

Summary
T lymphocytes are a critical component of the adaptive immune system mediating protection against infection and malignancy, but also implicated in many immune pathologies. Upon recognition of specific antigens T cells clonally expand, traffic to inflamed sites and acquire effector functions, such as the capacity to kill infected and malignantly transformed cells and secrete cytokines to coordinate the immune response. These processes have significant bioenergetic and biosynthetic demands, which are met by dynamic changes in T‐cell metabolism, specifically increases in glucose uptake and metabolism; mitochondrial function; amino acid uptake, and cholesterol and lipid synthesis. These metabolic changes are coordinate by key cellular kinases and transcription factors. Dysregulated T‐cell metabolism is associated with impaired immunity in chronic infection and cancer and conversely with excessive T‐cell activity in autoimmune and inflammatory pathologies. Here we review the key aspects of T‐cell metabolism relevant to their immune function, and discuss evidence for the potential to therapeutically modulate T‐cell metabolism in disease.
Keywords: T cells, cell activation, cell differentiation, cell proliferation, inflammation
Introduction
Upon recognition of their specific antigen via the T‐cell receptor (TCR) in the context of appropriate co‐stimulatory signals, T cells clonally expand and traffic to tissues, where they perform effector functions including direct killing of infected cells and secretion of cytokines coordinating local immune responses. Following antigen clearance, the majority of the expanded effector T‐cell pool contracts by apoptosis, but a residual population remains and forms the memory compartment. These memory cells comprise lymph‐node‐recirculating ‘central memory’ and tissue‐surveying ‘effector memory’ (EM) cells, as well as tissue‐resident memory populations – and all are equipped with superior capacity to respond to secondary antigen encounter. Two major lineages of T cells exist – CD8+ T cells, which, in addition to cytokine production, mediate direct target cell lysis, and CD4+ T cells, which largely secrete cytokines to shape and orchestrate the immune response. CD4+ T cells can be further subdivided into various inflammatory effector populations [e.g. T helper type 1 (Th1), Th2, Th17] eliciting different types of immune responses specific to the type of pathogen present; and regulatory T (Treg) cells that exert immune‐suppressive functions to prevent or control excessive or inappropriate inflammatory immune responses.
Activation, proliferation and differentiation of T cells are all supported by dynamic changes in their metabolism, which is the subject of this review. We have broken down the major aspects of metabolism into distinct modules, which will be discussed in turn: glycolysis; the tricarboxylic acid (TCA) cycle; mitochondrial oxidative phosphorylation (OXPHOS); amino acid metabolism; cholesterol and lipid metabolism and control of cellular metabolism. For each module, we discuss key biological features, relevance to T‐cell function, associations with disease and therapeutic potential.
Module 1: glycolysis
Upon antigen encounter, T cells significantly enhance rates of glucose uptake, through increased expression of the cell‐surface transporter Glut1.1 In parallel, excretion of lactate is augmented, indicating increased reduction of the end product of glycolysis, pyruvate, to lactate, rather than its oxidation via the mitochondrial TCA cycle to drive OXPHOS and ATP production (Fig. 1). This ‘Warburg Effect’ (aerobic glycolysis) was first described in cancer cells,2 and Otto Warburg postulated that the phenomenon was necessitated by defective mitochondrial function. However, we now understand that it is due to the requirement of rapidly proliferating cells for the biosynthetic precursor molecules produced during glycolysis (Fig. 1), rather than increasing ATP production. Indeed, increased mitochondrial ATP generation (yielding an increased ATP : AMP ratio) would inhibit glycolysis, therefore reduction and excretion of pyruvate as lactate maintains high glycolytic rates.3 Additionally, reduction of pyruvate to lactate generates NAD+, required for function of the glycolytic enzyme, glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH).
Figure 1.
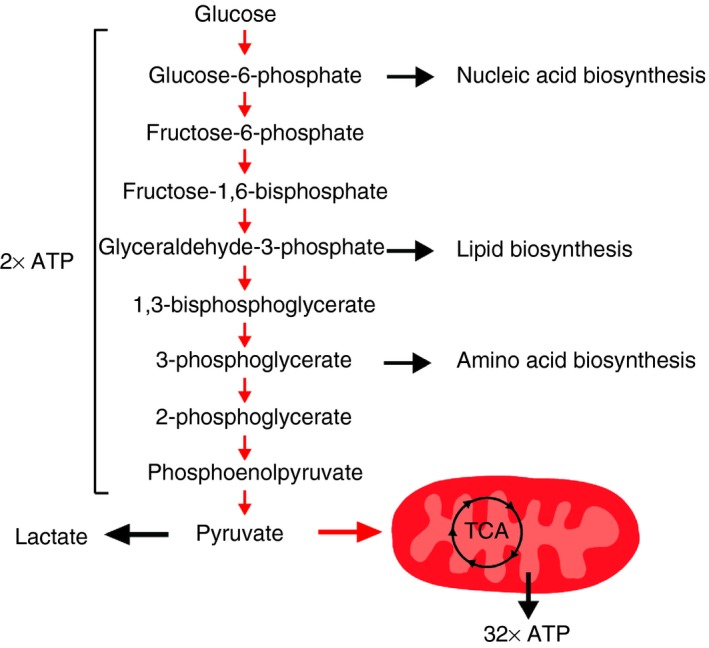
Glycolysis pathway. Glucose is converted to pyruvate through sequential enzymatic reactions occurring in the cytosol. Intermediates of this process can be further metabolised to yield precursors for synthesis of nucleic acids, lipids and amino acids, which are critically required for T‐cell clonal expansion. Pyruvate can be either oxidized in the mitochondria to drive the tricarboxylic acid (TCA) cycle; oxidative phosphorylation (OXPHOS) and ATP generation, or reduced to lactate and excreted. The latter is favoured in proliferating T cells to maintain high rates of glycolysis and precursor molecule generation.
