

Abstract
Background and Purpose
There is increasing evidence indicating that bromodomain and extra‐terminal domain (BET) proteins play a critical role in the regulation of immune and inflammatory responses; however, their contribution to vascular inflammation has not yet been elucidated. In this study, we investigated the effect of inhibiting BET bromodomain on vascular inflammation and the underlying mechanisms.
Experimental Approach
HUVECs were isolated from fresh umbilical cords. JQ1, a specific BET bromodomain inhibitor, and Brd shRNA were used to evaluate the regulation of the BET proteins in vascular inflammation. Leukocyte adhesion to HUVECs was measure by an adhesion assay. Western blot or immunohistochemical analysis was used to detect the protein expression. Real‐time PCR was used to evaluate mRNA expression. Leukocyte accumulation in vivo was determined by an acute lung inflammation model.
Key Results
BET bromodomain inhibition suppressed the expression of adhesion molecules induced by TNF‐α‐ or LPS, including ICAM‐1, VCAM‐1 and E‐selectin, and inhibited leukocyte adhesion to activated HUVEC monolayers. Treatment with JQ1 also attenuated the LPS‐induced accumulation of leukocytes and expression of endothelial adhesion molecules in the acute lung inflammation model in vivo. Furthermore, BET bromodomain inhibition reduced the activity of p38 and JNK MAPKs and NF‐κB in TNF‐α‐stimulated HUVECs. TNF‐α‐induced NF‐κB activation was also blocked by inhibitors of p38 (SB203580) or JNK (SP600125).
Conclusions and Implications
BET bromodomain is important for regulating endothelial inflammation. Strategies targeting endothelial BET bromodomain may provide a new therapeutic approach for controlling inflammatory‐related diseases.
Abbreviations
- BET
bromodomain and extra‐terminal domain
- ICAM‐1
intercellular adhesion molecule 1
- MPO
myeloperoxidase
- VCAM‐1
vascular cell adhesion molecule 1
Tables of Links
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
The vascular endothelium separates and connects the bloodstream and all extravascular tissues. Endothelial dysfunction is associated with the development of inflammatory disorders, including rheumatoid arthritis, tumours, atherosclerosis, inflammatory bowel disease and sepsis (Aird, 2007; Gareus et al., 2008; Roifman et al., 2009; Khan et al., 2010). An array of adhesion molecules, particularly vascular cell adhesion molecule 1 (VCAM‐1), intercellular adhesion molecule 1 (ICAM‐1) and E‐selectin, are up‐regulated in the vascular endothelium under inflammatory conditions, and play critical roles in regulating the early events of leukocyte adhesion to endothelial cells (Ley et al., 2007; Pober and Sessa, 2007).
Several signalling pathways play important roles in regulating endothelial inflammation. NF‐κB activation in the endothelium results in an increased expression of a series of inflammation‐related genes, such as those encoding adhesion molecules and pro‐inflammatory cytokines (Molestina et al., 2000; Kempe et al., 2005; Zhou et al., 2007). Targeting NF‐κB‐mediated activation of endothelial cells could provide a novel strategy for treating inflammation. The MAPK pathways consist of a family of highly conserved serine/threonine protein kinases that are believed to act as the regulators of key cellular processes including gene induction, cellular stress and inflammatory responses. There are three major classes of MAPKs in mammals, ERK, p38 and JNK (Johnson and Lapadat, 2002). TNFα‐induced expression of adhesion molecules in the endothelium has been shown to be mediated by MAPKs (Read et al., 1997; Hoefen and Berk, 2002; Nizamutdinova et al., 2007; Liang et al., 2011).
Bromodomain and extra‐terminal domain (BET) proteins (Brd2, Brd3, Brd4 and Brdt) are a family of epigenetic adaptors that bind to acetylated lysine residues, thereby connecting acetylated chromatin and gene transcription (Filippakopoulos et al., 2012). Brd proteins promote inducible gene transcription by the recruitment of transcriptional co‐activators including positive transcription elongation factor b (Hargreaves et al., 2009). Hence, BET bromodomain inhibition suppresses LPS‐induced expression of pro‐inflammatory cytokines in murine bone marrow‐derived macrophages (Nicodeme et al., 2010; Belkina et al., 2013). Brd4 promotes the expression of a series of NF‐κB‐mediated inflammatory genes in an acetylated lysine‐310‐dependent manner (Huang et al., 2009). These data indicate that BET bromodomain has the potential to affect inflammatory responses. However, the contribution of BET bromodomain to vascular inflammation remains to be defined. Therefore, in this study, we investigated the role of BET proteins in modulating endothelial inflammation and the underlying mechanisms.
Methods
Cell culture
HUVECs were isolated from fresh umbilical cords after treatment with collagenase. The cells were cultured in EBM2 media supplemented with 0.1% hEGF, 0.1% GA‐1000 (gentamicin, amphotericin‐B), 0.1% ascorbic acid, 0.1% R3‐IGF‐1, 0.1% heparin, 0.4% hFGF‐B, 0.1% VEGF, 0.04% hydrocortisone and 2% FBS (Lonza,CA, USA) and used at passages 4 to 6 when the cells reached 80–90% confluence. The cells were grown in 5% CO2 at 37°C, and the medium was replaced every 2 days. The isolation procedure of the HUVECs was approved by the Medical Ethics Committee of the First Affiliated Hospital, Sun Yat‐sen University, and was conducted in accordance with the recommendations of the Declaration of Helsinki. Informed consent was obtained for all of this study.
Endothelial‐leukocyte adhesion assay
Endothelial‐leukocyte adhesion was determined by the Cell Biolabs' CytoSelect™ Leukocyte‐endothelium Adhesion Assay. HUVECs grown in gelatin‐coated 24‐well plates at 80–90% confluence were treated with JQ1 for 8 h or were transfected with shRNA for 48 h. The cells were treated with 10 ng·mL−1 TNF‐α for 4 h and LeukoTracker™ solution‐loaded leukocytes were added to each well. Non‐adherent cells were removed after 1 h of incubation. The adherent leukocytes were counted under an inverted fluorescence microscope, and the results from five separate fields per well were averaged (cells counted by three blinded observers). The final wash was aspirated and Lysis Buffer was added to each well containing the cells, and fluorescence was read by using a fluorescence plate reader at 480/520 nm.
Model of acute lung inflammation
All animal care and experimental protocols complied with guidelines for protection of animals used for scientific purposes of Sun Yat‐sen University. Animal experiments were approved by the Animal Care and Ethics Committee, Sun Yat‐sen University, according to the Guide for the Care and Use of Laboratory Animals, which was published by the US National Institutes of Health. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
Male C57BL/6J mice (8 weeks old; average weight 25–29 g, obtained from Beijing Laboratory Animal Research Centre) were fed normal mouse chow and water ad libitum. All mice were reared and housed under standard conditions with air filtration (20 ± 2°C; 12 h light/12 h dark). The mice were randomly divided into three groups: control (10% DMSO, 0.2 mL), LPS and LPS + JQ1. Before inducing acute lung inflammation, JQ1 (50 mg·kg−1) was given by i.p. injection for 5 days, and then LPS (40 mg·kg−1, E. coli serotype 026:B6 endotoxin; Sigma‐Aldrich) was administered i.p. to induce lung injury. Five hours after LPS administration, all animals were anaesthetised with 110 mg·kg−1 ketamine and 4.8 mg·kg−1 xylazine, and then killed to collect lung tissues for further analysis. The tissues were fixed in 10% neutral buffered formalin, and the paraffin sections (5 μM) were used to perform immunohistochemical analysis and were stained with haemotoxylin and eosin (H&E); H&E staining was used to determine the lung injury score. The score was calculated by two blinded pathologists according to the following standards (Sun et al., 2012): no injury, score 0; mild‐to‐moderate injury, score 0.1 to 2.5; severe injury, score 2.6 to 4.0. An Amplex Red Peroxidase Assay Kit (Invitrogen) was used to detect myeloperoxidase (MPO) activity in homogenates of murine lung tissue.
