

Summary
The clinical benefits of short‐term therapy with glucocorticoids (GC) in patients with inflammatory bowel disease (IBD) are widely known. However, the effects of this treatment towards the re‐establishment of the regulatory network in IBD are not fully explored. We have evaluated the immunological effects of the abbreviated GC therapy in experimental colitis induced by 3% dextran sulphate sodium in C57BL/6 mice. Treatment with GC improved disease outcome, constrained circulating leucocytes and ameliorated intestinal inflammation. The control of the local inflammatory responses involved a reduction in the expression of interferon‐γ and interleukin‐1β, associated with augmented mRNA levels of peroxisome proliferator‐activated receptors (α and γ) in intestine. Furthermore, there was a reduction of CD4+ T cells producing interferon‐γ, together with an increased frequency of the putative regulatory population of T cells producing interleukin‐10, in spleen. These systemic alterations were accompanied by a decrease in the proliferative potential of splenocytes of mice treated in vivo with GC. Notably, treatment with GC also led to an increase in the frequency of the regulatory markers GITR, CTLA‐4, PD‐1, CD73 and FoxP3, more prominently in spleen. Taken together, our results pointed to a role of GC in the control of leucocyte responsiveness and re‐establishment of a regulatory system, which probably contributed to disease control and the restoration of immune balance. Finally, this is the first time that GC treatment was associated with the modulation of a broad number of regulatory markers in an experimental model of colitis.
Keywords: dextran sulphate sodium‐induced colitis, glucocorticoids, immune regulation, inflammatory bowel disease, regulatory cells
Introduction
The effect of glucocorticoids (GC) on the immune system was first described in 1924 when a relationship between adrenalectomy and thymus hyperplasia was portrayed in rats.1 Since then, GC have become one of the most frequently prescribed drugs to treat patients with inflammatory disorders such as asthma,2 rheumatoid arthritis3 or inflammatory bowel disease (IBD).4, 5 Among these immune disturbances, those related to the gastrointestinal tract, such as Crohn's disease and ulcerative colitis, are of special interest because they affect millions of people around the world and are responsible for various physical, nutritional and immunological disabilities. In this context, Crohn's disease and ulcerative colitis, generally known as IBD, are characterized by their chronic course with alternating episodes of disease activity, varying severity and clinical remission periods.6
Corticosteroids represent one of the most effective options to treat acute exacerbations of IBD and are recognized by their ability to induce remission in patients with moderate to severe disease.7, 8 These drugs down‐regulate key players in inflammation such as the nuclear factor‐κB, nuclear factor of activated T cells and activator protein‐1. These transcription factors are involved in the induction of a vast number of pro‐inflammatory cytokines, including interleukin‐1 (IL‐1), IL‐2, IL‐6, IL‐12, IL‐18, interferon‐γ (IFN‐γ) and tumour necrosis factor‐α (TNF‐α), which are related to disease worsening and progression.2, 9 Additionally, treatment with GC was also able to promote the differentiation of regulatory T (Treg) cells in a FoxP3‐dependent manner, besides inducing higher expression of mRNA for anti‐inflammatory proteins such as IL‐10 in asthma.10 In this context, when dexamethasone, a GC drug that represents one of the most potent immunosuppressive treatments currently available, is used in combination with vitamin D3, there is a great impact on the induction of IL‐10‐producing lymphocytes in vitro.11 Corticosteroids also inhibit the recruitment of immune cells and the expression of adhesion molecules in inflamed tissue.12 For such reasons, GCs have been used to treat IBD for over 50 years13 either alone or combined with other drugs such as thiopurines,14 biological therapies or aminosalicylates.15 Though some adverse effects and refractoriness were described, especially for long‐term therapy, the role of these drugs in the control of inflammatory disorders is of special relevance. Most importantly, the involvement of GC in the balance between effector and regulatory responses that drive the re‐establishment of immune tolerance cannot be underappreciated and may represent an important field of investigation.
Hence, this study aimed to investigate the impact of short‐term GC therapy on the regulation of host responses after breakdown of mucosal tolerance in an experimental model of intestinal inflammation.
Material and methods
Animal studies
All studies were performed in accordance with Institutional Animal Care and Use Committee of the University of Sao Paulo (Brazil). The procedures were approved under protocol 11.1.522.53.0. Male C57BL/6 mice, aged 6–8 weeks, weighing 20–25 g, were maintained under controlled temperature (25°), in specific pathogen‐free and standard controlled environmental conditions with a 12‐hr light/12‐hr dark cycle, with food and water ad libitum in the animal housing facility of the School of Pharmaceutical Sciences of Ribeirão Preto, University of São Paulo. The experiments were performed with five mice/group, as follows: Group 1; healthy control mice without colitis; Group 2, animals exposed to dextran sodium sulfate (DSS); Group 3, mice exposed to DSS and treated with dexamethasone.
DSS‐induced colitis, clinical assessment and macroscopic analysis
Colitis was induced by 3% DSS (MP Biomedicals, Illkirch, France; molecular weight: 36 000–50 000) added to the drinking water during 6 days for sample collection or continuously for survival evaluation. Besides water and food intake, in all experiments mice were evaluated daily for body weight change and clinical signs of disease to produce a clinical disease score. Each signal presented by the animal corresponded to one point and the sum of points for each mouse generated a clinical score. Macroscopic analysis of the colon on the day of euthanasia was performed to quantify the ‘post‐mortem score’. Either clinical or post‐mortem scores were determined as previously described by Sales‐Campos et al.16
Glucocorticoid treatment
Dexamethasone (Azium® Solução, Coopers Saúde Animal, Rio de Janeiro, Brazil) was diluted in saline and administered intraperitoneally, daily, at a dose of 1 mg/kg/day, according to previous dose–response experiments performed by the group (data not shown). Treatment with dexamethasone was given from the 3rd to the 5th day of colitis.
Intestinal permeability assay
Intestinal permeability assay was performed to assess barrier function using an FITC‐dextran method, as previously described.17 Briefly, food and water were withdrawn overnight, followed by oral administration of the permeability tracer in experimental mice (44 mg/100 g body weight of FITC‐labelled dextran, MW 4000; FD4; Sigma‐Aldrich, St Louis, MO). Serum was collected 4 hr later and fluorescence intensity was determined by reading the absorbance of the samples by spectrophotometry (excitation, 492 nm; emission, 525 nm). FITC‐dextran concentrations in sera were determined using a standard curve generated byserial dilution of FITC‐dextran, ranging from 2000 to 100 μg/ml.
Euthanasia and sample collection
Mice were killed on the 6th day of colitis and the colon, spleen and mesenteric lymph nodes (MLN) were collected for further analysis. The colon samples were divided into smaller fragments that were immersed in PBS/10% formaldehyde for paraffin embedding or immediately frozen in liquid nitrogen for the quantification of myeloperoxidase (MPO), eosinophil peroxidase (EPO) and N‐acetylglucosaminidase (NAG) activity by enzymatic assays. In some experiments, colon tissues as well as spleen and MLN were also immersed in culture medium RPMI‐1640 containing 5% fetal bovine serum for cell culture and flow cytometry assays.
Histopathological analysis
The large intestine samples were fixed in PBS/10% formaldehyde followed by standard histology procedures and paraffin embedding. Microtomy was performed to obtain 5‐μm thick sections that were later stained with haematoxylin and eosin for microscopic analysis of tissue inflammation.