Glycolysis occurs in the cytosol, where 10 enzymatic reactions convert glucose to pyruvate, producing multiple intermediary precursor molecules required by proliferating cells (Fig. 1).3 The intracellular signalling events mediating increased glycolytic activity following T‐cell activation have been delineated in some detail. Co‐stimulatory signalling via CD28 is critical, since ligation of the TCR (CD3) alone fails to up‐regulate Glut1 and glucose uptake.1, 4 Specifically, CD28 ligation leads to phosphatidylinositol‐3‐kinase (PI3K)‐dependent phosphorylation of Akt. Akt activity is sufficient and required for CD28‐dependent up‐regulation of glycolysis.1, 4 The molecular targets of phosphorylated Akt are not yet fully defined; however, it is proposed to augment glycolysis via mechanisms including inducing Glut1 trafficking to the cell surface; phosphorylation and augmentation of glycolytic enzyme activity and, importantly, activation of the kinase, mammalian target of rapamycin (mTOR).5 Akt phosphorylates tuberous sclerosis complex‐2, an inhibitor of mTOR, triggering its proteasomal degradation and thereby de‐repressing mTOR activity. The mTOR is a key regulator of cellular metabolism (discussed in more detail below), and is critical to the increased glycolysis observed in activated T cells.6, 7, 8 Additionally, transcription factors induced following T‐cell activation – and required for metabolic reprogramming – include the nuclear receptor oestrogen‐related receptor‐α (ERR‐α),9 myelocytomatosis oncogene (Myc),10, 11 hypoxia‐inducible factor (HIF)‐1α 6 and interferon regulatory factor 4 (IRF4).12
Use of glycolysis by T cells has implications beyond precursor molecule production. In murine memory CD4+ T cells, notch signalling was shown to drive glucose uptake by regulation of Glut1 expression, which was critical for long‐term survival of these cells in vivo. Inhibition of notch signalling promoted apoptosis, which could be rescued by provision of pyruvate.13 Similarly, we observed that EM human CD4+ T cells critically rely upon glycolysis to prevent apoptosis. Mechanistically we also linked this observation to provision of pyruvate, which maintained mitochondrial membrane potential and subsequent mitochondrial reactive oxygen species (mROS) levels.14
Glycolysis is also critical for gain of effector function in activated T cells, and particularly for secretion of interferon‐γ (IFN‐γ). The capacity to immediately up‐regulate glycolysis upon activation – associated with increased cytosolic GAPDH abundance – permits rapid IFN‐γ production by human EM CD8+ T cells.7 Consistently, CD4+ or CD8+ T cells activated in the absence of glucose demonstrate significantly impaired capacity to secrete IFN‐γ.7, 15 Mechanistically, this has been linked to chromatin remodelling at the IFNG locus,7 and activity of GAPDH, which, when not engaged in glycolysis, binds IFN‐γ mRNA via an AU‐rich region in its 3′ untranslated region and prevents its translation.15 Beyond IFN‐γ, glycolysis has been linked to other effector T‐cell functions. For example, CD4+ Th17 cells isolated from experimental models of multiple sclerosis demonstrate increased glycolytic enzyme expression, and Th17 cells differentiated in vitro are highly glycolytic.16 The role of glycolysis in the function of immune‐suppressive Treg cells remains controversial. Treg cells differentiated in vitro by treatment with transforming growth factor‐β demonstrate low glycolytic capacity compared with inflammatory subsets and no requirement of glycolysis for their suppressive function.17 However, Treg cells differentiated in vitro by suboptimal TCR stimulation do require glycolysis for expression of their hallmark transcription factor FoxP3, through a mechanism involving recruitment of the glycolytic enzyme, enolase‐1, to regulatory regions of the FOXP3 locus and control of variant splicing.18 Directly ex vivo, human Treg cells also appear to be highly glycolytic, and to require glycolysis for their proliferation and suppressive function.19
Given the importance of glycolysis for T‐cell survival and function, it is not surprising that alterations in glycolysis are reported in diseases associated with T‐cell dysfunction, and furthermore that manipulation of glycolysis in T cells represents a promising therapeutic target. Highly glycolytic T cells are described in human inflammatory and infectious diseases,20, 21 and in experimental models of systemic lupus erythematosus (SLE)22 and allogeneic transplantation.23 Indeed, inhibiting glycolysis in these models was beneficial. Furthermore, inhibition of glycolysis, or treatment with dicholoracetate, which favours conversion of pyruvate to acetyl‐CoA rather than lactate, impaired survival, proliferation and cytokine production of Th17 cells, and ameliorated disease progression in experimental autoimmune encephalitis models.16
Conversely, impaired glycolysis is reported in other pathologies, with significant functional implications for T cells. For example, CD4+ T cells from patients with rheumatoid arthritis demonstrate hypo‐metabolism of glucose, due to decreased expression of the enzyme 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase 3 (PFKFB3), which was associated with depleted cellular ROS levels and increased apoptosis. Indeed, survival could be rescued by ectopic PFKB3 expression.24 Reduced glycolytic rates can also be imposed on T cells by depletion of glucose in their environment. This was recently described in experimental tumour models, where highly glycolytic tumour cells impaired T‐cell immune function by glucose deprivation.25, 26, 27 Mechanistically, this was linked to decreased mTOR activity25; decreased abundance of a key glycolytic intermediate with T‐cell calcium signalling capacity,26 and dysregulated microRNA expression.27 Importantly, in these models, anti‐tumour T‐cell function could be restored by augmenting T‐cell expression of a key metabolic enzyme (phosphoenolpyruvate carboxykinase 1),26 raising promising potential for adoptive T‐cell therapies for cancer. Moreover, existing therapies – namely antibody blockade of the inhibitory co‐receptors programmed cell death protein 1 (PD‐1) and cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4) – also restored T‐cell glycolysis, and interlinked cytokine production and tumour clearance.25 In line with these observations, PD‐1 or CTLA‐4 ligation significantly inhibited glycolysis during T‐cell activation, via repression of PI3K‐Akt signalling.1, 28
Module 2: TCA cycle
In addition to the described changes in glycolysis, mitochondrial function is also augmented in activated T cells. Despite the shift in pyruvate metabolism towards lactate, increased glycolysis still augments pyruvate production, which along with fatty acids and glutamine, enters the mitochondria to fuel the TCA cycle, generating additional precursor biosynthetic molecules and driving OXPHOS and ATP production.
Pyruvate enters mitochondria via the mitochondrial pyruvate carrier, where it is first converted to acetyl‐CoA by pyruvate dehydrogenase, before entering into the TCA cycle. In further multiple steps, acetyl‐CoA is oxidized to carbon dioxide and water, yielding GTP and reducing the electron carriers NADH and FADH2, which, together, drive OXPHOS. Along this cycle, several metabolic intermediates are produced and used for anabolic processes (Fig. 2).3
Figure 2.