Retrovirus generation and transduction
Lentivirus‐ based shRNA vectors were constructed as described previously (Wang et al., 2012). The sequences for control, Brd2 or Brd4 shRNA oligonucleotides are presented in Table 1. These shRNAs were obtained from Shanghai GenePharma (Shanghai, China). To generate virus, 293T cells were transfected, using Lipofectamine 2000 (Invitrogen), with plasmids encoding shRNA targeting Brd2, Brd4 (1 μg·mL−1), or control together with CMV‐dR 8.2 packaging and CMV‐VSVG envelope plasmids. After 48 and 72 h transfection supernatants containing virus were collected and used to infect HUVECs in the presence of 8 μg·mL−1 polybrene.
Table 1.
The sequences of Brd shRNA oligonucleotides
| Oligo Sequence | ||
|---|---|---|
| Brd2 shRNA | Forward | AATTCCCTGCCTACAGGTTATGATTCTCGAGAATCATAACCTGTAGGCAGGGTTTTTAT |
| Reverse | AAAAACCCTGCCTACAGGTTATGATTCTCGAGAATCATAACCTGTAGGCAGGG | |
| Brd4 shRNA | Forward | AATTCCTGGAGATGACATAGTCTTACTCGAGTAAGACTATGTCATCTCCAGGTTTTTAT |
| Reverse | AAAAACCTGGAGATGACATAGTCTTACTCGAGTAAGACTATGTCATCTCCAGG | |
| Scrambled shRNA | Forward | AATTCCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGGTTTTTAT |
| Reverse | AAAAACCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG | |
Overexpression of Brd2 and Brd4 in HUVECs
Brd2 and Brd4 expression cassettes were cloned into FG12 between the XbaI and XhoI sites to construct the overexpression lentiviral vectors. The sequences of these primers are as follows: Brd2, forward 5′‐CTAGTCTAGAATGCTGCAAAAC GTGACTC‐3′, reverse 5′‐TCCGCTCGAGTTAGCCTGAGTCTGAATCAC‐3′; Brd4, forward 5′‐ CTAGTCTAGA ATGTCTGCGGAGAGCGGCC‐3′, reverse 5′‐TCCGCTCGAGTCAGGAGCAGC CA ACACAGC‐3′. The correct construct was confirmed by means of DNA sequencing. HEK‐293T cells were cotransfected with appropriate amounts of vector plasmid, the lentiviral packaging constructs pRSVREV and pMDLg/pRRE, and the VSV‐G expression plasmid pHCMVG. The viral supernatants were harvested on days 2 and 3 after transfection, filtered with 0.4 mm filters, concentrated by ultracentrifugation, and then resolved in MEM medium. For Brd2 and Brd4 overexpression, ECs (50% confluent) were incubated with Brd2, Brd4 or green fluorescent protein viral preparations in the presence of 8 mg·mL−1 polybrene (Santa Cruz Biotechnology, Dallas, Tex) overnight, and then replaced with fresh medium. The efficiency of the infection was determined by western blotting for protein expression.
RNA isolation and quantitative PCR
After the designated treatments with JQ1 or transfection with shRNA for 48 h and then stimulation with TNF‐α for 8 h, total RNAs were extracted using TRIzol (Sigma) and were reverse transcribed to cDNA by using miScript Reverse Transcription Kit. Quantitative real time PCR analysis for the expression of VCAM‐1, ICAM‐1, E‐selectin, IL‐6 and IL‐8 was performed on cDNA using QuantiTect SYBR Green RT‐PCR Kit on StepOnePlusTM Real Time PCR System (Applied Biosystems). Relative mRNA expression was normalized to the expression of GAPDH. The sequences of VCAM‐1, ICAM‐1, E‐selectin, IL‐6, IL‐8, Brd2 and Brd4 are listed in Table 2.
Table 2.
The sequences of RT‐PCR primers
| Sequences | ||
|---|---|---|
| IL‐6 | Forward | ACTCACCTCTTCAGAACGAATTG |
| Reverse | CCATCTTTGGAAGGTTCAGGTTG | |
| IL‐8 | Forward | ACTGAGAGTGATTGAGAGTGGAC |
| Reverse | AACCCTCTGCACCCAGTTTTC | |
| Brd2 | Forward | ACTCACCTCTTCAGAACGA ATTG |
| Reverse | CATCT TTGGAAGGTTCAGGTTG | |
| Brd3 | Forward | ACTGAGAGTGATTGAGAGTGGAC |
| Reverse | AACCCTCT GCACCCAGTTTTC | |
| Brd4 | Forward | CTCTGGAGTAATGTCACACCTCT |
| Reverse | TGTTGGTC CACCTTTCATCTTC | |
| Brdt | Forward | TGTAAAGAAACCTTCCTGCAA |
| Reverse | TTTAAAACACAGTATGCCCAA | |
| GAPDH | Forward | GCACCGTCAAGGCTGAGAAC |
| Reverse | TGGTGAAGACGCCAGTGGA | |
Detection of cell culture supernatant level of IL‐6 and IL‐8
After the designated treatments with JQ1 or transfection with shRNA for 48 h and then treatment with TNF‐α for 12 h, cell culture supernatants were collected for the determination of IL‐6 and IL‐8 by using elisa kit (R&D Systems, USA).
Western blot analysis
Cells were lysed with cell lysis buffer (CST) for 15 min on ice, and lysates were centrifuged at 10 000 × g for 15 min at 4°C. Supernatants were incubated with 2 × laemmli sample buffer (Sigma) at 100°C for 5 min. Equal amounts of the samples were then separated by SDS‐PAGE gel and transferred onto NC membranes and immunoblotted with the indicated antibodies: anti‐VCAM1 (Abcam), anti‐ICAM1 (Abcam), anti‐ E‐selectin (Abcam) anti‐JNK (Cell signalling), anti‐phospho‐JNK(Cell signalling), anti‐p38(Cell signalling), anti‐phospho‐p38(Cell signalling), anti‐ERK(Cell signalling), anti‐phospho‐ERK(Cell signalling), anti‐NF‐κB p65(Cell signalling), anti‐phospho‐IκBα (Cell signalling), anti‐IκBα (Cell signalling) and anti‐phospho‐IκB kinase (IKK) (Cell signalling).
Immunohistochemical analysis
Lung tissues were fixed overnight in 10% formalin, paraffin‐embedded, cut into sections of 5 μm thickness and placed on 3‐aminopropyltriethoxysilane‐coated slides. The sections were then deparaffinized with xylene and rehydrated with ethanol, followed by antigen retrieval using microwave heating. Endogenous peroxidase activity was suppressed by incubating the samples with 1% hydrogen peroxide for 30 min. The sections were incubated with primary antibodies against VCAM1 (mouse monoclonal to VCAM1, 1:100) or ICAM1 (mouse monoclonal to ICAM1, 1:100) or MRP14 (mouse monoclonal to MRP14, 1:100) (all from Abcam, Cambridge, UK), respectively, at 4°C overnight. After being washed with PBS, the sections were incubated with the respective HRP‐conjugated secondary antibodies, and substrates, and counterstained with haematoxylin.
Confocal laser scanning fluorescence microscopy
HUVECs on glass coverslips at 80–90% confluence were treated with JQ1 or DMSO for 8 h and then were stimulated with TNF‐α for 30 min. Then, they were fixed with paraformaldehyde and permeated with 0.1% TritonX‐100 in PBS. For detection of p65 NF‐κB, the cells were incubated with anti‐p65 NF‐κB antibody overnight and incubated with secondary antibodies for 1 h at room temperature. The cells were then incubated with DAPI and the coverslips were mounted on glass slides with antifade mounting media and examined using a confocal fluorescence microscopy (Zeiss LSM710).
MTT test for cell viability
HUVECs were pretreated for 24 h with JQ1 at different concentrations (50, 100 and 250 nM). The culture supernatants were removed, and the adherent cells were incubated for 30 min at 37°C with a solution of the 3‐(4,5‐dimethylthiazol‐2‐yl) ‐2,5‐diphenyltetrazolium (MTT) salt (1 mg·mL−1 in PBS). The dark blue crystals of formazan produced were dissolved in acidified isopropanol, and the amount of formazan quantified by subjecting the samples to a test wavelength of 570 nm and a reference wavelength of 620 nm.
Flow cytometry
HUVECs were fixed in 2% paraformaldehyde and were analysed by flow cytometry using a FACSan with CellQuest software (BD Bioscience, Oxford, United Kingdom). Cell apoptosis was assessed by labelling with annexin V (FITC) according to the manufacturer's instructions (BD Bioscience).
