

Abstract
Bim is a pro‐apoptotic Bcl‐2 family member of the BH3‐only protein subgroup. Expression levels of Bim determine apoptosis susceptibility in non‐malignant and in tumour cells. Bim protein expression is downregulated by proteasomal degradation following ERK‐dependent phosphorylation and ubiquitination. Here, we report the identification of a deubiquitinase, Usp27x, that binds Bim upon its ERK‐dependent phosphorylation and can upregulate its expression levels. Overexpression of Usp27x reduces ERK‐dependent Bim ubiquitination, stabilizes phosphorylated Bim, and induces apoptosis in PMA‐stimulated cells, as well as in tumour cells with a constitutively active Raf/ERK pathway. Loss of endogenous Usp27x enhances the Bim‐degrading activity of oncogenic Raf. Overexpression of Usp27x induces low levels of apoptosis in melanoma and non‐small cell lung cancer (NSCLC) cells and substantially enhances apoptosis induced in these cells by the inhibition of ERK signalling. Finally, deletion of Usp27x reduces apoptosis in NSCLC cells treated with an EGFR inhibitor. Thus, Usp27x can trigger via its proteolytic activity the deubiquitination of Bim and enhance its levels, counteracting the anti‐apoptotic effects of ERK activity, and therefore acts as a tumour suppressor.
Keywords: apoptosis, Bcl‐2, Bim, deubiquitinase, Usp27x
Subject Categories: Cancer; Post-translational Modifications, Proteolysis & Proteomics
Introduction
The Bcl‐2‐family protein Bim is an important member of the pro‐apoptotic group of BH3‐only proteins. Bim is widely expressed in numerous tissues 1, and deletion of Bim causes strong apoptosis defects in haematopoietic cells 2. Expression of Bim is lost in a number of human tumours. In B‐cell lymphomas, for instance silencing of the Bim promoter as well as homozygous gene deletions has been described 3; low expression levels in renal cell carcinoma have also been reported 4, 5. In a number of additional cancer types, the activity of the oncogenic kinases causes a reduction in Bim levels due to proteasomal degradation of the protein. This is the case for epithelial tumours expressing mutant Ras 6 and non‐small cell lung cancer (NSCLC) cells carrying activating mutations in the EGFR 7, 8, 9. The loss of Bim enhances the oncogenic potential of c‐myc in mice 10, supporting the view of Bim as a tumour suppressor.
The molecular function of Bim is the initiation of apoptosis. Bim can activate the effectors of mitochondrial apoptosis, Bax and Bak. This can very likely occur through direct interaction 11, 12 as well as by inactivating anti‐apoptotic Bcl‐2‐like proteins 13. The main determinant of Bim activity appears to be its expression level. Bim‐dependent apoptosis may be accompanied by transcriptional induction of Bim 14, 15. An additional, prominent pathway of Bim regulation is, however, its proteasomal degradation. During mitosis, Bim levels are reduced by proteasomal degradation, and this has been linked to phosphorylation by Aurora kinase 16 and ubiquitination by the anaphase‐promoting complex APCCdc20, functioning as an E3 ubiquitin ligase 17, although this proposed pathway would require a non‐canonical recognition of Bim since APCCdc20 normally recognizes a D box rather than phosphodegrons (see for instance 18).
Significantly, Bim degradation is also regulated through the oncogenic Raf‐ERK pathway. ERK1/2 can directly phosphorylate Bim, which promotes its proteasomal destruction 19, 20, 21. This ERK‐dependent phosphorylation event enables additional phosphorylation through the kinases RSK1/2, the subsequent ubiquitination by the ubiquitin E3‐ligase β‐TrCP and the degradation of Bim by the proteasome 22.
Ubiquitin ligases attach ubiquitin to target proteins. Ubiquitin may be linked as a single molecule but may also be attached in multiples, forming long chains that can be linked through various lysine residues on the ubiquitin peptide. Typically, K48‐linked polyubiquitin on a target protein can enhance proteasomal degradation of cellular proteins. A counter‐regulatory mechanism is the removal of ubiquitin chains by deubiquitinases (DUBs), reducing proteasomal recognition and thereby stabilizing the protein. Approximately 95 DUBs are encoded in the human genome that fall into five families, but only 79 of them are predicted to be active (for review see 23). The DUB family of ubiquitin‐specific peptidases (Usp) contains over 50 members, classified as cysteine proteases 24.
Given the clear regulation of Bim ubiquitination through major signalling pathways, it appears likely that a counter‐regulation through deubiquitination also exists. We here describe the identification of a DUB, Usp27x, that can deubiquitinate Bim in response to the Bim‐degrading activity of ERK‐signalling pathways and can sensitize human cancer cells to chemotherapeutics.
Results
Identification and validation of Usp27x as Bim‐interacting protein
To identify interaction partners regulating Bim activity, we performed immunoprecipitation (IP) experiments, followed by identification of co‐precipitated proteins using the method of stable isotope labelling with amino acids in cell culture (SILAC). For this, we expressed full‐length BimEL in Bax/Bak‐deficient murine embryonic fibroblasts (MEFs), with or without an N‐terminal triple‐HA tag. Cell lines were differentially labelled with normal or heavy amino acids, and lysates from heavy membrane fractions (enriched for mitochondria) were subjected to anti‐HA IP (cells expressing HA‐tagged BimEL were labelled with light amino acids, cells expressing untagged BimEL with heavy amino acids). Precipitates were combined, and differential content of individual proteins was assessed by mass spectrometry. By using cell lines identical in all respects other than the HA tag and specifically searching for proteins enriched in the HA‐associated IP, this was expected to minimize the non‐specific isolation of proteins.
The results showed the expected enrichment of Bim (about 27‐fold), enrichment for the known Bim‐interacting, anti‐apoptotic proteins Bcl‐2, Bcl‐XL and Mcl‐1 (12–28‐fold) as well as smaller enrichment of two TOM proteins (this has already been published 25). In the same experiment, we found a peptide corresponding to the DUB Usp27x (enriched about 183‐fold). The SILAC profiles of enriched proteins (normalized SILAC ratio given as fold‐change (FC) for Usp27x, Bcl‐2, Bim, Bcl‐X and Mcl‐1 are shown in Fig EV1A; see also Table EV1 for additional information (e.g. peptide sequences, scores and PEP values)). Usp27x is a DUB by structure but has unknown function and cellular targets. A recent study suggests a nuclear role in regulating turnover of a transcription factor, together with Usp22 and Usp51 26.
Figure EV1. Interaction of the Usp27x with BimEL .
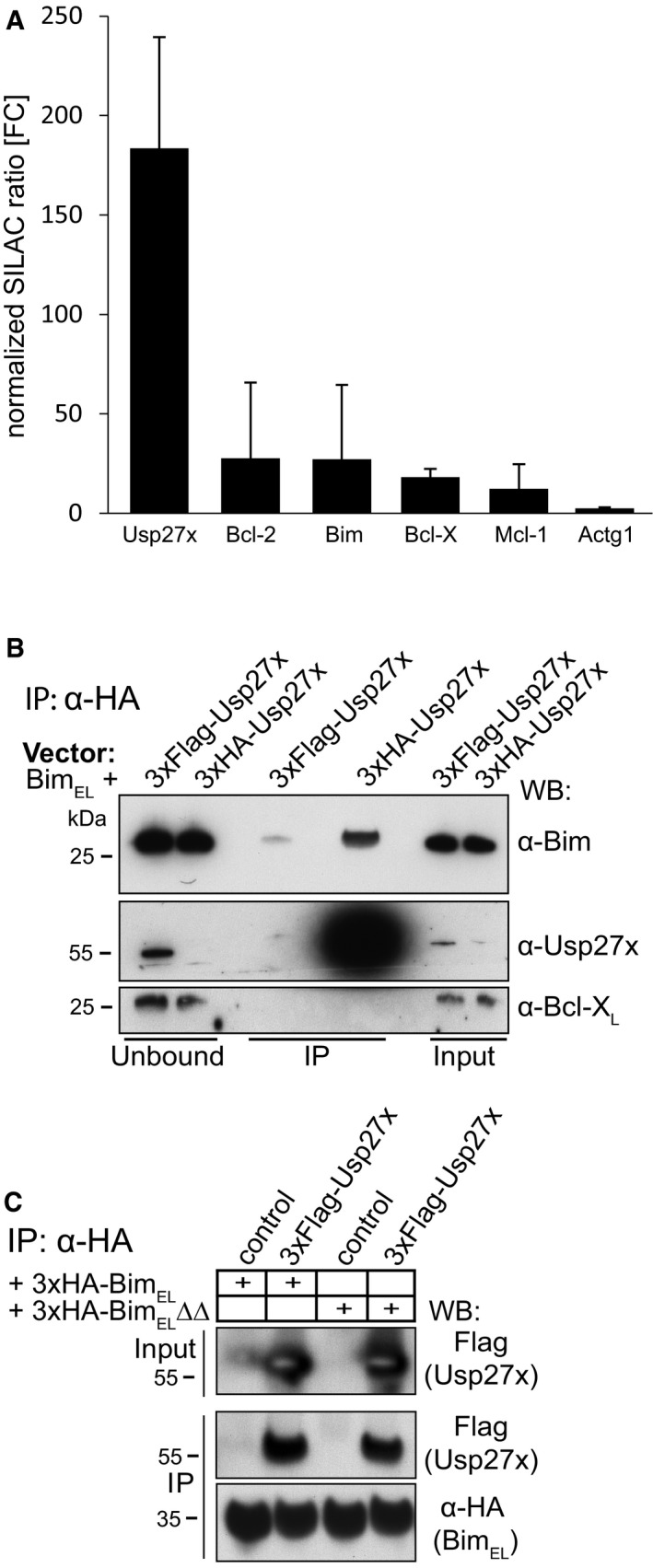
- Profile of enriched proteins in anti‐3xHA‐BIMEL immunoprecipitations of purified mitochondria from mouse embryonic fibroblasts (MEF Bax/Bak‐double knockout cells). Cells expressing 3xHA‐BimEL were labelled with light amino acids, whereas control cells (expressing only untagged BimEL) were labelled with heavy amino acids. Gamma actin (Actg1) served as a specificity control. Protein ratios are calculated from the respective peptide ratios. A minimum ratio count of two is needed to calculate a protein ratio. Error bars: protein quantification variability. Error bars indicate the coefficient of variability over all redundant quantifiable peptide signals. It is calculated as standard deviation of the fold enrichment of all peptide ratios (see also Table EV1). Identification of Usp27x as an interacting protein of BimEL was identified alongside the SILAC experiment published earlier. Enrichment of Bim and anti‐apoptotic proteins already appear in the earlier report 25.
- 293FT cells were transfected with expression constructs for untagged BimEL together with 3xFlag‐Usp27x or 3xHA‐Usp27x (all pMIG‐vector backbone). After cell lysis (1% Triton X‐100) and subsequent anti‐HA immunoprecipitation, the unbound fraction, input (50 μg each) and the IP‐eluate (IP, ˜60%) were run on SDS–PAGE. Flag‐ or HA‐tagged Usp27x were detected using an antibody directed against Usp27x (Abgent). See also Fig 1A. Similar results were seen in n = 3 experiments.
- Usp27x binds a mutant of Bim incapable of binding to anti‐apoptotic Bcl‐2 proteins. 293FT cells transfected with constructs encoding 3xFLAG‐Usp27x and 3xHA‐tagged BimEL (Fig 1B) or 3xHA‐tagged BimEL∆∆ (a mutant with two mutations in the BH3 domain, incapable of binding anti‐apoptotic Bcl‐2 proteins 50) were immunoprecipitated from whole‐cell extracts using anti‐HA resin. Bim and Usp27x were detected with anti‐HA and anti‐FLAG antibodies as indicated. Control, pEGFP‐N1 vector. The caspase inhibitor Q‐VD‐OPh (QVD) was added to the cultures to inhibit Bim‐induced apoptosis (see also Fig 1). Similar results were seen in n = 3 experiments.
We confirmed this interaction of BimEL with Usp27x in transfection experiments of 293FT cells with tagged proteins. Efficient co‐IP was seen in these experiments (Fig 1A). The interaction was stable both ways (i.e. precipitation of Bim recovered Usp27x and vice versa (Figs 1A and EV1B)). BimEL interacts with anti‐apoptotic Bcl‐2 proteins via its BH3 domain, and Bcl‐2, Bcl‐XL and Mcl‐1 had also been co‐purified with BimEL 25. We therefore tested whether the interaction of BimEL with Usp27x was direct or via an interaction of Bim with anti‐apoptotic proteins. When a version of BimEL with a mutation in the BH3 domain was expressed that cannot bind anti‐apoptotic Bcl‐2 proteins 27, there was still efficient binding to Usp27x (Figs 1B and EV1C) but binding to anti‐apoptotic proteins was absent (Mcl‐1) or strongly reduced (Bcl‐XL) in this complex. No binding of Bim to Bcl‐2 was observed in these cells (Fig 1B).
Figure 1. The deubiquitinase Usp27x interacts with BimEL .
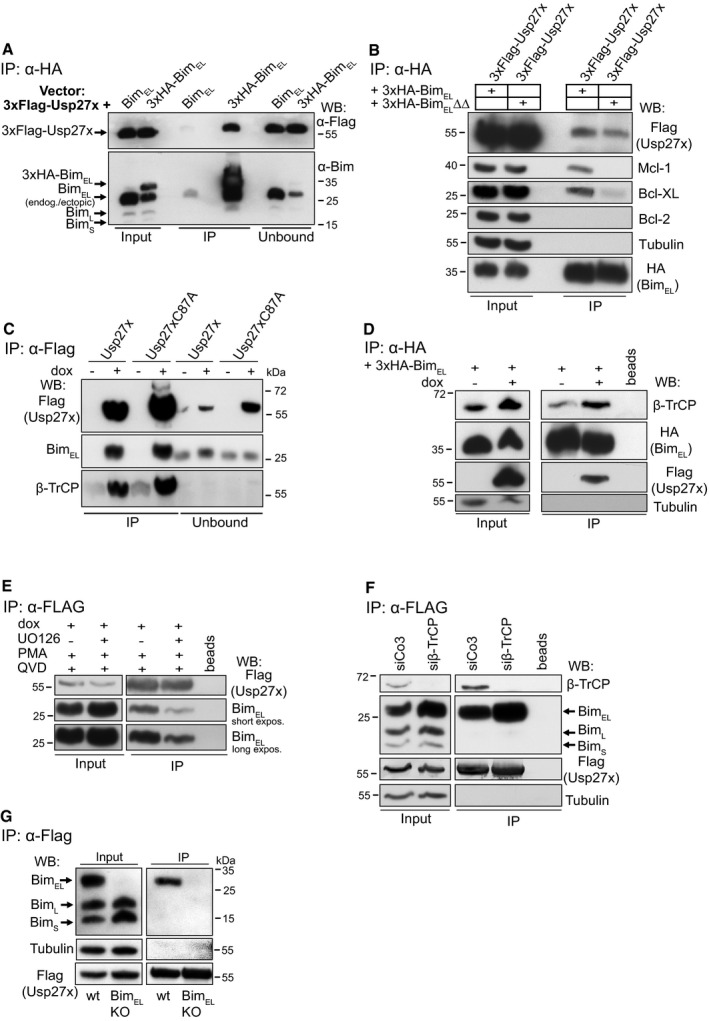
- 293FT cells were transfected with 3xFlag‐Usp27x (pFCMV7.1 vector backbone) together with a construct driving expression of untagged BimEL or 3xHA‐BimEL (both pMIG‐vector backbone). Cells were lysed, and 3xHA‐BimEL was immunoprecipitated with anti‐HA antibodies. Immunoprecipitation products were tested by Western blotting for the presence of Bim and FLAG‐Usp27x probing with antibodies against Bim or against the FLAG‐peptide. See also Fig EV1B. Western blots show representative of n ≥ 3 independent experiments.