MPO, EPO and NAG activities measurement
Colonic fragments were cut into small pieces, weighed and processed for MPO, EPO and NAG quantification as previously described.18 Briefly, for MPO assay, fragments were homogenized and erythrocytes were lysed. The pellet obtained after centrifugation was resuspended, followed by three freezing and thawing rounds. After centrifugation the supernatant was placed in 96‐well plates and revealed by addition of TMB Substrate Reagent Set (Tetramethylbenzidine – BD OptEIA™, San Diego, CA) at 37°. The reaction was stopped and readings were performed in a spectrophotometer at 450 nm. For EPO measurement, fragments were homogenized, the homogenate was centrifuged, pellet was resuspended, and freeze–thawed three times followed by centrifugation. Then, supernatant was incubated in 96‐well plates with OPD (o‐phenylenediamine dihidrochloride tablet sets, Sigma‐Aldrich) diluted in Tris–HCl and 0·05 mm H2O2 for 30 min at 37°. The reaction was stopped and absorbance was determined at 492 nm. For NAG assay, the supernatant from the MPO assay was used, and NAG activity was measured by the addition of 2·25 mm p‐nitrophenyl‐2‐acetamide‐d‐glucopyranoside, and 50 mm citrate buffer (pH 4·5). After incubating for 60 min at 37° in the dark, the reaction was stopped and absorbance was determined at 405 nm. The results were expressed as optical density per gram of tissue.
Real‐time PCR
Quantitative PCR was determined on gut homogenates containing TRIzol reagent (Invitrogen, Waltham, MA). Primers for IL‐1β (F, 5′‐TGACAGTGATGAGAATGACCTGTTC‐3′, R, 5′‐TTGGAAGCAGCCCTTCATCT‐3′), IFN‐γ (F, 5′‐CACCCTGATTACTACCTTC‐3′, R, 5′‐GGGTTGTTGACCTCAAAC‐3′), peroxisome proliferator‐activated receptor α (PPAR‐α) (F, 5′‐TCAATGCCTTAGAACTGGATGA‐3′, R, 5′‐CCGATCTCCACAGCAAATTATA‐3′) and PPAR‐γ (F, 5′‐TGAGATCATCTACACGATGCTG‐3′, R, 5′‐AGGAACTCCCTGGTCATGAA‐3′), were purchased from Sigma‐Aldrich. Total RNA from colon tissue specimens was extracted using a Promega kit and TRIzol (Promega, Madison, WI). For RT‐PCR, 1 μg of total RNA was reverse transcribed using the High‐capacity kit (Applied Biosystems, Madison, WI) following the manufacturer's instruction. The expression of target genes was evaluated using the GoTaq® qPCR Master Mix kit (Promega) in an SYBR Green system, according to the manufacturer's instructions. The differential expression of mRNA was determined using the formula 2–∆∆Ct, compared with the expression of the housekeeping gene β‐actin, according to the Applied Biosystems User's Bulletin #2 (P/N 4303859) published in 1997.
Isolation of leucocytes from spleen, MLN, intraepithelial compartment and lamina propria
Cells from spleen and MLNs were obtained and filtered through a 70‐μm cell strainer (BD Biosciences, Heidelberg, Germany). Red blood cells were lysed using ammonium–chloride–potassium) buffer. Colon leucocytes were isolated according to previous studies.19, 20 All cell suspensions were tested for viability by Trypan blue dye at 0·2% in a Neubauer chamber before being used in the culture and/or immunophenotyping experiments.
Flow cytometry
For phenotypic characterization of the leucocytes from spleen, MLN, lamina propria or intraepithelial lymphocytes, monoclonal antibodies (BD Pharmingen, San Diego, CA) conjugated to fluorochromes were used, according to the manufacturer's instructions. Cells were acquired on a DIVA Flow Cytometer (BD, San Jose, CA) and data obtained were analysed using flowjo version 7.6.3 software (TreeStar, San Carlos, CA). To identify Treg cells, our gating strategy comprised a first evaluation of leucocytes with lymphocyte characteristics (size and granulosity), by delineating a region in dot plots according to the FSC × SSC parameters. Then, the frequency of CD4+ CD25+ and CD4+ CD25− cells was related to the percentage of leucocytes derived from this first gate delineated in our analysis, which comprised the total putative lymphocyte population from spleen and MLN. The expression of the Treg cell markers resulted from histograms of the molecular fluorescence derived from the regions of the CD4+ CD25+ and CD4+ CD25− populations.
Detection of intracellular cytokines
Isolated spleen cells were cultured at a concentration of 5 × 105 cells/well in 96‐well plates and stimulated in vitro with PMA at 50 ng/ml and ionomycin at 500 ng/ml (both from Sigma‐Aldrich), in the presence of brefeldin A (BD GolgiPlug, San Diego, CA) for 4 hr, at 37° in 5% CO2. Cells were stained for surface markers, fixed, permeabilized and incubated with antibodies specific for intracellular cytokine detection, using staining (PBS containing fetal bovine serum 5% and azide sodium 0·09%), fixation (PBS/4% formaldehyde) and permeabilization buffers (PBS containing simulated body fluid 1%, azide sodium 0·1% and saponin 0·2%), before flow acquisition.
Proliferation assay
For proliferation assay, spleen cells from mice treated in vivo with GC (and controls) were isolated as described above and cultured at 5 × 105 cells/well in 96‐well plates in RPMI‐1640 for 72 hr, at 37° in 5% CO2, in the presence of Concanavalin A (Sigma‐Aldrich) at 3 μg/ml. Cells were labelled with 5,6‐carboxyfluorescein succinimidyl ester (Sigma‐Aldrich) at 5 μm, according to the manufacturer's instructions. Cells were acquired on a DIVA Flow Cytometer (BD Biosciences, San Jose, CA) and data obtained were analysed using flowjo version 7.6.3 software (TreeStar). The calculation of the proliferative index was performed using a specific tool in the software, which quantifies the cellular divisions of the responding leucocytes that divided at least once during the experimental period. The software automatically calculates it by dividing the total number of divisions by the quantity of divided cells.
Statistical analysis
In all variables the normal distribution and homogeneous variance were tested. When the distribution was normal and there was homogeneous variance we used the parametric one‐tailed unpaired Student's t‐test. In case of non‐Gaussian distribution, we used the non‐parametric Mann–Whitney one‐tailed test. Results were expressed as mean ± SEM. Differences were considered statistically significant when P < 0.05 (5%). All analyses were performed using the graphpad prism 5.0 software (GraphPad, San Diego, CA).
Results
Glucocorticoid treatment reduces disease clinical score and constrains the pool of circulating leucocytes
First, to verify the impact of exogenous GC on clinical disease outcome, mice were exposed to DSS and treated with dexamethasone, as described in the Material and methods. Animals treated with GC had lower disease clinical score (Fig. 1a), despite no differences in the weight loss when compared with untreated mice, on day 6 (Fig. 1b), which was a valuable time‐point for observation of important immune alterations in experimental colitis, as determined in a previous study.16 Then, as GCs are effective to reduce leucocyte recruitment, we next verified if the treatment was also able to modulate different circulating leucocyte populations. Indeed, although no effects were observed in the frequency of cells (Fig. 1d–f), the results from peripheral blood showed that after exposure of mice to DSS, GC was able to reduce the total number of leucocytes (Fig. 1c), including lymphocytes (Fig. 1g), monocytes (Fig. 1h) and neutrophils (Fig. 1i). These results reinforce the potential for GC in the control of inflammation by constraining the pool of circulating cells.
Figure 1.
