

Abstract
Background and Purpose
Peptides from venomous animals have long been important for understanding pain mechanisms and for the discovery of pain treatments. Here, we hypothesized that Phα1β, a peptide from the venom of the armed spider Phoneutria nigriventer, produces analgesia by blocking the TRPA1 channel.
Experimental Approach
Cultured rat dorsal root ganglion (DRG) neurons, human fetal lung fibroblasts (IMR90) or HEK293 cells expressing the human TRPA1 (hTRPA1‐HEK293), human TRPV1 (hTRPV1‐HEK293) or human TRPV4 channels (hTRPV4‐HEK293), were used for calcium imaging and electrophysiology. Nociceptive responses induced by TRPA1, TRPV1 or TRPV4 agonists or by bortezomib were investigated in mice.
Key Results
Phα1β selectively inhibited calcium responses and currents evoked by the TRPA1 agonist, allyl isothiocyanate (AITC), on hTRPA1‐HEK293, IMR90 fibroblasts and DRG neurons. Phα1β did not affect calcium responses evoked by selective TRPV1 (capsaicin) or TRPV4 (GSK 1016790A) agonists on the various cell types. Intrathecal (i.t.) and intraplantar (i.pl.) administration of low doses of Phα1β (up to 300 pmol per paw) attenuated acute nociception and mechanical and cold hyperalgesia evoked by AITC (i.t. or i.pl.), without affecting responses produced by capsaicin or hypotonic solution. Notably, Phα1β abated the TRPA1‐dependent neuropathic pain‐like responses induced by bortezomib. In vitro and in vivo inhibition of TRPA1 by Phα1β was reproduced by a recombinant form of the peptide, CTK 01512‐2.
Conclusions and Implications
Phα1β and CTK 01512–2 selectively target TRPA1, but not other TRP channels. This specific action underlines the potential of Phα1β and CTK 01512‐2 for pain treatment.
Abbreviations
- AITC
allyl isothiocyanate
- BTZ
bortezomib
- CIPN
chemotherapy‐induced peripheral neuropathy
- DRG
dorsal root ganglia
- i.pl.
intraplantar
- IMR90
human fetal lung fibroblasts
- PWT
paw withdrawal threshold
- VGCCs
voltage‐gated calcium channels
Tables of Links
| LIGANDS |
|---|
| AITC, allyl isothiocyanate |
| BTZ, bortezomib |
| Capsaicin |
| Capsazepine |
| GSK1016790A |
| HC030031 |
| HC067047 |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b)
Introduction
The poison of venomous animals is a rich source of potent and selective ion channel blockers with potential analgesic effects (Gomez et al., 2002; Estrada et al., 2007). Phα1β is a peptide purified from the venom of the armed spider Phoneutria nigriventer, that reversibly inhibits voltage‐gated calcium channels (VGCCs), with some selectivity for N‐type VGCCs (Vieira et al., 2005). This peptide exerts antinociceptive effects in several preclinical pain models, such as post‐surgical, neuropathic and cancer‐related pain (Souza et al., 2008; Rigo et al., 2013a,b). Phα1β seems to have a good tolerability (Rigo et al., 2013b), with a higher therapeutic index than the selective N‐type VGCC blocker, ω‐conotoxin MVIIA, suggesting that additional mechanisms besides VGCC blockade contribute to its antinociceptive effect.
The TRPA1 channel, a non‐selective cation channel activated by reactive endogenous and exogenous substances and noxious cold, is now considered to represent a major pain transduction pathway (Andrade et al., 2012, Nassini et al., 2014). TRPA1 channels are co‐expressed with other TRP channels, including the capsaicin receptor, (TRPV1 channels), in a specific subset of primary sensory neurons, with cell bodies located in trigeminal, vagal and dorsal root ganglia (DRG) (Story et al., 2003, Andrade et al., 2012). A variety of studies in classical rodent models indicated that TRPA1 channels contributed to signal acute nociception and hyperalgesia in models of inflammatory and neuropathic pain (Baraldi et al., 2010; Andrade et al., 2012; Nassini et al., 2014). Among several examples, in a model of neuropathic pain by chemotherapeutic agents, such as oxaliplatin (Nassini et al., 2011) or bortezomib (BTZ) (Trevisan et al., 2013), mechanical and cold hyperalgesia were found to be totally mediated by TRPA1 channels. Thus, these channels are potential targets for the development of new analgesic drugs.
Some animal venoms contain substances that target TRP channels, including APHC1, which antagonizes TRPV1, and the tarantula venom peptide, ProTx‐I, which inhibits TRPA1 channels (Andreev et al., 2008; Gui et al., 2014). Phα1β was found to reduce nociceptive responses evoked by capsaicin administration (Castro‐Junior et al., 2013). Notably, the reduction was obtained by using very high local doses of the toxin, while in vitro 2 μM Phα1β failed to inhibit TRPV1 channels. The present investigation aimed at identifying additional mechanisms, likely to be produced by small concentrations or doses of Phα1β, which might contribute to the analgesic properties of the toxin. We report here that Phα1β is a potent and selective antagonist of TRPA1 channels and does not target other TRP channels expressed by nociceptors.
Methods
Animals
All animal care and experimental procedures were carried out according to the current European Communities Council‐ECC guidelines for the care of laboratory animals, the Italian legislation (DL 116/92) application of the ECC directive (86/609/EEC) and ethical guidelines for investigations of experimental pain in conscious animals. All protocols were conducted under the University of Florence research permit #204/2012‐B. In addition, the number of animals and intensity of noxious stimuli used were the minimum necessary to demonstrate consistent effects of the treatments used. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Male C57BL/6 mice (20–25 g, Harlan Laboratories, Milan, Italy) or Sprague Dawley rats (150–200 g, male, Harlan Laboratories) were used inthese experiments. Animals were housed in a controlled‐temperature environment in individually ventilated cages: 10 per cage (mice) or five per cage (rats), with wood shaving bedding and nesting material, maintained at 22 ± 1°C. Animals were housed with a 12 h light/dark cycle (lights on at 0700 h) and fed with rodent chow (Global Diet 2018, Harlan, Lombardy, Italy) and tap water ad libitum. Animals were allowed to acclimatize to their housing environment for at least 7 days prior to experimentation and to the experimental room for 1 h before experiments.
General procedures
BTZ‐induced neuropathic pain in mice is considered a relevant model of neuropathic pain caused by chemotherapy treatment (Trevisan et al., 2013). In fact, chemotherapy‐induced peripheral neuropathy (CIPN) has emerged as a major complication of BTZ therapy in patients, which usually appears in the first courses of therapy with a number of sensory and painful symptoms, including a lower threshold to mechanical and cold stimuli (Cata et al., 2007). No effective treatment exists for BTZ‐evoked CIPN.
Allocation concealment was performed using a randomization procedure (http://www.randomizer.org/). Experimenters were blinded to the drug treatment when performing tests. No animals were excluded from the analysis. Each experiment was repeated two to three times (using two or three animals in each repetition) between 0800 and 1700 h.
For the in vivo experiments, the primary outcome was acute spontaneous nociception, and secondary outcomes were mechanical and cold hyperalgesia. The group size for each experiment was determined through sample size estimation (Armitage and Berry, 1987) based on the primary outcome. Expected standard deviation values were based on pilot results. The minimum effect size was considered as a prevention of at least 30% of the nociception response, for a significance level of 5%, with a test power of 90% and a two‐tailed hypothesis test. Thus, we estimated a group size of six mice for each experimental group.