CCK‐8 assay
Peripheral blood mononuclear cell (PBMCs) were seeded in 96‐well plates, deprived of serum, and then treated with JQ1 as described in the text. At the end of the incubation, CCK‐8 solution was added for 4 h and the absorbance at 450 nm (A 450 nm) was measured with a microplate reader.
Statistical analyses
The results are expressed as mean ± SEM. The present studies comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The experimental procedures or treatment and data analyses were carried out with randomization and blinding. For in vitro studies, we performed a minimum of five independent experiments, where individual data points were based on at least technical duplicates each. For statistical analysis, we used normalized data to reduce the variations in the baseline between independent experiments, with the exception of data for pro‐inflammatory cytokine secretion where the results were normalized to fold over control without TNF‐α or LPS. Student's t‐test was used to test compare the data from two groups. We compared ≥3 different groups with one way ANOVA, and if F reached significance two post hoc tests were used: the Dunnett post hoc test when comparing each group with control, or the Sidak post hoc test whenever a multiple group comparison was necessary. For in vivo studies, all groups were initially designed to contain 7 mice with a C57BL/6J background. We used nonparametric methods (Kruskal–Wallis test followed by Dunn's post hoc tests when F reached significance). P values less than 0.05 were considered statistically significant. Statistical analysis of data was performed by using GraphPad Prism 6.
Results
BET bromodomain inhibition suppresses TNF‐α‐induced expression of adhesion molecules
In our previous study (Huang et al., 2016), we examined the expression patterns of BET proteins in endothelial cells and found that Brd2 and Brd4 are highly expressed. To rule out non‐specific interference, we constructed 3 different sequences of shRNA oligonucleotides for Brd2 or Brd4. As shown in our previous study, transfection with all 3 shRNA oligonucleotides down‐regulated endogenous Brd2 or Brd4 protein expression; however, the inhibitory effect of Brd2 shRNA‐3 or Brd4 shRNA‐3 was the most prominent. Accordingly, Brd2 shRNA‐3 (Brd2 shRNA) or Brd4 shRNA‐3 (Brd4 shRNA) was used for subsequent experiments. In the present study, we also determined the inhibitory efficiency of Brd2 shRNA and Brd4 shRNA on their endogenous expression in HUVECs (Figure S1).
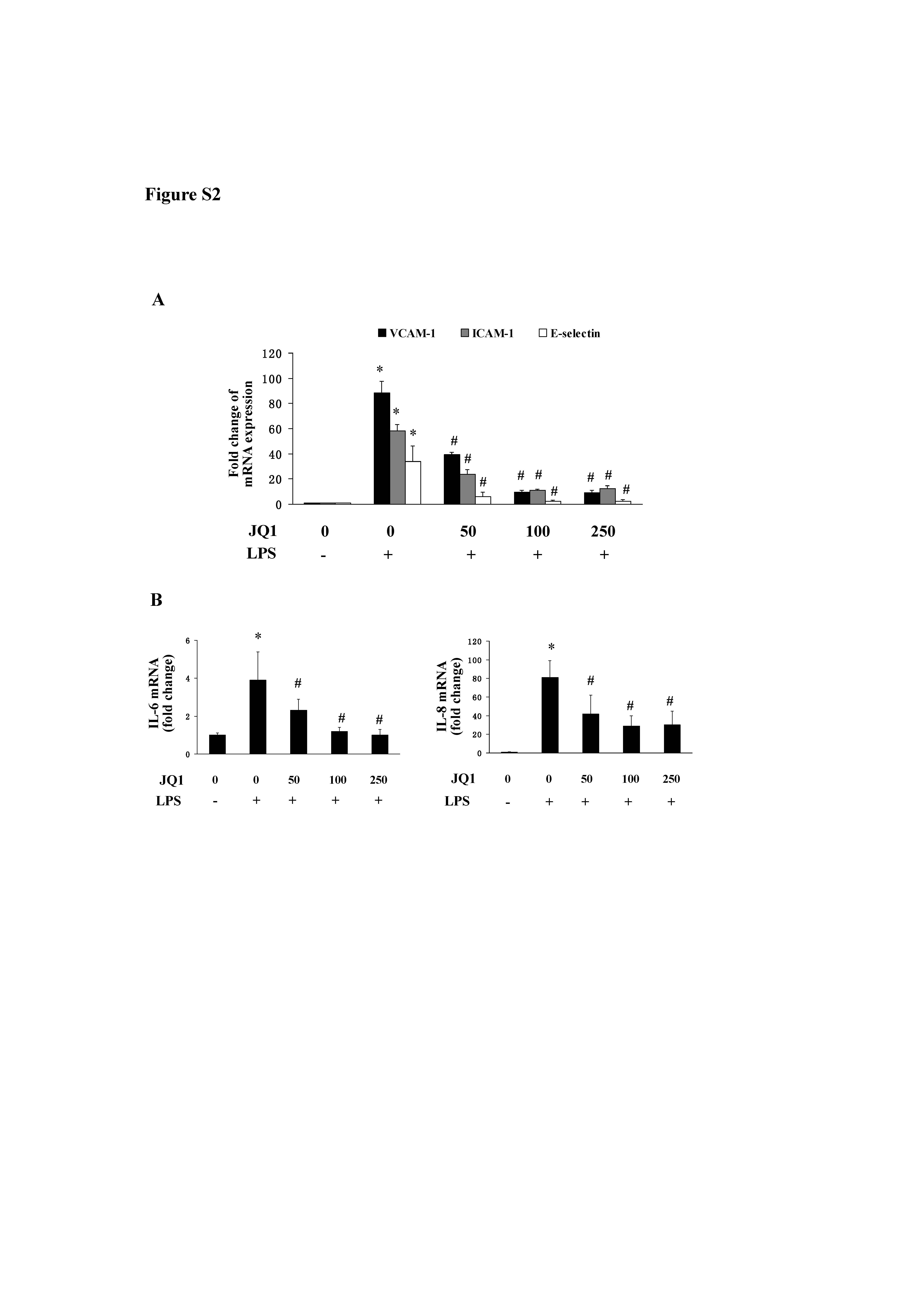
To evaluate the ability of BET protein to affect the activation of endothelial cells, we determined the effect of BET inhibition on the expression of adhesion molecules in TNF‐α‐stimulated HUVECs by using JQ1 or Brd shRNA transfection. JQ1 treatment suppressed TNF‐α‐induced gene expression of VCAM‐1, E‐selectin and ICAM‐1. Treatment with Brd2 or Brd4 shRNA also suppressed their expression respectively (Figure 1A and B). We further demonstrated that co‐transfection with Brd2 and Brd4 shRNA decreased the expression of these adhesion molecules to a higher degree compared with the transfection of either of them alone (Figure 1B). The protein expression of VCAM‐1, E‐selectin and ICAM‐1 was also decreased in cells treated with JQ1 compared with those treatment with DMSO alone (Figure 1C); moreover, the protein expression of these molecules was also inhibited by transfection with Brd2 shRNA or Brd4 shRNA (Figure 1D). Likewise, BET inhibition also reduced the mRNA expression of VCAM‐1 and ICAM‐1 in HUVECs in response to LPS treatment (Figure S2).
Figure 1.