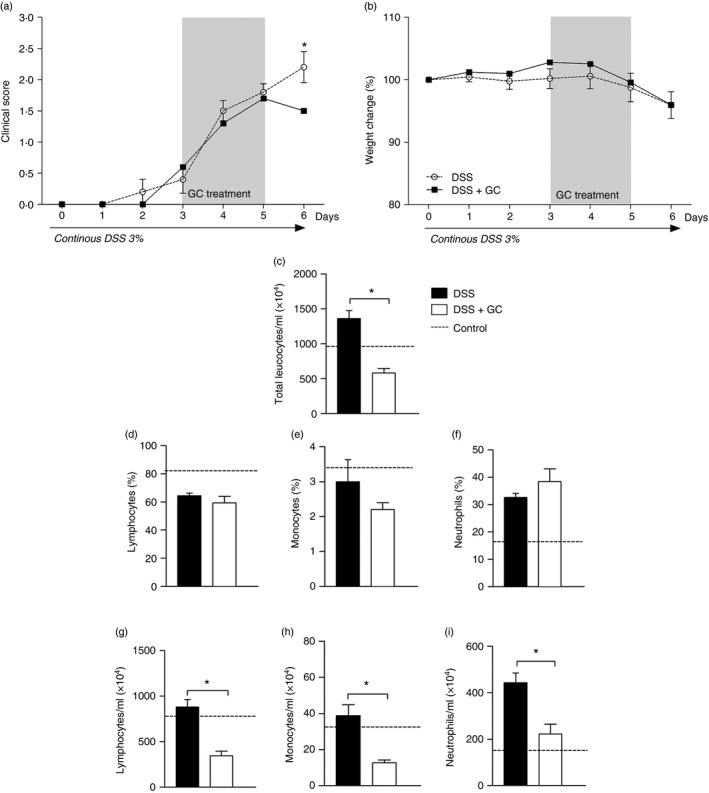
Glucocorticoid treatment reduces disease clinical score and constrains the pool of circulating leucocytes. C57BL/6 mice were exposed to dextran sulphate sodium (DSS) 3% and treated daily with dexamethasone as depicted in the grey rectangle. On day 6 mice were killed to obtain blood samples. (a) Clinical disease score. (b) Percentage of weight change. (c) Total leucocytes/ml. Frequencies of (d) lymphocytes, (e) monocytes and (f) neutrophils are expressed as percentage of cells. Absolute values of lymphocytes (g), monocytes (h) and neutrophils (i), are expressed as number of cells/ml. DSS: C57BL/6 mice exposed to DSS; DSS+GC: C57BL/6 mice exposed to DSS and treated with glucocorticoids. The dashed lines correspond to the group of C57BL/6 control mice not exposed to DSS. These results are representative of three independent experiments with five mice/group. Data were analysed using one‐tailed unpaired t‐tests in (c), (d), (f), (g) and Mann–Whitney one‐tailed tests in (a), (b), (e) (h), (i). *P < 0·05.
Glucocorticoid treatment is able to improve intestinal architecture and to modulate inflammatory infiltrate in intestine
As treatment with GC had an impact on the clinical signs of colitis and altered peripheral blood leucocytes, we next evaluated the histopathological changes induced in the gut after exposure to DSS and treatment with GC. As expected, control groups not exposed to DSS showed low cellularity of the lamina propria and no changes in the intestinal architecture (Fig. 2a, left panel). On the other hand, mice exposed to DSS had a huge accumulation of inflammatory cells, with the presence of both mononuclear and polymorphonuclear leucocytes, disruption of intestinal mucosa, muscle layer thickening, epithelial ulcerations and depletion of goblet cells. Furthermore, a loss of intestinal architecture was also observed (Fig. 2a, central panel). However, upon receiving treatment with exogenous GC, mice had a notable reduction in the inflammatory infiltrate with an early re‐establishment of the colonic architecture and re‐epithelialization at day 6. This amelioration was followed by the recovery of goblet cells, which probably accounted for restored mucus production in the gut (Fig. 2c, right panel).
Figure 2.
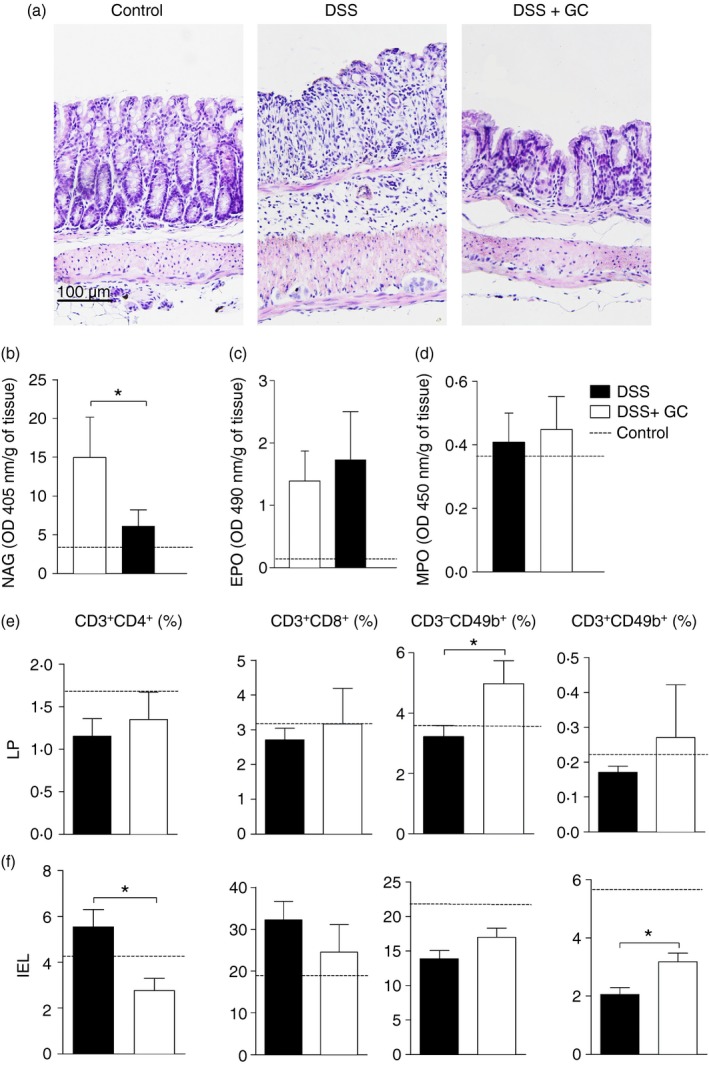
Treatment with glucocorticoid (GC) is able to improve intestinal architecture and to modulate inflammatory infiltrate in intestine. C57BL/6 mice were exposed to dextran sulphate sodium (DSS) 3% and treated with dexamethasone as described in the Material and methods. The colon was collected on the 6th day for histopathological analysis and cell immunophenotyping. (a) Left panel – control mice, not exposed to DSS; centre panel – DSS, mice exposed to DSS; right panel – DSS+GC, mice exposed to DSS and treated with glucocorticoids. (b) N‐acetylglucosaminidase (NAG), (c) eosinophil peroxidase (EPO) and (d) myeloperoxidase (MPO) activities. For phenotypic characterization, leucocytes were isolated from (e) lamina propria (LP) and (f) intraepithelial lymphocyte (IEL) compartments. After acquisition, samples were analysed by flowjo software. DSS: C57BL/6 mice exposed to DSS; DSS+GC: C57BL/6 mice exposed to DSS and treated with glucocorticoids. The dashed lines correspond to the group of C57BL/6 control mice not exposed to DSS. These results are representative of three independent experiments with five mice/group. Data were analysed using one‐tailed unpaired t‐tests in (c), (e) and Mann–Whitney one‐tailed tests in (b), (d). *P < 0·05.
Next, due to the differences observed in the local inflammatory infiltrate, we investigated the characteristics of these cells. Interestingly, treatment with GC was able to reduce the activity of the enzyme NAG (Fig. 2b), which may suggest an effect towards a decrease in the local macrophage population. However, no differences were observed for the local production of EPO (Fig. 2c) or MPO (Fig. 2d), which indirectly represents the activity of eosinophil and neutrophils in the intestine. To further investigate the phenotypic characteristics of cells in the colon inflammatory infiltrate, isolated leucocytes were labelled for flow cytometry assay as described in the Materials and methods . In vivo treatment with GC led to increased frequency of natural killer (CD3− CD49b+) cells in lamina propria (Fig. 2e and see Supplementary material, Fig. S1) and CD3+ CD49b+ cells in the intraepithelial lymphocyte compartment (Fig. 2f and see Supplementary material, Fig. S1). Additionally, this drug also reduced the frequency of CD3+ CD4+ T lymphocytes in intraepithelial lymphocytes (Fig. 2f), but no differences were observed in the other populations from lamina propria or intraepithelial lymphocytes (Fig. 2e,f). Taken together, these data indicated that GC treatment was able to regulate intestinal inflammatory infiltrate and improve tissue architecture, which was disrupted by colitis induction.