Drug treatments
Phα1β (10–300 pmol per site or pmol per paw), CTK 01512–2 (100 pmol per site or 300 pmol per paw), HC‐030031 (HC03, 30 nmol per site or 300 nmol per paw), ω‐conotoxin MVIIA (100 pmol per site or 300 pmol per paw) and allyl isothiocyanate (AITC, 0.01–3 nmol per site or 10 nmol per paw) were injected intrathecally (i.t.) in a volume of 5 μL per site (Hylden and Wilcox, 1980) or intraplantarly (i.pl.) in a volume of 10 μL per paw (Andrade et al., 2012). Hypotonic solution (0.27% NaCl), capsaicin (0.1 nmol per paw), capsazepine (1 nmol per paw) and HC‐067047 (300 nmol per paw) were injected i.pl. in a volume of 10 μL per paw. BTZ (1 mg·kg−1) was injected i.p. Phα1β and CTK 01512–2 were dissolved in PBS; HC‐030031, HC‐067047 and capsazepine were dissolved in DMSO 5%; BTZ and AITC were dissolved in DMSO 1%; capsaicin was dissolved in 0.005% ethanol; and NaCl 0.27% was dissolved in Milli‐Q water. The drug doses were based on pilot experiments and on previous studies (Andrade et al., 2012; Castro‐Junior et al., 2013; Trevisan et al., 2013; Rigo et al., 2013b; Nassini et al., 2015).
Cell culture and isolation of primary sensory neurons
Human fetal lung fibroblasts (IMR90; ATCC, Manassas, VA), used for the study of cells constitutively expressing the human TRPA1 channel, were cultured in DMEM supplemented with 10% FBS, 2 mM L‐glutamine, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin. Untransfected HEK293 cells (ATCC, Manassas, VA) and HEK293 cells stably transfected with the cDNA of human TRPA1 (hTRPA1‐HEK293), or with the cDNA of human TRPV1 (hTRPV1‐HEK293), or with the cDNA of human TRPV4 channels (hTRPV4‐HEK293), were cultured as previously described (Nassini et al., 2012). All cells were cultured in an atmosphere of 95% air and 5% CO2 at 37°C.
Primary sensory neurons were isolated from Sprague–Dawley rats and cultured as previously described (Materazzi et al., 2012). Briefly, ganglia were bilaterally excised under a dissection microscope and transferred to HBSS containing 2 mg·mL−1 of collagenase type 1A and 1 mg·mL−1 of trypsin for enzymic digestion (30 min, 37°C). Ganglia were then transferred to warmed DMEM containing 10% FBS, 10% horse serum, 2 mM·L‐glutamine, 100 U·mL−1 penicillin and 100 mg·mL−1 streptomycin and dissociated into single cells by several passages through a series of syringe needles (23–25 G). Medium and ganglia cells were filtered to remove debris and centrifuged. The pellet was suspended in DMEM with added 100 ng·mL−1 mouse‐NGF and 2.5 mM cytosine‐b‐D‐arabinofuranoside free base. Neurons were then plated on 25 mm‐diameter glass coverslips coated with poly‐L‐lysine (8.3 μM) and laminin (5 μM).
Calcium imaging assay
Intracellular calcium was measured in transfected and untransfected HEK293 and IMR90 cells, or in rat DRG neurons, as previously reported (Materazzi et al., 2013). Plated cells were loaded with 5 μM Fura‐2 AM‐ester (Alexis Biochemicals; Lausen, Switzerland) added to the buffer solution (37°C) containing the following (in mM): 2 CaCl2; 5.4 KCl; 0.4 MgSO4; 135 NaCl; 10 D‐glucose; 10 HEPES and 0.1% bovine serum albumin at pH 7.4. After loading (40 min), cells were washed and transferred to a chamber on the stage of a Nikon Eclipse TE‐2000 U microscope for recording. Cells were excited alternately at 340 and 380 nm to indicate relative intracellular calcium changes by the Ratio340/380 (R340/380) recorded with a dynamic image analysis system (Laboratory Automation 2.0; RCSoftware, Florence, Italy). To evoke a TRPA1 channel‐dependent calcium response, cells and neurons were challenged with the selective agonist, AITC (1–10 μM). Buffer solution containing DMSO 0.3% was used as vehicle. Capsaicin (0.1 μM) was used to identify TRPV1‐expressing neurons. Cells or neurons were pre‐exposed to Phα1β, CTK 01512–2, HC‐030031 or vehicle (DMSO 0.3%) before (10 min) the acute addition of the TRPA1 channel agonist. Results are expressed as the percentage of the increase in R340/380 over baseline, normalized to the maximum effect induced by ionomycin (5 μM) added at the end of each experiment (% Change in R340/380), or as the percentage of the inhibitory effect on the calcium response evoked by AITC (% AITC response) for constructing the concentration–response curves in the presence of PDs.
Electrophysiology
Whole‐cell patch‐clamp recordings were performed in IMR90 cells on a poly‐Llysine‐coated 13 mm‐diameter glass coverslips (Nassini et al., 2015). Each coverslip was transferred to a recording chamber (1 mL volume) mounted on the platform of an inverted microscope (Olympus CKX41, Milan, Italy) and superfused at a flow rate of 2 mL·min−1 with a standard extracellular solution containing (in mM): HEPES 10, D‐glucose 10, NaCl 147, KCl 4, MgCl2 1 and CaCl2 2 (pH adjusted to 7.4 with NaOH). Borosilicate glass electrodes (Harvard Apparatus, Holliston, MA, USA) were pulled with a Sutter Instruments puller (model P‐87) to a final tip resistance of 4–7 MΩ. Pipette solution used contained the following (in mM): K‐gluconate 134, KCl 10, EGTA 11 and HEPES 10 (pH adjusted to 7.4 with KOH). Data were acquired with an Axopatch 200B amplifier (Axon Instruments, CA, USA), stored and analysed with a pClamp 9.2 software (Axon Instruments, CA, USA). Cells were voltage‐clamped at a holding potential of −60 mV, and currents were detected as inward currents activated on cell superfusion with AITC (30 and 100 μM) or KCl (90 mM) in the absence or presence of CTK 01512–2 (3 μM) or HC‐030031 (50 μM). Cell membrane capacitance was calculated in each cell throughout the experiment by integrating the capacitive currents elicited by a ±10mV voltage pulse. Peak currents activated by each compound were normalized to cell membrane capacitance and expressed as mean of the current density (pA/pF) in averaged results.
Behavioural studies
For behavioural experiments, after habituation, C57BL/6 mice were randomized into treatment groups, and an investigator blinded to treatments recorded the responses. In a first series of experiments, C57BL/6 mice were previously (10 min) treated with Phα1β (30–300 pmol per paw), CTK 01512–2 (300 pmol per paw) or their vehicle (PBS), via i.pl. injection (10 μL). Nociceptive response, as well as mechanical and cold hyperalgesia induced by i.pl. injection of the TRPA1 agonist, AITC (10 nmol per paw), were recorded. Mechanical and cold hyperalgesia were assessed from 0.25 to 2 h after i.pl. AITC treatment. The nociceptive response (total time spent in lifting/licking) or the mechanical and cold hyperalgesia produced by AITC in naïve mice was not affected by the vehicle (PBS). Some C57BL/6 mice were pretreated with HC‐030031 (TRPA1 antagonist, 300 nmol per paw, 10 min before) as a control. In addition, to test the ability of the peptide to selectively inhibit TRPA1 channels, the effect of pretreatment with Phα1β (30–300 pmol per paw, 10 min before) was evaluated on the nociceptive response and mechanical hyperalgesia induced by i.pl. (10 μL) capsaicin (TRPV1 channel agonist, 0.1 nmol per paw), hypotonic solution (TRPV4 channel stimulator, 0.27% NaCl) or their vehicles (0.005% ethanol and 0.9% NaCl, respectively) 0.25 h after capsaicin or 0.27% NaCl injection. Some mice were pretreated with capsazepine (TRPV1 channel antagonist, 1 nmol per paw, 10 min before capsaicin) and HC‐067047 (TRPV4 channel antagonist, 300 nmol per paw, 10 min before 0.27% NaCl) or their vehicle (DMSO 5%).
In another set of experiments, we assessed the hyperalgesia evoked by i.t. injection of AITC. Mechanical and cold hyperalgesia induced by AITC (0.01–3 nmol per site) were measured at 0.5 to 6 h after treatment. Next, the effects of both Phα1β and CTK 01512–2 were evaluated on mechanical and cold hyperalgesia induced by i.t. injection (5 μL per site) of the TRPA1 channel agonist, AITC (1 nmol per site). Mice were treated 10 min before AITC treatment with Phα1β (10–100 pmol per site, i.t.), CTK 01512–2 (100 pmol per site, i.t.), HC‐030031 (30 nmol per site, i.t.) or their vehicle, and 0.5 to 2 h after treatment, the mechanical and cold hyperalgesia were measured.