- Usp27x binds a mutant of Bim incapable of binding to anti‐apoptotic Bcl‐2 proteins. 293FT cells transfected with constructs encoding 3xFLAG‐Usp27x and 3xHA‐tagged BimEL (see A, 2 μg each) or 3xHA‐tagged BimEL∆∆ (a mutant with two mutations in the BH3 domain, incapable of binding anti‐apoptotic Bcl‐2 proteins 50) were immunoprecipitated from whole‐cell extracts using anti‐HA resin. Bim and Usp27x were detected with anti‐HA and anti‐FLAG antibodies as indicated; anti‐apoptotic proteins: Mcl‐1, Bcl‐XL, Bcl‐2. The caspase inhibitor Q‐VD‐OPh (QVD) was added to the cultures described in (A) and (B) to inhibit Bim‐induced apoptosis. Western blots are representative of n = 3 independent experiments.
- Usp27x interacts with endogenous BimEL independently of its catalytic activity. 293FT cells either carrying 3xFlag‐Usp27x (293FT‐TetR‐3xFlag‐Usp27x) or the catalytically inactive mutant 3xFlag‐Usp27xC87A under the control of the Tet repressor (TetR) were treated for 24 h with doxycycline (dox) to induce expression of Usp27x or Usp27xC87A. In all conditions, PMA (to induce Bim ubiquitination, 16.2 nM) and Q‐VD‐OPh (to inhibit apoptosis, 10 μM, see Fig 3) were added at the time of Usp27x induction. MG132 (to prevent Bim degradation, 40 μM) was added in all conditions 4 h prior to cell lysis. 3xFlag‐tagged Usp27x or Usp27xC87A was immunoprecipitated from whole‐cell lysates using anti‐Flag resin. Interaction with BimEL or β‐TrCP was detected by Western blotting using anti‐Bim or anti‐β‐TrCP antibodies. Western blots are representative of n ≥ 3 independent experiments.
- Usp27x expression does not inhibit interaction of BimEL to β‐TrCP. 293FT‐TetR‐3xFlag‐Usp27x cells were transfected with pMIG‐3xHA‐BIMEL in the presence of both PMA and QVD. At the same time, dox (to induce 3xFlag‐Usp27x) was added as indicated and 20 h later cells were treated with MG132 (40 μM) for additional 4 h. Cells were lysed and 3xHA‐BimEL was immunoprecipitated as described above. As a control, HA matrix was used without lysate to rule out any unspecific signal that might come from the immobilized anti‐HA antibody (beads). Western blots show representative of n = 2 independent experiments.
- Binding of Usp27x to BimEL can be blocked by the MEK inhibitor UO126. 293FT‐TetR‐3xFlag‐Usp27x cells (see C) were treated for 24 h with dox to induce expression of Usp27x. In all conditions, PMA (16.2 nM) and Q‐VD‐OPh (10 μM) were added at the time of Usp27x induction. As a control, cells were pre‐treated with UO126 (10 μM) for 30 min to block the PMA‐stimulated ERK pathway before the addition of dox plus PMA plus QVD. 3xFlag‐tagged Usp27x was immunoprecipitated from whole‐cell lysates using anti‐Flag resin. Interaction with BimEL was detected by Western blotting using anti‐Bim antibodies. As a control, Flag‐matrix (beads) was used alongside the IP under same conditions but without addition of protein lysates. Western blots are representative of n = 4 independent experiments (see also Fig EV2B).
- Binding of Usp27x to BimEL does not require β‐TrCP. 293FT‐TetR‐3xFlag‐Usp27x cells were transfected with control siRNA (siCo3) or siRNA specific for β‐TrCP. Forty‐eight hours later, cells were stimulated with PMA plus dox plus QVD for additional 24 h. After cell lysis, Flag‐Usp27x was immunoprecipitated using anti‐Flag‐matrix, and BimEL and β‐TrCP bound to Usp27x were identified by Western blotting. Blots are representative of n = 2 independent experiments.
- Usp27x binds specifically to BimEL. 293FT‐TetR‐3xFlag‐Usp27x cells or 293FT‐3xFlag‐Usp27x cells with a specific deletion of the BimEL protein were treated with dox, PMA and QVD as in (C). Cells were lysed, and lysates were subjected to anti‐Flag immunoprecipitation. 3xFlag‐Usp27x was detected using anti‐FLAG antibodies. Blots are representative of n = 3 independent experiments.
As an enzymatically inactive control, we generated a point mutant of Usp27x, where the cysteine in the predicted catalytic triad was mutated (Usp27xC87A), based on homology with other known Usps 28. We generated 293FT‐TetR (TetR, Tet repressor) cells carrying a tetracycline‐inducible construct for the expression of Usp27x wt or mutant. Both Usp27x wt and inactive Usp27x mutant bound endogenous BimEL with similar efficiency when cells were stimulated with PMA to induce Bim ubiquitination (Fig 1C). We noticed that the known E3‐ubiquitin‐ligase β‐TrCP was also isolated in the same complex. Induction of Usp27x did not decrease (even appeared to increase slightly) the amount of β‐TrCP on Bim (Fig 1D). This is consistent with the idea that Usp27x is active in the same degradation pathway as β‐TrCP.
Since Usp27xC87A precipitated Bim as well as wt Usp27x, proteolytic activity appears not to be required for binding of Usp27x to Bim. However, inhibition of MEK using the inhibitor UO126 in PMA‐stimulated cells reduced the association of Bim and Usp27x (Figs 1E and EV2B). Similar results were obtained when co‐localization of Bim, and Usp27x was assessed by proximity‐ligation assay (PLA) 29.
Figure EV2. Usp27x is in close proximity to Bim.
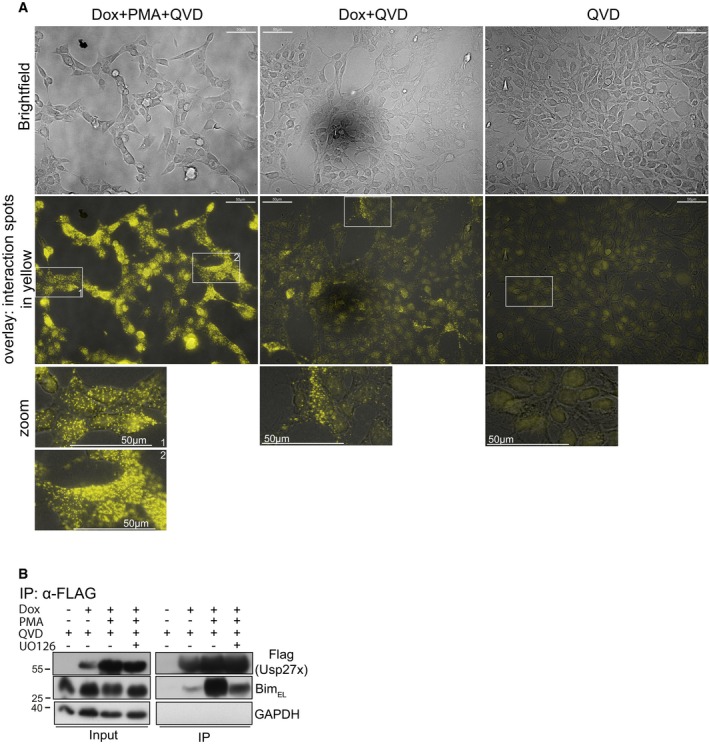
- In situ identification of complexes between Usp27x and BimEL molecules by proximity‐ligation assay (PLA). 293FT cells carrying dox‐inducible 3xFlag‐Usp27x (293FT‐3xFlag‐Usp27x) were stimulated as indicated for 24 h, fixed and permeabilized. Yellow dots show interaction spots between endogenous Bim and inducible 3xFlag‐Usp27x. For dox+PMA+QVD, two different zoom regions (1, 2) are shown from the overlay. Data are representative of n = 2 independent experiments. Scale bar, 50 μm.
- Immunoprecipitation of 3xFlagUsp27x. The same cell line and conditions as in (A) were used. In one sample, UO126 was added 30 min before stimulation. Twenty‐four hours after stimulation, cells were lysed and 3xFlag‐Usp27x was immunoprecipitated using anti‐Flag‐resin. Detection of Usp27x was done using anti‐Flag antibody, and Bim was detected using a Bim‐specific antibody. GAPDH served as a loading control. Data are representative of n = 4 independent experiments.
A PLA signal depends on the two proteins being co‐localized within 30 nm distance 29. A weak PLA signal was observed in unstimulated cells overexpressing Usp27x, but the signal was strongly increased upon PMA stimulation (Fig EV2A), indicating a recruitment of Usp27x to phosphorylated Bim. A similar pattern was observed by immunoprecipitation (Fig EV2B).
We could observe some Bim phosphorylation in 293FT cells even in the absence of MEK/ERK activity (see below, Fig 2C). Bim phosphorylation may therefore be a prerequisite for binding of Usp27x.
Figure 2. Expression of Usp27x stabilizes the expression of the phosphorylated form of BimEL and reduces Bim ubiquitination in PMA‐stimulated 293FT cells.
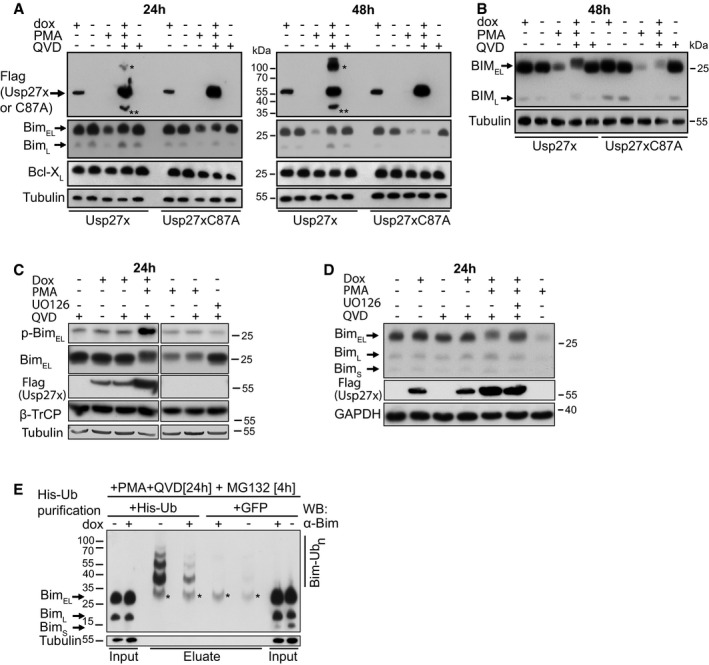
- Usp27x inhibits the PMA‐induced degradation of BimEL in 293FT cells. 293FT‐TetR‐3xFlagUsp27x or TetR‐3xFlag‐Usp27xC87A cells were treated with dox to induce 3xFlag‐Usp27x or Usp27xC87A, or with PMA (to induce BimEL degradation, 16.2 nM), or with the combination of dox, PMA and QVD (to inhibit apoptosis, 10 μM) as indicated for 24 (left) or 48 h (right). Cell lysates were analysed by Western blotting. The blots are representative of n = 4 similar experiments. See also Fig EV5B. The asterisk (*) indicates a probable modified form of wild‐type 3xFlag‐Usp27x (such as dimer, ubiquitinated Usp27x or a stable complex of Usp27x with another interacting partner); this form is only seen if high expression levels of catalytically active Usp27x are reached (see Fig EV5A where Usp27x was transfected into 293FT cells) or if wt Usp27x is enriched during immunoprecipitation. Double asterisk (**) indicates a probable degradation product of 3xFlag‐Usp27x. These forms of Usp27x were not seen in every experiment.
- 293FT‐TetR‐3xFlagUsp27x or TetR‐3xFlag‐Usp27xC87A cells treated as in (A) were analysed using conditions of higher resolution. A band very likely corresponding to phosphorylated BimEL is detectable. Similar results were obtained in n = 2 separate experiments.
- Usp27x stabilizes expression of p‐BimEL (Ser69) in Bim‐degrading conditions without reducing β‐TrCP levels. 293FT‐TetR‐3xFlagUsp27x were treated with dox, PMA (16.2 nM), UO126 (10 μM) or QVD (10 μM) as indicated for 24 h, and levels of phosphorylated BimEL at serine 69 were analysed by Western blotting using a phospho‐Bim (Ser69)‐specific antibody. Scans are from the same membrane and exposure times. Similar results were obtained in n = 2 separate experiments.
- UO126 reduces phosphorylation‐associated shift of BimEL. 293FT‐TetR‐3xFlagUsp27x were treated for 24 h with the indicated drug combinations, and Bim was detected by Western blotting. UO126 was added 30 min prior other stimulation. Similar results were obtained in n = 3 separate experiments (see Fig EV5D).
- Usp27x‐expression reduces ubiquitination of Bim. 293FT‐TetR‐3xFlagUsp27x cells were transfected with a vector coding for 6His‐ubiquitin (left) or GFP (right). After 24 h, cells were treated with PMA and QVD, and 3xFlag‐Usp27x was induced by dox at the same time. After additional 20 h, cells were treated for 4 h with MG132 (40 μM) to block proteasomal degradation of ubiquitinated proteins. Cells were lysed under denaturing conditions, and His‐ubiquitin‐labelled proteins or proteins from only GFP‐expressing cells were purified by Ni2+‐NTA affinity chromatography. Ubiquitinated Bim (Bim‐Ubn) was detected by Western blot using anti‐Bim antibodies (left). Similar results were obtained in n = 3 separate experiments (another experiment including a Coomassie stain is shown in Fig EV3E). Asterisk (*) indicates non‐His‐ubiquitinated BimEL that was bound unspecifically to the Ni2+‐agarose beads.