Glucocorticoid acts synergistically with PPAR (α and γ) to dampen inflammation during experimental colitis
As GC treatment was able to constrain inflammation and ameliorate intestinal morphology, we next assessed its impact on the expression of key molecules with divergent roles in the development of intestinal inflammation. Hence, treatment reduced the expression of the inflammatory cytokines IFN‐γ (Fig. 3a) and IL‐1β (Fig. 3b) while enhancing the expression of the nuclear receptors PPAR‐α and PPAR‐γ (Fig. 3c,d, respectively), which exert a potential anti‐inflammatory activity in gut homeostasis. In addition, the role of short‐term GC administration in the amelioration of intestinal inflammation was reinforced by the observation of diminished serum levels of FITC‐dextran (Fig. 3e), which points to decreased intestinal permeability following treatment, accompanied by a reduced post‐mortem score (Fig. 3f), as evaluated by macroscopic analysis of the gut. Altogether, these results suggested that the differential modulation of molecules with opposing roles in the development of intestinal inflammation may have contributed to local colitis control in mice treated with GC.
Figure 3.
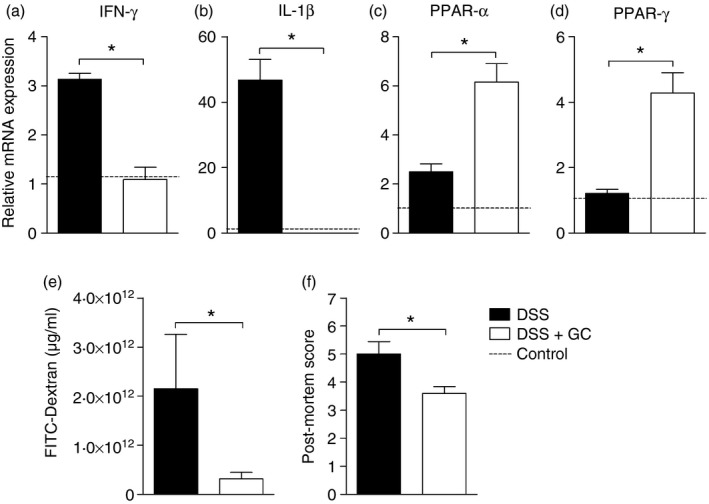
Glucocorticoid induces peroxisome proliferator‐activated receptor (PPAR) expression and attenuates inflammation during experimental colitis. C57BL/6 mice were exposed to dextran sulphate sodium (DSS) 3% and treated with dexamethasone as described in the Material and methods. The colon was collected on the 6th day of colitis for real‐time PCR assay. In (a) relative expression of interferon‐γ (IFN‐γ), (b) interleukin‐1β (IL‐1β), (c) PPAR) ‐α and (d) PPAR‐γ (d). (e) Concentration of FITC‐Dextran expressed as μg/ml. (f) Post‐mortem score. DSS: C57BL/6 mice exposed to DSS; DSS+GC: C57BL/6 mice exposed to DSS and treated with glucocorticoids. The dashed lines correspond to the group of C57BL/6 control mice not exposed to DSS. These results are representative of three independent experiments with five mice/group. Data were analysed using one‐tailed unpaired t‐tests in (c) and Mann–Whitney one‐tailed tests in (a), (b), (d), (e), (f). *P < 0·05.
Short‐term therapy with GC modulates leucocyte responsiveness during colitis
Therapy with GC was able to improve and down‐regulate the colitogenic response in the gut, so we next investigated the impact of this treatment in the modulation of different spleen cell populations that could trigger inflammation after breakdown of the intestinal mucosal barrier. In fact, the results showed that the treatment was able to reduce the frequency of CD8+ T cells and increase the natural killer (CD49b+) population (Fig. 4b,c, respectively), whereas no differences were observed in the natural killer T cells (CD3+ CD49b+; Fig. 4d) or CD3+ CD4+ lymphocytes (Fig. 4a). However, the function of CD4 T cells seemed to be altered by GC therapy, as there was reduced frequency of inflammatory CD4 T lymphocytes producing IFN‐γ (Fig. 4e) and a notable augment in the percentage of the putative regulatory population of CD4+ T cells producing IL‐10 (Fig. 4h). No differences were observed in the frequency of CD4+ T cells producing IL‐17 or IL‐4 (Fig. 4f,g, respectively). Most interestingly, splenocytes from mice with colitis treated in vivo with GC had a decreased ex vivo proliferative potential, a condition that was reversed by exposure to a mitogenic stimulus, in vitro (Fig. 4i). Altogether, these data indicated that, besides the known molecular mechanisms of GC action, this drug might modulate inflammation by altering leucocyte responsiveness and inducing regulatory responses during colitis onset.
Figure 4.
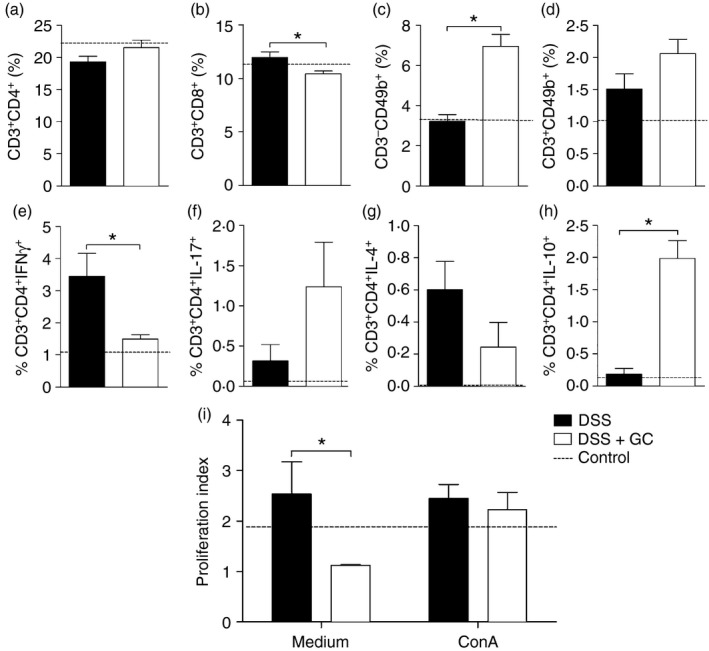
Short‐term therapy with glucocorticoid modulates leucocyte responsiveness during colitis onset. C57BL/6 mice were exposed to dextran sulphate sodium (DSS) 3% and treated with dexamethasone as described in the Material and methods. The spleen was collected on the 6th day for immunophenotyping experiments. In (a) CD3+ CD4+ cells, (b) CD3+ CD8+ cells, (c) CD49b+ and (d) CD3+ CD49b+, evaluated by flow cytometry. Spleen cell function was evaluated by the frequency of CD4+ T cells producing (e) interferon‐γ (IFN‐γ), (f) interleukin‐17 (IL‐17), (g) IL‐4 and (h) IL‐10. The proliferation index (i) was determined by cytometry analysis of splenocytes from mice with colitis treated or not with glucocorticoids in vivo and cultured ex vivo in the absence or presence of concanavalin A (ConA). DSS: C57BL/6 mice exposed to DSS; DSS+GC: C57BL/6 mice exposed to DSS and treated with glucocorticoids. The dashed lines in (a–h) correspond to the group of C57BL/6 control mice not exposed to DSS and in (i) to the cells from these control mice cultured without re‐stimuli in vitro. These results are representative of three independent experiments with five mice/group. Data were analysed using one‐tailed unpaired t‐tests in (a), (b), (c), (d), (g) and Mann–Whitney one‐tailed tests in (e), (f), (h), (i). *P < 0·05.