A single administration of BTZ (1 mg·kg−1, i.p.) has previously been shown to produce persistent (15–20 days) mechanical and cold hyperalgesia in mice (Trevisan et al., 2013). At day 7 after BTZ injection (1 mg·kg−1, i.p.), mechanical and cold hyperalgesia were measured from 0.5 to 4 h after treatment with Phα1β (10–100 pmol per site, i.t.), CTK 01512–2 (100 pmol per site, i.t. or 300 pmol per paw), HC‐030031 (30 nmol per site, i.t. or 300 nmol per paw), ω‐conotoxin MVIIA (100 pmol per site, i.t. 300 pmol per paw) or their vehicle.
Behavioural tests
Acute spontaneous nociception test
Immediately after i.pl. injection with AITC, capsaicin and 0.27% NaCl, mice were placed inside a plexiglass chamber, and the total time spent in lifting/licking of the injected hind paw was recorded for 10 min, as an indicative parameter of nociception (Trevisani et al., 2007). The i.pl. injection with vehicles of AITC, capsaicin and 0.27% NaCl produced nociceptive behaviour for a maximum of 2 s.
Von Frey test
Mechanical nociceptive pain was assessed through the measurement of paw withdrawal threshold (PWT) using the ‘Up‐and‐Down’ paradigm, as previously described (Tonello et al., 2014). Mechanical nociceptive threshold was determined before (basal level threshold) and after different treatments. The 50% mechanical PWT (in g) response was then calculated from resulting scores as previously described by Dixon (1980).
Cold stimulation
Cold hyperalgesia was assessed in mice by measuring the acute nocifensive response to the acetone‐evoked evaporative cooling as previously reported (Trevisan et al., 2013). Briefly, a droplet (50 μL) of acetone, formed on the flat‐tip needle of a syringe, was gently touched to the plantar surface of the mouse hind paw, and the time spent in elevation and licking of the plantar region over a 60 s period was measured.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The results are expressed as the means ± SEM, with the exception of the ED50 or IC50 values, which were reported as the geometric mean accompanied by the corresponding 95% CIs. The values of IC50 and ED50 for concentration and dose–response curves were calculated with non‐linear regression using a sigmoidal dose–response curve. The percentages of maximum effect (Emax) and maximum inhibition (Imax) were reported as the means ± SEM for each individual experiment in relation to the control values. Differences among three or more groups at one time point were analysed by one‐way ANOVA followed by Newman–Keuls' post hoc test. Differences among three or more groups at time course curves were analysed by two‐way ANOVA (treatment and time as factors, time as repeated measure) followed by Bonferroni's post hoc test. P values less than 0.05 were considered significant. To meet the ANOVA assumptions, the mechanical hyperalgesia data were log transformed prior to statistical analysis. Statistical analysis was performed using GraphPad Software 5.0 (GraphPadSoftware, San Diego, CA, USA).
Materials
Native Phα1β was purified as previously described and had the following amino acid sequence: ACIPRGEICT DDCECCGCDN QCYCPPGSSL GIFKCSCAHA NKYFCNRKKE KCKKA (Cordeiro et al., 1993). The recombinant forms of Phα1β (CTK 01512‐2) and ω‐conotoxin MVIIA were purchased from Giotto Biotech S.r.l. (Florence, Italy) and Latoxan (Portes lès Valence, France). The stock solutions of drugs were prepared with PBS (pH 7.4) in siliconized plastic tubes, maintained at −20°C and diluted to the desired concentration just before use. The TRPA1 channel‐selective antagonist HC‐030031 was synthesized as previously described (Andre et al., 2008), and BTZ was purchased from LC Laboratories. Unless otherwise indicated, all reagents were from Sigma (St Louis, MO, USA) and were dissolved in appropriate vehicle solutions.
Results
Phα1β is a selective antagonist of TRPA1 channels
The ability of Phα1β or CTK 01512–2 to affect calcium responses evoked by stimulation of TRPA1 channels was investigated in hTRPA1‐HEK293 cells. Similar to the TRPA1 channel antagonist HC‐030031, Phα1β and CTK 01512–2 inhibited responses evoked by AITC (Figure 1A and Table 1). In hTRPV1‐HEK293 cells, calcium responses evoked by the selective TRPV1 agonist, capsaicin, were abated by the selective channel antagonist, capsazepine, but were unaffected by Phα1β or CTK 01512–2 (Figure 1B). Likewise, calcium responses evoked by the selective TRPV4 channel agonist, GSK1016790A, were abated by the corresponding channel antagonist, HC‐067047, in hTRPV4‐HEK293 cells, but were unaffected by Phα1β or CTK 01512–2 (Figure 1B).
Figure 1.
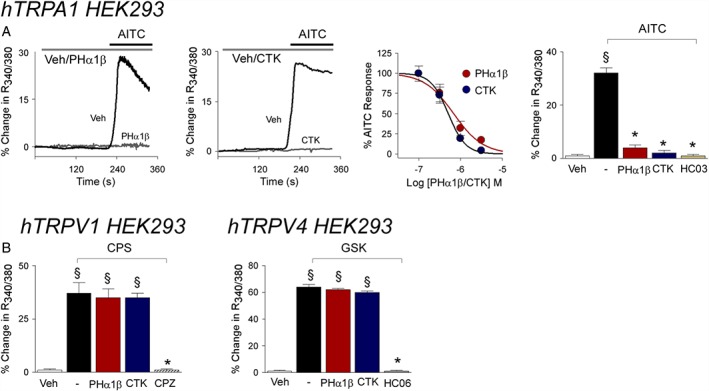
Phα1β and its recombinant form (CTK 01512–2) selectively inhibit the calcium response evoked by stimulation of human TRPA1 channels. (A) Typical traces of the inhibitory effect of pre‐exposure (10 min) to Phα1β (3 μM) and CTK 01512–2 (3 μM) on the calcium response evoked by the TRPA1 channel agonist, AITC (30 μM), in HEK293 cells transfected with the cDNA for human TRPA1 channels (hTRPA1‐HEK293). Concentration–response curve of the inhibitory effect of Phα1β and CTK 01512–2 on the calcium response evoked by AITC (AITC concentrations are 30 μM). Phα1β (3 μM) and CTK 01512–2 (3 μM) and the selective TRPA1 channel antagonist HC‐030031 (HC03, 30 μM) inhibit calcium response evoked in hTRPA1‐HEK293 cells by AITC. (B) Phα1β (3 μM) and CTK 01512–2 (3 μM) do not affect responses evoked by capsaicin (CPS, 0.1 μM) in HEK293 cells transfected with the cDNA for human TRPV1 channels (hTRPV1‐HEK293) and by GSK1016790A (GSK, 50 nM) in HEK293 cells transfected with the cDNA for human TRPV4 channels (hTRPV4‐HEK293). Values are mean ± SEM of n > 25 cells from at least three different experiments for each condition. § P < 0.05, significantly different from vehicle (Veh); *P < 0.05, significantly different from AITC.
Table 1.
The concentration inhibiting 50% of the maximum response (with corresponding CIs) of Phα1β and CTK 01512–2 on calcium responses evoked by AITC
| Cell type | Phα1β (nM) | CTK 01512–2 (nM) |
|---|---|---|
| TRPA1‐HEK293 | 681 (472–983) | 506 (414–619) |
| IMR90 | 40 (22–74) | 28 (15–51) |
| DRG neurons | 32 (21–47) | 34 (11–101) |
The ability of Phα1β or CTK 01512–2 to antagonize TRPA1 channels was also studied in IMR90 fibroblasts, a cell line from which TRPA1 channels were originally cloned (Jaquemar et al., 1999), and in rat DRG neurons that constitutively express TRPV1, TRPV4 and TRPA1 channels. Pre‐exposure to Phα1β, CTK 01512–2 or HC‐030031 inhibited responses evoked by AITC in IMR90 cells and in rat DRG neurons (Figure 2 and Table 1). Neither Phα1β nor CTK 01512–2 produced any stimulatory effect nor affected any responses to other excitatory stimuli, such as the activating peptide of the human proteinase activated receptor‐2 (hPAR2‐AP) in IMR90 cells, or the selective TRPV1 channel agonist, capsaicin, in DRG neurons, thus indicating selectivity (Figure 2).