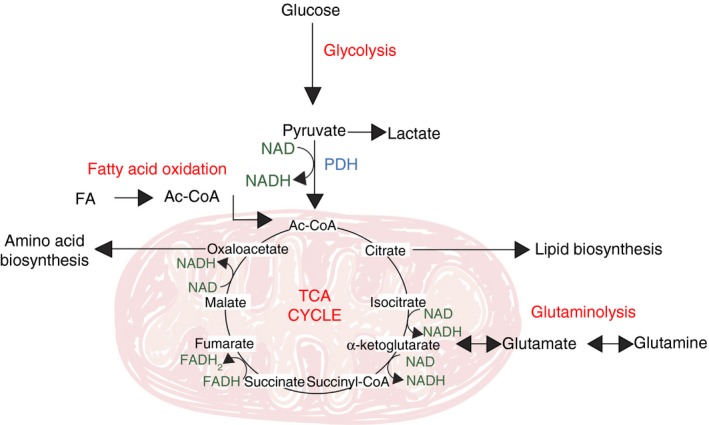
The tricarboxylic acid (TCA) cycle. Glucose‐derived pyruvate, once converted in the mitochondria to acetyl‐CoA by pyruvate dehydrogenase (PDH), enters the TCA cycle to yield biosynthetic intermediates (citrate, α‐ketoglutarate and oxaloacetate) and reduced electron carriers (NADH and FADH 2) that drive oxidative phosphorylation (OXPHOS) by the electron transport chain. Fatty acids and glutamine can also fuel the TCA cycle, following fatty acid oxidation and glutaminolysis, respectively.
The TCA cycle not only oxidizes glucose‐derived acetyl‐CoA, but also integrates the metabolism of glucose, fatty acids and another critical nutrient for T cells, glutamine. Fatty acids, once converted to acyl‐CoA, pass the outer and inner mitochondrial membranes by concerted activity of carnitine palmitoyltransferase 1 and 2 (CPT1 and 2), and carnitine‐acylcarnitine translocase. In the mitochondrial matrix, the process of β‐oxidation breaks down the long carbon chains into a series of two‐carbon acetate units, which, when combined with coenzyme A, form acetyl‐CoA (Fig. 2). Glutamine enters the TCA cycle following its cytosolic conversion to glutamate by glutaminase, which is followed by mitochondrial entry of glutamate through the mitochondrial glutamate transporter and finally conversion of glutamate to α‐ketoglutarate (Fig. 2).
The importance of oxidative metabolism of glucose, fatty acids and glutamine for T‐cell function has been examined in several studies. As discussed above, when glucose availability or metabolism is restricted in quiescent T cells, provision of pyruvate critically maintains survival.13, 14 Pyruvate oxidation further increases upon T‐cell activation16 and inhibitors of pyruvate dehydrogenase (pyruvate dehydrogenase kinases) are concomitantly down‐regulated.29 T‐cell activation, proliferation and interleukin‐2 (IL‐2) production are significantly impaired under glucose‐limiting conditions, but rescued by provision of pyruvate or galactose (which forces respiration over aerobic glycolysis),15, 30 indicating that mitochondrial substrate oxidation plays a significant role in these processes.
Fatty acid oxidation (FAO) is also reported to be critical to T‐cell function. Specifically, it was shown that CD4+ Treg cells, differentiated in vitro, had a high capacity for FAO compared with inflammatory CD4+ T‐cell subsets, and that the CPT1 inhibitor, etomoxir, impaired Treg cell differentiation and suppressive function in vitro, without impacting inflammatory subsets.17 Conversely, in human ex vivo populations, non‐Treg CD4+ T cells demonstrated increased FAO capacity than Treg cells. However, Treg cells required FAO for their suppressive capacity, whereas non‐Treg function was FAO‐independent.19 FAO has also been linked to superior mitochondrial capacity and longevity of memory CD4+ and CD8+ T cells – as further discussed below.
Glutamine availability is critical for T‐cell survival, proliferation and effector function upon activation.31 Activated T cells dramatically increase glutamine uptake, through increased expression of glutamine transporters,31, 32 and concomitantly increase expression of enzymes involved in glutaminolysis.32 These changes are instructed by CD28‐ERK signalling, and induction of Myc expression is required.10 Mechanistically, glutamine is required for full mTOR activation,31 which is probably related to its role facilitating direct mTOR complex I (mTORC1) activation by other amino acids (as discussed below).33, 34 Additionally, glutamine critically fuels the TCA cycle, particularly when glucose availability is limited, maintaining abundance of key intermediates such as pyruvate and citrate.35
Another substrate that can enter the TCA cycle, following conversion to acetyl‐CoA, is acetate. We recently observed this pathway to have important implications for T‐cell effector functions. Specifically, upon infection, systemic acetate levels increased. Upon uptake into CD8+ memory T cells, acetate entered the TCA cycle and expanded the citrate‐derived acetyl‐CoA pool. This promoted post‐translational acetylation of GAPDH, increasing its efficiency and interlinked IFN‐γ production. Consistently, acetate‐exposed memory CD8+ T cells mediated superior protection in a Listeria infection model.36
Increased T‐cell glucose oxidative capacity is reported in human inflammatory diseases including SLE,22, 37 and in experimental models of SLE22, 29 and allograft.38 In SLE models, increased glucose metabolism was successfully targeted to ameliorate disease, by combined inhibition of glycolysis and mitochondrial oxidation,22, 29 whereas further promotion of glucose oxidation with dichloroacetate favoured inflammatory T‐cell differentiation in vitro and conferred no protection in vivo.29 Increased FAO capacity of T cells in pathology has also been reported –specifically in an experimental model of graft‐versus‐host disease – and indeed was also targeted successfully by in vivo treatment with the CPT1 inhibitor etomoxir.39 Finally, the importance of glutamine metabolism for T‐cell activation and function has been exploited therapeutically in an experimental skin transplantation model, where pharmacological inhibition of glutaminolysis, either alone or in combination with inhibition of glycolysis and/or mitochondrial respiration, promoted graft survival.23
Module 3: oxidative phosphorylation
As well as generating precursor molecules for biosynthesis, a key function of the TCA cycle is to reduce the electron carriers NAD+ and FADH to NADH and FADH2, respectively. Subsequent oxidation of these molecules drives activity of the mitochondrial electron transport chain to yield ATP (Fig. 3). Another important product of OXPHOS is mROS, produced at complexes I and III.
Figure 3.