GC treatment up‐regulates different regulatory markers in lymphoid organs
To further investigate the importance of GC in the re‐establishment of the regulatory network that controls excessive inflammation, we next assessed the impact of the therapy on the frequency of regulatory cells and their markers in spleen and MLN. Though GC reduced cellularity in these lymphoid organs (see Supplementary material, Fig. S2), there were important changes in the composition of cell subtypes infiltrated in the spleen and MLN. Indeed, we observed enhanced frequency of both splenic CD4+ CD25− and CD4+ CD25+ T lymphocytes (Fig. 5a‐R1 and R2). This effect was also detected for the putative population of Treg cells in MLN, besides a reduction in the frequency of CD4+ CD25− in that organ (Fig. 5a‐R1). Indeed, treatment with GC augmented the percentage of almost all regulatory markers either in spleen or MLN, especially for the splenic CD4+ CD25+ cells that could partially account for some of the population of natural Treg cells (Fig. 5a‐R2). In this organ, the activity of GC was more prominent and was confirmed by the induction of augmented GITR (Fig. 5b), CTLA‐4 (Fig. 5c), CD73 (Fig. 5e) and FoxP3 (Fig. 5f) in both CD4+ CD25− and CD4+ CD25+ populations, besides an increase in the CD4+ CD25+ PD‐1+ cells (Fig. 5d). Similarly, treatment with GC was also able to boost the frequency of regulatory markers in the MLN. However, in this case, the improvement was not to the same extent as that observed in spleen. Taken together, these data suggested a remarkable action of GC on specific populations of regulatory cells, which might have contributed to the re‐establishment of immune tolerance and disease amelioration after the short‐term therapy.
Figure 5.
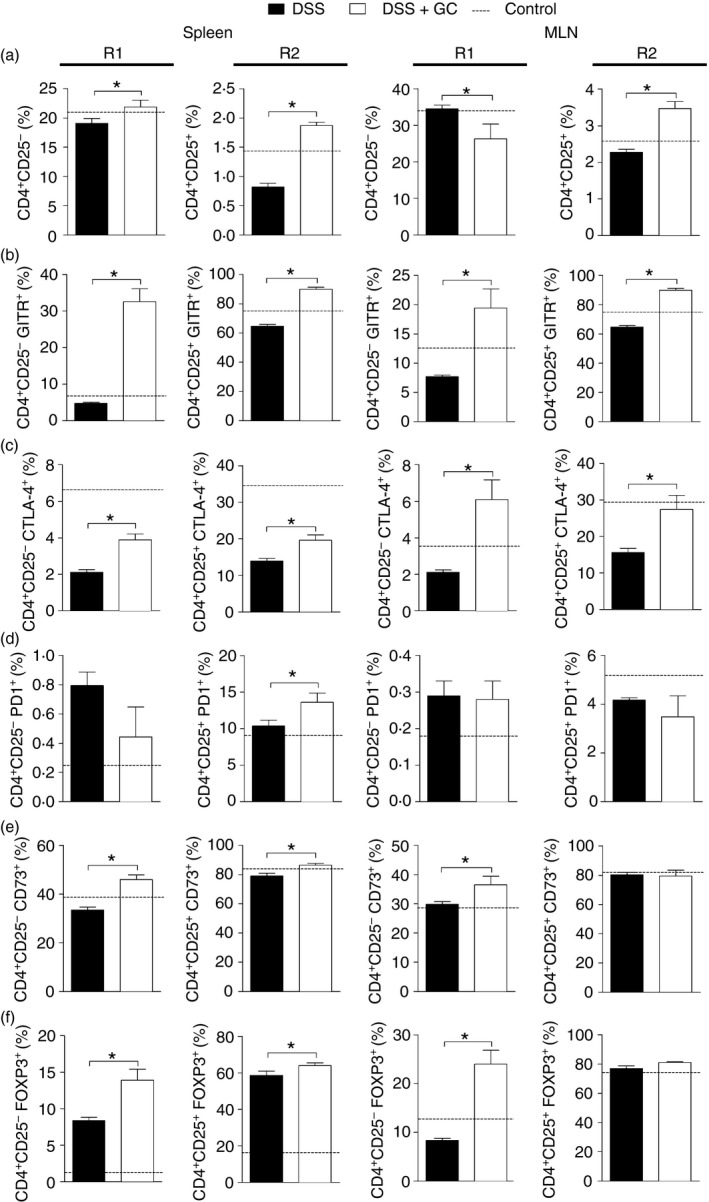
Glucocorticoid treatment up‐regulates different regulatory markers in lymphoid organs. C57BL/6 mice were exposed to dextran sulphate sodium (DSS) 3% and treated with dexamethasone as described in the Material and methods. The spleen and mesenteric lymph nodes (MLN) were collected on the 6th day and processed for regulatory T‐cell staining. In (a) leucocytes from spleen and MLN. R1 and R2 depict the gating strategy for CD4+ CD25− and CD4+ CD25+ cells, respectively. (b) CD4+ CD25− GITR + and CD4+ CD25+ GITR + leucocytes from spleen and MLN. (c) Frequency of CD4+ CD25− CTLA4+ and CD4+ CD25+ CTLA4+ cells from spleen and MLN. (d) Percentage of CD4+ CD25− PD‐1+ and CD4+ CD25+ PD‐1+ cells from spleen and MLN. (e) Frequency of CD4+ CD25− CD73+ and CD4+ CD25+ CD73+ cells from spleen and MLN. (f) Percentage of CD4+ CD25− FoxP3+ and CD4+ CD25+ FoxP3+ leucocytes from spleen and MLN. DSS: C57BL/6 mice exposed to DSS; DSS+GC: C57BL/6 mice exposed to DSS and treated with glucocorticoids. The dashed lines correspond to the group of C57BL/6 control mice not exposed to DSS. These results are representative of two independent experiments with five mice/group. Data were analysed using one‐tailed unpaired t tests in (a) for CD4+ CD25− cells from spleen and for CD4+ CD25+ cells from MLN; (b) for CD4+ CD25+ GITR + cells from spleen; (c) for CD4+ CD25− CTLA4+ and CD4+ CD25+ CTLA4+ cells from spleen; (d) for CD4+ CD25+ PD‐1+ cells from spleen and CD4+ CD25− PD‐1+ cells from spleen and MLN; (e) CD4+ CD25− CD73+ cells from spleen and CD4+ CD25+ CD73+ cells from spleen and MLN; (f) CD4+ CD25+ FoxP3+ cells from spleen. In the other figures the Mann–Whitney one‐tailed tests were used. *P < 0·05.
Discussion
The results presented here showed that short‐term treatment with GC plays an important role in the modulation of immune responses during experimental colitis. Notably, this therapy guided the ongoing inflammatory responses towards immune regulation by the induction of different regulatory markers. This assumption was based on the fact that mice treated with GC had an increase in the frequency of regulatory cells in association with a reduction in inflammatory components of immunity, which may have accounted for the improvement of intestinal architecture and clinical disease outcome.
The beneficial effects of GC in colitis control were followed by a decrease in different populations of circulating leucocytes, pointing to the widely known immunosuppressive effects of these drugs. In this context, GC have successfully been used to treat IBD, especially because of their role in reducing inflammation, so driving disease remission.7 Moreover, due to GC properties in dampening immune responses, it is reasonable to assume that their effects in constraining lymphocyte proliferation may be related to apoptosis21, 22, 23 and down‐regulation of key players in inflammation such as the nuclear factor‐κB, nuclear factor of activated T cells and activator protein‐1,2, 9, 24, 25, 26 besides boosting regulatory components such as Treg cells or anti‐inflammatory proteins like IL‐10.10 Additionally, GCs are also able to induce lymphocyte anergy or hyporesponsiveness by different mechanisms. Among them, this drug induces down‐regulation of tyrosine kinases required for T‐cell activation27 and reduces phosphorylation of T‐cell receptor‐associated substrates such as ζ chain, the ZAP70 kinase and the transmembrane adapter molecule, all required for T‐cell activation.22
Owing to the importance of lymphoid organs in promoting or controlling immune responses and to the effects of treatment with GC towards immunity, it is reasonable to assume that this therapy may also interfere with immune balance and then disease progression. The effects of GC in constraining the T helper type 1 response or inflammatory cytokines such as IFN‐γ and TNF‐α besides decreasing the T cytotoxic or natural killer cell effector activities, are already described and may represent one of the major functions of this class of drugs over immunity.26, 28, 29 Similarly, our results showed that mice treated with GC had a reduction in the frequency of T cytotoxic lymphocytes and CD4+ T cells producing IFN‐γ, besides an augmented percentage of T cells releasing the anti‐inflammatory cytokine IL‐10. The dual role of GC therapy in the inhibition of inflammation by re‐establishing the immune balance, and so reducing inflammatory players and boosting anti‐inflammatory components, was already proven. Therefore, its ability to induce regulatory cytokines such as IL‐10 or transforming growth factor‐β,30, 31 besides the down‐regulation of key transcription factors in the development of inflammation,32 can reinforce the GC potential to dampen inflammatory responses and control disease progression. The modulation of these different cell populations and the cytokines produced by them may also have contributed to the amelioration observed in intestinal architecture. Furthermore, we cannot underestimate the impact of a systemic increase in the regulatory components in the spleen, over a local reduction in the expression of the inflammatory cytokines IL‐1β and IFN‐γ in intestine. This scenario also contributed to the amelioration observed in GC‐treated mice.