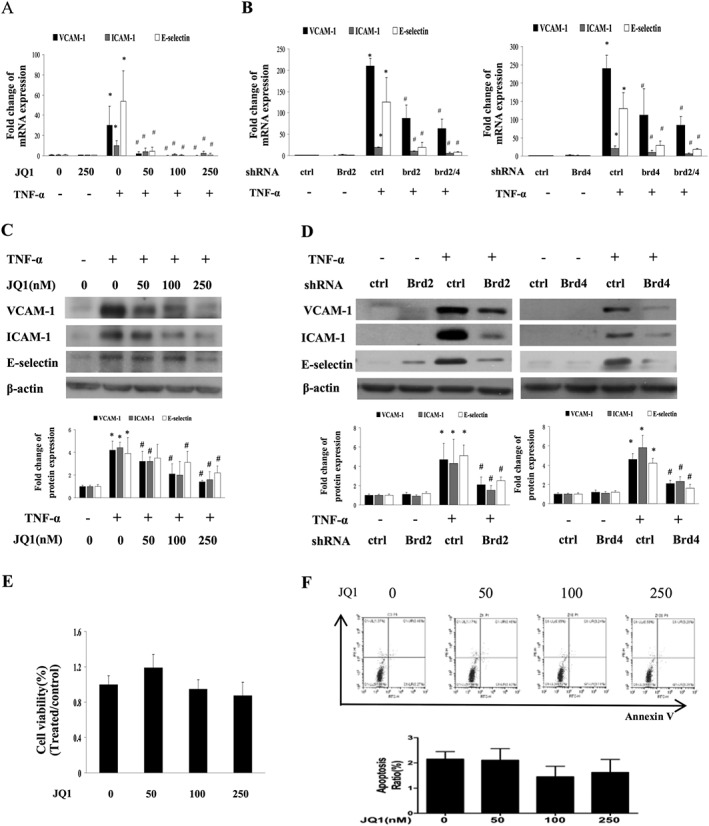
Effect of BET bromodomain inhibition on TNF‐α‐induced expression of adhesion molecules. (A and B) Real time qPCR analysis of VCAM‐1, ICAM‐1 and E‐selectin mRNA expression from five independent experiments. HUVECs were treated with JQ1, or transfected with shRNA for Brd2, Brd4 or control, or co‐transfected with Brd2 shRNA or Brd4 shRNA, and then stimulated with TNF‐α (10 ng·mL−1) for 8 h. The results were normalized to GAPDH and expressed as fold change over mean control. Data are representative of five independent experiments (mean ± SEM; *P < 0.05 vs DMSO treatment (control) or control shRNA, # P < 0.05 vs TNF‐α treatment; P values were obtained by one‐way ANOVA). (C and D) Western blot analysis of VCAM‐1, ICAM‐1 and E‐selectin in HUVECs treated with different concentrations of JQ1 (C) or transfected with shRNA for Brd2, Brd4 or control (D) after treatment with 10 ng·mL−1 TNF‐α for 12 h. Data are a combination of densitometric analyses from five independent experiments (mean ± SEM; *P < 0.05 vs control or control shRNA, # P < 0.05 vs TNF‐α treatment; P values were obtained by one‐way ANOVA.) (E) Effect of JQ1 on viability of HUVECs. HUVECs were treated with JQ1 for 48 h. HUVECs’ viability was detected by MTT test. Data are a combination from five independent experiments, (mean ± SEM, one‐way ANOVA test). (F) Effect of JQ1 on apoptosis of HUVECs. HUVECs were treated with JQ1 for 48 h. Annexin V labelling was used to determine cell apoptosis by flow cytometry. Data are shown are (upper panel) representative or (lower panel) combined results from five independent experiments (mean ± SEM, one‐way ANOVA test).
To determine the toxic effect of JQ1, the viability or apoptosis of HUVECs was evaluated by the MTT test or annexin V labelling assay. Treatment with JQ1 at concentrations of 50, 100 and 250 nM did not affect cell viability (Figure 1E) and apoptosis (Figure 1F), suggesting that the effects of JQ1 in our experiments were not associated with cellular cytotoxic effects.
BET bromodomain inhibition reduced leukocyte adhesion to activated HUVEC monolayers
Adhesion molecules are critical for the initiation, promotion and maintenance of leukocyte adhesion to the vascular endothelium; therefore, we performed an in vitro cell adhesion assay to evaluate the effect of BET inhibition on the interaction between leukocytes and HUVECs. As shown in Figure 2A and B, treatment with TNF‐α increased the adhesion of human PBMC to HUVECs compared with the control group; however, this increase was suppressed by treatment with JQ1 or Brd2 shRNA or Brd4 shRNA. We also demonstrated that co‐transfection with Brd2 shRNA and Brd4 shRNA decreased the adhesion of PBMC to HUVECs to a higher degree than treatment with Brd2 or Brd4 shRNA alone. Furthermore, the decreased adhesion was restored by the overexpresssion of Brd2 or Brd4 in their previously knocked‐down cells (Figure 2C). In addition, we determined whether JQ1 affected leukocyte viability so as to rule out the possibility that JQ1‐induced reduction of PBMC adhesion to HUVECs is associated with reduced leukocyte viability. Our results showed that treatment with JQ1 for 8 h did not affect leukocyte viability (Figure 2D). Taken together, our findings suggest that BET protein is involved in the pro‐inflammatory stimuli‐induced expression of adhesion molecules and adhesion of leukocytes to activated HUVECs.
Figure 2.

Effect of BET bromodomain inhibition on TNF‐α‐induced leukocyte adhesion to activated EC monolayers. (A and B) Effect of BET inhibition on the adhesion of PBMCs to TNF‐α–activated HUVECs. Upper panels: representative photo images of PBMCs adhering to HUVECs treated with JQ1 (A) or transfected with shRNA for Brd2, Brd4 or control, or co‐transfected with shRNA for Brd2 and Brd4 (B) in the presence of TNF‐α (10 ng·mL−1). Scale bars: 100 μm. The lower panels show data for the number and fluorescence values of adhered cells from five independent experiments (mean ± SEM; *P < 0.05 vs control or control shRNA, # P < 0.05 vs TNF‐α treatment; P values were obtained by one way ANOVA.). (C) Effect of overexpresssion of Brd2 or Brd4 on adhesion of PBMCs to TNF‐α‐activated HUVECs. Plasmids containing Brd2 or Brd4 (Oe plasmid) were transfected in their previously knocked‐down HUVECs, and then the cells were treated with TNF‐α (10 ng·mL−1) for 4 h. The data shown are from five independent experiments (mean ± SEM; *P < 0.05 vs control or control plasmid, # P < 0.05 vs TNF‐α treatment; $ P < 0.05 vs Brd2 or Brd4 shRNA, one‐way ANOVA test). (D) Effect of JQ1 on viability of PBMCs. PBMCs were treated with JQ1 for 8 h. PBMCs’ viability was detected by CCK8 test from five independent experiments (mean ± SEM, one‐way ANOVA test).
BET inhibition reduced the production of pro‐inflammatory cytokines in HUVECs
Pro‐inflammatory cytokines, including IL‐6 and IL‐8, play important roles in endothelial inflammation. We found that TNF‐α‐induced mRNA expression and secretion of IL‐6 and IL‐8 was reduced by JQ1 treatment in HUVECs (Figure 3A and B). Moreover, the mRNA expression and secretion of IL‐6 and IL‐8 were reduced in TNF‐α‐stimulated cells transfected with Brd2 shRNA or Brd4 shRNA. Co‐transfection with Brd2 and Brd4 shRNA also reduced mRNA expression and secretion of IL‐6 and IL‐8 to a higher degree than transfection with Brd2 or Brd4 shRNA alone.
Figure 3.

Effects of BET inhibition on the production of pro‐inflammatory cytokines in HUVECs. HUVECs were treated with DMSO (as the control), or different concentrations of JQ1 for 8 h or transfected with shRNA for Brd2, Brd4 or control, and then incubated with or without 10 ng·mL−1 TNF‐α for 12 h (for IL‐6 and IL‐8 mRNA) or 24 h (for IL‐6 and IL‐8 secretion). (A–B) Effect of BET inhibition on the expression of IL‐6 and IL‐8. Gene expression of IL‐6 and IL‐8 was determined by quantitative real‐time PCR (A). Data were normalized to GAPDH. The levels of the IL‐6 and IL‐8 in cultured cell supernatants were measured by elisa (B). Data were obtained from five independent experiments (mean ± SEM; *P < 0.05 vs control or control shRNA, # P < 0.05 vs TNF‐α treatment; one‐way ANOVA test).
BET inhibition regulates NF‐κB signal pathways in activated HUVECs
In response to pro‐inflammatory stimuli, NF‐κB pathway plays a critical role in regulating inflammatory responses in ECs (Molestina et al., 2000). In inflammatory conditions, optimal classical activation of NF‐κB requires the activation of the IKK complex, which results in the phosphorylation, ubiquitination and degradation of the inhibitor of NF‐κB (IκB), and phosphorylation of p65 (Perkins, 2007). To determine whether BET proteins are involved in the activation of NF‐κB in TNF‐α‐stimulated HUVECs, the cells were incubated with JQ1 for 8 h and then stimulated with TNF‐α for 30 min. As shown in Figure 5A, JQ1 treatment blocked TNF‐α‐induced phosphorylation of IKKβ. JQ1 treatment also suppressed the phosphorylation and degradation of IκBα in TNF‐α‐stimulated cells (Figure 5B). We further found that TNF‐α‐induced nuclear accumulation of p65 protein was reduced in HUVECs treated with JQ1 (Figure 4C–F). Transfection with Brd2 or Brd4 shRNA also reduced the TNFα‐induced phosphorylation of IKK (Figure 4G). JQ1 also suppressed LPS‐induced increase in IKK activity and nuclear translocation of p65 in HUVECs (Figure S3). These results suggest that BET bromodomain has an important role in regulating endothelial NF‐κB activation under inflammatory conditions.