Bim phosphorylation is also required for the recruitment of β‐TrCP 22; however, RNAi against β‐TrCP did not reduce the association of Bim and Usp27x (Fig 1F). The signal for the recruitment of both E3 ligase and deubiquitinase to Bim therefore appears to be its phosphorylation.
Bim is expressed in three main isoforms, which are generated by splicing from one precursor mRNA 30. The isoform BimEL is most highly expressed in most tissues, and the one exon only translated in the largest isoform BimEL but not in BimL or BimS is the target of phosphorylation and ubiquitination in the Raf‐ERK pathway (the degron comprises amino acids 91–100 in human BimEL 22). We therefore hypothesized that the binding of Usp27x to BimEL also occurred in the BimEL‐specific exon (aa 42–101). To test this hypothesis, we used the 293FT‐TetR‐Usp27x cell line above and generated from this a cell line where the expression of BimEL was specifically disrupted by placing a mutation into the BimEL exon using CRISPR/Cas9. As shown in Fig 1G, expression of BimEL was specifically disrupted as expected in these cells. This mutation abrogated co‐IP of endogenous BimEL with Usp27x, confirming that the interaction occurs in the BimEL‐specific exon (Fig 1G).
Purified, recombinant Usp27x showed proteolytic activity towards both K48 and K63 ubiquitin linkages (Fig EV3A). In vitro‐translated Usp27x (Fig EV3B) also cleaved both K48‐ and K63‐linked di‐ubiquitin, while the Usp27xC87A mutant had almost no activity (Fig EV3C; see Fig EV3D for cleavage specificity towards differently quenched substrates). In the absence of co‐factors/posttranslational modifications, Usp27x therefore can cleave both ubiquitin linkages, a specificity that has been reported for numerous DUBs 31.
Figure EV3. Usp27x cleaves di‐ubiquitin molecules.
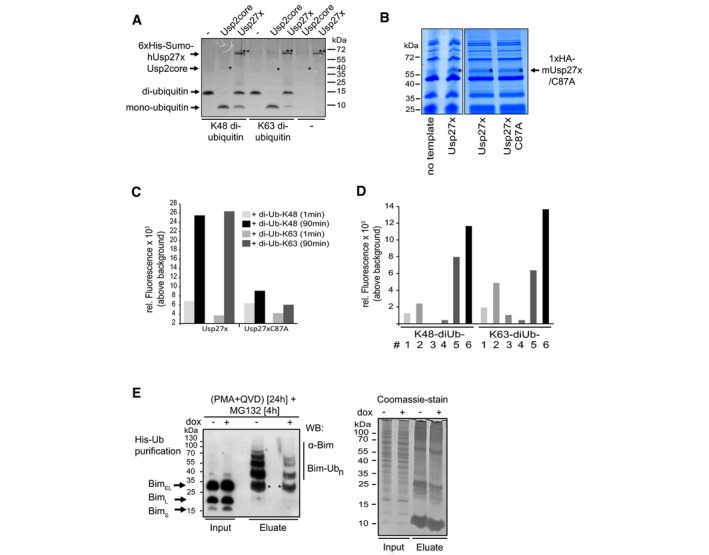
- Human 6xHis‐Sumo‐Usp27x was purified from E. coli. Differently linked di‐ubiquitin molecules (K48‐, K63‐linked di‐ubiquitin; 1 μg each) were incubated for 1 h at 37°C with 0.1 μM Usp2core (* positive control, ˜41 kDa) or 2.9 μM of the affinity‐purified 6xHis‐Sumo‐hUsp27x (**, ˜63 kDa). Usp27x was able to cleave K48‐ and K63‐linked di‐ubiquitin molecules into mono‐ubiquitin as visualized by Coomassie Blue staining. Similar results were observed in n = 2 separate experiments.
- Coomassie‐stained gels of soluble fractions (4 μl) from a PureExpress preparation without a template DNA (showing only proteins of the in vitro PureExpress translational E. coli system) or of in vitro‐translated 1xHA‐Usp27x (left gel) or the products of translation of 1xHA‐Usp27x and its catalytically inactive mutant 1xHA‐Usp27xC87A (right gel; Usp27x bands are marked with an asterisk). Reactions were used for (C). n = 2.
- Usp27x or Usp27xC87A were in vitro‐translated with the PureExpress system (see B). Four microlitres of soluble fractions of the reactions were directly used for assays of proteolytic activity. As substrates, K48‐linked IQF‐di‐UbK48‐6 or K63‐linked IQF‐di‐UbK63‐6 (200 nM each) (the 6 in K48‐6 and K63‐6 refers to the substrate variant where the quencher and TAMRA dye is linked at specific positions; these substrates were selected based on the preliminary experiment with the label at various positions (see Fig EV3D)) were added, and reactions were incubated for 1–90 min at 23°C. As readout for cleavage of K48/K63‐linked di‐ubiquitin, the relative fluorescence generated by dequenching was measured. Bars indicate mean values of duplicate measurements in one experiment.
- Four microlitres of soluble fractions (from the reactions shown in A) was directly used for the activity assay. Six different K48‐linked IQF‐diUb substrates and six K63‐linked IQF‐diUb substrates (substrates #1‐6; 200 nM each) were incubated with 1xHA‐Usp27x for 60 min at 23°C. As readout for cleavage of K48/K63‐linked di‐ubiquitin, the relative fluorescence was measured using a Tecan M200 fluorescence reader. IQF‐di‐UbK48‐6 and IQF‐di‐UbK63‐6 substrates showed the best performance and were further used for activity assays (see Fig EV3C). Bars indicate mean values of an experiment measured in duplicate.
- Usp27x expression reduces ubiquitination of Bim. 293FT‐TetR‐3xFlagUsp27x cells were transfected with a vector coding for 6His‐ubiquitin. After 24 h, cells were treated with PMA and QVD, and 3xFlag‐Usp27x was induced by dox at the same time. After additional 20 h, cells were treated for 4 h with MG132 (40 μM) to block proteasomal degradation of ubiquitinated proteins. Cells were lysed under denaturing conditions, and His‐ubiquitin‐labelled proteins were purified by Ni2+‐NTA affinity chromatography. Ubiquitinated Bim (Bim‐Ubn) was detected by Western blot using anti‐Bim antibodies (left). Asterisk (*) indicates non‐His‐ubiquitinated Bim that binds unspecifically to the Ni2+‐agarose beads (see Fig 2E). Equal loading was verified by a Coomassie‐stained SDS–PAGE from same protein extracts (right). Similar results were obtained in n = 3 separate experiments.
The Usp family member most closely related to Usp27x is Usp22, a DUB with functions in regulating the nuclear SAGA complex 32, and Usp27x has recently been shown to act together with Usp22 in regulating transcription factor stability 26. We tested the subcellular localization of Usp27x by fusing it to GFP. Upon induction in a cell line stably carrying GFP‐Usp27x, GFP fluorescence was found both in the nucleus and the cytosol (Fig EV4), while Usp22 appeared to be localized almost exclusively to the nucleus as reported previously (Fig EV4; 33). The very small amount of Usp22 in the cytosol may be due to an effect of the GFP tag or the high levels of overexpression. Usp27x may therefore have a nuclear function (most likely together with Usp22) but also targets non‐nuclear proteins such as Bim.
Figure EV4. Subcellular localization of Usp27x and Usp22 as analysed by fluorescence microscopy.
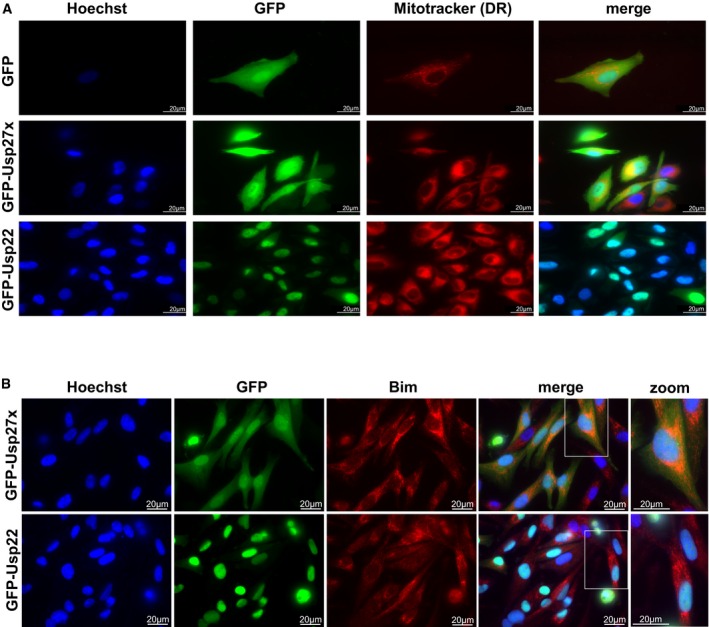
- 1205Lu melanoma cells carrying dox‐inducible GFP, GFP‐Usp27x or GFP‐Usp22 were treated for 24 h with dox. Subcellular localization of GFP or GFP‐fusion proteins (green) was visualized by microscopy. Nuclei were stained with Hoechst (blue), and mitochondria were identified by Mitotracker DeepRed FM staining (red). Scale bar, 20 μm. n = 2. In the merged image for GFP‐Usp22, the Mitotracker signal is not shown. GFP‐Usp27x showed very similar localization in HCC827‐GFP‐Usp27x cells (not shown).
- 1205Lu‐GFP‐Usp27x and 1205Lu‐GFP‐Usp22 were treated for 48 h with dox. Subcellular localization of GFP‐fusion proteins (green) was visualized by microscopy. Nuclei were stained with Hoechst (blue), and Bim was detected by intracellular staining for endogenous Bim (red). A higher magnification of merged figures is shown to the right (zoom). Scale bar, 20 μm. n = 2. Similar results were seen in every visual field analysed.
Usp27x‐expression can cause deubiquitination of Bim and stabilize Bim, antagonizing ERK‐induced Bim degradation
The binding of Usp27x to Bim and its deubiquitinating activity suggested that Bim is a target of Usp27x‐dependent deubiquitination. The Raf‐ERK pathway is known to stimulate ubiquitination and proteasomal degradation of Bim. We therefore tested whether Usp27x was able to counter‐regulate this activity and to stabilize Bim when its degradation is enhanced by the Raf‐ERK pathway.
We first tested this by expressing Usp27x under an inducible promoter in 293FT cells. As controls, the inactive mutant Usp27xC87A or GFP alone was used. Cells were treated with PMA, which (among other effects) activates the ERK pathway. As expected, PMA stimulation reduced Bim expression over the course of 24 h; after 48 h, Bim was hardly detectable (Fig 2A). Caspase inhibitor was added because the overexpression of Usp27x had a pro‐apoptotic effect (see below, Fig 3). Expression of Usp27x prevented this loss of Bim to a substantial degree, while expression of the mutant or GFP alone had no effect (Figs 2A and EV5C). The same was seen when the cells were stained for Bim expression and analysed by flow cytometry (Fig EV5B). Bim expression was not increased by experimental expression of Usp27x in the absence of PMA (Fig 2A–D), or even when higher expression levels of Usp27x was reached using transient transfection (Fig EV5A).
Figure 3. Usp27x expression sensitizes 293FT cells to apoptosis induction by stimulation with PMA .
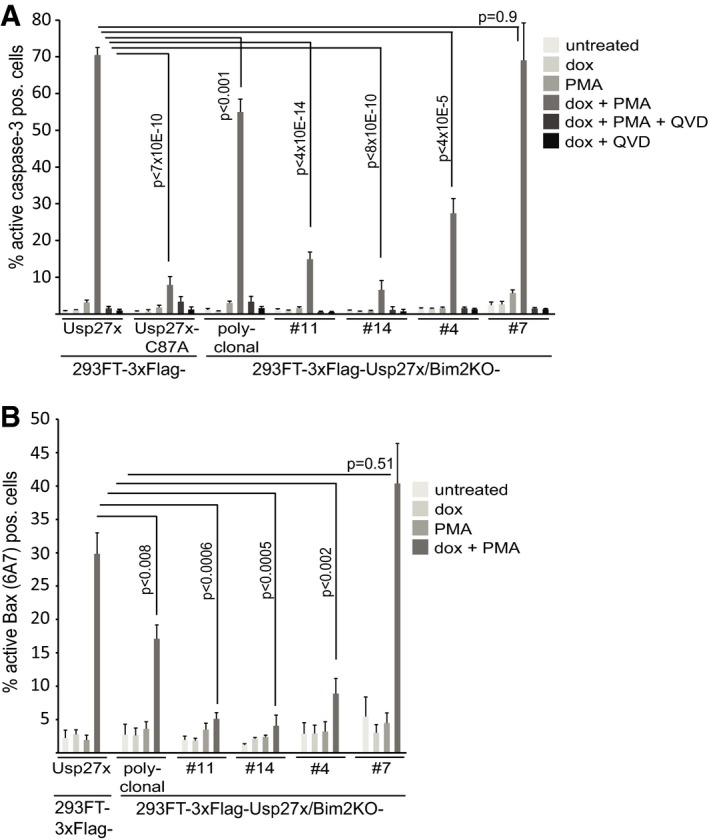
- 293FT‐TetR‐3xFlag‐Usp27x or 3xFlag‐Usp27xC87A, a polyclonal derivative where the Bim locus had been targeted by CRISPR/Cas9 and four single clones obtained by serial dilution from the polyclonal line, was treated as indicated (PMA 16.2 nM, QVD 10 μM). Apoptosis was measured after 24 h of treatment by staining for active caspase‐3, followed by flow cytometric analysis. Data (means ± SEM) are from n = 14 (3xFlag‐Usp27x line and 3xFlag‐Usp27xBim2KO polyclonal line), n = 5 (3xFlag‐Usp27xC87A and 3xFlag‐Usp27xBim2KO clone #14), n = 7 (3xFlag‐Usp27xBim2KO clone #11) or n = 4 (3xFlag‐Usp27xBim2KO clone #4 and #7) separate experiments. P‐values (t‐test) for statistically significant differences are shown.
- The same cells as in (A) (except Usp27xC87A mutant) were treated as in (A) and stained for active Bax. Data (means ± SEM) are from n = 8 (3xFlag‐Usp27x line and 3xFlag‐Usp27xBim2KO polyclonal line), n = 5 (3xFlag‐Usp27xBim2KO clone #11 and #14) or n = 4 (3xFlag‐Usp27xBim2KO clone #4 and #7) separate experiments. P‐values (t‐test) for statistically significant differences are shown. Expression levels of Bim, β‐TrCP and Flag‐Usp27x of the different cell lines are shown in Fig EV5E.