Treatment of IBD represents a great challenge due to the lack of responsiveness of some patients and to the presence of recurrent side effects. This could be at least in part attributed to abnormal activity of some genes associated to GC‐response/resistance in patients with IBD, such as multidrug resistance 1 (MDR1)33 and NACHT domain, LRR domain, and pyrin domain‐containing protein 1 (NALP1),34 among others. Furthermore, because of the pleiotropic effects of GC therapy, some important adverse events including immunosuppression, hypertension, delayed wound repair and metabolic disturbances may occur and can explain why this therapy cannot be used for long periods.35 However, though prolonged therapy with GCs in patients with IBD is still controversial and current therapies are not curative, the short‐term use of this drug may be of great importance to control disease flares and acute inflammation.4
The effectiveness of GC therapy is attributed not only to their anti‐inflammatory properties per se but also to their ability to interact and activate a cascade of anti‐inflammatory players such as PPARs. Of note, PPARs are transcriptional factors composed of three major subtypes: PPAR‐α, PPAR‐β/δ and PPAR‐γ. Their physiological ligands are polyunsaturated fatty acids and eicosanoids.36 The synergism between PPAR‐α and dexamethasone was demonstrated in an experimental model of lung inflammation,37 in which the anti‐inflammatory activity of dexamethasone was reduced in PPAR‐α knockout mice when compared with wild‐type controls. Furthermore, PPAR‐γ presents an anti‐inflammatory role in the gut, as their activation led to inhibition of the inflammatory response and was associated with a better clinical outcome in different models of experimental colitis.38, 39, 40 In accordance, a recent study demonstrated that extra‐adrenal production of glucocorticoids by colonic epithelial cells induced the expression of PPAR‐γ. However, this up‐regulation was abrogated in patients with ulcerative colitis,41 which suggests the importance of these receptors in the counter‐inflammatory mechanisms necessary to control disease worsening. Hence, as our results showed that treatment with GC induced up‐regulation of both PPAR‐α and PPAR‐γ, we believe that this modulation is one of the key elements contributing to the amelioration of treated mice.
One of the key aspects regarding immune balance is the role of the Treg cell populations in the re‐establishment of mucosal homeostasis. Treg cells are known by their ability to modulate the pathogenesis of many immune‐mediated diseases, such as IBD.42 The systemic treatment with this drug was shown to improve the frequency of Treg cells and the expression of FoxP3 in asthmatic subjects.10 The up‐regulation of Treg cells was also observed in patients with systemic lupus erythematosus receiving GC therapy.43 Similarly, treatment of patients with severe hepatitis B with GC was able to increase the population of Treg cells besides reducing T helper type 17 cells, which contributed to the re‐establishment of immune balance and disease amelioration in those individuals.44 In general, during exaggerated inflammatory responses, such as in IBD, FoxP3 is not functional or expressed,45, 46 so reinforcing its relevance in the immune modulation and prevention of autoimmunity. On the other hand, the stimulation of naive CD4+ CD25− T cells in the periphery can induce the expression of FoxP3 in these leucocytes, which acquire suppressor capabilities.47, 48, 49 The increase of FoxP3 mainly in the CD4+ CD25− population, after in vivo GC treatment, suggests that, in our study, this drug may have led to the differentiation of inducible Treg cells to control the ongoing intestinal inflammation, besides the natural CD4+ CD25+ Foxp3+ population in spleen and MLN.
Further markers are also related to the activity of Treg cells. Among them, glucocorticoid‐induced TNF receptor‐related protein (GITR or CD357), a co‐stimulatory receptor belonging to the TNF receptor superfamily, is one of the most important regulatory markers. In experimental models of inflammation or autoimmunity, such as in colitis, the role of this receptor in protecting mice from developing the disease is already shown.50 One of the main mechanisms regarding GITR function is related to its ability to enhance the suppressive activity of Treg cells besides inducing proliferation of this cell population.51 CD73 is another marker of Treg cells, which is expressed on the surface of T lymphocytes. Its main function is to inhibit the proliferation of effector cells due to the conversion of extracellular 5‐AMP to adenosine.52 Furthermore, due to the importance of CD73 in controlling this catabolic cell machinery, and the consequences of this process for the outcome of different pathological conditions, the modulation of this molecule has been suggested as a prominent target to control infections, autoimmune diseases and cancer.53, 54, 55 Programmed death‐1 (PD‐1) mediates inhibitory signals depending on the strength of the T‐cell receptor signalling, with greater inhibition delivered at low levels of T‐cell receptor stimulation. The effects can be observed either on cytokine production, with significant reduction in IFN‐γ, TNF‐α and IL‐2 production,56, 57 or on cell death, by inhibiting surviving factors such as Bcl‐xL.58 In addition, PD‐1 can also restrain effector cell function through the inhibition of the expression of transcription factors such as GATA‐3 and Tbet,59 associated with T helper type 2 and T helper 1 phenotypes, respectively. Regarding CTLA‐4, it is a cell surface receptor, member of the CD28 family. Though it shares the same ligands with CD28, CTLA‐4 is able to inhibit T‐cell proliferation and constrain immune response.60, 61 Hence, it is reasonable to believe that induction of regulatory markers may explain the colitis improvement, as the effects of this hormone in driving Treg cell differentiation in vivo and in vitro were already known.62, 63 Most importantly, our results pointed to a broad effect of GC treatment towards different immune‐regulatory markers, which may have a pivotal role in the re‐establishment of immune homeostasis and disease amelioration.
Taken together, our data showed that short‐term treatment with GC plays an important role in the control of local inflammation during the development of intestinal inflammation. One of the key aspects regarding the control of inflammation is related to the role of therapy in the up‐regulation of Treg cells and their functionality, in lymphoid organs, which together with other counter‐inflammatory molecules had a positive impact on disease outcome. To our knowledge, though the role of GC therapy over regulatory components was already demonstrated, this is the first time that a remarkable effect of this drug is shown towards distinct regulatory markers in colitis. As the re‐establishment of immune balance to constrain progression of intestinal inflammation is of great importance, therapies aimed at inducing regulatory responses can represent a valuable alternative in the treatment inflammatory disorders such as IBD. Finally, this study demonstrates new features regarding the effects of GC in the immune tolerance and control of inflammation during short‐term therapy of colitis.
Disclosures
The authors declare no conflict of interest.
Supporting information
Figure S1. Gating strategy for quantification of CD3− CD49b+ (natural killer; NK) cells and CD3+ CD49b+ natural killer T (NKT) cells in lamina propria (LP) and intraepithelial lymphocytes (IEL) compartment.
Figure S2. Total cell numbers obtained from spleen and mesenteric lymph nodes (MLN) of mice exposed to dextran sodium sulphate (DSS) treated or not with glucocorticoids (GC), as described in the Material and methods.
Acknowledgements
The authors would like to thank Fabiana Rosseto for helping with flow cytometry acquisition data. This study was supported by research funding from Núcleo de Apoio à Pesquisa em Doenças Inflamatórias [NAPDIN] under grant agreement no. 11.1.21625.01.0; Fundação de Amparo à Pesquisa de São Paulo (FAPESP), under agreements no. 2010/20162‐7 and 2012/00984‐8; Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq 141050/2013‐6, 482390/2013‐1 and 311882/2013‐7).