Figure 2.
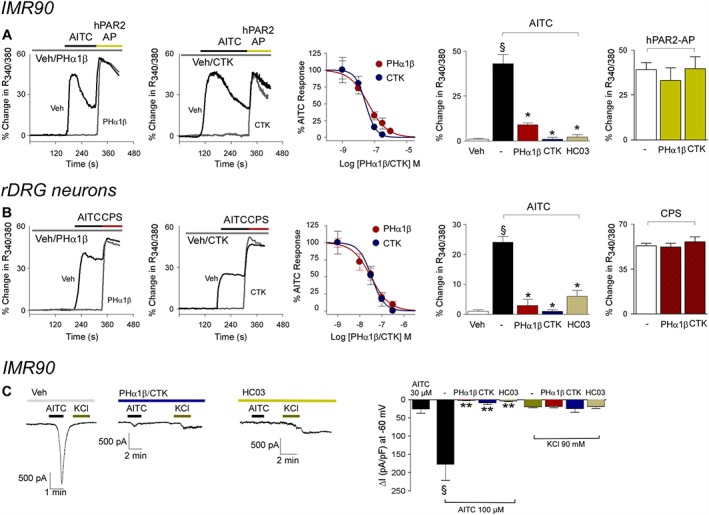
Phα1β and its recombinant form (CTK 01512–2) selectively inhibit calcium response or currents evoked by stimulation of TRPA1 channels. (A) Typical traces of the inhibitory effect of pre‐exposure (10 min) to the Phα1β (300 nM) and CTK 01512–2 (300 nM) on the calcium response evoked by the TRPA1 channel agonist, AITC (10 μM), in cultured IMR90 cells. Concentration–response curves of the inhibitory effect of Phα1β and CTK 01512–2 on the calcium response evoked by AITC in IMR90 cells (AITC concentrations are 1 μM). Phα1β (300 nM) and CTK 01512–2 (300 nM) and the selective TRPA1 channel antagonist HC‐030031 (HC03, 30 μM) inhibit calcium responses evoked in IMR90 cells by AITC. Phα1β (300 nM) and CTK 01512–2 (300 nM) do not affect responses evoked by the activating peptide of the human PAR2 receptor (hPAR2‐AP, 100 μM) in IMR90 cells. TRP, trypsin. (B) Typical traces of the inhibitory effect of pre‐exposure (10 min) to Phα1β (300 nM) and CTK 01512–2 (300 nM) on the calcium response evoked by AITC (10 μM), in cultured rat DRG (rDRG) neurons. Concentration–response curves of the inhibitory effect of Phα1β and CTK 01512–2 on the calcium response evoked by AITC in rDRG neurons (AITC concentrations are 10 μM). Phα1β (300 nM) and CTK 01512–2 (300 nM) and HC03 (30 μM) inhibit calcium response evoked in rDRG neurons by AITC. Phα1β (300 nM) and CTK 01512–2 (300 nM) do not affect responses evoked by capsaicin (CPS, 0.1 μM) in rDRG neurons. Values are mean ± SEM of n > 25 cells from at least three different experiments for each condition. § P < 0.05, significantly different from vehicle (Veh); *P < 0.05, significantly different from AITC. (C) Typical traces and pooled data obtained by whole‐cell patch‐clamp recordings in cultured IMR90 exposed to the selective TRPA1 channel agonist AITC. The inward currents evoked at −60 mV by AITC (100 μM), but not those evoked by KCl (90 mM), are attenuated by the selective TRPA1 channel antagonist, HC‐030031 (HC03; 50 μM), Phα1β or CTK 01512–2 (3 μM). Values are the mean ± SEM of at least five independent experiments. §P < 0.05, significantly different from AITC 30 μM; **P < 0.05, significantly different from AITC 100 μM alone.
The ability of Phα1β or CTK 01512–2 to affect inward currents evoked by TRPA1 channel stimulation was investigated in IMR90 cells. In such cells, inward currents evoked by AITC were reduced by HC‐030031, Phα1β or CTK 01512–2 (Figure 2C). HC‐030031, Phα1β or CTK 01512–2 did not affect the inward currents evoked by high KCl, indicating selectivity.
Phα1β selectively blocks nocifensive responses evoked by reactive TRPA1 channel agonist
Next, we studied whether Phα1β or CTK 01512–2 produced in vivo antinociceptive and antihyperalgesic effects via TRPA1channel antagonism. Phα1β administration (i.pl.) dose‐dependently reduced spontaneous nociception evoked by i.pl. AITC injection (Figure 3A) with an ED50 (CI) of 42 (32–54) pmol per paw. Administration (i.pl.) of CTK 01512–2 also reduced the spontaneous nociception (Figure 3A and Table 2). AITC induced spontaneous nociception, mechanical hyperalgesia and cold‐hyperalgesia (Figure 3A–C and Table 2). Moreover, Phα1β and CTK 01512–2 reduced mechanical and cold hypersensitivities evoked by i.pl injection of AITC (Figure 3B–C and Table 2). Phα1β did not affect TRPV1‐ or TRPV4 channel‐mediated spontaneous nociception response and mechanical hyperalgesia evoked by i.pl. capsaicin or a hypotonic stimulus (which is known to activate TRPV4 channels) (Benemei et al., 2015), respectively (Figure 3D–E), thus indicating in vivo selectivity.
Figure 3.
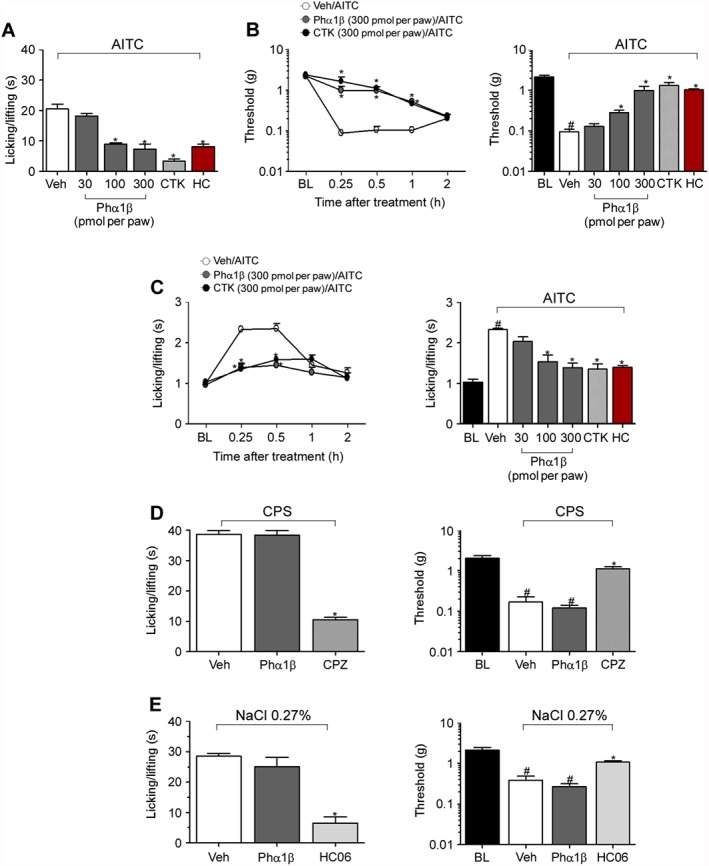
Phα1β and its recombinant form (CTK 01512–2) selectively block nocifensor responses evoked by reactive TRPA1 channel agonist. (A–C) Effect of increasing doses of i.pl. administration of Phα1β (30–300 pmol per paw) or CTK 01512–2 (300 pmol per paw) and the selective TRPA1 channel antagonist, HC‐030031 (HC, 300 nmol per paw) on the nociceptive response evoked by the i.pl. injection (10 μL, i.pl.) of AITC (10 nmol per paw) in C57BL/6 mice. (B–C) Time course and dose–response curve (0.25 h after AITC treatment) of Phα1β (30–300 pmol per paw) or CTK 01512–2 (300 pmol per paw) and HC‐030031 (HC, 300 nmol per paw) on mechanical (B) and cold (C) hyperalgesia evoked by the i.pl. injection of AITC (10 nmol per paw) in C57BL/6 mice. (D) Effect of i.pl. administration of Phα1β or CTK 01512–2 and the selective TRPV1 channel antagonist, capsazepine (CPZ, 1 nmol per paw) on the nociceptive response and mechanical hyperalgesia evoked by the injection (10 μL, i.pl.) of capsaicin (CPS, 0.1 nmol per paw) in C57BL/6 mice. (E) Effect of i.pl. administration of Phα1β or CTK 01512–2 and the selective TRPV4 channel antagonist, HC‐067047 (HC06, 300 nmol per paw) on the nociceptive response and mechanical hyperalgesia evoked by the injection (10 μL, i.pl.) of hypotonic solution (0.27% NaCl per paw) in C57BL/6 mice. Each column represents the mean of six mice, and vertical lines show the SEM. Statistical analysis was performed using one‐way or two‐way ANOVA followed by Student–Newman–Keuls or by Bonferroni's post hoc test respectively. *P < 0.05, significantly different from Veh; # P < 0.05, significantly different from BL values; § P < 0.05, significantly different from Phα1β (300 pmol per paw).