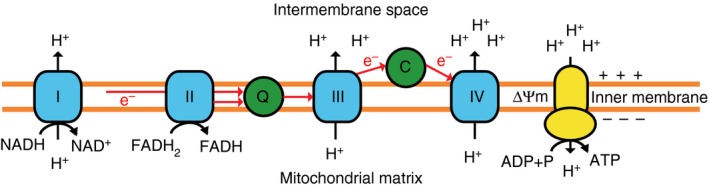
Oxidative phosphorylation (OXPHOS) by the electron transport chain (ETC). The ETC consists of five multi‐subunit complexes, which are located within the inner mitochondrial membrane. Complexes I and II accept electrons from reduced NADH and FADH2, respectively, and pass them, via Coenzyme Q (Q), to Complex III and subsequently via cytochrome c (C) to complex IV. Complex IV finally transfers the electrons to molecular oxygen as final electron acceptor to reduce oxygen to water. The redox energy generated through the electron transfer can be used by complexes I, III and IV to pump protons (H+) across the mitochondrial inner membrane into the inter‐membrane space, building up an electrochemical proton gradient across the mitochondrial inner membrane. This membrane potential (Δϕm) can then be used by Complex V (ATP‐Synthase) to generate adenosine triphosphate (ATP) from adenosine diphosphate (ADP) and phosphate.
Increased activity of the TCA cycle in activated T cells augments OXPHOS. TCR‐induced calcium signalling supports this metabolic change, potentially by increasing calcium‐dependent TCA enzyme activity. A direct consequence of increased OXPHOS is elevated production of mROS, which is critical to T‐cell proliferation and effector function. The mROS induce IL‐2 production via NFAT activation, and T cells lacking the capacity to produce mROS at complex III fail to proliferate and mediate inflammation in vivo.30 The importance of mROS for CD8+ T‐cell function was further confirmed by identification of the protein lymphocyte expansion molecule (LEM), which regulates expression of the mitochondrial electron transport chain complex, and consequent mROS production, and is crucial for CD8+ T‐cell proliferation, cytotoxicity, memory generation and control of lymphocytic choriomeningitis virus infection in vivo.40
In addition to increased function, there is also evidence for mitochondrial biogenesis following T‐cell activation, reflected by increases in mitochondrial mass and mitochondrial DNA abundance.41 How this process is regulated in T cells is largely unknown and warrants further investigation. Biogenesis of mitochondria is a complex and highly regulated process. Mitochondria are semi‐autonomous organelles containing their own circular genome, mitochondrial DNA, encoding 13 proteins with essential function in respiratory complexes. The remaining majority of electron transport chain complex proteins are encoded by nuclear DNA. Co‐ordinated expression of these protein complexes is achieved by hierarchical activity of a panel of transcription factors, including nuclear respiratory factors NRF1 and NRF2, oestrogen‐related receptors (ERR‐α, ERR‐β, ERR‐γ), mitochondrial transcription factor A (TFAM), and the peroxisome proliferator‐activated receptor γ co‐activator 1‐α (PGC‐1α). Of note, ERR‐α and TFAM expression are required for normal T‐cell activation.9, 42
Although it has yet to be defined how and when mitochondrial biogenesis occurs in T cells, it is established that antigen‐experienced memory CD4+ and CD8+ T cells have an increased mitochondrial mass, structural complexity and functional capacity compared with naive cells, with significant implications for their immune functions.7, 14, 43 For example, memory CD4+ and CD8+ T cells demonstrate higher spare respiratory capacity (SRC) than antigen‐naive cells.7, 14, 43 The SRC represents the reserve mitochondrial capacity available to the cell for energy production in response to increased work or metabolic stress, such as hypoxia. The EM CD4+ T cells retain residual SRC under hypoxia, whereas in naive cells SRC is entirely consumed. This enables EM CD4+ T cells to maintain ATP levels, survival, migration and cytokine production under hypoxia, whereas naive T cells fail to maintain ATP turnover and activate intrinsic apoptosis pathways.14 Hence, EM CD4+ T cells are adapted to their role surveying metabolically challenging tissue environments for antigen. Mechanistically, SRC has been linked to increased capacity for FAO and specifically increased expression of CPT1. Indeed, CPT1 over‐expression increases SRC and memory T‐cell generation and survival in vivo,43 whereas impaired FAO is associated with the inverse.44 Interestingly, it appears that memory T cells – unlike naive cells – do not obtain the lipids required for increased FAO from their external environment, but rather use glucose‐derived carbon to synthesize triacylglycerols in the endoplasmic reticulum. These lipids then undergo lysosomal acid lipase‐mediated lipolysis.45 Consistent with this model, memory CD8+ T cells also demonstrate up‐regulation of the glycerol channel aquaporin 9, facilitating uptake of glycerol required for triacylglycerol synthesis and storage.46 Expression of both lysosomal acid lipase and aquaproin 9 – co‐ordinated by activity of the cytokines IL‐15 and IL‐7, respectively (which promote memory T‐cell formation) – is required for optimal memory T‐cell generation and survival in vivo.45, 46
In addition to equipping memory T cells for longevity, their superior mitochondrial capacity is also linked to rapid recall responses of memory cells. Increased proliferation of memory T cells upon secondary antigen encounter is dependent upon mitochondrial ATP‐synthase activity. Additionally, it appears that mitochondrial ATP is required for memory T cells to up‐regulate glycolysis upon activation, since pharmacological dissociation of the ATP‐dependent glycolytic enzyme hexokinase from the mitochondria has been reported to prevent this glycolytic switch.47
Mitochondria play important roles in T cells beyond maintaining cellular bioenergetics. For example, cross‐talk of mitochondria with other cellular organelles also appears to be critical for normal T‐cell function. T cells lacking TFAM – demonstrating reduced mtDNA transcription and OXPHOS capacity – also show dysfunctional lysosomal calcium mobilization, endopeptidase activity and lipid degradation, which is associated with defective autophagosome–lysosome fusion. Functionally, this translated to increased CD4+ Th1 cell differentiation in vitro and increased in vivo pathogenicity of these cells in a colitis model.42 Mitochondria also play an important role in calcium signalling. During T‐cell activation, calcium enters across the plasma membrane through the opening of calcium release‐activated calcium ((CRAC)/ORAI1) channels at the immune synapse. Mitochondria take up inflowing calcium very efficiently, so acting as calcium buffers preventing calcium‐dependent inactivation of these channels. Notably, it has been observed that mitochondria translocate to the immune synapse where they locally regulate calcium flux.48