Figure 5.
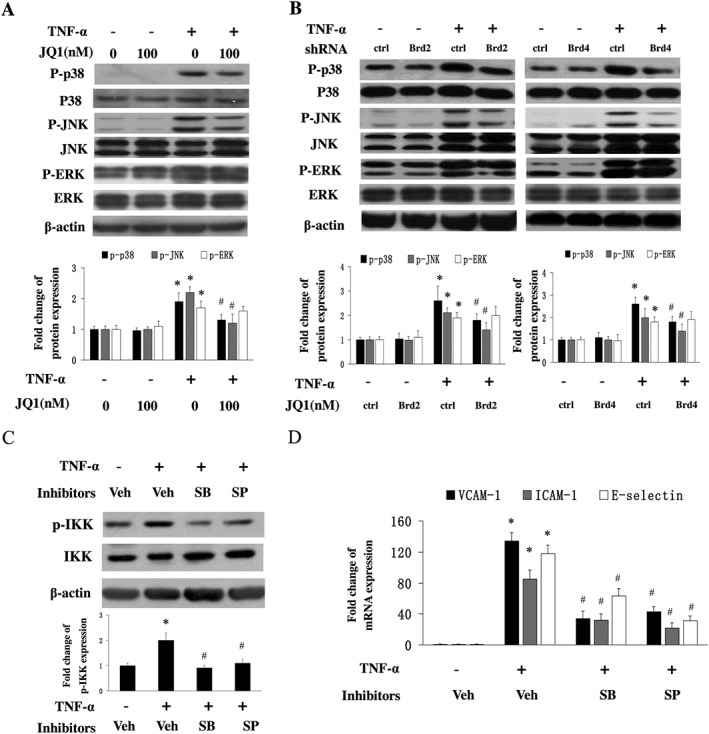
Effect of BET inhibition on the activation of the MAPK signalling pathway in HUVECs. (A and B) HUVECs were pretreated with DMSO or JQ1 for 8 h followed by TNF‐α (10 ng·mL−1) for 10 min. The expression of phosphorylated p38, JNK and ERK was measured by western blot analysis. The results of semiquantitative densitometry of protein expression are shown (lower panel) from five independent experiments (mean ± SEM; *P < 0.05 vs DMSO or control shRNA, # P < 0.05 vs TNF‐α alone; one‐way ANOVA test). (C) Effects of p38 and JNK inhibitors on the activity of IKK. HUVECs were treated with DMSO (as vehicle, Veh) or p38 inhibitor SB203580 (SB) or JNK inhibitor SP600125 (SP) for 3 h followed by TNF‐α (10 ng·mL−1) for 15 min. The results of semiquantitative densitometry of phosphorylated IKK expression are shown in the lower panel from five independent experiments (mean ± SEM; *P < 0.05 vs DMSO, # P < 0.05 vs TNF‐α alone; one‐way ANOVA test). (D) Effect of p38 or JNK inhibitor on the expression of adhesion molecules. HUVECs were treated with DMSO or SB203580 or SP600125 for 3 h followed by TNF‐α (10 ng·mL−1) for 8 h. Real time qPCR analysis of the expression of adhesion molecules mRNA from 5 independent experiments (mean ± SD; *P < 0.05 vs DMSO, # P < 0.05 vs TNF‐α alone; one‐way ANOVA test).
Figure 4.
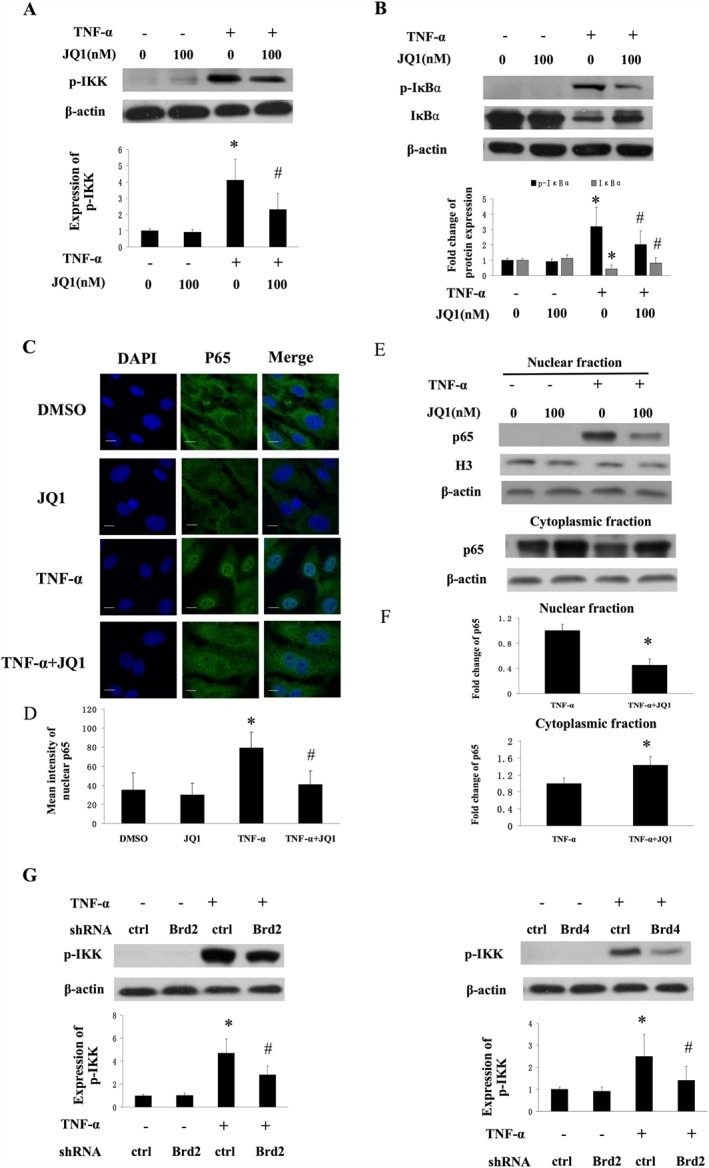
Effect of BET inhibition on TNF‐α‐induced NF‐κB activation in HUVECs. (A and B) Western blot analysis of phosphorylated IKK or phosphorylated and degradated IκBα in HUVECs treated with 10 ng·mL−1 TNF‐α for 15 min in the presence of DMSO or JQ1. Data represent relative quantities obtained by densitometry (lower panel) from five independent experiments (mean ± SEM; *P < 0.05 vs DMSO, # P < 0.05 vs TNF‐α treatment; one‐way ANOVA test). (C and D) Effect of JQ1 on nuclear translocation of p65. HUVECs were treated with TNF‐α (10 ng·mL−1) for 30 min in the presence or absence of JQ1. The representative images show immunofluorescence staining analysis of p65 localization (green). Nuclei were stained with DAPI. (D ) Mean intensity of nuclear p65 protein from 5 independent experiments (mean ± SEM; *P < 0.05 vs DMSO, # P < 0.05 vs TNF‐α alone; one‐way ANOVA test). (E and F) The expression of p65 protein, detected by western blot analysis, in nuclear and cytoplasmic fractions isolated from HUVECs. Results of semiquantitative densitometry of nuclear p65 protein expression are shown in (D), from five independent experiments (mean ± SEM; *P < 0.05 vs TNF‐α alone, Student's t‐test). (G) Western blot analysis of TNF‐α‐induced phosphorylation of IKKβ in HUVECs transfected with Brd2 shRNA or Brd4 shRNA or control shRNA in the presence or absence of TNF‐α stimulation. Results of semiquantitative densitometry of protein expression are shown in the right panel, from five independent experiments (mean ± SEM; *P < 0.05 vs control shRNA, # P < 0.05 vs TNF‐α treatment; one‐way ANOVA test).