Table 2.
Inhibitory effect of Phα1β, CTK 01512–2 and HC‐030031 on AITC‐induced nociception
| Intraplantar route | Phα1β (300 pmol per paw) | CTK 01512–2 (300 pmol per paw) | HC‐030031 (300 nmol per paw) |
|---|---|---|---|
| AITC‐induced spontaneous nociception (%) | 64 | 84 | 60 |
| AITC‐induced mechanical hyperalgesia (%) | 71 | 82 | 77 |
| AITC‐induced cold hyperalgesia (%) | 72 | 75 | 72 |
| Intrathecal route | Phα1β (100 pmol per site) | CTK 01512–2 (100 pmol per site) | HC‐030031 (30 nmol per site) |
|---|---|---|---|
| AITC‐induced mechanical hyperalgesia (%) | 94 | 94 | 94 |
| AITC‐induced cold hyperalgesia (%) | 91 | 94 | 97 |
The i.t. administration of AITC (1 nmol per site) induced mechanical and cold hyperalgesia that lasted 4 h after administration (Figure 4A–B). AITC (0.1–3 nmol per site, i.t.) caused a dose‐dependent mechanical and cold hyperalgesia with ED50 (CI) of 0.2 (0.1–0.3) and 0.4 (0.2–0.6) nmol per site respectively. Intrathecal administration of Phα1β (10–100 pmol per site, i.t.) reduced in a dose‐dependent manner the mechanical and cold hyperalgesia evoked by AITC (1 nmol per site, i.t.), with an ED50 (CI) of 28 (20–38) or 19 (15–24) pmol per site respectively (Figure 4C–D and Table 2). Similarly, CTK 01512–2 (100 pmol per site, i.t.) reverted mechanical and cold hyperalgesia induced by AITC (Figure 4C–D and Table 2). As expected, HC‐030031 (30 nmol per site) attenuated AITC‐induced mechanical and cold hyperalgesia (Figure 4C–D and Table 2).
Figure 4.
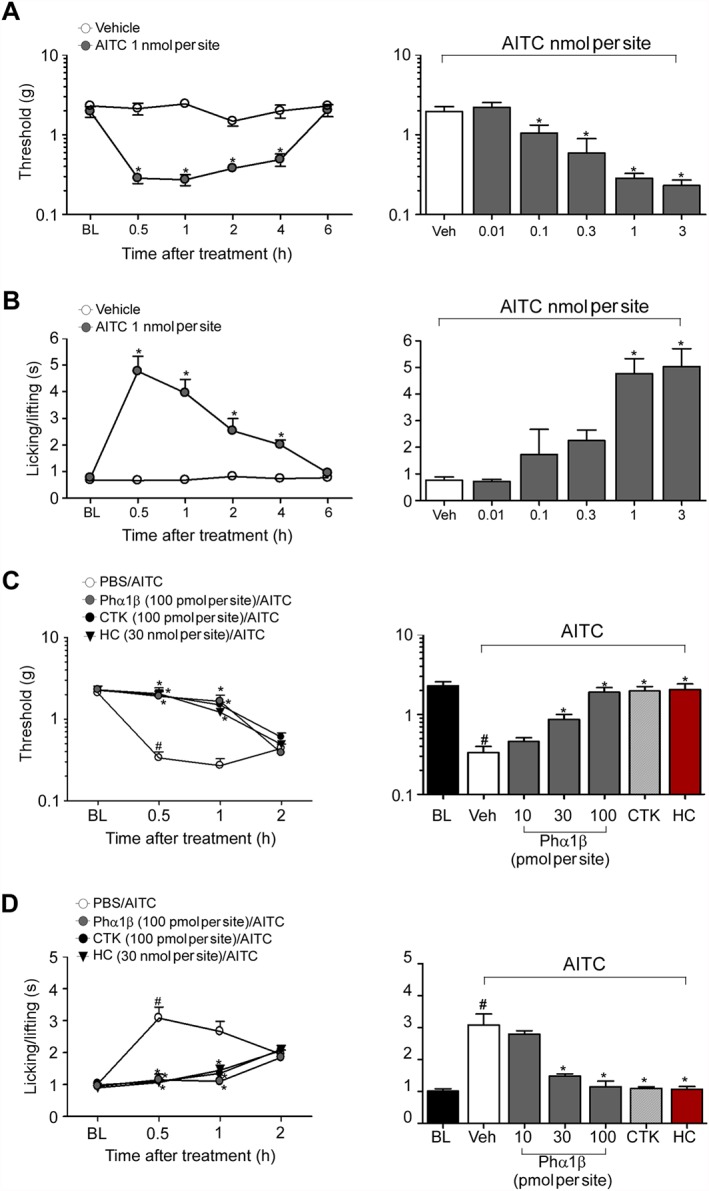
Phα1β and its recombinant form (CTK 01512–2) block the hyperalgesia evoked by i.t. injection of AITC, a TRPA1 channel agonist. (A–B) Time course and dose–response curve of the effect of intrathecal (5 μL, i.t.) administration of AITC (0.01–3 nmol per site) on mechanical (A) and cold (B) nociception. (C–D) Time course and dose–response curve of i.t. administration of Phα1β (10–100 pmol per site), CTK 01512–2 (100 pmol per site) and the selective TRPA1 channel antagonist, HC‐030031 (HC, 30 nmol per site) on mechanical (C) and cold (D) hyperalgesia evoked by the injection (5 μL, i.t.) of AITC (1 nmol per site) in C57BL/6 mice. Dose–response curves were performed 0.5 h after AITC treatment. Each point and column represents the mean of six mice, and vertical lines show the SEM. Statistical analysis was performed using one‐way or two‐way ANOVA followed by Student–Newman–Keuls post hoc test or by Bonferroni's post hoc test respectively. *P < 0.05, significantly different from Veh; # P < 0.05, significantly different from BL values.
Phα1β reduced TRPA1 channel‐dependent hyperalgesia in a model of neuropathic pain induced by the chemotherapeutic agent BTZ
In mice, mechanical and cold hyperalgesia produced by the administration of BTZ have been reported, and we confirm here (Figure 5) that they are entirely dependent on activation of TRPA1 channels (Trevisan et al., 2013). In our model, Phα1β markedly attenuated mechanical and cold hyperalgesia induced by BTZ (Figure 5A–D). Phα1β (i.t., 10–100 pmol per site) markedly reduced mechanical and cold hyperalgesia (92 and 100% inhibition at a dose of 100 pmol per site respectively) with an ED50 (CI) of 19 (13–27) pmol per site and 11 (8–16) pmol per site respectively (Figure 5A–B). CTK 01512–2 (i.t., 100 pmol per site or i.pl., 300 pmol per paw) also reduced mechanical (94 and 100% inhibition respectively) and cold (97 and 100% inhibition respectively) hyperalgesia induced by i.pl. BTZ (Figure 5A–D). The inhibitor of neuronal VGCC, ω‐conotoxin MVIIA, did not affect BTZ‐induced hyperalgesia when given by i.pl. (Figure 5C–D) or i.t. (Figure S1) injection.