Given the importance of mitochondria for T‐cell activation, differentiation and function, it is not surprising that deficient mitochondrial function is associated with impaired T‐cell activity. For example, T‐cell senescence is associated with decreased mitochondrial content and function. EMRA (effector memory CD45RA‐expressing) T cells have significantly less proliferative capacity – despite potent effector functions – and are postulated to be an end‐stage differentiated/senescent T‐cell population accumulating with age and in certain diseases. EMRA cells demonstrate increased mitochondrial and cytoplasmic ROS, pointing to dysfunctional mitochondria. Indeed EMRA cells have fewer mitochondria and decreased mitochondrial mass and SRC when compared with central and effector memory T cells.49 Improving mitochondrial capacity to improve T‐cell longevity, memory formation and function is an attractive possibility. Effectively, promotion of mitochondrial fusion and cristae remodelling by over‐expression of optic atrophy 1 (Opa1), which regulates inner mitochondrial membrane fusion, promotes memory T‐cell generation in vivo and generates cells with increased anti‐tumour capacity.50 Additionally, modulation of expression of the regulatory methylation‐controlled J protein enhances mitochondrial respiratory capacity of CD8+ T cells, associated with increased IFN‐γ production and anti‐viral activity.51
Inversely, elevated OXPHOS capacity, a hyperpolarized mitochondrial membrane and increased ROS production are associated with inappropriate T‐cell activity in experimental models of graft‐versus‐host disease and SLE, as well as in SLE patients. In graft‐versus‐host disease, elevated ROS levels of alloreactive cells were targeted therapeutically, using an inhibitor of F1F0‐ATPase (complex V), which further increased ROS production and induced their selective apoptosis.38 In SLE models, on the other hand, direct inhibition of mitochondrial complex I with the anti‐diabetic drug metformin, was able to control disease activity.22
Module 4: amino acid metabolism
In addition to glucose and glutamine, T cells also increase their uptake of large neutral amino acids upon activation.8, 52 The importance of large neutral amino acids to this process was demonstrated by studies where the system L transporter Slc7a5 (LAT1), transporting phenylalanine, tyrosine, leucine, arginine and tryptophan, was genetically deleted in murine T cells.52 LAT1‐deficient CD4+ T cells were unable to proliferate and to differentiate in vitro into Th1 or Th17 cells, whereas Treg cell differentiation was not affected. Similarly, CD8+ T‐cell expansion and differentiation were significantly impaired both in vitro and in vivo. One important cellular function of large neutral amino acids, particularly leucine and arginine, is the signalling of amino acid sufficiency to mTOR complex 1 (mTORC1) – allowing its activation. This pathway has been characterized in depth in recent years and involves the RAS‐related GTP‐binding protein (Rag) family of small GTPases. Specifically, in the presence of amino acids, Rag A/B‐dependent translocation of mTORC1 to the lysosomal membrane occurs, bringing it into proximity with its activator Rheb.53, 54 The Rags themselves are located to the lysosome via interaction with the pentameric ‘Ragulator complex’, comprising the ‘late endosomal/lysosomal adaptor and MAPK and mTOR activator’ proteins 1–5 (LAMTOR 1–5).54, 55 Upstream molecular events permitting Rag A/B activation by specific amino acids have more recently been elucidated. A critical hub in this process appears to be the protein GATOR2, which positively regulates Rag A/B activity. GATOR2 itself is inhibited via interaction with either Sestrin2 or CASTOR1 – interactions that are competitively disrupted by the presence of leucine or arginine, respectively.56, 57, 58 Interestingly, during T‐cell activation both amino acid uptake and elements of this molecular pathway are enhanced, events which, in combination, strongly promote mTORC1 activity. Specifically, TCR ligation and CD28‐mediated co‐stimulation of CD4+ T cells induces their production and cleavage of complement C3, yielding C3a and C3b. In turn, C3b, in an autocrine manner, ligates its cell‐surface receptor CD46, driving transcriptional events including increased expression of LAMTOR5. Accordingly, both mTORC1 recruitment to the lysosome and activity are enhanced in activated T cells. These events are critical for CD4+ T‐cell differentiation into Th1 effector cells, which is abrogated in hereditary or experimentally induced CD46 deficiency, LAMTOR5 deficiency or by pharmacological mTORC1 inhibition.8 More recently other important observations have linked AA‐sensing capacity to T‐cell fate. Interestingly, it appears that during proliferation of CD8+ T cells, LAT1 and its heterodimer CD98 are asymmetrically inherited by daughter cells. Cells inheriting more of this transporter demonstrated higher mTORC1 lysosomal recruitment and activity as well as higher c‐Myc expression. Functionally this translated into increased glycolytic and mitochondrial respiratory capacity, improved proliferation and effector cytokine production.59, 60
Similarly to the scenario described for glucose, depletion of critical amino acids can impair T‐cell responses. This has been assessed specifically for arginine. Myeloid cells, including polymorphonuclear leucocytes, macrophages and myeloid‐derived suppressor cells, as well as epithelial cells, all express arginase and can release this enzyme into the extracellular environment – leading to decreased local and systemic arginine levels.61, 62, 63, 64 Co‐culture and in vivo experiments indicate that these cells thereby impair T‐cell proliferation and cytokine production in an arginase‐dependent manner, through a mechanism involving down‐regulation of the CD3ζ chain.61, 65, 66 This phenomenon is implicated in pathophysiological suppression of T‐cell responses in chronic infection61, 62, 63 and cancer,66 and additionally in immune regulation at immune privileged sites such as the cornea,64 or during pregnancy.67
Module 5: cholesterol and lipid metabolism
T‐cell clonal expansion requires significant synthesis of cellular and organelle membranes, which is supported by alterations in metabolism of lipids, including cholesterol. Activated CD8+ T cells increase expression of many lipogenic enzymes. This process is transcriptionally mediated by the sterol regulatory element‐binding protein (SREBP) transcription factors, and regulated by activity of PI3K and mTOR. Loss of SREBP activity impairs blasting and cell cycle entry of T cells, which is largely rescued by provision of exogenous cholesterol. In vivo, CD8+ T cells lacking SREBP activity demonstrated poor clonal expansion and effector cytokine function during viral infection.68