BET inhibition affects the p38 and JNK MAPK signal in HUVECs
Since several MAPKs have been implicated in regulating endothelial inflammation, we determined whether BET inhibition affects the activation of MAPK signalling pathways. TNF‐α treatment resulted in an increased phosphorylation of p38, JNK and ERK; however, JQ1 treatment decreased TNF‐α‐induced phosphorylation of p38 and JNK, but not ERK (Figure 5A). We also observed similar results from HUVECs transfected with Brd2 shRNA or Brd4 shRNA (Figure 5B). These data suggest that p38 and JNK are involved in the effects mediated by BET proteins on the inflammatory response in HUVECs.
Previous studies have shown that the MAPK pathway is involved in regulating NF‐κB activation (Liang et al., 2011; Liu et al., 2012). Therefore, we investigated whether a MAPK inhibitor affected the activity of IKK, a critical upstream protein of NF‐κB pathway. As shown in Figure 5C, inhibitors of p38 and JNK reduced the activity of IKK. We also found that both of the inhibitors decreased the TNF‐α‐induced expression of VCAM‐1 ICAM‐1 and E‐selectin in HUVECs (Figure 5D). Our findings suggest that BET inhibition prevents the activation of NF‐κB in endothelial cells by, at least in part, decreasing upstream MAPK activity.
BET bromodomain inhibition suppresses leukocyte infiltration in vivo
The preceding results showing the effects of BET inhibition on vascular leakiness (Huang et al., 2016), and adhesion prompted us to investigate whether BET proteins play a role in leukocyte infiltration through permeable endothelium in vivo.
Acute inflammation is an essential host response mediated by the infiltration of circulating leukocytes from the peripheral blood to remove pathogens. We determined the effect of JQ1 administration on the acute inflammatory response in murine lungs challenged with LPS. It has been demonstrated that JQ1 powerfully inhibits BET bromodomain function and displays excellent effects when administered once‐daily (Filippakopoulos et al., 2011). Also no obvious toxicity was observed in adult mice injected with JQ1 50–100 mg−1·kg−1·day−1. Therefore, in the present study, the mice were administered 50 mg−1·kg−1 of JQ1 once a day. LPS treatment resulted in a robust increase in leukocyte accumulation in lungs 5 h after its administration; however, JQ1 administration attenuated this LPS‐induced leukocyte accumulation (Figure 6A). The expression of migration inhibitory factor‐related protein 14 (MRP14), a marker neutrophils and monocytes, was decreased in mice treated with JQ1 (Figure 6 F and G). The activity of MPO, a peroxidase enzyme abundantly present in neutrophils, was decreased in JQ1‐treated animals (Figure 6E). Furthermore, in vivo administration of JQ1 decreased the elevated expression of VCAM‐1 and ICAM‐1 in lung tissue of mice treated with LPS (Figure 6A and C). In addition, we observed that JQ1 treatment did not affect weight and survival of mice, and no animals died after LPS administration (data not shown). Our findings indicate that BET proteins might play an important role in regulating endotoxin‐induced vascular inflammation and leukocyte accumulation.
Figure 6.

Effect of BET bromodomain inhibitor JQ1 on leukocyte infiltration in vivo. (A) Effect of JQ1 on the acute inflammatory response in murine lungs challenged with LPS. Mice were administered DMSO (as control) or JQ1, i.p., for 5 days and then treated with or without LPS (40 mg·kg−1, i.p.) for 5 h. The lung tissues were collected for staining of H&E (upper panel), VCAM‐1 (middle panel) and ICAM1 (lower panel). Scale bars: 50 μm. (B) LPS‐induced lung injury was determined by lung injury scoring (n = 7 mice per group). Kruskal–Wallis test was used for the statistical analysis of injury scores (*P < 0.05 vs DMSO, # P < 0.05 vs LPS alone). (C) Quantification of VCAM‐1 staining in lung tissue. Data represent mean ± SEM from seven independent experiments (*P < 0.05 vs normal, # P < 0.05 vs LPS alone; Kruskal–Wallis test). (D and E) Lung tissues were collected for detection of VCAM‐1 and ICAM‐1 protein expression by western blot analysis. Results of semiquantitative densitometry of protein expression are shown from seven independent experiments (*P < 0.05 vs normal (DMSO), # P < 0.05 vs LPS alone; Kruskal–Wallis test). (F) The lung tissues were collected and stained for MRP14. (G) Quantification of MRP14 staining in lung tissues. Data represent mean ± SEM, *P < 0.05 versus normal, # P < 0.05 versus LPS alone (Kruskal–Wallis test). (H) Lung tissues were collected and evaluated for MPO activity (n = 7 mice per group). Values represent mean ± SEM (*P < 0.05 versus normal; # P < 0.05 versus LPS; Kruskal–Wallis test).
Discussion
In the present study, we demonstrate that BET bromodomain is involved in endothelial inflammatory responses by regulating the expression of VCAM‐1, ICAM‐1, E‐selectin, IL‐6, IL‐8 and the adhesion of leukocytes to HUVECs. Furthermore, BET inhibition attenuated IKK‐mediated activation of the NF‐κB pathway in TNFα‐treated endothelial cells. It was also shown that BET inhibition suppresses the activation of p38 and JNK MAPKs. TNF‐α‐induced NF‐κB activation was also blocked by inhibitors of p38 or JNK inhibitor. Our findings identify BET bromodomain as a new epigenetic modulator of vascular inflammation.
ECs play a critical role in maintaining the homeostasis of the vascular endothelium, particularly in the modulations of leukocyte trafficking and vascular permeability. The adhesion molecules mediate the attachment of leukocytes to the vascular endothelium and transendothelial migration and are critical early stages in the responses to inflammatory stimuli and associated diseases (Berlin et al., 1995; Kansas, 1996; Ley et al., 2007). Our findings that BET inhibition powerfully suppresses the expression of endothelial adhesion molecules and pro‐inflammatory cytokines and the attachment of leukocytes to endothelial cells suggest that BET bromodomain might is involved in regulating the early events of vascular inflammation. Since the induction of VCAM‐1, ICAM‐1, E‐selectin, IL‐6 and IL‐8 in ECs is mainly modulated by the NF‐κB signalling pathway (Hou et al., 1994; Manning et al., 1995), our results showing that suppression of BET inhibited NF‐κB activation further support the notion that BET bromodomain is essential for controlling vascular inflammation. In line with these findings, recent studies indicate that BET bromodomain suppression modulates the transcription of NF‐κB target genes in macrophages (Nicodeme et al., 2010), renal tubular epithelial cells (Zhang et al., 2012), tumour cells (Wu et al., 2013; Zou et al., 2014) and rheumatoid fibroblast‐like synoviocytes (Xiao et al., 2016).
In addition to the NF‐κB pathway, the MAPK pathway is important in regulating the expression of endothelial adhesion molecules (Read et al., 1997; Hoefen and Berk, 2002; Liang et al., 2011). In this study, we found that treatment with the BET inhibitor JQ1 or specific shRNA for Brd2 or Brd4 reduced the TNFα‐induced activation of p38 and JNK, but not ERK in HUVECs, suggesting the effects of BET bromodomain on endothelial inflammation are mediated by the activation of p38 and JNK. However, in contrast to our findings, a recent study showed that Brd2 is involved in regulating ERK1/2 phosphorylation but does not affect the activities of JNK and p38MAPK in 3T3‐L1 cells (Zang et al., 2013). This suggests a the effects of BET bromodomain on the MAPK pathway are cell type‐specific.
We hypothesized initially that BET bromodomain contributes to vascular inflammation owing to its direct regulation of the transcriptional activity of NF‐κB, as found in recent studies in HIV‐associated kidney disease (Zhang et al., 2012) and macrophages (Nicodeme et al., 2010). However, in contrast to findings from a recent report (Brown et al., 2014), we demonstrated, unexpectedly, that BET inhibition reduces the phosphorylation of IKK, phosphorylation and degradation of IκBα and nuclear translocation of p65 in TNF‐α‐stimulated HUVECs. These data indicate that the nuclear Brd proteins modulate NF‐κB activity by, at least partly, interfering in the early event of cytoplasmic IKK activation. Indeed, recent studies have also shown that BET inhibition reduces IKK activity in rheumatoid fibroblast‐like synoviocytes (Xiao et al., 2016) and diffuse large beta cell lymphoma (Ceribelli et al., 2014).