Figure 5.
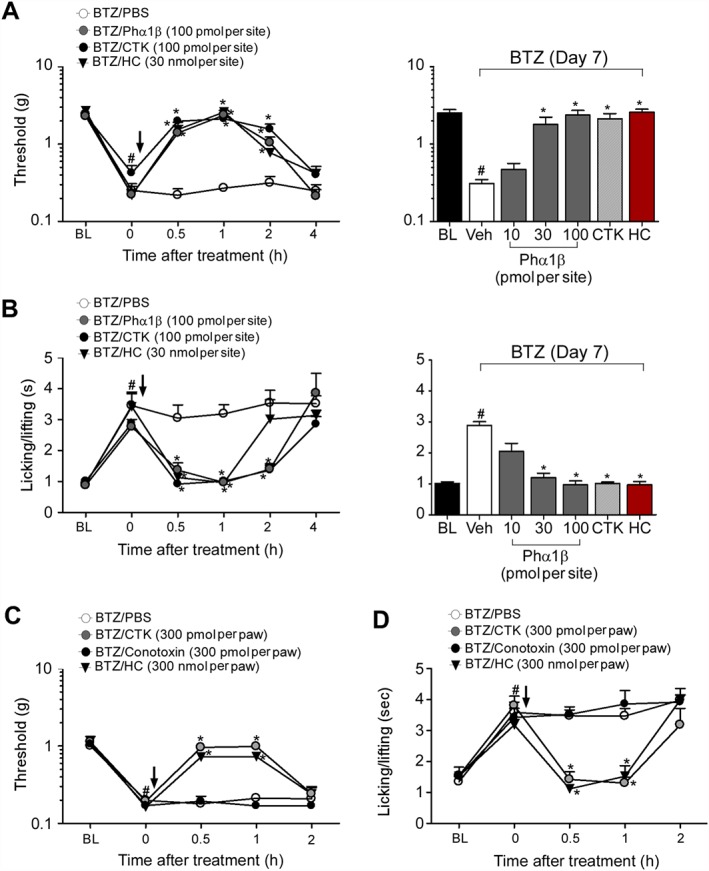
Phα1β and its recombinant form (CTK 01512–2) reduce TRPA1 channel‐dependent hyperalgesia in a model of neuropathic pain induced by BTZ. At day 7 after treatment with BTZ (1 mg·kg−1, i.p.), mechanical hyperalgesia and cold hyperalgesia are increased (BL, basal level threshold at day 0 before BTZ). Time course and dose–response curve of i.t. administration of Phα1β (10–100 pmol per site), CTK 01512–2 (100 pmol per site) and the selective TRPA1 channel antagonist, HC‐030031 (HC, 30 nmol per site) on mechanical (A) and cold (B) hyperalgesia evoked by BTZ (1 mg·kg−1, i.p.) at day 7 after treatment. Time course of the effect of the i.pl. administration of CTK 01512–2 (300 pmol per paw), ω‐conotoxin MVIIA (300 pmol per paw) and HC‐030031 (HC, 300 nmol per paw) on mechanical (C) and cold (D) hyperalgesia evoked by BTZ (1 mg·kg−1, i.p.) at day 7 after treatment. Dose–response curves were performed 0.5 h after AITC treatment. Each point and column represents the mean of six mice, and vertical lines show the SEM. Statistical analysis was performed using one‐way or two‐way ANOVA followed by Student–Newman–Keuls post hoc test or by Bonferroni's post hoc test respectively. *P < 0.05, significantly different from BTZ/PBS; # P < 0.05, significantly different from BL values.
Discussion
In the present study, we show that Phα1β and CTK 01512–2 behave as TRPA1 channel antagonists. Phα1β showed similar efficacy, but a higher potency than the widely used, low MW antagonist, HC‐030031 (McNamara et al., 2007) in inhibiting the AITC‐induced calcium response. Of note, the potency of Phα1β in antagonizing TRPA1 channels was higher than that of the tarantula venom peptide ProTx‐I (Gui et al., 2014). Phα1β potency was lower in the recombinant human TRPA1 channels than in the native rat (DRG neurons) or human (IMR90 fibroblasts) channels. The reason for the difference cannot be the species as the toxin behaves similarly in the rat and human native channels, but rather the recombinant versus the native nature of the TRPA1 channels. Although the underlying mechanism for the difference remains unknown, similar findings have been reported previously (Shapiro et al., 2013; Nassini et al., 2015). One possible hypothesis is that the constitutive form of TRPA1 channels, but apparently not recombinant TRPA1 channels, is coexpressed with accompanying proteins, such as the Tmem100, which may change the affinity of ligands to the channel (Weng et al., 2015). The failure of Phα1β or CTK 01512–2 to affect TRPV1 or TRPV4 channel‐mediated responses, or responses evoked by KCl and hPAR2‐AP, indicated in vitro selectivity for the TRPA1 channels.
The ability of Phα1β or CTK 01512–2 to selectively target TRPA1 channels in vitro was paralleled by the capacity of the two compounds to act as antagonists of these channels in vivo. This effect was obtained via both peripheral (i.pl.) and central (i.t.) routes of administration, as the drug may target channel‐enriched sites, such as the peripheral and central (within the dorsal spinal cord) terminals of nociceptors. The high potency of i.pl. Phα1β to attenuate AITC‐mediated acute nociception and mechanical and cold hyperalgesia derives from the observation that maximum inhibition was attained by a dose of Phα1β about 200 times lower than that of HC‐030031. Selectivity for TRPA1 channels of Phα1β in in vivo experiments is underlined by the fact that the peptide did not affect TRPV1‐ or TRPV4 channel‐mediated acute nociception and mechanical and cold hypersensitivities. In a previous publication (Castro‐Junior et al., 2013), we reported that a high dose of Phα1β (3 nmol per paw), but not lower doses (1 and 0.3 nmol per paw), reduced spontaneous nociception and delayed hyperalgesia induced by i.pl. capsaicin (5 nmol per paw) in rats. Present results support these previous findings (Castro‐Junior et al., 2013), confirming that low doses of the spider toxin peptide Phα1β (1 or 0.3 nmol per paw) did not affect capsaicin‐induced pain‐like responses.
Although few studies have investigated the role of the TRPA1 channel in pain transmission in the spinal cord, the ability of the channel to activate nociceptive signals at this central level has received some support (Raisinghani et al., 2011; Koivisto et al., 2014). Accordingly, we evaluated Phα1β in a central paradigm of nociceptive transmission, using the responses induced by i.t. AITC administration. AITC‐evoked mechanical and cold hyperalgesia is consistent with recent findings that a TRPA1 channel agonist induces mechanical hyperalgesia when injected i.t. in rodents (Raisinghani et al., 2011; Klafke et al., 2012). Consistent with this finding, HC‐030031 and centrally administered Phα1β produced a robust analgesic effect against the i.t. AITC‐mediated mechanical and cold hyperalgesia. Notably, the effect of i.t. Phα1β lasted for a longer period of time than that observed after i.pl. administration. A reduced protease activity in the spinal cord as compared with peripheral tissues (King, 2011; Diao and Meibohm, 2013) might be responsible for such a prolonged half‐life of the peptide and the consequent antinociceptive and antihyperalgesic action of Phα1β. These findings confirm the antagonistic action of Phα1β towards TRPA1 channel‐mediated responses and suggest an additional route of administration for potential clinical applications (Smith et al., 2008; King, 2011) of this venom‐derived drug.