Figure EV5. Usp27x inhibits PMA‐induced degradation of Bim.
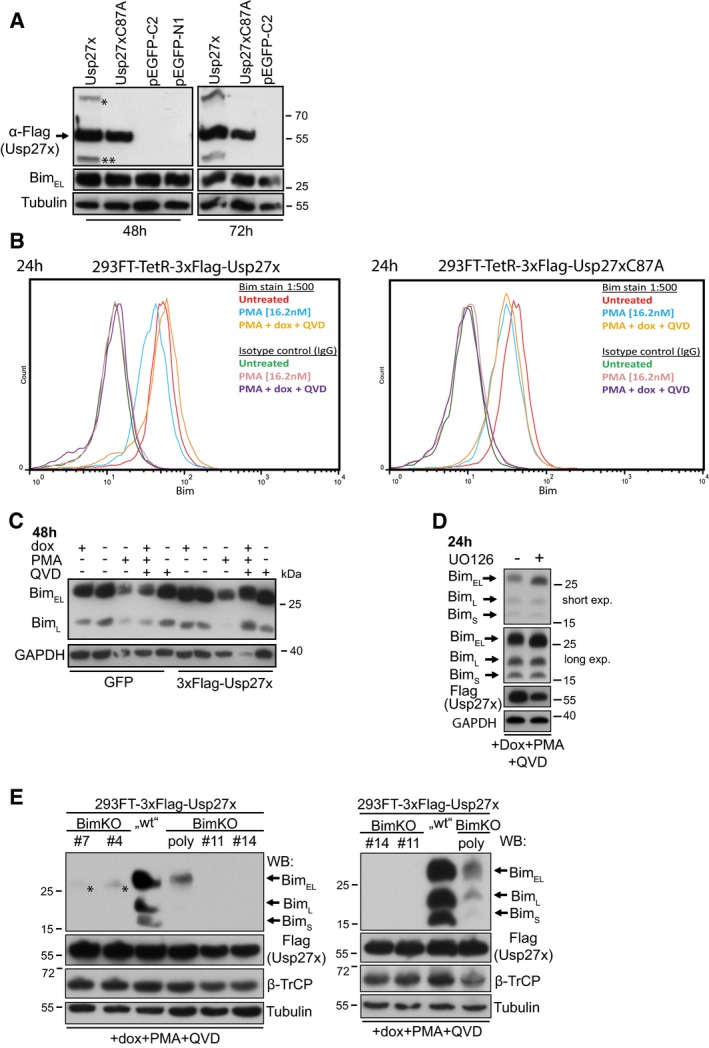
- Expression of Usp27x does not increase Bim levels in the absence of PMA. 293FT cells were transfected with pCMV‐7.1‐3xFlag‐Usp27x, pCMV‐7.1‐3xFlag‐Usp27xC87A or with two different EGFP vectors (pEGFP‐C2, pEGFP‐N1). Forty‐eight (left) or 72 h (right) later, cells were collected and lysed and the protein level of BimEL was analysed by immunoblotting. Asterisk (*) and double asterisk (**) represent Usp27x bands of unknown origin (compare Fig 2A). Similar results were obtained in n = 2 separate experiments.
- 293FT cells carrying dox‐inducible 3xFlag‐Usp27x (left) or 3xFlag‐Usp27xC87A (right) were untreated or treated for 24 h as indicated. Cells were fixed, stained with anti‐Bim antibodies or isotype control and secondary antibodies and analysed by flow cytometry. See also Fig 2A and B. n = 2.
- 293FT cells with dox‐inducible GFP or 3xFlag‐Usp27x were treated as indicated for 48 h prior to cell lysis. Bim was detected by Western blotting. Similar results were obtained in n = 3 separate experiments.
- The MEK inhibitor UO126 blocks phosphorylation of BimEL. 293FT‐TetR‐3xFlagUsp27x were treated for 24 h with dox in the presence of PMA and QVD. As control, the same cells were additionally pre‐treated with UO126 (10 μM) for 30 min before stimulation. Shown are the immunoblots of Bim, Flag‐Usp27x and as a loading control GAPDH. Similar results were obtained in n = 3 separate experiments (see also Fig 2B and D).
- Expression analysis of Bim and Flag‐Usp27x in 293FT‐3xFlag‐Usp27x cells versus Bim‐knockout cell lines. 293FT‐TetR‐3xFlag‐Usp27x, one polyclonal Bim‐knockout (Bim2KO) cell line established from 293FT‐3xFlag‐Usp27x cells using CRISPR/Cas9 and four additional single clones (#4, #7, #11, #14) obtained by serial dilution from the polyclonal Bim2KO cell line were treated for 24 h with dox plus PMA (16.2 nM) and QVD (10 μM). Expression of Bim, Usp27x (Flag) and β‐TrCP was analysed by immunoblotting. For apoptosis measurements, see Fig 3. Asterisk (*) indicates a signal obtained with the Bim antibody (recognizes the N‐terminus of Bim (around proline 25)) only present in clones #4 and #7. This band runs slightly lower than full‐length BimEL. Right immunoblot shows a second experiment (n = 2). Of note, the relative expression levels for Bim between the cell lines were the same in non‐stimulated cells (data not shown).
Although binding of Usp27x was confined to the isoform BimEL (Fig 1G), the splice variant BimL also appeared to be somewhat regulated by Usp27x (Fig 2A). Since Bim is often involved in binding to Bcl‐2‐family proteins, this may be indicative of specific protection through such varying complexes.
At higher resolution SDS–PAGE, it was apparent that in PMA‐stimulated cells expressing Usp27x, Bim ran at slightly higher molecular weight, consistent with its ERK‐dependent phosphorylation (Fig 2B). Phosphorylation is the signal for Bim ubiquitination, and the predicted function of Usp27x is to remove the ubiquitin chains attached by β‐TrCP. The expected result is the appearance of higher levels of phosphorylated Bim, as indeed seen in these experiments (Fig 2B and C). Addition of the MEK inhibitor UO126 caused a shift to the lower band (Figs 2D and EV5D).
With an antibody specific for Bim Ser69 phosphorylation (which is one known ERK‐dependent phosphorylation event), small levels of phospho‐Bim were detectable at steady state. PMA treatment in the presence of Usp27x overexpression strongly increased this phosphorylation as expected (Fig 2C). Again, Usp27x counteracted the PMA‐dependent decrease in Bim and caused the accumulation of phospho‐Bim but had no effect in the absence of PMA stimulation (Fig 2C).
To measure the Bim‐deubiquitinating activity of Usp27x more directly, 293FT cells carrying inducible Usp27x were transfected with 6His‐ubiquitin (or with an EGFP control) vector. Usp27x was induced or not; ubiquitinated proteins were isolated with Ni2+‐NTA agarose through the 6His‐tag and probed for the presence of Bim. Substantial amounts of ubiquitinated Bim could be recovered from PMA‐stimulated cells, and this amount was greatly reduced when the cells were overexpressing Usp27x (Figs 2E and EV3E). We also found some non‐ubiquitinated Bim bound non‐specifically to the Ni2+‐NTA beads, because Bim signals also appeared when an EGFP control vector was used instead of the His‐ubiquitin vector. Nevertheless, ubiquitinated Bim was only seen when co‐expressing His‐ubiquitin. Overexpression of Usp27x thus reduces the amounts of ubiquitinated Bim and can stabilize Bim in PMA‐stimulated cells, counteracting the enhanced turnover of Bim that is triggered by ERK activity.
Although Usp27x‐expressing cells treated with PMA had Bim levels comparable to untreated cells, there were high levels of apoptosis during this treatment. Catalytically inactive Usp27x failed to sensitize the cells to PMA (Fig 3A and B). We targeted Bim by CRISPR/Cas9 (Fig EV5E) and found a relatively small but significant protection in a polyclonal cell line carrying the Bim deletion (although some Bim expression was still observed Fig EV5E). Of four randomly selected clones from this polyclonal line, three were strongly protected against Usp27x/PMA, while one showed no protection at all, compared to the maternal, Bim‐containing line (Fig 3). This indicates that Bim does play some role but also that other, Bim‐independent mechanisms are triggered by PMA treatment.
The Ras‐Raf‐ERK pathway is a major oncogenic signalling pathway that is constitutively activated by genomic mutations in a number of human tumour entities such as malignant melanoma and non‐small cell lung cancer (NSCLC). Melanoma cells often harbour a typical mutation in the BRAF gene (V600E) that causes its constitutive activity and leads to the downstream activation of ERK 34. Human melanoma cells often express only low levels of Bim protein 35, and inhibition of ERK enhances the levels of Bim in melanoma cells in culture 36, suggesting that continuous ERK‐dependent phosphorylation is required to keep Bim at a low expression level in melanoma (see Fig EV6F for a comparison of Bim levels in 293FT cells and 1205Lu melanoma cells).
Figure EV6. Usp27x stabilizes Bim.
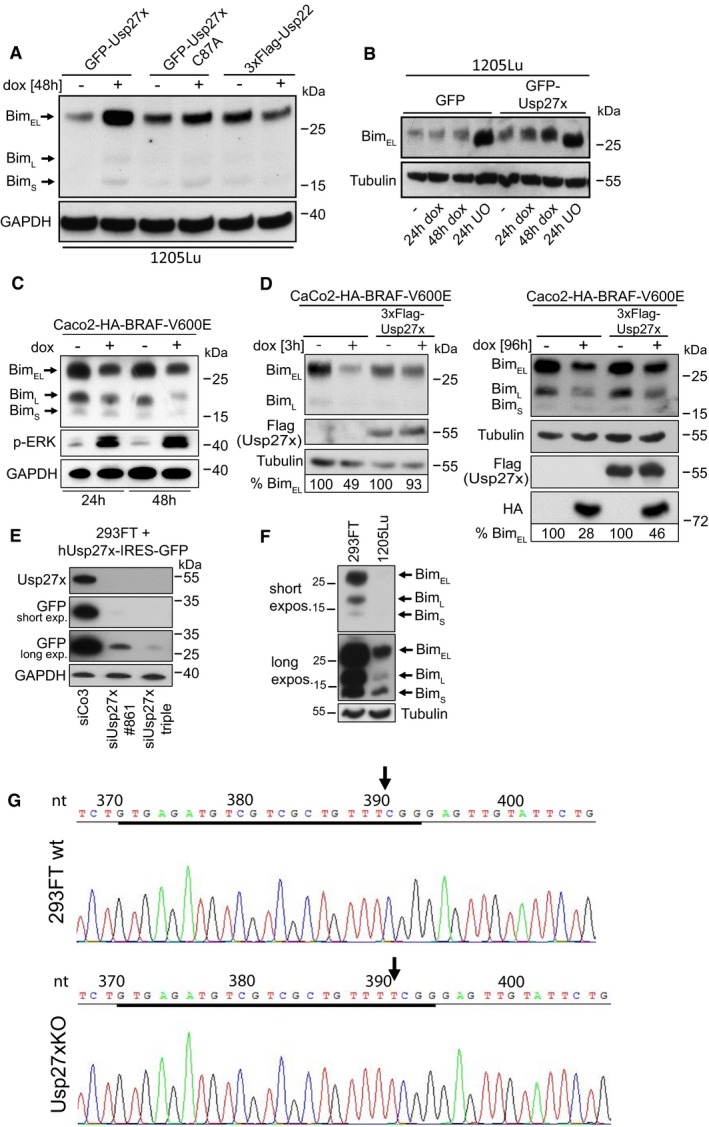
- Overexpression of Usp27x but not Usp22 stabilized endogenous BimEL in 1205Lu melanoma cells. 1205Lu cells carrying inducible constructs were treated with dox to induce GFP‐Usp27x, GFP‐Usp27xC87A or 3xFlag‐tagged Usp22 for 48 h. Bim was detected by Western blotting. Similar results were obtained in n = 2 separate experiments.
- 1205Lu cells with inducible constructs were treated as indicated to induce GFP or GFP‐Usp27x. The MEK inhibitor UO126 was used to block ERK activity. Of note, BimEL protein shifted to a lower molecular size when cells were treated with UO126, presumably as an effect of dephosphorylation. Similar results were obtained in n = 2 separate experiments.
- Caco2 colon carcinoma cells carrying dox‐inducible HA‐BRAF‐V600E were treated as indicated. ERK phosphorylation was measured by Western blotting. Note the loss of Bim protein upon induction of HA‐BRAF‐V600E (n = 2).
- 3xFlagUsp27x inhibits Bim loss upon BRAF‐V600E induction. Maternal Caco2‐HA‐BRAF V600E cells or the same cells stably expressing constitutive 3xFlag‐Usp27x were treated with dox for 3 (left) or 96 h (right) to induce BRAF‐V600E (two separate experiments). Bim levels were measured by Western blotting. For each condition, BimEL levels were quantified from the shown immunoblot and normalized to the tubulin signal. Per cent BimEL gives the expression relative to the starting point set to 100%. Similar results were obtained in n = 3 separate experiments with varying induction times. See also Fig 4D.
- Validation of Usp27x‐specific siRNAs. 293FT cells were transfected with siRNAs targeting human Usp27x or with control siRNA. Knockdown of Usp27x was either tested by using one single siRNA against Usp27x (#861) or by using a combination of three siRNAs targeting Usp27x (#495, #498, #861). After 24 h, cells were transfected with a construct driving expression of human Usp27x and expression of GFP off an IRES. Usp27x and GFP were detected 24 h later by Western blotting (n = 2). The combination of three siRNAs was used for the later knockdown experiments (see Fig 5A).
- Comparison of Bim expression levels between 293FT and 1205Lu. Western blot of whole‐cell lysates obtained from 293FT cells and from 1205Lu melanoma cells. For both cell lines, 50 μg protein was run by SDS–PAGE, and Bim was identified using antibody against Bim. Tubulin served as a loading control. Similar results were obtained in n = 2 separate experiments.
- Validation of 293FT‐Usp27xKO (clone 2/10). Sequencing results (n = 1) indicate a thymidine (T) insertion a nucleotide (nt) 391 (arrow). This insertion leads to the addition of 14 different amino acids after aa 130 followed by a stop codon. Therefore, this clone expresses a truncated version of Usp27x (wt has 438 aa) deleting part of the catalytic activity/triad (involves aa C87A (nucleophile), and the predicted sites H380 (proton acceptor) and aa D396/D397 in Usp27x 28).