At the single cell level, regulation of cholesterol metabolism has important implications for TCR clustering and signalling. Total cellular, and specifically plasma membrane cholesterol levels increase markedly in activated CD8+ T cells, associated with augmented expression of enzymes involved in cholesterol biosynthesis and decreased expression of those involved in its efflux. However, key enzymes mediating esterification of cholesterol for storage are also increased in activated CD8+ T cells – acetyl‐CoA acetyltransferase (Acat) 1 and 2. Inhibition or deletion of these enzymes further increased CD8+ T‐cell cholesterol levels, and in parallel enhanced TCR clustering and proximal signalling. Consequently, CD8+ T‐cell cytotoxicity, cytokine production and anti‐tumour activity were enhanced.69
Regulatory T cells appear to be particularly dependent upon heightened cholesterol metabolism for proper function – driven by high constitutive mTORC1 activity – since their suppressive function in vitro can be abrogated by treatment with inhibitors of 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase, the rate‐controlling enzyme of the cholesterol synthesis pathway. This was associated with reduced Treg cell proliferation, and impaired expression of surface CTLA‐4 and inducible T‐cell co‐stimulator, which mediate cell‐contact dependent suppressive mechanisms.70
In contrast to cholesterol synthesis, Treg cells appear to be less dependent than effector CD4+ T cells on fatty acid synthesis from glucose‐derived carbon. Indeed, compared to Treg cells, Th17 cells more abundantly express key enzymes involved in the conversion of glucose‐derived pyruvate into citrate for fatty acid synthesis. Consistently, Th17 cells incorporate significantly more glucose‐derived pyruvate into fatty acids than Treg cells, whereas Treg cells demonstrate significant capacity to take up exogenous fatty acids. Furthermore, inhibition of acetyl‐CoA carboxylase 1, which performs a key step in fatty acid synthesis, directed the fate of CD4+ T cells away from a Th17 phenotype, and towards Treg cells, both in vitro and in vivo, ameliorating experimental autoimmune encephalitis.71
There is evidence for dysregulated T‐cell lipid metabolism in disease. For example, in SLE, increased expression of the liver X receptor β (LXRβ), which controls cellular lipid metabolism, in CD4+ T cells, was found to associate with increased cell membrane glycosphingolipid abundance. These cells further demonstrated aberrant TCR signalling, increased proliferation and altered cytokine expression, all of which could be corrected by inhibition of glycosphingolipid biosynthesis.72 Also, in an experimental model of non‐alcoholic fatty liver disease, accumulation of hepatic free fatty acids was shown to be associated with increased apoptosis of T cells – specifically CD4+ T cells – and increased frequency of hepatocellular carcinoma. Of the accumulated free fatty acids, linoleic acid demonstrated significant capacity to induce CD4+ T‐cell apoptosis in vitro, which mechanistically was linked to induction of increased mitochondrial OXPHOS and FAO capacity, and mitochondrial ROS‐dependent cell death.73
Module 6: control of cellular metabolism
As described here, adaption of specific metabolic phenotypes is critical for T‐cell immune functions. Regulation of metabolism therefore critically guides T‐cell functional plasticity. A key metabolic regulator of T cells is mTOR, which integrates immune and metabolic cues, such as antigens (via PI3K–Akt signalling) and nutrients (via amino acid sensing), and phosphorylates downstream targets to control mRNA translation and protein degradation. This activity directs T‐cell metabolic reprogramming and ultimately acquisition of effector function. The mTOR‐deficient T cells, or T cells treated with the mTOR inhibitor rapamycin, fail to engage metabolic reprogramming upon activation, and become anergic, rather then proliferating and gaining effector functions. Rapamycin is therefore used therapeutically in many T‐cell‐mediated pathologies.74 The role of mTOR in T‐cell biology was recently clarified by unbiased proteomic analyses. These confirmed a high activity of mTOR in activated CD8+ T cells, and more interestingly revealed that mTORC1 inhibition with rapamycin decreased protein abundance of a specific subset of proteins, rather than having a global effect. These proteins included CD8+ T‐cell effector molecules such as IFN‐γ and cell‐lytic granzymes, glucose transporters and enzymes involved in glycolysis and cholesterol metabolism. Proteins associated with mitochondrial OXPHOS and glutamine metabolism were, however, increased following rapamycin treatment, indicating selectivity of the role of mTORC1 in T‐cell metabolism.75 These findings will probably have implications for the use of rapamycin therapeutically, particularly since it appears that the balance of short‐term effector versus long‐lived memory CD8+ T cells can be influenced by specifically regulating glycolytic activity.76 Indeed, pharmacological or small interfering RNA targeting of mTORC1 has already been reported to increase memory CD8+ T‐cell responses in vaccination and tumour models.77, 78
AMP‐activated protein kinase (AMPK) counterbalances mTOR as a regulator of cellular metabolism. Rising AMP concentrations, an indicator of excess energy expenditure, activate AMPK, which promotes ATP production by phosphorylating targets in glycolytic and FAO pathways, and limits ATP‐consuming processes by inhibiting catabolic processes; protein translation, and importantly inhibiting mTORC1. The liver kinase B1 (LKB1) is a positive regulator of AMPK, and T cells deficient in LKB1 exhibit augmented glycolytic metabolism associated with increased T‐cell activation and production of inflammatory cytokines,79 pointing to the importance of this axis in regulating T‐cell function. Furthermore, AMPK plays an important role regulating metabolic plasticity of T cells under nutrient poor conditions. Under glucose‐replete conditions, quiescent T cells adopt aerobic glycolysis upon activation. However, an environment with reduced nutrient availability leads to metabolic reprogramming dictated by AMPK activity. Cells engage glutamine‐dependent OXPHOS and IFN‐γ mRNA translation is suppressed to meet bioenergetic requirements and viability.35 The capacity for AMPK to regulate inflammatory T‐cell function has been therapeutically targeted using AMPK activators such as metformin and 5‐aminoimidazole‐4‐carboxamide ribonucleoside in disease models including experimental autoimmune encephalitis and inflammatory bowel disease.80, 81
Summary
Recent developments in our understanding of how T‐cell immune function is inextricably linked to their metabolic phenotype have significantly advanced our comprehension of T‐cell biology in health and disease. Furthermore, these developments have revealed significant potential to therapeutically modulate T‐cell metabolism and interlinked immune function in the treatment of diseases characterized by insufficient or excessive T‐cell function. As metabolism is fundamental to all cellular processes, the next obstacle will be to specifically target these processes in T cells; however, advances in targeted therapeutics and adoptive cellular therapies raise exciting potential to address this challenge.