Previous studies have indicated that MAPK pathways are involved in the modulation of NF‐κB activation in TNF‐α‐stimulated endothelial cells (Liang et al., 2011; Liu et al., 2012; Olejarz et al., 2014), therefore, in the present study, we determined whether the effects of BET inhibition on activation of the IKK/NF‐κB signal is mediated by MAPK pathways. We found that p38 and JNK inhibitors decreased the activity of IKK and reduced the TNF‐α‐induced expression of VCAM‐1, ICAM‐1 and E‐selectin in HUVECs. These observations suggest that BET bromodomain regulates IKK‐mediated NF‐κB activation through, at least in part, the p38 and JNK MAPK pathways. However, the detailed mechanism(s) of action for the inhibitory effect induced by the suppression of BET on MAPK signalling remains unclear. BET bromodomains localize genome‐wide to promoter and enhancer regions (Hargreaves et al., 2009; Chapuy et al., 2013). It is well documented that several extracellular stimuli including inflammatory cytokines can induce the activation of MAPK pathways, which occurs in the cytoplasm of endothelial cells. However, BET proteins are highly enriched in nuclear super enhancers, and JQ1 disrupts the formation of the chromatin complexes, essential for the expression of many genes, by interfering with the binding of BET bromodomain to acetylated histones. This implies that nuclear located BET proteins regulate cytoplasmic activation of the MAPK pathway, most likely, in an indirect manner in TNF‐α‐stimulated ECs. That is to say, the effect of inhibiting BET on the MAPK pathway may be through a decrease in the transcription of genes encoding signalling proteins that function in the phosphorylation of p38 and JNK or their upstream proteins such as MAPKK and MKK. Another possibility is that BET proteins may mediate ‘inside‐out’ signalling from the nucleus to the cytoplasm to modulate activation of the MAPK pathways. In addition, we cannot eliminate the possibility that BET proteins might shuttle out of the nucleus and interact directly with acetylated signal proteins that control the activation of MAPKs in the cytoplasm of ECs. However, this possibility seems to be unlikely because we did not find that TNF‐α‐induced the translocation of Brd4 from the nucleus to cytoplasm (data not shown). In the future, it would be interesting to explore further the mechanism by which the nuclear BET bromodomain activates the cytoplasmic MAPK signalling pathway.
In addition, we observed that JQ1 treatment resulted in a slight but not significant reduction in the expression of adhesion molecules and cytokines, and leukocyte adhesion in the absence of TNF‐α or LPS; these data indicate that JQ1 does not affect the function of unstimulated ECs. However, the possibility that these observations might be due to the low baseline expression of adhesion molecules and cytokines cannot be ruled out.
Conclusions
We have identified BET bromodomain as a target that regulates endothelial inflammation by modulating the MAPK/NF‐κB signalling pathway. Strategies targeting BET bromodomain might provide a new therapeutic approach for controlling inflammatory‐related diseases.
Author contributions
H.S.X., X.Y.Y, G.Q.C and L.Q.L. conceived and designed the experiments. M.C.H., S.Z., Y.Y.Z and M.H.S. performed most of the biological experiments. M.C.H., Q.Q. and Y.J.X. analysed the data. H.S.X. wrote the main manuscript text. All the authors reviewed the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Targeted depletion of Brd2 and Brd4 in HUVECs. HUVECs were transfected with shRNA for Brd2 (shBrd2) or Brd4 (shBrd4) oligonucleotide or control (shC). After 72 h of transfection, cells were lysed, and mRNA (A) and protein (B) expression of Brd2 or Brd4 was assessed by real time qPCR and Western blot analysis respectively. Values for mRNA expression represent mean ± SEM from 5 independent experiments. *P < 0.05 vs control shRNA, Student t‐test).
Figure S2 Effect of BET bromodomain inhibition on LPS‐induced expression of adhesion molecules and proinflammatory cytokines. A and B, HUVECs were pretreated with JQ1, and then incubated with 10 ng/mL LPS for 8 h. The mRNA expression of adhesion molecules (A) and proinflammatory cytokines (B) were determined by Real time qPCR. Data were obtained from 5 independent experiments (mean ± SEM; *P < 0.05 vs control, # P < 0.05 vs TNF‐α treatment; one‐way ANOVA test).
Figure S3 Effect of BET bromodomain suppression on LPS‐induced activation of NF‐κB signal by HUVECs. A, HUVECs were treated with DMSO or JQ1 (100 nM) for 8 h followed by LPS (10 ng/mL) for 15 min. The phosphorylated IκB kinase (IKK) expression was measured by Western blot analysis. Semiquantitative densitometry of p‐IKK expression is shown in right panel from 5 independent experiments (mean ± SEM; *P < 0.05 vs control, # P < 0.05 vs TNF‐α treatment; one‐way ANOVA test). B and C, Effect of JQ1 on nuclear translocation of p65. HUVECs were treated with 10 ng/mL LPS for 30 min in the presence or absence of JQ1 (100 nM). The images show immunofluorescence staining analysis of p65 localization (green). Nuclei were stained with 40,6‐diamidino‐2‐phenylindole (DAPI). Panel C shows mean intensity of nuclear p65 protein (mean ± SEM) from 5 independent experiments, *P < 0.05 vs DMSO, # P < 0.05 vs LPS (one‐way ANOVA test).
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Acknowledgements
The authors would like to thank Jinjin Fan for her technical assistance. The cDNA plasmids containing Brd2 and Brd4 were kindly provided by Prof. Jiazhun Han (Xiamen University, Xiamen, China). This study is supported by grants from National Natural Science Foundation of China (81373182, 81373194, U1401222 and 81671591), Guangdong Natural Science Foundation (S2011020002358 and S2013010015363), and Guangdong Project of Science and Technology (2012B031800375).
Huang, M. , Zeng, S. , Zou, Y. , Shi, M. , Qiu, Q. , Xiao, Y. , Chen, G. , Yang, X. , Liang, L. , and Xu, H. (2017) The suppression of bromodomain and extra‐terminal domain inhibits vascular inflammation by blocking NF‐κB and MAPK activation. British Journal of Pharmacology, 174: 101–115. doi: 10.1111/bph.13657.