We generated melanoma cells carrying inducible GFP‐Usp27x on the basis of two BRAF‐V600E‐positive human melanoma cell lines. Induction of Usp27x in these cells caused the increase in (probably phosphorylated) Bim protein levels (Figs 4A and B, and EV6A and B), while Usp27xC87A had no effect in the cell line tested (Figs 4A, left panel, and EV6A). As a further control, we expressed the closely related DUB Usp22 (identity score to Usp27x 75%) fused to GFP in 1205Lu melanoma cells; this had no effect on Bim expression (Fig 4A, right panel). Another 1205Lu cell line expressing a doxycycline‐inducible 3xFlag‐Usp22 construct again showed no stabilization compared to Usp27x (Fig EV6A). The MEK inhibitor UO126 enhanced Bim expression (Fig EV6B), confirming that the low Bim levels in 1205Lu cells are a consequence of constitutive Raf‐ERK activity. These data indicate that Usp27x is able to stabilize Bim in melanoma cells when its degradation is triggered by the continuous activity of the Raf‐ERK pathway.
Figure 4. Usp27x but not Usp22 can counter Bim destabilization through the Raf‐ERK pathway.
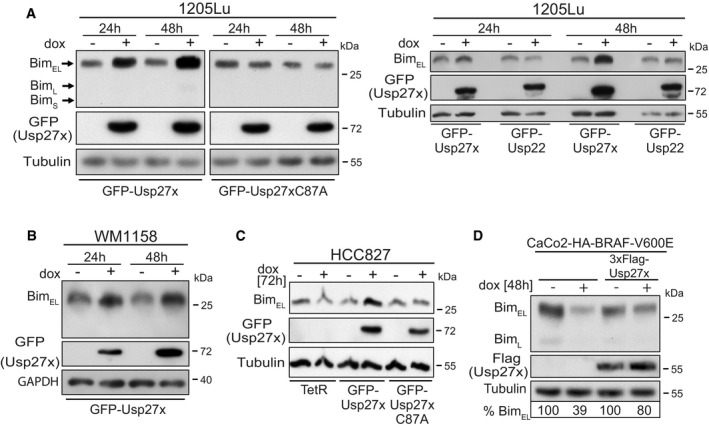
- Lines based on the human melanoma cell line 1205Lu (BRAF‐V600E‐positive) were made to carry GFP‐Usp27x, GFP‐Usp27xC87A or GFP‐Usp22 under the control of a doxycycline (dox)‐inducible promoter. GFP‐Usp27x, GFP‐Usp27xC87A or GFP‐Usp22 was induced with dox for 24 or 48 h. Levels of Bim were determined by Western blotting. The two left panels are from the same membrane and exposure times. Similar results were obtained in n = 4 separate experiments (left) and n = 3 experiments (right). See also Fig EV6A where 3xFlag‐Usp22 is also shown to be unable to increase Bim levels. GFP‐Usp27x, GFP‐Usp27xC87A or GFP‐Usp22 expression was detected using an antibody against GFP.
- A cell line derived from the BRAF‐V600E‐positive human melanoma line WM1158 was generated to carry inducible GFP‐Usp27x. GFP‐Usp27x was induced, and Bim was detected as in (A). Similar results were obtained in n = 3 separate experiments. GFP‐Usp27x expression was detected using an antibody against GFP.
- Derivatives of the NSCLC cell line HCC827 (expressing constitutively active EGFR) were generated that only express the Tet repressor (TetR) or carrying in addition either GFP‐Usp27x or GFP‐Usp27xC87A. Cells were treated with dox for 72 h. Bim was detected by Western blotting. Similar results were obtained in n = 2 separate experiments. GFP‐Usp27x or GFP‐Usp27xC87A expression was detected using an antibody against GFP.
- Caco2 cells carrying a dox‐inducible HA‐BRAF‐V600E 37 (left two lanes) or the same cells stably expressing 3xFlag‐Usp27x were treated with dox for 48 h to induce expression of HA‐BRAF‐V600E. Bim was detected by Western blotting. The amount of BimEL was quantified from the shown immunoblot using the expression levels of the uninduced control cells set to 100% (normalized to the tubulin signal for each condition). Similar results were obtained in n = 3 separate experiments with varying induction times. See also Fig EV6D.
In NSCLC, a different upstream alteration also causes the constitutive activity of ERK. In these cells, a genomic mutation leads to the constitutive activity of the EGF receptor, which downregulates Bim through the ERK pathway 7, 8, 9. When we expressed Usp27x in an NSCLC line carrying mutant EGFR (HCC827), we likewise found an increase in Bim levels, which was not seen for Usp27xC87A or in HCC827 cells only expressing the Tet repressor (TetR, Fig 4C). This again indicates that Usp27x is able to counteract the Bim downregulation that is found as a consequence of oncogenic ERK signalling.
In the Caco2 colon carcinoma cell line, the inducible expression of BRAF‐V600E leads to ERK phosphorylation (Fig EV6C) 37. When BRAF‐V600E was induced, a reduction in Bim levels was observed, an effect that was seen after a few hours and that persisted over at least 96 h (Fig EV6C and D). However, constitutive overexpression of Usp27x rescued Bim expression (Figs 4D and EV6D).
Caco2 cells are notable for their relatively high endogenous expression of Usp27x mRNA (http://www.nextbio.com/b/search/ov/USP27X?type=feature&id=103162; the link works following free registration at the site). We therefore took the approach of targeting Usp27x by RNAi in these cells. Since we were unable to detect endogenous Usp27x protein with commercially available Usp27x antibodies, we tested the efficiency of the siRNA in cells overexpressing Usp27x (Fig EV6E). Indeed, when Caco2 cells had been transfected with siRNA specific for Usp27x, induction of BRAF‐V600E caused the loss of Bim protein (Fig 5A), indicating that endogenous Usp27x is active in stabilizing Bim in these cells.
Figure 5. Endogenous Usp27x stabilizes Bim in Caco2 and 293FT cells.
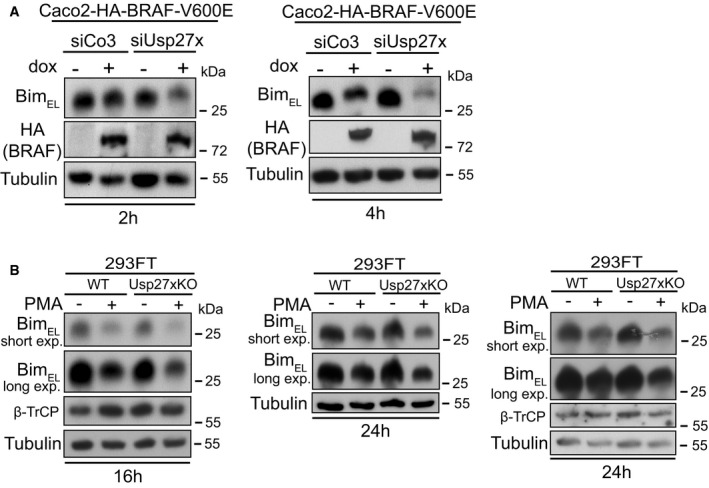
- Loss of endogenous Usp27x enhances destabilization of BimEL in response to BRAF‐V600E. Caco2 cells carrying inducible HA‐BRAF‐V600E were transfected with control siRNA (siCo3) or siRNA directed against Usp27x (combination of three siRNAs targeting Usp27x mRNA) for 24 h prior to BRAF‐V600E expression with dox for 2 (left) or 4 h (right). BimEL was detected by Western blotting. Similar results were also observed in n = 2 more experiments after 3 h induction (not shown, see Fig EV6E for siRNA efficacy).
- 293FT cells deficient for Usp27x show enhanced destabilization of BimEL in response to PMA. 293FT wt cells or 293FT‐Usp27xKO (clone 2/10, generated using CRISPR/Cas9) were treated with PMA (16.2 nM) for 16 or 24 h to induce BimEL degradation. Western blots show protein levels of n = 3 independently performed experiments (similar results were obtained in one more experiment, not shown). See Fig EV6G for verification of the Usp27xKO cell line by DNA sequence analysis.
In addition to these gain‐of‐function approaches, we generated a 293FT cell clone genomically deficient for Usp27x using CRISPR/Cas9 (Fig EV6G). In this clone, PMA had a stronger Bim‐degrading effect than in the maternal line (Fig 5B shows three separate experiments), suggesting that the endogenous expression levels of Usp27x in 293FT cells contributed to Bim stability during PMA treatment.
When de novo protein synthesis was inhibited with the translation inhibitor cycloheximide, we found that the stability of Bim was enhanced by overexpression of Usp27x in situations where Bim is phosphorylated and degraded via the proteasome. This was the case for PMA‐stimulated 293FT cells (Fig 6A), BRAF‐V600E melanoma cells (Fig 6B) as well as (although not as strongly) for NSCLC cells (Fig 6C) (in NSCLC ERK is active and Bim has a high turnover due to expression of mutant EGFR 8). This effect may be diluted by the inhibition of synthesis of other Bim/Usp27x‐regulating components (e.g. Usp27x seems to undergo phosphorylation and ubiquitination that might influence its activity 38, 39). However, the results again support the view that Usp27x stabilizes Bim by reducing its ubiquitination.
Figure 6. Usp27x stabilizes Bim protein levels in the ERK‐degradation pathway in 293FT cells, 1205Lu melanoma and HCC827 NSCLC cells.
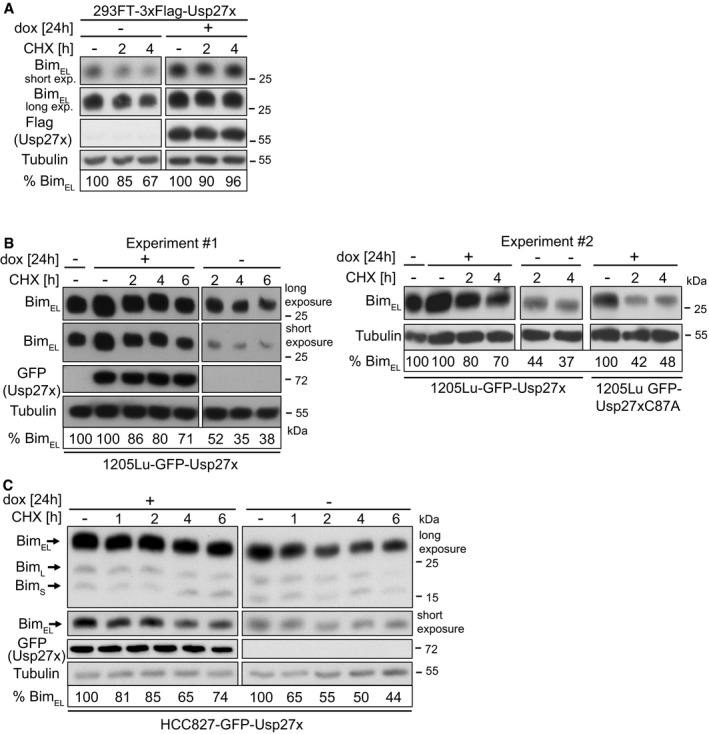
- 293FT‐TetR‐3xFlagUsp27x cells were treated with PMA (16.2 nM) plus QVD (10 μM) and 3xFlag‐Usp27x was induced by dox at the same time for 24 h followed by addition of cycloheximide (CHX). Cells were harvested at the indicated time points, and levels of BimEL were determined by Western blotting. Similar results were obtained in n = 3 separate experiments. Cut membranes shown were from one membrane with same exposure time. For each condition, BimEL levels were quantified and normalized to the tubulin signal. Per cent BimEL gives the expression relative to the starting point.
- 1205Lu melanoma cells carrying dox‐inducible GFP‐Usp27x were treated with dox for 24 h. Cycloheximide (CHX, 1 μg/ml) was then added. Cells were harvested at the indicated time points, and levels of BimEL were determined by Western blotting. GFP‐Usp27x was detected with anti‐GFP antibodies. Similar results were obtained in n = 3 separate experiments. A second experiment is shown in the right panel also including 1205Lu melanoma carrying dox‐inducible GFP‐Usp27xC87A mutant. For each condition, BimEL levels were quantified and normalized to the tubulin signal. Per cent BimEL gives the expression relative to the starting point set to 100%. The starting point for the non‐induced Usp27x/C87A cells (‐dox) is shown in lane 1 for experiment one and two (‐dox, ‐CHX). Cut membranes shown were from one membrane with same exposure time. Same results were obtained for WM1158 melanoma cells (data not shown).
- HCC827 cells were induced to express GFP‐Usp27x with dox as indicated. Twenty‐four hours later, cycloheximide (CHX, 1 μg/ml) was added for the indicated time. BimEL was detected after the indicated times of treatment by Western blotting. For each condition, BimEL levels were quantified and normalized to the tubulin signal. Per cent BimEL gives the expression relative to the starting point. Similar results were obtained in n = 2 separate experiments. Cut membranes shown were from one membrane with same exposure time.
Usp27x has pro‐apoptotic activity when the Raf‐ERK pathway is inhibited
Since Usp27x can enhance the levels of pro‐apoptotic Bim, we examined the effect of Usp27x levels on apoptosis induction in the cells where we had found that Usp27x reversed the Bim loss through oncogenic Raf‐ERK signalling. In 1205Lu melanoma cells, expression of Usp27x had a significant but very small pro‐apoptotic effect (Figs 7A and EV7A and C). Inhibition of the ERK pathway with UO126 induced higher levels of apoptosis (Figs 7A and EV7A and B). However, inhibition of ERK signalling in cells overexpressing Usp27x had a substantial pro‐apoptotic effect, while the Usp27xC87A mutant or GFP alone had no such activity (Figs 7A and EV7A and B). Loss of Bim provided some protection in 1205Lu cells against apoptosis induced by UO126 in the presence of Usp27x (Fig EV7A). The BRAF inhibitor vemurafenib had a very small pro‐apoptotic effect in 1205Lu cells, and this effect was also enhanced by Usp27x expression (Fig EV7C).
Figure 7. Usp27x sensitizes 1205Lu melanoma and HCC827 NSCLC cells to apoptosis induction by inhibition of the Raf‐ERK pathway.
