma aminobutyric acid, reverses systemic acute inflammation and oxidative stress in mice. Inflammation 37: 1826–1836. [DOI] [PubMed] [Google Scholar]
- Dinarello CA (2007). Historical insights into cytokines. Eur J Immunol 37 (Suppl 1): S34–S45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doth AH, Hansson PT, Jensen MP, Taylor RS (2010). The burden of neuropathic pain: a systematic review and meta‐analysis of health utilities. Pain 149: 338–344. [DOI] [PubMed] [Google Scholar]
- Drel VR, Pacher P, Vareniuk I, Pavlov I, Ilnytska O, Lyzogubov VV et al. (2007). A peroxynitrite decomposition catalyst counteracts sensory neuropathy in streptozotocin‐diabetic mice. Eur J Pharmacol 569: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH et al. (2015). Pharmacotherapy for neuropathic pain in adults: a systematic review and meta‐analysis. Lancet Neurol 14: 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeotti N, Sanna MD, Ghelardini C (2013). Pleiotropic effect of histamine H4 receptor modulation in the central nervous system. Neuropharmacology 71: 141–147. [DOI] [PubMed] [Google Scholar]
- Guedes RP, Bosco LD, Teixeira CM, Araújo AS, Llesuy S, Belló‐Klein A et al. (2006). Neuropathic pain modifies antioxidant activity in rat spinal cord. Neurochem Res 31: 603–609. [DOI] [PubMed] [Google Scholar]
- Guedes RP, Araujo AS, Janner D, Bello‐Klein A, Ribeiro MF, Partata WA (2008). Increase in reactive oxygen species and activation of Akt signaling pathway in neuropathic pain. Cell Mol Neurobiol 28: 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988). A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32: 77–88. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Dawson TM, Dawson VL (2004). Nuclear and mitochondrial conversations in cell death: PARP‐1 and AIF signaling. Trends Pharmacol Sci 25: 259–264. [DOI] [PubMed] [Google Scholar]
- Hsieh GC, Chandran P, Salyers AK, Pai M, Zhu CZ, Wensink EJ et al. (2010). H4 receptor antagonism exhibits anti‐nociceptive effects in inflammatory and neuropathic pain models in rats. Pharmacol Biochem Behav 95: 41–50. [DOI] [PubMed] [Google Scholar]
- Ji XT, Qian NS, Zhang T, Li JM, Li XK, Wang P et al. (2013). Spinal astrocytic activation contributes to mechanical allodynia in a rat chemotherapy‐induced neuropathic pain model. PLoS One 8: e60733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutel M, Akdis M, Akdis CA (2009). Histamine, histamine receptors and their role in immune pathology. Clin Exp Allergy 39: 1786–1800. [DOI] [PubMed] [Google Scholar]
- Kandhare AD, Raygude KS, Ghosh P, Ghule AE, Bodhankar SL (2012). Therapeutic role of curcumin in prevention of biochemical and behavioral aberration induced by alcoholic neuropathy in laboratory animals. Neurosci Lett 511: 18–22. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley PC, Van Litsenberg C, Cappelleri JC, Carroll D (2013). The burden associated with neuropathic pain in Western Europe. J Med Econ 16: 85–95. [DOI] [PubMed] [Google Scholar]
- Leurs R, Chazot PL, Shenton FC, Lim HD, de Esch IJ (2009). Molecular and biochemical pharmacology of the histamine H4 receptor. Br J Pharmacol 157: 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao YH, Zhang GH, Jia D, Wang P, Qian NS, He F et al. (2011). Spinal astrocytic activation contributes to mechanical allodynia in a mouse model type 2 diabetes. Brain Res 1368: 324–335. [DOI] [PubMed] [Google Scholar]
- Lodovici M, Casalini C, Cariaggi R, Michelucci L, Dolara P (2000). Levels of 8‐hydroxydeoxyguanosine as a marker of DNA damage in human leukocytes. Free Radic Biol Med 28: 13–17. [DOI] [PubMed] [Google Scholar]
- Low PA, Nickander KK, Trischler HJ (1997). The roles of oxidative stress and antioxidant treatment in experimental diabetic neuropathy. Diabetes 46 (Suppl 2): S38–S42. [DOI] [PubMed] [Google Scholar]
- Masini E, Bani D, Vannacci A, Pierpaoli S, Mannaioni PF, Comhair SA et al. (2005). Reduction of antigen‐induced respiratory abnormalities and airway inflammation in sensitized guinea pigs by a superoxide dismutase mimetic. Free Radic Biol Med 39: 520–531. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Itadani H, Hidaka Y, Ohta M, Tanaka K (2000). Molecular cloning and characterization of a new human histamine receptor, H4R. Biochem Biophys Res Commun 279: 615–620. [DOI] [PubMed] [Google Scholar]
- Nazıroglu M, Dikici DM, Dursun S (2012). Role of oxidative stress and Ca2 + signaling on molecular pathways of neuropathic pain in diabetes: focus on TRP channels. Neurochem Res 37: 2065–2075. [DOI] [PubMed] [Google Scholar]