References
- Aird WC (2007). Endothelium as a therapeutic target in sepsis. Curr Drug Targets 8: 501–507. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina AC, Nikolajczyk BS, Denis GV (2013). BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol 190: 3670–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin C, Bargatze RF, Campbell JJ, von Andrian UH, Szabo MC, Hasslen SR et al. (1995). Alpha 4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell 80: 413–422. [DOI] [PubMed] [Google Scholar]
- Brown JD, Lin CY, Duan Q, Griffin G, Federation AJ, Paranal RM et al. (2014). NF‐kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell 56: 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceribelli M, Kelly PN, Shaffer AL, Wright GW, Xiao W, Yang Y et al. (2014). Blockade of oncogenic IkappaB kinase activity in diffuse large B‐cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc Natl Acad Sci U S A 111: 11365–11370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J et al. (2013). Discovery and characterization of super‐enhancer‐associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24: 777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte‐Lovejoy D et al. (2012). Histone recognition and large‐scale structural analysis of the human bromodomain family. Cell 149: 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O et al. (2011). Selective inhibition of BET bromodomains. Nature 468: 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R et al. (2008). Endothelial cell‐specific NF‐kappaB inhibition protects mice from atherosclerosis. Cell Metab 8: 372–383. [DOI] [PubMed] [Google Scholar]
- Hargreaves DC, Horng T, Medzhitov R (2009). Control of inducible gene expression by signal‐dependent transcriptional elongation. Cell 138: 129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefen RJ, Berk BC (2002). The role of MAP kinases in endothelial activation. Vascul Pharmacol 38: 271–273. [DOI] [PubMed] [Google Scholar]
- Hou J, Baichwal V, Cao Z (1994). Regulatory elements and transcription factors controlling basal and cytokine‐induced expression of the gene encoding intercellular adhesion molecule 1. Proc Natl Acad Sci U S A 91: 11641–11645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Yang XD, Zhou MM, Ozato K, Chen LF (2009). Brd4 coactivates transcriptional activation of NF‐kappaB via specific binding to acetylated RelA. Mol Cell Biol 29: 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Qiu Q, Xiao Y, Zeng S, Zhan M, Shi M et al. (2016). BET bromodomain suppression inhibits VEGF‐induced angiogenesis and vascular permeability by blocking VEGFR2‐mediated activation of PAK1 and eNOS. Sci Rep 6: 23770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R (2002). Mitogen‐activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298: 1911–1912. [DOI] [PubMed] [Google Scholar]
- Kansas GS (1996). Selectins and their ligands: current concepts and controversies. Blood 88: 3259–3287. [PubMed] [Google Scholar]
- Kempe S, Kestler H, Lasar A, Wirth T (2005). NF‐kappaB controls the global pro‐inflammatory response in endothelial cells: evidence for the regulation of a pro‐atherogenic program. Nucleic Acids Res 33: 5308–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan F, Galarraga B, Belch JJ (2010). The role of endothelial function and its assessment in rheumatoid arthritis. Nat Rev Rheumatol 6: 253–261. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7: 678–689. [DOI] [PubMed] [Google Scholar]
- Liang CJ, Wang SH, Chen YH, Chang SS, Hwang TL, Leu YL et al. (2011). Viscolin reduces VCAM‐1 expression in TNF‐alpha‐treated endothelial cells via the JNK/NF‐kappaB and ROS pathway. Free Radic Biol Med 51: 1337–1346. [DOI] [PubMed] [Google Scholar]
- Liu X, Pan L, Wang X, Gong Q, Zhu YZ (2012). Leonurine protects against tumor necrosis factor‐a‐mediated inflammation in human umbilical vein endothelial cells. Atherosclerosis 222: 34–42. [DOI] [PubMed] [Google Scholar]
- Manning AM, Bell FP, Rosenbloom CL, Chosay JG, Simmons CA, Northrup JL et al. (1995). NF‐kappa B is activated during acute inflammation in vivo in association with elevated endothelial cell adhesion molecule gene expression and leukocyte recruitment. J Inflamm 45: 283–296. [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molestina RE, Miller RD, Lentsch AB, Ramirez JA, Summersgill JT (2000). Requirement for NF‐kappaB in transcriptional activation of monocyte chemotactic protein 1 by Chlamydia pneumoniae in human endothelial cells. Infect Immun 68: 4282–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW et al. (2010). Suppression of inflammation by a synthetic histone mimic. Nature 468: 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizamutdinova IT, Oh HM, Min YN, Park SH, Lee MJ, Kim JS et al. (2007). Paeonol suppresses intercellular adhesion molecule‐1 expression in tumor necrosis factor‐alpha‐stimulated human umbilical vein endothelial cells by blocking p38, ERK and nuclear factor‐kappaB signaling pathways. Int Immunopharmacol 7: 343–350. [DOI] [PubMed] [Google Scholar]
- Olejarz W, Bryk D, Zapolska‐Downar D, Małecki M, Stachurska A, Sitkiewicz D (2014). Mycophenolic acid attenuates the tumour necrosis factor‐α‐mediated proinflammatory response in endothelial cells by blocking the MAPK/NF‐κB and ROS pathways. Eur J Clin Invest 44: 54–64. [DOI] [PubMed] [Google Scholar]
- Perkins ND (2007). Integrating cell‐signalling pathways with NF‐kappaB and IKK function. Nat Rev Mol Cell Biol 8: 49–62. [DOI] [PubMed] [Google Scholar]
- Pober JS, Sessa WC (2007). Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 7: 803–815. [DOI] [PubMed] [Google Scholar]
- Read MA, Whitley MZ, Gupta S, Pierce JW, Best J, Davis RJ et al. (1997). Tumor necrosis factor alpha‐induced E‐selectin expression is activated by the nuclear factor‐kappaB and c‐JUN N‐terminal kinase/p38 mitogen‐activated protein kinase pathways. J Biol Chem 272: 2753–2761. [DOI] [PubMed] [Google Scholar]
- Roifman I, Sun YC, Fedwick JP, Panaccione R, Buret AG, Liu H et al. (2009). Evidence of endothelial dysfunction in patients with inflammatory bowel disease. Clin Gastroenterol Hepatol 7: 175–182. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl. Acids Res. 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L et al. (2012). MicroRNA‐181b regulates NF‐κB‐mediated vascular inflammation. J Clin Invest 122: 1973–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wu J, Liu X, He H, Ding F, Yang H et al. (2012). Identification of short hairpin RNA targeting foot‐and‐mouth disease virus with transgenic bovine fetal epithelium cells. PLoS One 7: e42356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Qi J, Bradner JE, Xiao G, Chen LF (2013). Bromodomain and extraterminal (BET) protein inhibition suppresses human T cell leukemia virus 1 (HTLV‐1) Tax protein‐mediated tumorigenesis by inhibiting nuclear factor kappaB (NF‐kappaB) signaling. J Biol Chem 288: 36094–36105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Liang L, Huang M, Qiu Q, Zeng S, Shi M et al. (2016). Bromodomain and extra‐terminal domain bromodomain inhibition prevents synovial inflammation via blocking IκB kinase‐dependent NF‐κB activation in rheumatoid fibroblast‐like synoviocytes. Rheumatology (Oxford) 55: 173–184. [DOI] [PubMed] [Google Scholar]
- Zang K, Wang J, Dong M, Sun R, Wang Y, Huang Y et al. (2013). Brd2 inhibits adipogenesis via the ERK1/2 signaling pathway in 3 T3‐L1 adipocytes. PLoS One 8: e78536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Liu R, Zhong Y, Plotnikov AN, Zhang W, Zeng L et al. (2012). Down‐regulation of NF‐kappaB transcriptional activity in HIV‐associated kidney disease by BRD4 inhibition. J Biol Chem 287: 28840–28851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Connell MC, MacEwan DJ (2007). TNFR1‐induced NF‐kappaB, but not ERK, p38MAPK or JNK activation, mediates TNF‐induced ICAM‐1 and VCAM‐1 expression on endothelial cells. Cell Signal 19: 1238–1248. [DOI] [PubMed] [Google Scholar]
- Zou Z, Huang B, Wu X, Zhang H, Qi J, Bradner J et al. (2014). Brd4 maintains constitutively active NF‐kappaB in cancer cells by binding to acetylated RelA. Oncogene 33: 2395–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Targeted depletion of Brd2 and Brd4 in HUVECs. HUVECs were transfected with shRNA for Brd2 (shBrd2) or Brd4 (shBrd4) oligonucleotide or control (shC). After 72 h of transfection, cells were lysed, and mRNA (A) and protein (B) expression of Brd2 or Brd4 was assessed by real time qPCR and Western blot analysis respectively. Values for mRNA expression represent mean ± SEM from 5 independent experiments. *P < 0.05 vs control shRNA, Student t‐test).
Figure S2 Effect of BET bromodomain inhibition on LPS‐induced expression of adhesion molecules and proinflammatory cytokines. A and B, HUVECs were pretreated with JQ1, and then incubated with 10 ng/mL LPS for 8 h. The mRNA expression of adhesion molecules (A) and proinflammatory cytokines (B) were determined by Real time qPCR. Data were obtained from 5 independent experiments (mean ± SEM; *P < 0.05 vs control, # P < 0.05 vs TNF‐α treatment; one‐way ANOVA test).
Figure S3 Effect of BET bromodomain suppression on LPS‐induced activation of NF‐κB signal by HUVECs. A, HUVECs were treated with DMSO or JQ1 (100 nM) for 8 h followed by LPS (10 ng/mL) for 15 min. The phosphorylated IκB kinase (IKK) expression was measured by Western blot analysis. Semiquantitative densitometry of p‐IKK expression is shown in right panel from 5 independent experiments (mean ± SEM; *P < 0.05 vs control, # P < 0.05 vs TNF‐α treatment; one‐way ANOVA test). B and C, Effect of JQ1 on nuclear translocation of p65. HUVECs were treated with 10 ng/mL LPS for 30 min in the presence or absence of JQ1 (100 nM). The images show immunofluorescence staining analysis of p65 localization (green). Nuclei were stained with 40,6‐diamidino‐2‐phenylindole (DAPI). Panel C shows mean intensity of nuclear p65 protein (mean ± SEM) from 5 independent experiments, *P < 0.05 vs DMSO, # P < 0.05 vs LPS (one‐way ANOVA test).
Supporting info item
Supporting info item
Supporting info item