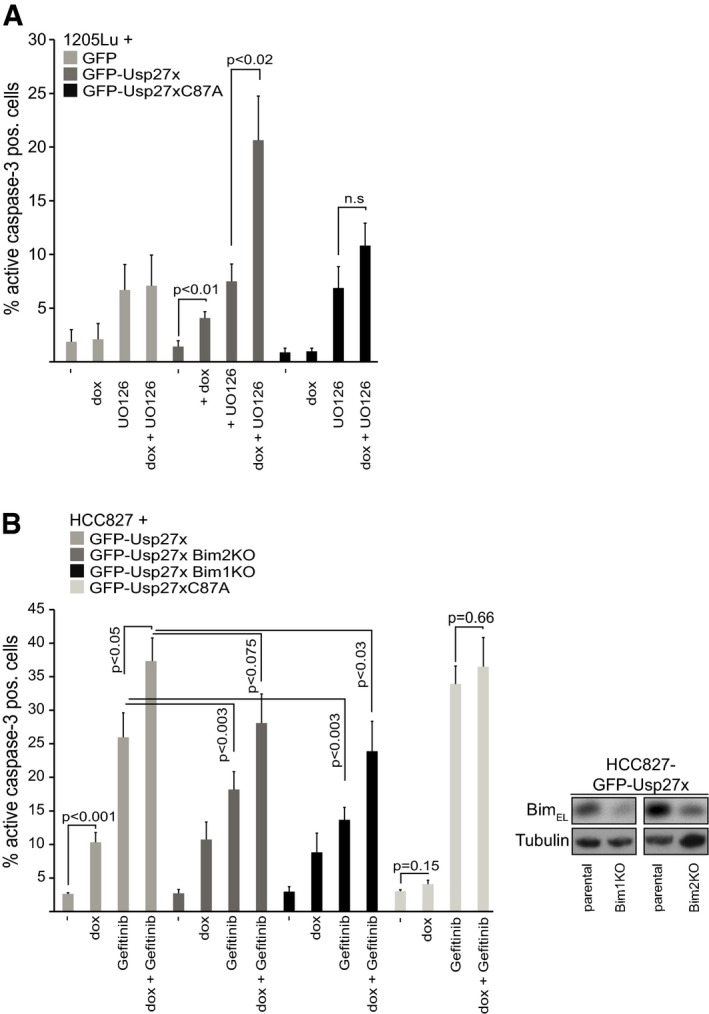
- 1205Lu melanoma cells carrying dox‐inducible GFP, GFP‐Usp27x or GFP‐Usp27xC87A were treated with doxycycline (dox) as indicated. After 48 h, UO126 (10 μM) was added for another 48 h. Apoptosis was measured by staining for active caspase‐3, followed by flow cytometric analysis. Data (means ± SEM) are from n = 3 (GFP) or from n ≥ 6 separate experiments (GFP‐Usp27x and GFP‐Usp27xC87A). P‐values (t‐test) for statistically significant differences are shown. N.s., P > 0.05. The addition of QVD inhibited cells from active caspase‐3‐positive staining (not shown). A stain for active Bax is shown in Fig EV7B.
- HCC827 NSCLC cells carrying dox‐inducible GFP‐Usp27xC87A, GFP‐Usp27x or two separate lines established from these GFP‐Usp27x cells, where the Bim locus had been targeted for deletion using CRISPR/Cas9 (Bim‐KO), were treated with combinations of dox and the EGFR inhibitor gefitinib (10 μM) as indicated. Apoptosis was measured after 72 h of treatment by staining for active caspase‐3, followed by flow cytometric analysis. Data (means ± SEM) are from n ≥ 15 (maternal GFP‐Usp27x line) or from n ≥ 5 (for Bim1KO) or from n ≥ 6 (for Bim2KO) or n = 4 (for GFP‐Usp27xC87A) separate experiments. P‐values (t‐test) for statistically significant differences are shown. Again, the addition of QVD inhibited cells from active caspase‐3‐positive staining (not shown). The inset shows Bim‐knockout efficiency (n = 2).
Figure EV7. Usp27x sensitizes 1205Lu melanoma cells to apoptosis induction by inhibition of the Raf‐ERK pathway.
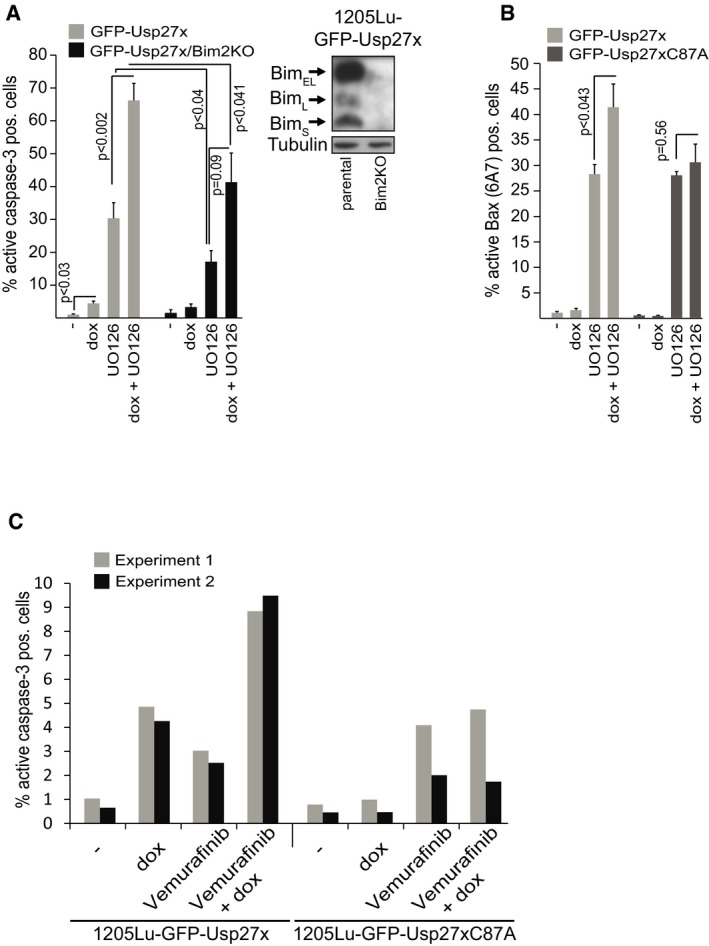
- 1205Lu melanoma cells carrying dox‐inducible GFP‐Usp27x and the same cells where Bim had been targeted using CRISPR/Cas9 (1205Lu‐GFP‐Usp27x/Bim2KO) were treated with dox as indicated. After 48 h, UO126 (10 μM) was added for another 48 h. Apoptosis was measured by staining for active caspase‐3, followed by flow cytometric analysis. All data (means ± SEM) are from n = 5 separate experiments. P‐values (t‐test) for statistically significant differences are shown. The addition of QVD blocked the appearance of active caspase‐3 staining (not shown). Inset shows knockout efficiency of Bim in 1205Lu‐GFP‐Usp27x melanoma cells (n = 2).
- GFP‐Usp27x‐ or GFP‐Usp27xC87A‐expressing 1205Lu melanoma cells were treated as in (A) and percentage of cells with active Bax was measured by FACS using a conformation‐specific antibody (6A7 clone) that recognizes only the activated form of Bax. All data (means ± SEM) are from n = 5 (GFP‐Usp27x) or n = 3 (GFP‐Usp27xC87A) separate experiments. P‐values (t‐test) for statistically significant differences are shown.
- Dox‐inducible GFP‐Usp27x or GFP‐Usp27xC87A were induced in 1205Lu melanoma cells as indicated. After 48 h, cells were treated with vemurafenib as indicated for additional 24 h. Apoptosis was measured by staining for active caspase‐3 and flow cytometry. Bars show percentage of active caspase‐3‐positive cells from n = 2 independently performed experiments.
We also tested for the relevance of Bim for this form of apoptosis by targeting the Bim locus in the NSCLC line HCC827 with two different gRNAs using CRISPR/Cas9. Although the knockout was incomplete, we achieved a good reduction in Bim levels in the two polyclonal lines tested here (Fig 7B). In these cells, the inhibition of EGFR kinase activity with gefitinib induces Bim and apoptosis 8. Usp27x, either on its own or together with gefitinib, caused significant apoptosis in HCC827 cells. Apoptosis induced by either gefitinib (as reported before) as well as by the combination Usp27x/gefitinib was reduced in cells with reduced Bim levels (Fig 7B). No enhanced apoptosis was observed when mutant GFP‐Usp27xC87A protein was expressed either alone or in combination with gefitinib (Fig 7B).
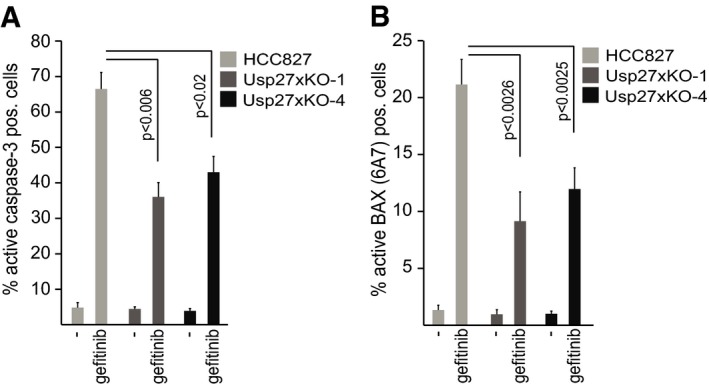
HCC827 cells express significant amounts of Usp27x mRNA (https://genevestigator.com/gv). We had observed that Usp27x overexpression in HCC827 cells leads to increased apoptosis when combined with gefitinib. We therefore analysed whether the loss of Usp27x decreased apoptosis induction in HCC827 cells by gefitinib. Usp27x was targeted in HCC827 cells using CRISPR/Cas9 and two different guide RNAs (gUsp27x‐1 and gUsp27x‐4). Polyclonal cell lines carrying either mutation clearly showed less apoptosis induction upon gefitinib treatment as measured by staining for active caspase‐3 (Fig 8A) and staining for the active form of Bax (Fig 8B). High levels of Usp27x thus have a pro‐apoptotic effect at least in some cells, although this effect is probably at least not exclusively dependent on Bim.
Figure 8. Loss of Usp27x reduces gefitinib‐induced apoptosis in HCC827 NSCLC cells.
-
A, BHCC827 NSCLC cells and two separate polyclonal lines established from these cells, where the Usp27x locus had been targeted for deletion using CRISPR/Cas9 with two different guide RNAs against Usp27x (Usp27xKO‐1 and Usp27xKO‐4), were treated with gefitinib (10 μM) for 48 h. Apoptosis was measured by staining for active caspase‐3 (A) or by staining for activated Bax (B; antibody: 6A7 clone) followed by flow cytometric analysis. Data shown (means ± SEM) are from n = 4 separate experiments. P‐values (t‐test) for statistically significant differences are shown.
Discussion
We here report discovery and characterization of the DUB Usp27x as a deubiquitinase that can reduce the levels of Bim ubiquitination and stabilize Bim in response to the Raf‐ERK‐degradation signal. High levels of Usp27x had pro‐apoptotic activity in a melanoma and a NSCLC cell line (see above) as well as in 293FT cells.
The activity of the Bcl‐2 family is probably mostly regulated through the expression levels of the individual members although the number of proteins makes it difficult to control and understand their relative contribution. Transcriptional regulation is common for some genes, such as the BH3‐only proteins Puma and Noxa 40. Expression of anti‐apoptotic Mcl‐1 can be substantially regulated by adjustment of its turnover, and a number of ubiquitin ligases as well as an Mcl‐1 DUB have been discovered 41. Regulation of Noxa by the DUB UCHL‐1 has further been described 42. UCHL‐1 cleaves linear ubiquitin linkages and its expression is normally restricted to neuronal cells, but UCHL‐1 may be overexpressed in human cancer cells 42. As far as we know, no other DUB of a pro‐apoptotic protein has been identified.
Bim is degraded in response to a major oncogenic pathway, and it is likely that both ubiquitination and deubiquitination of Bim in this pathway will affect survival and affect tumour development. Usp27x was found in a complex with both Bim and the Bim‐ubiquitin‐ligase β‐TrCP. However, the expression level of either E3 ligase or deubiquitinase did not seem to alter the recruitment of the other to Bim, as indicated by the experiments of over‐ and underexpression. Both β‐TrCP 22 and Usp27x (see above) were recruited more efficiently to Bim upon its phosphorylation. The activity of the ERK pathway therefore appears to be a stimulus for the regulation of both Bim destabilization and stabilization. The outcome of this activity may then depend on the expression levels or activity of E3 ligase and deubiquitinase. Since Bim degradation during mitosis, regulated by Aurora kinase, has also been suggested 17, it is possible that Usp27x also associated with Aurora kinase and the APCCdc20 complex.
Usp27x mRNA is detectable in most normal tissues as well as, at varying levels, in many tumour cell lines and tissues (see free online expression databases such as genevisible.com (https://genevestigator.com/gv/). Bim can at least in some situations act as a tumour suppressor. By enhancing the expression of Bim levels, Usp27x may have similar function.
In the cellular models tested, Usp27x had pro‐apoptotic function, as would be expected from an enzyme increasing the expression of the pro‐apoptotic protein Bim. This may, however, not be quite as straightforward since a high activity of Usp27x may specifically increase the amount of phosphorylated Bim. ERK‐dependent Bim phosphorylation leads to reduced binding of Bim to anti‐apoptotic Bcl‐2‐family proteins 43. Since the binding of Bim to Bcl‐2‐like proteins may on the one hand inhibit apoptosis (since Bim is sequestered and not free to activate Bax/Bak), or on the other hand induce apoptosis (since it may prevent Bcl‐2‐like proteins from inhibiting Bax/Bak; both scenarios have been demonstrated 44), it is difficult to predict what the net result of Bim phosphorylation in a cell will be.
Interestingly, however, apoptosis was not strictly Bim dependent: cells with reduced Bim expression still showed a pro‐apoptotic effect of Usp27x expression. This may indicate that Usp27x has other, unidentified targets. Although this would not explain the pro‐apoptotic activity, one additional target is the transcription factor Hes1; here, Usp27x acts together with Usp22 and Usp51, presumably in the nucleus, to regulate Hes1‐expression. Furthermore, in the nucleus, Usp27x might interact with Usp22 (interaction of these two proteins has been observed earlier) 45. However, DUBs do typically have a number of different targets that may affect different biological processes 45. A case in point is Usp9x, which has been identified as a Mcl‐1‐specific DUB 46 but has found to act Mcl‐1 independently as a suppressor of pancreatic cancer 47 and to regulate T‐cell receptor signalling 48 as well as the alkylation damage response 49. Which other potential targets of Usp27x may be relevant for apoptosis remains to be clarified.
In summary, we have identified Usp27x as an enzyme regulating deubiquitination and stability of Bim, capable of antagonizing the Raf‐ERK Bim‐degradation pathway. Expression of Usp27x is a likely co‐determinant of Bim levels and of apoptosis sensitivity and may act as a tumour suppressor.