- Negi G, Kumar A, Sharma SS (2010). Concurrent targeting of nitrosative stress–PARP pathway corrects functional, behavioral and biochemical deficits in experimental diabetic neuropathy. Biochem Biophys Res Commun 391: 102–106. [DOI] [PubMed] [Google Scholar]
- Nie F, Wang J, Su D, Shi Y, Chen J, Wang H et al. (2013). Abnormal activation of complement C3 in the spinal dorsal horn is closely associated with progression of neuropathic pain. Int J Mol Med 31: 1333–1342. [DOI] [PubMed] [Google Scholar]
- Oda T, Morikawa N, Saito Y, Masuho Y, Matsumoto S (2000). Molecular cloning and characterization of a novel type of histamine receptor preferentially expressed in leukocytes. J Biol Chem 275: 36781–36786. [DOI] [PubMed] [Google Scholar]
- Ohtori S, Takahashi K, Moriya H, Myers RR (2004). TNF‐alpha and TNF‐alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine 29: 1082–1088. [DOI] [PubMed] [Google Scholar]
- Okoro T, Tafazal SI, Longworth S, Sell PJ (2010). Tumor necrosis alpha blocking agent (etanercept): a triple blind randomized controlled trial of its use in treatment of sciatica. J Spinal Disord Tech 23: 74–77. [DOI] [PubMed] [Google Scholar]
- Sacerdote P, Franchi S, Moretti S, Castelli M, Procacci P, Magnaghi V et al. (2013). Cytokine modulation is necessary for efficacious treatment of experimental neuropathic pain. J Neuroimmune Pharmacol 8: 202–211. [DOI] [PubMed] [Google Scholar]
- Saifi GM, Szigeti K, Snipes GJ, Garcia CA, Lupski JR (2003). Molecular mechanisms, diagnosis, and rational approaches to management of and therapy for Charcot–Marie–Tooth disease and related peripheral neuropathies. J Invest Med 51: 261–283. [DOI] [PubMed] [Google Scholar]
- Salvemini D, Little JW, Doyle T, Neumann WL (2011). Roles of reactive oxygen and nitrogen species in pain. Free Radic Biol Med 51: 951–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna MD, Quattrone A, Mello T, Ghelardini C, Galeotti N (2014). Increased expression of the RNA‐binding protein HuD promotes neuroregeneration in antiretroviral neuropathy. Exp Neurol 261: 343–353. [DOI] [PubMed] [Google Scholar]
- Sanna MD, Ghelardini C, Galeotti N (2015). Activation of JNK pathway in spinal astrocytes contributes to acute ultra‐low dose morphine thermal hyperalgesia. Pain 156: 1265–1275. [DOI] [PubMed] [Google Scholar]
- Sanna MD, Stark H, Lucarini L, Ghelardini C, Masini E, Galeotti N (2015b). Histamine H4 receptor activation alleviates neuropathic pain through differential regulation of ERK, JNK and P38 MAPK phosphorylation. Pain 156: 2492–2504. [DOI] [PubMed] [Google Scholar]
- Schloss JM, Colosimo M, Airey C, Masci PP, Linnane AW, Vitetta L (2013). Nutraceuticals and chemotherapy induced peripheral neuropathy (CIPN): a systematic review. Clin Nutr 32: 888–893. [DOI] [PubMed] [Google Scholar]
- Seifert R, Schneider EH, Dove S, Brunskole I, Neumann D, Strasser A et al. (2011). Paradoxical stimulatory effects of the “standard” histamine H4‐receptor antagonist JNJ7777120: the H4 receptor joins the club of 7 transmembrane domain receptors exhibiting functional selectivity. Mol Pharmacol 79: 631–638. [DOI] [PubMed] [Google Scholar]
- Smith FM, Haskelberg H, Tracey DJ, Moalem‐Taylor G (2007). Role of histamine H3 and H4 receptors in mechanical hyperalgesia following peripheral nerve injury. Neuroimmunomodulation 14: 317–325. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strakhova MI, Nikkel A, Manelli AM, Hsieh G, Esbenshade TA, Brioni JD et al. (2009). Localization of histamine H4 receptors in the central nervous system of human and rat. Brain Res 1250: 41–48. [DOI] [PubMed] [Google Scholar]
- Sui BD, Xu TQ, Liu JW, Wei W, Zheng CX, Guo BL et al. (2013). Understanding the role of mitochondria in the pathogenesis of chronic pain. Postgrad Med J 89: 709–174. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Masini E, Mazzocca C, Cuzzocrea S, Ciampa A, Suzuki H et al. (2004). Inhibition of poly(ADP‐ribose) polymerase prevents allergen‐induced asthma‐like reaction in sensitized Guinea pigs. J Pharmacol Exp Ther 311: 1241–1248. [DOI] [PubMed] [Google Scholar]
- Vranken JH (2012). Elucidation of pathophysiology and treatment of neuropathic pain. Cent Nerv Syst Agents Med Chem 12: 304–314. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Inagaki S, Nomura T, Takagi H (1992). Differential regulation of manganese and copper/zinc superoxide dismutases by facial nerve transection. Brain Res 582: 342–345. [DOI] [PubMed] [Google Scholar]
- Zelko IN, Mariani TJ, Folz RJ (2002). Superoxide dismutase multigene family: a comparison of the CuZn‐SOD (SOD1), Mn‐SOD (SOD2), and EC‐SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med 33: 337–349. [DOI] [PubMed] [Google Scholar]