References
- 1. Jaffe HL. The influence of the suprarenal gland on the thymus: III. Stimulation of the growth of the thymus gland following double suprarenalectomy in young Rats. J Exp Med 1924; 40:753–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barnes PJ. How corticosteroids control inflammation: quintiles Prize Lecture 2005. Br J Pharmacol 2006; 148:245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cutolo M. Glucocorticoids and chronotherapy in rheumatoid arthritis. RMD Open 2016; 2:e000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sales‐Campos H, Basso PJ, Alves VB, Fonseca MT, Bonfa G, Nardini V, et al Classical and recent advances in the treatment of inflammatory bowel diseases. Braz J Med Biol Res 2015; 48:96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maranville JC, Micic D, Hanauer SB, Di Rienzo A, Kupfer SS. In vitro sensitivity assays and clinical response to glucocorticoids in patients with inflammatory bowel disease. J Crohns Colitis 2014; 8:1539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Basso PJ, Fonseca MT, Bonfa G, Alves VB, Sales‐Campos H, Nardini V, et al Association among genetic predisposition, gut microbiota, and host immune response in the etiopathogenesis of inflammatory bowel disease. Braz J Med Biol Res 2014; 47:727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ford AC, Bernstein CN, Khan KJ, Abreu MT, Marshall JK, Talley NJ, et al Glucocorticosteroid therapy in inflammatory bowel disease: systematic review and meta‐analysis. Am J Gastroenterol 2011; 106:590–9; quiz 600. [DOI] [PubMed] [Google Scholar]
- 8. Cohen RD. How should we treat severe acute steroid‐refractory ulcerative colitis? Inflamm Bowel Dis 2009; 15:150–1. [DOI] [PubMed] [Google Scholar]
- 9. De Bosscher K, Haegeman G. Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol 2009; 23:281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karagiannidis C, Akdis M, Holopainen P, Woolley NJ, Hense G, Ruckert B, et al Glucocorticoids upregulate FOXP3 expression and regulatory T cells in asthma. J Allergy Clin Immunol 2004; 114:1425–33. [DOI] [PubMed] [Google Scholar]
- 11. Barrat FJ, Cua DJ, Boonstra A, Richards DF, Crain C, Savelkoul HF, et al In vitro generation of interleukin 10‐producing regulatory CD4+ T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)‐ and Th2‐inducing cytokines. J Exp Med 2002; 195:603–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jones SC, Banks RE, Haidar A, Gearing AJ, Hemingway IK, Ibbotson SH, et al Adhesion molecules in inflammatory bowel disease. Gut 1995; 36:724–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baron JH, Connell AM, Kanaghinis TG, Lennard‐Jones JE, Jones AF. Out‐patient treatment of ulcerative colitis. Comparison between three doses of oral prednisone. Br Med J 1962; 2:441–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Terdiman JP, Gruss CB, Heidelbaugh JJ, Sultan S, Falck‐Ytter YT, Practice AGAIC , et al American Gastroenterological Association Institute guideline on the use of thiopurines, methotrexate, and anti‐TNF‐α biologic drugs for the induction and maintenance of remission in inflammatory Crohn's disease. Gastroenterology 2013; 145:1459–63. [DOI] [PubMed] [Google Scholar]
- 15. Krajcovicova A, Hlavaty T, Killinger Z, Miznerova E, Toth J, Letkovsky J, et al Combination therapy with an immunomodulator and anti‐TNFα agent improves bone mineral density in IBD patients. J Crohns Colitis 2014; 8:1693–701. [DOI] [PubMed] [Google Scholar]
- 16. Sales‐Campos H, de Souza PR, Basso PJ, Ramos AD, Nardini V, Chica JE, et al Aedes aegypti salivary gland extract ameliorates experimental inflammatory bowel disease. Int Immunopharmacol 2015; 26:13–22. [DOI] [PubMed] [Google Scholar]
- 17. Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, et al Hypoxia‐inducible factor 1‐dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med 2001; 193:1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bento AF, Leite DF, Claudino RF, Hara DB, Leal PC, Calixto JB. The selective nonpeptide CXCR2 antagonist SB225002 ameliorates acute experimental colitis in mice. J Leukoc Biol 2008; 84:1213–21. [DOI] [PubMed] [Google Scholar]
- 19. Arstila T, Arstila TP, Calbo S, Selz F, Malassis‐Seris M, Vassalli P, et al Identical T cell clones are located within the mouse gut epithelium and lamina propria and circulate in the thoracic duct lymph. J Exp Med 2000; 191:823–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cardoso CR, Teixeira G, Provinciatto PR, Godoi DF, Ferreira BR, Milanezi CM, et al Modulation of mucosal immunity in a murine model of food‐induced intestinal inflammation. Clin Exp Allergy 2008; 38:338–49. [DOI] [PubMed] [Google Scholar]
- 21. Zacharchuk CM, Mercep M, Chakraborti PK, Simons SS Jr, Ashwell JD. Programmed T lymphocyte death. Cell activation‐ and steroid‐induced pathways are mutually antagonistic. J Immunol 1990; 145:4037–45. [PubMed] [Google Scholar]
- 22. Van Laethem F, Baus E, Smyth LA, Andris F, Bex F, Urbain J, et al Glucocorticoids attenuate T cell receptor signaling. J Exp Med 2001; 193:803–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gonzalo JA, Gonzalez‐Garcia A, Martinez C, Kroemer G. Glucocorticoid‐mediated control of the activation and clonal deletion of peripheral T cells in vivo . J Exp Med 1993; 177:1239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ferreira ZS, Bothorel B, Markus RP, Simonneaux V. Plasma corticosterone elevation inhibits the activation of nuclear factor κB (NFKB) in the Syrian hamster pineal gland. Stress 2012; 15:339–47. [DOI] [PubMed] [Google Scholar]
- 25. Bonnet S, Paulin R, Sutendra G, Dromparis P, Roy M, Watson KO, et al Dehydroepiandrosterone reverses systemic vascular remodeling through the inhibition of the Akt/GSK3‐β/NFAT axis. Circulation 2009; 120:1231–40. [DOI] [PubMed] [Google Scholar]
- 26. King EM, Chivers JE, Rider CF, Minnich A, Giembycz MA, Newton R. Glucocorticoid repression of inflammatory gene expression shows differential responsiveness by transactivation‐ and transrepression‐dependent mechanisms. PLoS One 2013; 8:e53936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Desbarats J, You‐Ten KE, Lapp WS. Levels of p56lck and p59fyn are reduced by a glucocorticoid‐dependent mechanism in graft‐versus‐host reaction‐induced T cell anergy. Cell Immunol 1995; 163:10–8. [DOI] [PubMed] [Google Scholar]
- 28. Franchimont D, Galon J, Gadina M, Visconti R, Zhou Y, Aringer M, et al Inhibition of Th1 immune response by glucocorticoids: dexamethasone selectively inhibits IL‐12‐induced Stat4 phosphorylation in T lymphocytes. J Immunol 2000; 164:1768–74. [DOI] [PubMed] [Google Scholar]
- 29. Brooks AK, Lawson MA, Smith RA, Janda TM, Kelley KW, McCusker RH. Interactions between inflammatory mediators and corticosteroids regulate transcription of genes within the Kynurenine Pathway in the mouse hippocampus. J Neuroinflammation 2016; 13:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Elenkov IJ, Papanicolaou DA, Wilder RL, Chrousos GP. Modulatory effects of glucocorticoids and catecholamines on human interleukin‐12 and interleukin‐10 production: clinical implications. Proc Assoc Am Physicians 1996; 108:374–81. [PubMed] [Google Scholar]
- 31. Franchimont D, Martens H, Hagelstein MT, Louis E, Dewe W, Chrousos GP, et al Tumor necrosis factor α decreases, and interleukin‐10 increases, the sensitivity of human monocytes to dexamethasone: potential regulation of the glucocorticoid receptor. J Clin Endocrinol Metab 1999; 84:2834–9. [DOI] [PubMed] [Google Scholar]