Materials and Methods
Cell lines, culture conditions
Mouse embryonic fibroblasts (MEFs) deficient for Bax and Bak overexpressing either 3xHA‐BimEL or untagged BimEL have been described 25. 293FT cells (Invitrogen) and human colon carcinoma cells expressing doxycycline‐inducible HA‐tagged BRAF‐V600E 37 were cultured in DMEM with 10% FCS (Gibco) and 1% (v/v) penicillin/streptomycin (Gibco). 1205Lu and WM1158 human metastatic melanoma cells carrying the BRAF‐V600E mutation were obtained from Dr. Meenhard Herlyn, Wistar Institute, Philadelphia. The HCC827 cell line was kindly provided by Dr. Mark S. Cragg 8 and was cultured in RPMI‐1640 medium (Gibco) supplemented with 10% FCS and 1% (v/v) penicillin/streptomycin. Lentivirally transduced cells were selected using hygromycin B (Invitrogen, HEK293FT: 300 μg/ml; HCC827: 1,000 μg/ml; 1205Lu: 750 μg/ml and WM1158: 500 μg/ml) and/or puromycin (Invivogen, 5 μg/ml). Gefitinib and vemurafenib were from Selleckchem, PMA from Sigma, UO126 (in solution #662009) from Merck Calbiochem and Q‐VD‐OPh from SM Biochemicals. Doxycycline (dox, Sigma) was always used in a 1 μg/ml concentration.
SILAC labelling of Bax/Bak‐double KO MEFs and MS sample and analysis
Usp27x was found as an interacting partner of BimEL in an experiment published earlier 25. In brief, Bax/Bak‐double KO cells expressing 3xHA‐BimEL were labelled with L‐arginine (Arg0) and L‐lysine (Lys0) (light amino acids) and BimEL cells were labelled with the heavy amino acids L‐lysine–U‐13C6‐15N2 (Lys8) and L‐arginine–U‐13C6‐15N4 (Arg10). 3xHA‐BimEL was immunoprecipitated. Cells expressing untagged BimEL were subjected to the same protocol. The anti‐HA matrix was collected and washed with 25 ml of lysis buffer. Elution was done in the presence of 3×SDS–Laemmli buffer at 95°C for 5 min (3 × 50 μl). After elution, the samples were mixed and identification of BimEL interacting proteins was done by LC‐MS/MS as described earlier 25.
Construction of expression vectors and generation of cell lines
Retroviral constructs (pMIG‐GW) of murine BimEL or N‐terminal triple‐HA‐tagged 3xHA‐BimEL or 3xHA‐BimEL∆∆ 50 were generated as described 27. All three BimEL constructs were splice site mutants of BimEL permitting only translation of BimEL 27. Murine full‐length Usp27x (mUsp27x; UniProt Q8CEG8; 438 amino acids; Gene ID: 54651) and full‐length murine Usp22 (mUsp22; UniProt Q5DU02; 525 amino acids; Gene ID: 216825) were PCR‐amplified from cDNA isolated from mouse lung. Human full‐length Usp27x (hUsp27x; UniProt A6NNY8; 438 amino acids; Gene ID: 389856) was PCR‐amplified from HeLa cDNA. Murine Usp27x was cloned either into p3xFlag‐CMV7.1 (Sigma) to obtain pCMV‐7.1‐3xFlag‐Usp27x or into pENTR‐SD‐D‐Topo using TOPO cloning (Life Technologies) with either a 3xFlag tag or a 3xHA tag fused with the N‐terminus of full‐length mUsp27x to obtain pENTR‐SD‐D‐3xFlag‐Usp27x and pENTR‐SD‐D‐3xHA‐Usp27x. pENTR‐SD‐D‐3xFlag‐Usp22 (murine) was constructed as described for Usp27x. Murine GFP‐Usp27x or GFP‐Usp22 (GFP fused to the N‐terminus of full‐length Usp27x or Usp22) was generated using two‐step PCR and was subsequently introduced again into pENTR‐SD‐D‐Topo vector to generate pENTR‐SD‐D‐GFP‐Usp27x (or GFP‐Usp22). Mutagenesis of the cysteine at position 87 in murine Usp27x to alanine (mUsp27xC87A) was done using the QuickChange II site‐directed mutagenesis kit (Agilent Technologies). 3xFlag‐tagged, or 3xHA‐tagged, or GFP‐tagged murine Usp27x and untagged human Usp27x were cloned using Gateway (GW) cloning (Life Technologies) to generate retroviral pMIG‐GW‐3xFlag‐Usp27x and pMIG‐3xHA‐Usp27x or pMIG‐GW‐hUsp27x.
For inducible expression, genes were cloned into the doxycycline‐regulated (Teton) lentiviral construct pF‐CMV‐to‐GW‐SV40‐puro by Gateway cloning. The initial lentiviral expression vector pF‐CMV‐to‐GW‐SV40‐puro vector was created from pF 5x UAS MCS SV40 PURO vector 51 with PacI and NheI to remove the 5xUAS. The CMV promoter and gateway cassette (GW) including the V5 tag were amplified by PCR from the Invitrogen vector pLENTI4/TO/GW/V5/DEST and cloned into the PacI and NheI sites. To generate the TetR lentiviral construct (pF‐CMV‐TetR‐PGK‐Hygro), the CMV promoter and TetR were amplified together by PCR from the invitrogen vector pLENTI6/TR and cloned into the PacI and NheI sites of pFU GEV16 PGK HYGRO vector 51. Cell lines were first transduced with the TetR lentivirus, selected with hygromycin B (7–10 days) and subsequently transduced with the lentiviral expression systems following selection with puromycin. To generate Caco2‐HA‐BRAF‐V600E cells that additionally express 3xFlag‐mUsp27x from the constitutive pEF1α promoter, 3xFlag‐mUsp27x was first cloned into pF‐hEF1α‐GW‐SV40 PURO by gateway cloning and was then used for lentiviral production and transduction of Caco2 cells. Production of lentiviral particles was done by transfecting 293FT cells together with packaging vectors pMD2.G and psPAX2 (Dr. Didier Trono, Lausanne) using either FugeneHD (Promega) or PEI.
N‐terminal 6xHis‐ and SUMO‐tagged human Usp27x was cloned into the prokaryotic expression vector pE‐SUMOstar_Amp (vector backbone from Life Sensors Cat. #1106) using BsaI/XbaI restriction of the hUsp27x insert and BsaI cutting of the pE‐SUMOstar_Amp vector and subsequent ligation to obtain pE‐SUMOstar‐hUsp27x (6xHis‐Sumo‐hUsp27x). This vector was used to purify human Usp27x from E. coli by Ni2+NTA agarose (see Purification of 6xHis‐Sumo‐hUsp27x). Usp27x template vectors for in vitro translation using the PureExpress system of murine 1xHA‐Usp27x or its catalytically inactive mutant (1xHA‐Usp27xC87A) were generated by cleavage of pPE‐DHFR vector (New England Biolabs) with NdeI/XhoI and ligation of 1xHA‐tagged Usp27x (or Usp27xC87A) with a stop codon and NdeI/XhoI overhangs.
Immunoprecipitation
Experiments were done in the presence of Q‐VD‐OPh (QVD) (10 μM). After cell lysis (20 mM Tris/HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1× Protease Inhibitor Mix (Roche), 1% Triton X‐100 (1% digitonin was used in Fig 1A), IP was performed with antibodies to the HA tag (30 μl anti‐HA affinity matrix, Roche) using 1.5 mg of whole‐cell lysates after pre‐clearing with proteinG agarose (Roche). The anti‐HA matrix was collected and washed with 20 ml of lysis buffer. Elution was done with 3×SDS–Laemmli buffer (5 min 95°C). Anti‐Flag‐immunoprecipitation was done using 30 μl anti‐Flag‐M2‐affinity resin/sample (Sigma), and 1.5 mg of 1% Triton X‐100 lysates was incubated for anti‐Flag‐IP, washed and eluted as described for HA immunoprecipitation.
Purification of His‐ubiquitin proteins from 293FT cells
3.5 × 106 293FT‐TetR‐3xFlag‐Usp27x cells (10‐cm culture plate) were transfected with 10 μg 6His‐ubiquitin vector (or with an EGFP control vector). After 24 h, cells were treated with PMA (16.2 nM) and QVD (10 μM) ± dox. After additional 20 h, cells were treated for 4 h with MG132 (40 μM). Cells were collected, washed twice with ice‐cold PBS and lysed under denaturing conditions (6 M guanidinium‐HCl, 0.1 M NaH2PO4, 10 mM Tris, 5 mM imidazole, 1× protease inhibitor, 1 mM PMSF, 30 mM NEM, 0.01 M β‐mercaptoethanol, adjusted to pH 8.0), and His‐ubiquitin‐labelled proteins (2.5 mg) were purified by Ni2+‐NTA affinity chromatography. The beads were washed once with 1 ml lysis buffer, once with 5 ml wash buffer (8 M urea, 0.1 M NaH2PO4, 10 mM Tris, 5 mM imidazole, 0.01 M β‐mercaptoethanol, adjusted to pH 6.8) and twice with 5 ml wash buffer plus 0.1% Triton X‐100. Proteins were eluted in 75 μl buffer (250 mM imidazole, 150 mM Tris, 10% glycerol, 0.72 M β‐mercaptoethanol, 4.5% SDS, adjusted to pH 6.8).
Purification of 6xHis‐Sumo‐hUsp27x
Single colonies of transformed BL21‐CodonPlus(DE3)‐RIPL‐competent cells were induced using 1 mM IPTG for 4 h at 21°C. E. coli were lysed in lysis buffer (sonication in 50 mM NaH2PO4, 300 mM NaCl pH 8.0, 10% glycerol, 0.8% CHAPS, 1× PI; lysozyme, benzonase, 10 mM imidazole) and 6His‐Sumo‐Usp27x protein was purified on Ni2+‐NTA matrix at 4°C. The matrix was washed (buffer: 50 mM NaH2PO4, 300 mM NaCl pH 8.0, 10% glycerol, 0.8% CHAPS; 1× PI, 100 mM imidazole), and Usp27x was eluted (same buffer plus 250 mM imidazole) and concentrated. Concentrated protein was dialysed. Protein concentration was estimated by NanoDrop.
In vitro translation of 1xHA‐tagged murine Usp27x/Usp27xC87A
In vitro translation was done using PureExpress protein synthesis kit (New England Biolabs Cat. #E6800) using pPE‐1xHA‐mUsp27x or pPE‐1xHA‐mUsp27xC87A (250 ng vector, each) for 2 h at 37°C. Activity assays were directly done with soluble protein.
Catalytic activity assay of hUsp27x
K48‐linked, K63‐linked di‐ubiquitin and Usp2core protein were from Life Sensors (Cat. #SI4802, #SI6302, #DB501). 0.1 μM of Usp2core and 2.9 μM of 6xHis‐Sumo‐hUsp27x were incubated with 1 μg di‐ubiquitin in a buffer consisting of 25 mM NaH2PO4, 150 mM NaCl/pH 8.0, 10% glycerol, 10 mM DTT, 0.05% CHAPS in a total volume of 20 μl for 1 h at 37°C. Cleavage of di‐ubiquitin molecules into monomeric ubiquitin was analysed by SDS–PAGE and Coomassie Blue staining. To measure quantitative activity, we used Internally Quenched Fluorescence – Diubiquitin (IQF‐DiUb) substrates (a dequenching assay, Life Sensors). Six K48‐ and K63‐linked IQF substrates labelled at different position were tested (K48‐1‐2‐3‐4‐5‐6 or K63‐1‐2‐3‐4‐5‐6; Cat. #DU0201; three different quencher positions and two TAMRA positions yield six different diubiquitins). For Fig EV3C, the best IQF‐diUb (namely DiUbK48‐6 (cat. #DU4806) and DiUbK63‐6 (Cat. #DU6306) were used.
Generation of Bim−/− and Usp27xKO cell lines using CRISPR‐Cas9
For CRISPR design, we used a web‐based server (http://crispr.mit.edu/). One gRNA against Bim (5′‐GACAATTGCAGCCTGCGGAGAGG‐3′; Bim2KO) was used targeting a region in exon2 (ENST00000393256) present in all three Bim isoforms. A second used gRNA (5′‐AATCCTGAAGGCAATCACGGAGG‐3′; Bim1KO) targets specifically only BimEL. gRNA against human Usp27x (5′‐TCTTAAACCGATCGTAAAGCTGG‐3′; Usp27x‐1, targets a sequence starting 234 nt downstream from the ATG start (antisense‐strand)) and (5′‐GTGAGATGTCGTCGCTGTTTCGG; Usp27x‐4; target sequence 371 bp downstream from the ATG start (sense‐strand)). LentiCRISPR v2 (a kind gift from Dr. Feng Zhang 52; Addgene plasmid # 52961) was used for cloning the gRNAs. Cells (HCC827, 1205Lu‐TetR‐GFP‐Usp27x, HCC827‐TetR‐GFP‐Usp27x and 293FT‐TetR‐3xFlag‐Usp27x) were transduced with CRISPR‐Cas9‐lentivirus and cultivated for at least 4 weeks in the presence of puromycin. 1205Lu‐TetR‐GFP‐Usp27x, HCC827‐TetR‐GFP‐Usp27x and HEK‐293FT‐TetR‐3xFlag‐Usp27x cells were already resistant to puromycin and could not be selected. Individual clones of 293FT‐TetR‐3xUsp27x/Bim2KO were obtained by serial dilution. To obtain the 293FT‐Usp27xKO (clone 2/10) cell line, a two‐vector‐based system where gRNA‐Usp27x‐4 was cloned into MLM3636 was used. The Cas9 expression vector (JDS246) was transfected together with MLM3636 and a GFP vector into 293FT, and cells were sorted for high‐GFP‐positive cells. Single cells were seeded into a 96‐well plate. Deletion of Usp27x was verified by sequencing.
Gene silencing with RNAi
Sequences of specific siRNAs were as follows: human hUsp27x #495: 5′‐GCAAGGAGAAGCTTTGAAA‐3′, hUsp27x #498: 5′‐AGGAGAAGCTTTGAAATTA‐3′; hUsp27x #861: 5′‐TCATGTGCCCTATAAGTTA‐3′; siCo3 (control and protocol see 53). si β‐TrCP: 5′‐GUGGAAUUUGUGGAACAUC‐3′ 22.
Western blotting
Antibodies used for Western blotting were as follows: β‐TrCP (D13F10, #4394), Bcl‐XL (54H6, #2764), Bim (C34C5, #2933), phospho‐Bim (Ser69)‐specific antibody (D7E11, #4585), Flag (#2368), GFP (D5.1, #2956), phospho‐p44/42 MAPK (Erk1/2, #4370), HA‐tag antibody (C29F4, #3724), which were obtained from Cell Signaling. Flag (M2 clone, F1804), and tubulin (both Sigma), GAPDH (C6C5, Millipore), Usp27x (Abgent, does only recognize overexpressed Usp27x and not endogenous Usp27x in our hands). All antibodies were used as suggested by the manufacturers. Signals were detected using horseradish peroxidase‐conjugated secondary antibodies (anti‐mouse (Dianova) or anti‐rabbit (Sigma) IgG) and enhanced chemiluminescence (GE Healthcare).