Funding
This study received funds from the Swiss National Science Foundation (310030_154059 and CRSII_160766), the Gebert‐Rüf Foundation (GER‐058/14) and the Swiss Cancer League (KFS‐3773‐08‐2015).
Disclosures
The authors have no conflict of interest to declare.
References
- 1. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR et al The CD28 signaling pathway regulates glucose metabolism. Immunity 2002; 16:769–77. [DOI] [PubMed] [Google Scholar]
- 2. Warburg O. On the origin of cancer cells. Science 1956; 123:309–14. [DOI] [PubMed] [Google Scholar]
- 3. Lunt SY, Vander MG. Heiden, aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Ann Rev Cell Dev Biol 2011; 27:441–64. [DOI] [PubMed] [Google Scholar]
- 4. Jacobs SR, Herman CE, MacIver NJ, Wofford JA, Wieman HL, Hammen JJ et al Glucose uptake is limiting in T cell activation and requires CD28‐mediated Akt‐dependent and independent pathways. J Immunol 2008; 180:4476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol 2004; 172:4661–5. [DOI] [PubMed] [Google Scholar]
- 6. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR et al HIF1α‐dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 2011; 208:1367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G et al Rapid effector function of memory CD8+ T cells requires an immediate‐early glycolytic switch. Nat Immunol 2013; 14:1064–72. [DOI] [PubMed] [Google Scholar]
- 8. Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA et al Complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. Immunity 2015; 42:1033–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Michalek RD, Gerriets VA, Nichols AG, Inoue M, Kazmin D, Chang C‐Y et al Estrogen‐related receptor‐α is a metabolic regulator of effector T‐cell activation and differentiation. Proc Natl Acad Sci USA 2011; 108:18348–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D et al The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011; 35:871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Preston GC, Sinclair LV, Kaskar A, Hukelmann JL, Navarro MN, Ferrero I et al Single cell tuning of Myc expression by antigen receptor signal strength and interleukin‐2 in T lymphocytes. EMBO J 2015; 34:2008–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S et al The transcription factor IRF4 is essential for TCR affinity‐mediated metabolic programming and clonal expansion of T cells. Nat Immunol 2013; 14:1155–65. [DOI] [PubMed] [Google Scholar]
- 13. Maekawa Y, Ishifune C, Tsukumo S‐I, Hozumi K, Yagita H, Yasutomo K. Notch controls the survival of memory CD4+ T cells by regulating glucose uptake. Nat Med 2014; 21:55–61. [DOI] [PubMed] [Google Scholar]
- 14. Dimeloe S, Mehling M, Frick C, Loeliger J, Bantug GR, Sauder U et al The immune‐metabolic basis of effector memory CD4+ T cell function under hypoxic conditions. J Immunol 2016; 196:106–14. [DOI] [PubMed] [Google Scholar]
- 15. Chang C‐H, Curtis JD, Maggi LB, Faubert B, Villarino AV, Sullivan DO et al Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013; 153:1239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O et al Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest 2015; 125:194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF et al Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 2011; 186:3299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C et al Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol 2015; 16:1174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M et al The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity 2016; 44:406–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ostroukhova M, Goplen N, Karim MZ, Michalec L, Guo L, Liang Q et al The role of low‐level lactate production in airway inflammation in asthma. Am J Physiol Lung Cell Mol Physiol 2012; 302:L300–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Palmer CS, Ostrowski M, Gouillou M, Tsai L, Yu D, Zhou J et al Increased glucose metabolic activity is associated with CD4+ T‐cell activation and depletion during chronic HIV infection. AIDS 2013; 28:297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yin Y, Choi S‐C, Xu Z, Perry DJ, Seay H, Croker BP et al Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med 2015; 7:274ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee C‐F, Lo Y‐C, Cheng C‐H, Furtmüller GJ, Oh B, Andrade‐Oliveira V et al Preventing allograft rejection by targeting immune metabolism. Cell Rep 2015; 13:760–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med 2013; 210:2119–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang C‐H, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD et al Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015; 162:1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ho P‐C, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R et al Phosphoenolpyruvate is a metabolic checkpoint of anti‐tumor T cell responses. Cell 2015; 162:1217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L et al Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol 2016; 17:95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN et al PD‐1 alters T‐cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun 2015; 6:6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yin Y, Choi S‐C, Xu Z, Zeumer L, Kanda N, Croker BP et al Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol 2015; 196:80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA et al Mitochondria are required for antigen‐specific T cell activation through reactive oxygen species signaling. Immunity 2013; 38:225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakaya M, Xiao Y, Zhou X, Chang J‐H, Chang M, Cheng X et al Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014; 40:692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A et al Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol 2010; 185:1037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B et al Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009; 136:521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Durán RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E et al Glutaminolysis activates Rag‐mTORC1 signaling. Mol Cell 2012; 47:349–58. [DOI] [PubMed] [Google Scholar]
- 35. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia‐Vazquez G, Yurchenko E et al The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo . Immunity 2015; 42:41–54. [DOI] [PubMed] [Google Scholar]
- 36. Balmer ML, Ma EH, Bantug GR, Grählert J, Pfister S, Glatter T et al Memory CD8+ T cells require increased concentrations of acetate induced by stress for optimal function. Immunity 2016; 44:1312–24. [DOI] [PubMed] [Google Scholar]
- 37. Wahl DR, Petersen B, Warner R, Richardson BC, Glick GD, Opipari AW. Characterization of the metabolic phenotype of chronically activated lymphocytes. Lupus 2010; 19:1492–501. [DOI] [PubMed] [Google Scholar]
- 38. Gatza E, Wahl DR, Opipari AW, Sundberg TB, Reddy P, Liu C et al Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft‐versus‐host disease. Sci Transl Med 2011; 3:67ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Byersdorfer CA, Tkachev V, Opipari AW, Goodell S, Swanson J, Sandquist S et al Effector T cells require fatty acid metabolism during murine graft‐versus‐host disease. Blood 2013; 122:3230–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Okoye I, Wang L, Pallmer K, Richter K, Ichimura T, Haas R et al T cell metabolism. The protein LEM promotes CD8+ T cell immunity through effects on mitochondrial respiration. Science 2015; 348:995–1001. [DOI] [PubMed] [Google Scholar]