The effect of Phα1β was assessed in a mouse model of CIPN, i.e., that evoked by a single injection of BTZ which had been previously identified as entirely dependent on activation of TRPA1 channels (Trevisan et al., 2013). Administration of Phα1β (both i.pl. and i.t.) inhibited mechanical and cold hyperalgesia evoked by BTZ, with a profile in efficacy and duration similar to that produced by HC‐030031, thus providing further, although indirect, indication that Phα1β evokes analgesia in the BTZ model of CIPN by targeting TRPA1 channels. However, in vitro concentrations of Phα1β required to block TRPA1 channels are similar to those required to block VGCC (Vieira et al., 2005), suggesting that, in vivo, the actions of Phα1β could depend on VGCC inhibition. In agreement with a previous report (Kitamura et al., 2014), we failed to observe any protective effect by either i.pl. or i.t. administration of ω‐conotoxin MVIIA in BTZ‐induced nociceptive hypersensitivities at a dose that was able to produce antinociception (Castro‐Junior et al., 2013; Rigo et al., 2013a,b). As both Phα1β and ω‐conotoxin MVIIA (both i.t.) have previously been found to reverse mechanical hyperalgesia in a model of post‐operative pain (de Souza et al., 2011), whereas a specific TRPA1 channel antagonist (i.t.) was ineffective in such pain models (Wei et al., 2012), the analgesic mechanism of action of Phα1β appears to depend on the pain model under investigation. This analgesic action is likely to derive from inhibition of VGCC in a post‐operative pain model and from antagonism of TRPA1 channels in a model of CIPN.
The low yield of Phα1β from spider venom is a limitation to the use of the native peptide as an analgesic agent (Gomez et al., 2002). A possible alternative is the production of a recombinant peptide, such as CTK 01512–2. CTK 01512–2, showing efficacy and potency similar to that of the native peptide, in terms of inhibiting calcium responses evoked by stimulation of TRPA1 channels in vitro, and robust antinociceptive/antihyperalgesic effects against AITC‐mediated pain‐like behaviours, represents a similarly useful and more easily available drug than the native Phα1β. Poisons of venomous animals have long been an important source of new drugs, including analgesic molecules (Gomez et al., 2002; Estrada et al., 2007). Phα1β, the peptide purified from the venom of the armed spider Phoneutria nigriventer , which previously has been shown to exhibit antinociceptive effects (Souza et al., 2008; Rigo et al., 2013a,b), and its recombinant form (CTK 01512–2) have now been identified as selective and potent TRPA1 channel antagonists with antihyperalgesic effects in a relevant model of neuropathic pain. These findings, in addition to reinforcing the role of TRPA1 channels in pain transmission, suggest Phα1β and CTK 01512–2 as novel strategies for the treatment of painful conditions where TRPA1 channels might be involved. The dual activity of Phα1β on both TRPA1 channels and VGCC may represent a potential advantage of the two drugs that could result in broader activity in human pain conditions. However, the ability to target TRPA1 channels and VGCC might also increase the chance to cause adverse reactions.
Author contributions
R.T., R.N., S.M., P.G., J.F., M.V.G., C.C.J. and S.B. designed the experiments and interpreted the results. C.F., S.M., F.D.L., E.C. and R.T. performed the in vitro experiments. R.T., R.N., M.C.G., J.F. and I.M.M. performed the in vivo experiments.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effect of ω‐conotoxin MVIIA in a model of neuropathic pain induced by bortezomib. Bortezomib (BTZ, 1 mg kg‐1, i.p.) at day 7 after treatment increases mechanical hyperalgesia and cold hyperalgesia. Time course of ω‐conotoxin MVIIA (100 pmol per site) by intrathecal (i.t.) administration does not affect BTZ‐evoked pain‐like responses. Each point represents the mean of 6 mice, and vertical lines show the SEM. Statistical analysis was performed using two‐way ANOVA, followed by Bonferroni's post hoc test. #P < 0.05, significantly different from BL values.
Supporting info item
Supporting info item
Acknowledgements
We thank Prof. A.H. Morice, (University of Hull, Hull, UK) for HEK293 cells stably transfected with the cDNA for human TRPA1 (hTRPA1‐HEK293), Prof. M.J. Gunthorpe (GlaxoSmithKline, Harlow, UK) for HEK293 stably transfected with the cDNA for human TRPV1 (hTRPV1‐HEK293) and Prof. N.W. Bunnett (Monash Institute of Pharmaceutical Sciences, Parkville, Australia) for HEK293 stably transfected with the cDNA for human TRPV4 channels (hTRPV4‐HEK293). This work was supported by Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior ‐ CAPES (Brasília, Distrito Federal, Brazil)(AUX‐PE Toxinologia) and Conselho Nacional de Desenvolvimento Científico – CNPq (Brasília, Distrito Federal, Brazil). We also acknowledge the receipt of fellowships from CAPES – Proc. no. 2622‐14‐9.
Tonello, R. , Fusi, C. , Materazzi, S. , Marone, I. M. , De Logu, F. , Benemei, S. , Gonçalves, M. C. , Coppi, E. , Castro‐Junior, C. J. , Gomez, M. V. , Geppetti, P. , Ferreira, J. , and Nassini, R. (2017) The peptide Phα1β, from spider venom, acts as a TRPA1 channel antagonist with antinociceptive effects in mice. British Journal of Pharmacology, 174: 57–69. doi: 10.1111/bph.13652.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade EL, Meotti FC, Calixto JB (2012). TRPA1 antagonists as potential analgesic drugs. Pharmacol Ther 133: 189–204. [DOI] [PubMed] [Google Scholar]
- Andre E, Campi B, Materazzi S, Trevisani M, Amadesi S, Massi D et al. (2008). Cigarette smoke‐induced neurogenic inflammation is mediated by alpha,beta‐unsaturated aldehydes and the TRPA1 receptor in rodents. J Clin Invest 118: 2574–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreev YA, Kozlov SA, Koshelev SG, Ivanova EA, Monastyrnaya MM, Kozlovskaya EP et al. (2008). Analgesic compound from sea anemone Heteractis crispa is the first polypeptide inhibitor of vanilloid receptor 1 (TRPV1). J Biol Chem 283: 23914–23921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage P, Berry GG (1987). Statistical methods in medical research, 2nd edn. Blackwell Scientific: Oxford, Boston: ; Chicago, Ill.: Distributors, USA, Year Book Medical Publishers. [Google Scholar]
- Baraldi PG, Preti D, Materazzi S, Geppetti P (2010). Transient receptor potential ankyrin 1 (TRPA1) channel as emerging target for novel analgesics and antiinflammatory agents. J Med Chem 53: 5085–5107. [DOI] [PubMed] [Google Scholar]
- Benemei S, Patacchini R, Trevisani M, Geppetti P (2015). TRP channel. Curr Opin Pharmacol 22: 18–23. [DOI] [PubMed] [Google Scholar]
- Castro‐Junior CJ, Milano J, Souza AH, Silva JF, Rigo FK, Dalmolin G et al. (2013). Phα1β toxin prevents capsaicin‐induced nociceptive behavior and mechanical hypersensitivity without acting on TRPV1 channels. Neuropharmacology 71: 237–246. [DOI] [PubMed] [Google Scholar]
- Cata JP, Weng HR, Burton AW, Villareal H, Giralt S, Dougherty PM (2007). Quantitative sensory findings in patients with bortezomib‐induced pain. J Pain 8: 296–306. [DOI] [PubMed] [Google Scholar]
- Cordeiro MN, de Figueiredo SG, Valentim AC, Diniz CR, von Eickstedt VR, Gilroy J et al. (1993). Purification and amino acid sequences of six Tx3 type neurotoxins from the venom of the Brazilian ‘armed’ spider Phoneutria nigriventer (Keys). Toxicon 31: 35–42. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza AH, Lima MC, Drewes CC, da Silva JF, Torres KC, Pereira EM et al. (2011). Antiallodynic effect and side effects of Phα1β, a neurotoxin from the spider Phoneutria nigriventer: comparison with ω‐conotoxin MVIIA and morphine. Toxicon 58: 626–633. [DOI] [PubMed] [Google Scholar]