- 32. Adcock IM, Caramori G. Cross‐talk between pro‐inflammatory transcription factors and glucocorticoids. Immunol Cell Biol 2001; 79:376–84. [DOI] [PubMed] [Google Scholar]
- 33. Yang QF, Chen BL, Zhang QS, Zhu ZH, Hu B, He Y, et al Contribution of MDR1 gene polymorphisms on IBD predisposition and response to glucocorticoids in IBD in a Chinese population. J Dig Dis 2015; 16:22–30. [DOI] [PubMed] [Google Scholar]
- 34. De Iudicibus S, Stocco G, Martelossi S, Londero M, Ebner E, Pontillo A, et al Genetic predictors of glucocorticoid response in pediatric patients with inflammatory bowel diseases. J Clin Gastroenterol 2011; 45:e1–7. [DOI] [PubMed] [Google Scholar]
- 35. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. N Engl J Med 2005; 353:1711–23. [DOI] [PubMed] [Google Scholar]
- 36. Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, et al Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator‐activated receptors α and γ . Proc Natl Acad Sci USA 1997; 94:4318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cuzzocrea S, Bruscoli S, Mazzon E, Crisafulli C, Donato V, Di Paola R, et al Peroxisome proliferator‐activated receptor‐α contributes to the anti‐inflammatory activity of glucocorticoids. Mol Pharmacol 2008; 73:323–37. [DOI] [PubMed] [Google Scholar]
- 38. Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, et al A novel therapy for colitis utilizing PPAR‐γ ligands to inhibit the epithelial inflammatory response. J Clin Invest 1999; 104:383–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, et al Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator‐activated receptor γ (PPARγ) heterodimer. A basis for new therapeutic strategies. J Exp Med 2001; 193:827–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rousseaux C, Lefebvre B, Dubuquoy L, Lefebvre P, Romano O, Auwerx J, et al Intestinal antiinflammatory effect of 5‐aminosalicylic acid is dependent on peroxisome proliferator‐activated receptor‐γ . J Exp Med 2005; 201:1205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bouguen G, Langlois A, Djouina M, Branche J, Koriche D, Dewaeles E, et al Intestinal steroidogenesis controls PPARγ expression in the colon and is impaired during ulcerative colitis. Gut 2015; 64:901–10. [DOI] [PubMed] [Google Scholar]
- 42. Sakaguchi S, Wing K, Onishi Y, Prieto‐Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int Immunol 2009; 21:1105–11. [DOI] [PubMed] [Google Scholar]
- 43. Suarez A, Lopez P, Gomez J, Gutierrez C. Enrichment of CD4+ CD25high T cell population in patients with systemic lupus erythematosus treated with glucocorticoids. Ann Rheum Dis 2006; 65:1512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu ZH, Huang XP, Sun W, Zhu YL, Cui JJ, Chen W, et al T helper cell dysregulation with hepatitis B and rebalance with glucocorticoids. World J Gastroenterol 2014; 20:18354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Okou DT, Mondal K, Faubion WA, Kobrynski LJ, Denson LA, Mulle JG, et al Exome sequencing identifies a novel FOXP3 mutation in a 2‐generation family with inflammatory bowel disease. J Pediatr Gastroenterol Nutr 2014; 58:561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun M, He C, Cong Y, Liu Z. Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol 2015; 5:969–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Curotto de Lafaille MA, Lino AC, Kutchukhidze N, Lafaille JJ. CD25− T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J Immunol 2004; 173:7259–68. [DOI] [PubMed] [Google Scholar]
- 48. Valzasina B, Piconese S, Guiducci C, Colombo MP. Tumor‐induced expansion of regulatory T cells by conversion of CD4+ CD25− lymphocytes is thymus and proliferation independent. Cancer Res 2006; 66:4488–95. [DOI] [PubMed] [Google Scholar]
- 49. Kang X, Zhang X, Liu Z, Xu H, Wang T, He L, et al Granulocytic myeloid‐derived suppressor cells maintain feto‐maternal tolerance by inducing Foxp3 expression in CD4+ CD25− T cells by activation of the TGF‐β/β‐catenin pathway. Mol Hum Reprod 2016; 22:499–511. [DOI] [PubMed] [Google Scholar]
- 50. Uraushihara K, Kanai T, Ko K, Totsuka T, Makita S, Iiyama R, et al Regulation of murine inflammatory bowel disease by CD25+ and CD25− CD4+ glucocorticoid‐induced TNF receptor family‐related gene+ regulatory T cells. J Immunol 2003; 171:708–16. [DOI] [PubMed] [Google Scholar]
- 51. Placke T, Kopp HG, Salih HR. Glucocorticoid‐induced TNFR‐related (GITR) protein and its ligand in antitumor immunity: functional role and therapeutic modulation. Clin Dev Immunol 2010; 2010:239083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′‐adenosine monophosphate to adenosine. J Immunol 2006; 177:6780–6. [DOI] [PubMed] [Google Scholar]
- 53. Deaglio S, Robson SC. Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv Pharmacol 2011; 61:301–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bonner F, Borg N, Burghoff S, Schrader J. Resident cardiac immune cells and expression of the ectonucleotidase enzymes CD39 and CD73 after ischemic injury. PLoS One 2012; 7:e34730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang L, Tang S, Wang Y, Xu S, Yu J, Zhi X, et al Ecto‐5′‐nucleotidase (CD73) promotes tumor angiogenesis. Clin Exp Metastasis 2013; 30:671–80. [DOI] [PubMed] [Google Scholar]
- 56. Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, et al PD‐1:PD‐L inhibitory pathway affects both CD4+ and CD8+ T cells and is overcome by IL‐2. Eur J Immunol 2002; 32:634–43. [DOI] [PubMed] [Google Scholar]
- 57. Yamamoto R, Nishikori M, Kitawaki T, Sakai T, Hishizawa M, Tashima M, et al PD‐1‐PD‐1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood 2008; 111:3220–4. [DOI] [PubMed] [Google Scholar]
- 58. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP‐1 and SHP‐2 associate with immunoreceptor tyrosine‐based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004; 173:945–54. [DOI] [PubMed] [Google Scholar]
- 59. Nurieva R, Thomas S, Nguyen T, Martin‐Orozco N, Wang Y, Kaja MK, et al T‐cell tolerance or function is determined by combinatorial costimulatory signals. EMBO J 2006; 25:2623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Teft WA, Kirchhof MG, Madrenas J. A molecular perspective of CTLA‐4 function. Annu Rev Immunol 2006; 24:65–97. [DOI] [PubMed] [Google Scholar]
- 61. Reikvam DH, Perminow G, Lyckander LG, Gran JM, Brandtzaeg P, Vatn M, et al Increase of regulatory T cells in ileal mucosa of untreated pediatric Crohn's disease patients. Scand J Gastroenterol 2011; 46:550–60. [DOI] [PubMed] [Google Scholar]
- 62. Mao R, Xiao W, Liu H, Chen B, Yi B, Kraj P, et al Systematic evaluation of 640 FDA drugs for their effect on CD4+Foxp3+ regulatory T cells using a novel cell‐based high throughput screening assay. Biochem Pharmacol 2013; 85:1513–24. [DOI] [PubMed] [Google Scholar]
- 63. Calmette J, Ellouze M, Tran T, Karaki S, Ronin E, Capel F, et al Glucocorticoid‐induced leucine zipper enhanced expression in dendritic cells is sufficient to drive regulatory T cells expansion in vivo . J Immunol 2014; 193:5863–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Gating strategy for quantification of CD3− CD49b+ (natural killer; NK) cells and CD3+ CD49b+ natural killer T (NKT) cells in lamina propria (LP) and intraepithelial lymphocytes (IEL) compartment.
Figure S2. Total cell numbers obtained from spleen and mesenteric lymph nodes (MLN) of mice exposed to dextran sodium sulphate (DSS) treated or not with glucocorticoids (GC), as described in the Material and methods.