- 41. D'Souza AD, Parikh N, Kaech SM, Shadel GS. Convergence of multiple signaling pathways is required to coordinately up‐regulate mtDNA and mitochondrial biogenesis during T cell activation. Mitochondrion 2007; 7:374–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baixauli F, Acín‐Pérez R, Villarroya‐Beltrí C, Mazzeo C, Nuñez‐Andrade N, Gabandé‐Rodriguez E et al Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab 2015; 22:485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van der Windt GJW, Everts B, Chang C‐H, Curtis JD, Freitas TC, Amiel E et al Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012; 36:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L‐S et al Enhancing CD8 T‐cell memory by modulating fatty acid metabolism. Nature 2009; 460:103–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Sullivan D, van der Windt GJW, Huang SC‐C, Curtis JD, Chang C‐H, Buck MD et al Memory CD8+ T cells use cell‐intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 2014; 41:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cui G, Staron MM, Gray SM, Ho P‐C, Amezquita RA, Wu J et al IL‐7‐induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell 2015; 161:750–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van der Windt GJW, O'Sullivan D, Everts B, Huang SC‐C, Buck MD, Curtis JD et al CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci USA 2013; 110:14336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schwindling C, Quintana A, Krause E, Hoth M. Mitochondria positioning controls local calcium influx in T cells. J Immunol 2010; 184:184–90. [DOI] [PubMed] [Google Scholar]
- 49. Henson SM, Lanna A, Riddell NE, Franzese O, Macaulay R, Griffiths SJ et al p38 signaling inhibits mTORC1‐independent autophagy in senescent human CD8⁺ T cells. J Clin Invest 2014; 124:4004–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Buck MD, O'Sullivan D, Klein GR, Curtis JD, Chang C‐H, Sanin DE et al Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 2016; 166:63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Champagne DP, Hatle KM, Fortner KA, D'Alessandro A, Thornton TM, Yang R et al Fine‐tuning of CD8+ T cell mitochondrial metabolism by the respiratory chain repressor MCJ dictates protection to influenza virus. Immunity 2016; 44:1299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sinclair LV, Rolf J, Emslie E, Shi Y‐B, Taylor PM, Cantrell DA. Control of amino‐acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol 2013; 14:500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Long X, Ortiz‐Vega S, Lin Y, Avruch J. Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J Biol Chem 2005; 280:23433–6. [DOI] [PubMed] [Google Scholar]
- 54. Sancak Y, Bar‐Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator‐Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010; 141:290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bar‐Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012; 150:1196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saxton RA, Knockenhauer KE, Wolfson RL, Chantranupong L, Pacold ME, Wang T et al Structural basis for leucine sensing by the Sestrin2‐mTORC1 pathway. Science 2016; 351:53–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR et al Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016; 351:43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA et al The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 2016; 165:153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Verbist KC, Guy CS, Milasta S, Liedmann S, Kamiński MM, Wang R et al Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature 2016; 532:389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pollizzi KN, Sun I‐H, Patel CH, Lo Y‐C, Oh M‐H, Waickman AT et al Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8+ T cell differentiation. Nat Immunol 2016; 17:704–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Munder M, Schneider H, Luckner C, Giese T, Langhans C‐D, Fuentes JM et al Suppression of T‐cell functions by human granulocyte arginase. Blood 2006; 108:1627–34. [DOI] [PubMed] [Google Scholar]
- 62. Pallett LJ, Gill US, Quaglia A, Sinclair LV, Jover‐Cobos M, Schurich A et al Metabolic regulation of hepatitis B immunopathology by myeloid‐derived suppressor cells. Nat Med 2015; 21:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Modolell M, Choi B‐S, Ryan RO, Hancock M, Titus RG, Abebe T et al Local suppression of T cell responses by arginase‐induced l‐arginine depletion in nonhealing leishmaniasis. PLoS Negl Trop Dis 2009; 3:e480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fu H, Khan A, Coe D, Zaher S, Chai J‐G, Kropf P et al Arginine depletion as a mechanism for the immune privilege of corneal allografts. Eur J Immunol 2011; 41:2997–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Munder M, Engelhardt M, Knies D, Medenhoff S, Wabnitz G, Luckner‐Minden C et al Cytotoxicity of tumor antigen specific human T cells is unimpaired by arginine depletion. PLoS ONE 2013; 8:e63521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB et al Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T‐cell receptor expression and antigen‐specific T‐cell responses. Cancer Res 2004; 64:5839–49. [DOI] [PubMed] [Google Scholar]
- 67. Kropf P, Baud D, Marshall SE, Munder M, Mosley A, Fuentes JM et al Arginase activity mediates reversible T cell hyporesponsiveness in human pregnancy. Eur J Immunol 2007; 37:935–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP et al Sterol regulatory element‐binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol 2013; 14:489–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X et al Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016; 531:651–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish Treg‐cell function. Nature 2013; 499:485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K et al De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med 2014; 20:1327–33. [DOI] [PubMed] [Google Scholar]
- 72. McDonald G, Deepak S, Miguel L, Hall CJ, Isenberg DA, Magee AI et al Normalizing glycosphingolipids restores function in CD4+ T cells from lupus patients. J Clin Invest 2014; 124:712–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ma C, Kesarwala AH, Eggert T, Medina‐Echeverz J, Kleiner DE, Jin P et al NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016; 531:253–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity 2010; 33:301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hukelmann JL, Anderson KE, Sinclair LV, Grzes KM, Murillo AB, Hawkins PT et al The cytotoxic T cell proteome and its shaping by the kinase mTOR. Nat Immunol 2016; 17:104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z et al Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 2013; 123:4479–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Berezhnoy A, Castro I, Levay A, Malek TR, Gilboa E. Aptamer‐targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Invest 2014; 124:188–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Turner AP, Shaffer VO, Araki K, Martens C, Turner PL, Gangappa S et al Sirolimus enhances the magnitude and quality of viral‐specific CD8+ T‐cell responses to vaccinia virus vaccination in rhesus macaques. Am J Transplant 2011; 11:613–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC et al The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol 2011; 187:4187–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nath N, Khan M, Paintlia MK, Singh I, Hoda MN, Giri S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol 2009; 182:8005–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bai A, Yong M, Ma AG, Ma Y, Weiss CR, Guan Q et al Novel anti‐inflammatory action of 5‐aminoimidazole‐4‐carboxamide ribonucleoside with protective effect in dextran sulfate sodium‐induced acute and chronic colitis. J Pharmacol Exp Ther 2010; 333:717–25. [DOI] [PubMed] [Google Scholar]