- Diao L, Meibohm B (2013). Pharmacokinetics and pharmacokinetic‐pharmacodynamic correlations of therapeutic peptides. Clin Pharmacokinet 52: 855–868. [DOI] [PubMed] [Google Scholar]
- Dixon WJ (1980). Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 20: 441–462. [DOI] [PubMed] [Google Scholar]
- Estrada G, Villegas E, Corzo G (2007). Spider venoms: a rich source of acylpolyamines and peptides as new leads for CNS drugs. Nat Prod Rep 24: 145–161. [DOI] [PubMed] [Google Scholar]
- Gomez MV, Kalapothakis E, Guatimosim C, Prado MA (2002). Phoneutria nigriventer venom: a cocktail of toxins that affect ion channels. Cell Mol Neurobiol 22: 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui J, Liu B, Cao G, Lipchik AM, Perez M, Dekan Z et al. (2014). A tarantula‐venom peptide antagonizes the TRPA1 nociceptor ion channel by binding to the S1‐S4 gating domain. Curr Biol 24: 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL (1980). Intrathecal morphine in mice: a new technique. Eur J Pharmacol 67: 313–316. [DOI] [PubMed] [Google Scholar]
- Jaquemar D, Schenker T, Trueb B (1999). An ankyrin‐like protein with transmembrane domains is specifically lost after oncogenic transformation of human fibroblasts. J Biol Chem 274: 7325–7333. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GF (2011). Venoms as a platform for human drugs: translating toxins into therapeutics. Expert Opin Biol Ther 11: 1469–1484. [DOI] [PubMed] [Google Scholar]
- Kitamura R, Andoh T, Mizoguchi S, Saito Y, Takahata H, Kuraishi Y (2014). Gabapentin inhibits bortezomib‐induced mechanical allodynia through supraspinal action in mice. J Pharmacol Sci 124: 502–510. [DOI] [PubMed] [Google Scholar]
- Klafke JZ, da Silva MA, Trevisan G, Rossato MF, da Silva CR, Guerra GP et al. (2012). Involvement of the glutamatergic system in the nociception induced intrathecally for a TRPA1 agonist in rats. Neuroscience 222: 136–146. [DOI] [PubMed] [Google Scholar]
- Koivisto A, Chapman H, Jalava N, Korjamo T, Saarnilehto M, Lindstedt K et al. (2014). TRPA1: a transducer and amplifier of pain and inflammation. Basic Clin Pharmacol Toxicol 114: 50–55. [DOI] [PubMed] [Google Scholar]
- Materazzi S, Fusi C, Benemei S, Pedretti P, Patacchini R, Nilius B et al. (2012). TRPA1 and TRPV4 mediate paclitaxel‐induced peripheral neuropathy in mice via a glutathione‐sensitive mechanism. Pflugers Arch 463: 561–569. [DOI] [PubMed] [Google Scholar]
- Materazzi S, Benemei S, Fusi C, Gualdani R, De Siena G, Vastani N et al. (2013). Parthenolide inhibits nociception and neurogenic vasodilatation in the trigeminovascular system by targeting the TRPA1 channel. Pain 154: 2750–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara CR, Mandel‐Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M (2007). TRPA1 mediates formalin‐induced pain. Proc Natl Acad Sci U S A 104: 13525–13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassini R, Gees M, Harrison S, De Siena G, Materazzi S, Moretto N et al. (2011). Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain 152: 1621–1631. [DOI] [PubMed] [Google Scholar]
- Nassini R, Pedretti P, Moretto N, Fusi C, Carnini C, Facchinetti F et al. (2012). Transient Receptor Potential Ankyrin 1 channel localized to non‐neuronal airway cells promotes non‐neurogenic inflammation. PLoS One 7: 42454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassini R, Materazzi S, Benemei S, Geppetti P (2014). The TRPA1 channel in inflammatory and neuropathic pain and migraine. Rev Physiol Biochem Pharmacol 167: 1–43. [DOI] [PubMed] [Google Scholar]
- Nassini R, Fusi C, Materazzi S, Coppi E, Tuccinardi T, Marone IM et al. (2015). The TRPA1 channel mediates the analgesic action of dipyrone and pyrazolone derivatives. Br J Pharmacol 172: 3397–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisinghani M, Zhong L, Jeffry JA, Bishnoi M, Pabbidi RM, Pimentel F et al. (2011). Activation characteristics of transient receptor potential ankyrin 1 and its role in nociception. Am J Physiol Cell Physiol 301: C587–C600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigo FK, Dalmolin GD, Trevisan G, Tonello R, Silva MA, Rossato MF et al. (2013a). Effect of ω‐conotoxin MVIIA and Phα1β on paclitaxel‐induced acute and chronic pain. Pharmacol Biochem Behav 114‐115: 16–22. [DOI] [PubMed] [Google Scholar]
- Rigo FK, Trevisan G, Rosa F, Dalmolin GD, Otuki MF, Cueto AP et al. (2013b). Spider peptide Phα1β induces analgesic effect in a model of cancer pain. Cancer Sci 104: 1226–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro D, Deering‐Rice CE, Romero EG, Hughen RW, Light AR, Veranth JM et al. (2013). Activation of transient receptor potential ankyrin‐1 (TRPA1) in lung cells by wood smoke particulate material. Chem Res Toxicol 26: 750–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HS, Deer TR, Staats PS, Singh V, Sehgal N, Cordner H (2008). Intrathecal drug delivery. Pain Physician 11: S89–S104. [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza AH, Ferreira J, Cordeiro Mdo N, Vieira LB, De Castro CJ, Trevisan G et al. (2008). Analgesic effect in rodents of native and recombinant Ph alpha 1beta toxin, a high‐voltage‐activated calcium channel blocker isolated from armed spider venom. Pain 140: 115–126. [DOI] [PubMed] [Google Scholar]
- Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR et al. (2003). ANKTM1, a TRP‐like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112: 819–829. [DOI] [PubMed] [Google Scholar]
- Tonello R, Rigo F, Gewehr C, Trevisan G, Pereira EM, Gomez MV et al. (2014). Action of Phα1β, a peptide from the venom of the spider Phoneutria nigriventer, on the analgesic and adverse effects caused by morphine in mice. J Pain 15: 619–631. [DOI] [PubMed] [Google Scholar]
- Trevisan G, Materazzi S, Fusi C, Altomare A, Aldini G, Lodovici M et al. (2013). Novel Therapeutic Strategy to Prevent Chemotherapy‐Induced Persistent Sensory Neuropathy By TRPA1 Blockade. Cancer Res 73: 3120–3131. [DOI] [PubMed] [Google Scholar]
- Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B et al. (2007). 4‐Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A 104: 13519–13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira LB, Kushmerick C, Hildebrand ME, Garcia E, Stea A, Cordeiro MN et al. (2005). Inhibition of high voltage activated calcium channels by spider toxin PnTx3‐6. J Pharmacol Exp Ther 314: 1370–1377. [DOI] [PubMed] [Google Scholar]
- Wei H, Karimaa M, Korjamo T, Koivisto A, Pertovaara A (2012). Transient receptor potential ankyrin 1 ion channel contributes to guarding pain and mechanical hypersensitivity in a rat model of postoperative pain. Anesthesiology 117: 137–148. [DOI] [PubMed] [Google Scholar]
- Weng HJ, Patel KN, Jeske NA, Bierbower SM, Zou W, Tiwari V et al. (2015). Tmem100 Is a Regulator of TRPA1‐TRPV1 Complex and Contributes to Persistent Pain. Neuron 85: 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of ω‐conotoxin MVIIA in a model of neuropathic pain induced by bortezomib. Bortezomib (BTZ, 1 mg kg‐1, i.p.) at day 7 after treatment increases mechanical hyperalgesia and cold hyperalgesia. Time course of ω‐conotoxin MVIIA (100 pmol per site) by intrathecal (i.t.) administration does not affect BTZ‐evoked pain‐like responses. Each point represents the mean of 6 mice, and vertical lines show the SEM. Statistical analysis was performed using two‐way ANOVA, followed by Bonferroni's post hoc test. #P < 0.05, significantly different from BL values.
Supporting info item
Supporting info item