Bim stability assay
Stability of endogenous BimEL was analysed in inducible 1205Lu‐GFP‐Usp27x and HCC827‐GFP‐Usp27x cells without or with dox for 24 h followed by addition of cycloheximide (1 μg/ml, CHX, Sigma) for the indicated time points. In the inducible 293FT‐TetR‐3xFlag‐Usp27x cells, Bim was destabilized using PMA (16.2 nM) followed by cycloheximide treatment (10 μg/ml) for 2–4 h.
In situ proximity‐ligation assay
Proximity‐ligation assay (PLA) was performed to detect the interaction between USP27X and endogenous Bim using the Duolink in situ kit (OLINK, Sweden). For this, 293FT cells with dox‐inducible 3x‐Flag‐Usp27x were seeded on poly‐L‐lysine (0.1%, Sigma‐Aldrich)‐coated glass coverslips in 24‐well cell culture plates. After 24 h of dox treatment, cells were fixed using 4% PFA, permeabilized with 0.1% Triton X‐100/PBS and followed by incubation with 5% BSA/PBS. Staining was done with α‐Bim (1:200) and α‐Flag (1:800, Roche, M2 clone, F1804) in 5% BSA/0.5% saponin/PBS. Thereafter, incubation with oligonucleotide‐conjugated secondary antibodies, ligation and amplification were performed according to the manufacturer's instructions. Images were acquired in a Keyence BZ‐9000 fluorescence microscope under similar conditions of magnification and exposure time (excitation and emission wavelengths were 554 nm and 579 nm).
Immunofluorescence
1205Lu cells were seeded in 24‐well plates onto coverslips, and GFP, GFPUsp27x or GFP‐Usp22 was induced by dox for 24–48 h. For microscopy, cells were incubated with 100 nM Mitotracker Deep Red FM (Invitrogen) for 30 min before fixing the cells in 3.7% formalin for 15 min. Samples were stained with Hoechst 33342 (1 μg/ml, Sigma) for 10 min before being mounted in Permafluor (Thermo Fisher). The samples were analysed with a BZ 9000E microscope (Keyence), and photographs were processed using the BZ II Analyzer software 1.42 (Keyence). For intracellular staining of Bim, cells were fixed using 4% PFA, permeabilized with 0.1% Triton X‐100/PBS and blocked with 5% BSA/PBS. Staining with primary antibody was done for 1 h with α‐Bim (1:100) in 5% BSA/0.5% saponin/PBS. Cells were washed and incubated for 1 h in fluorescently labelled secondary antibody. Cell nuclei were stained with Hoechst as above for 10 min, and the cover slips were mounted in Permafluor medium. Images were acquired as above.
Detection of apoptosis
Cell lines were seeded in 6‐well plates (2 × 105 cells) and stimulated for 24 h as indicated. Inducible 1205Lu melanoma cells were seeded in 6‐well plates and treated with dox to induce GFP‐Usp27x or GFP‐Usp27xC87A (not shown). Forty‐eight hours later, cells were treated with or without UO126 (10 μM) for additional 48 h or vemurafenib (1 μM) for 24 h. Inducible HCC827‐GFP‐Usp27x cells or cells transduced with CRISPR‐Cas9 lentivirus targeting Bim (HCC827‐GFP‐Usp27x Bim2 KO) or targeting only BimEL (HCC827‐GFP‐Usp27x Bim1 KO) were treated for 72 h with dox alone or with gefitinib (10 μM). HCC827 cells deficient for Usp27x were treated for 48 h with gefitinib.
Cells were washed in PBS, fixed and stained with monoclonal anti‐active caspase‐3 antibody (Abcam, dilution 1:500 or BD Pharmingen 1:500) or with an antibody specific for active Bax (6A7 clone, Sigma, dilution 1:300). Flow cytometry was performed using a FACSCalibur (Becton Dickinson).
Statistics/quantification
For statistical analysis, two‐tailed Student's t‐test was used to assess the significance of mean values. Bars show mean values of all experiments (± SEM). Differences were considered significant at a P‐value of 0.05 or less. Quantification of the shown immunoblots was done using the LabImage1D (INTAS) software.
Author contributions
AW, JD and GH conceived and designed the experiments. AW, MH, CA, PKS and JD performed the experiments. AW, MH, JD and GH analysed the data. AW and GH wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
This work was supported by a grant from the Deutsche Krebshilfe, Dr. Mildred Scheel Stiftung, to A.W. and G.H. (#111480).
EMBO Reports (2016) 17: 724–738
References
- 1. O'Reilly LA, Cullen L, Visvader J, Lindeman GJ, Print C, Bath ML, Huang DC, Strasser A (2000) The proapoptotic BH3‐only protein bim is expressed in hematopoietic, epithelial, neuronal, and germ cells. Am J Pathol 157: 449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A (1999) Proapoptotic Bcl‐2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286: 1735–1738 [DOI] [PubMed] [Google Scholar]
- 3. Mestre‐Escorihuela C, Rubio‐Moscardo F, Richter JA, Siebert R, Climent J, Fresquet V, Beltran E, Agirre X, Marugan I, Marin M et al (2007) Homozygous deletions localize novel tumor suppressor genes in B‐cell lymphomas. Blood 109: 271–280 [DOI] [PubMed] [Google Scholar]
- 4. Zantl N, Weirich G, Zall H, Seiffert BM, Fischer SF, Kirschnek S, Hartmann C, Fritsch RM, Gillissen B, Daniel PT et al (2007) Frequent loss of expression of the pro‐apoptotic protein Bim in renal cell carcinoma: evidence for contribution to apoptosis resistance. Oncogene 26: 7038–7048 [DOI] [PubMed] [Google Scholar]
- 5. Sturm I, Stephan C, Gillissen B, Siebert R, Janz M, Radetzki S, Jung K, Loening S, Dorken B, Daniel PT (2006) Loss of the tissue‐specific proapoptotic BH3‐only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ 13: 619–627 [DOI] [PubMed] [Google Scholar]
- 6. Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves‐Neira W, Bouillet P, Villunger A, Adams JM, White E (2005) Key roles of BIM‐driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell 7: 227–238 [DOI] [PubMed] [Google Scholar]
- 7. Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, Tenen DG, Kobayashi S (2007) BIM mediates EGFR tyrosine kinase inhibitor‐induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med 4: e315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A (2007) Gefitinib‐induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med 4: e316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gong Y, Somwar R, Politi K, Balak M, Chmielecki J, Jiang X, Pao W (2007) Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR‐dependent lung adenocarcinomas. PLoS Med 4: e294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Egle A, Harris AW, Bouillet P, Cory S (2004) Bim is a suppressor of Myc‐induced mouse B cell leukemia. Proc Natl Acad Sci USA 101: 6164–6169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ et al (2013) Bax crystal structures reveal how bh3 domains activate bax and nucleate its oligomerization to induce apoptosis. Cell 152: 519–531 [DOI] [PubMed] [Google Scholar]
- 12. Leshchiner ES, Braun CR, Bird GH, Walensky LD (2013) Direct activation of full‐length proapoptotic BAK. Proc Natl Acad Sci USA 110: E986–E995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC (2005) Proapoptotic Bak is sequestered by Mcl‐1 and Bcl‐xL, but not Bcl‐2, until displaced by BH3‐only proteins. Genes Dev 19: 1294–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, Medema RH (2002) The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL‐2. J Immunol 168: 5024–5031 [DOI] [PubMed] [Google Scholar]
- 15. Gilley J, Coffer PJ, Ham J (2003) FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol 162: 613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moustafa‐Kamal M, Gamache I, Lu Y, Li S, Teodoro JG (2013) BimEL is phosphorylated at mitosis by Aurora A and targeted for degradation by betaTrCP1. Cell Death Differ 20: 1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wan L, Tan M, Yang J, Inuzuka H, Dai X, Wu T, Liu J, Shaik S, Chen G, Deng J et al (2014) APC(Cdc20) suppresses apoptosis through targeting Bim for ubiquitination and destruction. Dev Cell 29: 377–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu H (2007) Cdc20: a WD40 activator for a cell cycle degradation machine. Mol Cell 27: 3–16 [DOI] [PubMed] [Google Scholar]
- 19. Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P (2003) Phosphorylation of Bim‐EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene 22: 6785–6793 [DOI] [PubMed] [Google Scholar]
- 20. Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ (2003) Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome‐dependent degradation of the BH3‐only protein, Bim. J Biol Chem 278: 18811–18816 [DOI] [PubMed] [Google Scholar]
- 21. Hubner A, Barrett T, Flavell RA, Davis RJ (2008) Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell 30: 415–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dehan E, Bassermann F, Guardavaccaro D, Vasiliver‐Shamis G, Cohen M, Lowes KN, Dustin M, Huang DC, Taunton J, Pagano M (2009) betaTrCP‐ and Rsk1/2‐mediated degradation of BimEL inhibits apoptosis. Mol Cell 33: 109–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Komander D, Clague MJ, Urbe S (2009) Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 10: 550–563 [DOI] [PubMed] [Google Scholar]
- 24. Grabbe C, Husnjak K, Dikic I (2011) The spatial and temporal organization of ubiquitin networks. Nat Rev Mol Cell Biol 12: 295–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frank DO, Dengjel J, Wilfling F, Kozjak‐Pavlovic V, Hacker G, Weber A (2015) The pro‐apoptotic BH3‐only protein Bim interacts with components of the translocase of the outer mitochondrial membrane (TOM). PLoS One 10: e0123341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobayashi T, Iwamoto Y, Takashima K, Isomura A, Kosodo Y, Kawakami K, Nishioka T, Kaibuchi K, Kageyama R (2015) Deubiquitinating enzymes regulate Hes1 stability and neuronal differentiation. FEBS J 282: 2475–2487 [DOI] [PubMed] [Google Scholar]
- 27. Wilfling F, Weber A, Potthoff S, Vogtle FN, Meisinger C, Paschen SA, Hacker G (2012) BH3‐only proteins are tail‐anchored in the outer mitochondrial membrane and can initiate the activation of Bax. Cell Death Differ 19: 1328–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quesada V, Diaz‐Perales A, Gutierrez‐Fernandez A, Garabaya C, Cal S, Lopez‐Otin C (2004) Cloning and enzymatic analysis of 22 novel human ubiquitin‐specific proteases. Biochem Biophys Res Commun 314: 54–62 [DOI] [PubMed] [Google Scholar]
- 29. Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG et al (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3: 995–1000 [DOI] [PubMed] [Google Scholar]
- 30. Bouillet P, Zhang LC, Huang DC, Webb GC, Bottema CD, Shore P, Eyre HJ, Sutherland GR, Adams JM (2001) Gene structure alternative splicing, and chromosomal localization of pro‐apoptotic Bcl‐2 relative Bim. Mamm Genome 12: 163–168 [DOI] [PubMed] [Google Scholar]
- 31. Ritorto MS, Ewan R, Perez‐Oliva AB, Knebel A, Buhrlage SJ, Wightman M, Kelly SM, Wood NT, Virdee S, Gray NS et al (2014) Screening of DUB activity and specificity by MALDI‐TOF mass spectrometry. Nat Commun 5: 4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang XY, Varthi M, Sykes SM, Phillips C, Warzecha C, Zhu W, Wyce A, Thorne AW, Berger SL, McMahon SB (2008) The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell‐cycle progression. Mol Cell 29: 102–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xiong J, Wang Y, Gong Z, Liu J, Li W (2014) Identification of a functional nuclear localization signal within the human USP22 protein. Biochem Biophys Res Commun 449: 14–18 [DOI] [PubMed] [Google Scholar]
- 34. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W et al (2002) Mutations of the BRAF gene in human cancer. Nature 417: 949–954 [DOI] [PubMed] [Google Scholar]
- 35. Dai DL, Wang Y, Liu M, Martinka M, Li G (2008) Bim expression is reduced in human cutaneous melanomas. J Invest Dermatol 128: 403–407 [DOI] [PubMed] [Google Scholar]
- 36. Sheridan C, Brumatti G, Martin SJ (2008) Oncogenic B‐RafV600E inhibits apoptosis and promotes ERK‐dependent inactivation of Bad and Bim. J Biol Chem 283: 22128–22135 [DOI] [PubMed] [Google Scholar]
- 37. Roring M, Herr R, Fiala GJ, Heilmann K, Braun S, Eisenhardt AE, Halbach S, Capper D, von Deimling A, Schamel WW et al (2012) Distinct requirement for an intact dimer interface in wild‐type, V600E and kinase‐dead B‐Raf signalling. EMBO J 31: 2629–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C (2011) A proteome‐wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics 10: M111 013284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post‐translational modifications in man and mouse. Nucleic Acids Res 40: D261–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Happo L, Strasser A, Cory S (2012) BH3‐only proteins in apoptosis at a glance. J Cell Sci 125: 1081–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mojsa B, Lassot I, Desagher S (2014) Mcl‐1 ubiquitination: unique regulation of an essential survival protein. Cells 3: 418–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brinkmann K, Zigrino P, Witt A, Schell M, Ackermann L, Broxtermann P, Schull S, Andree M, Coutelle O, Yazdanpanah B et al (2013) Ubiquitin C‐terminal hydrolase‐L1 potentiates cancer chemosensitivity by stabilizing NOXA. Cell Rep 3: 881–891 [DOI] [PubMed] [Google Scholar]
- 43. Ewings KE, Hadfield‐Moorhouse K, Wiggins CM, Wickenden JA, Balmanno K, Gilley R, Degenhardt K, White E, Cook SJ (2007) ERK1/2‐dependent phosphorylation of BimEL promotes its rapid dissociation from Mcl‐1 and Bcl‐xL. EMBO J 26: 2856–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Llambi F, Moldoveanu T, Tait SW, Bouchier‐Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR (2011) A unified model of mammalian BCL‐2 protein family interactions at the mitochondria. Mol Cell 44: 517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sowa ME, Bennett EJ, Gygi SP, Harper JW (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138: 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O'Rourke K, Bazan F, Eastham‐Anderson J et al (2010) Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463: 103–107 [DOI] [PubMed] [Google Scholar]
- 47. Perez‐Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, Silverstein KA, Grutzmann R, Aust D, Rummele P et al (2012) The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 486: 266–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Naik E, Webster JD, DeVoss J, Liu J, Suriben R, Dixit VM (2014) Regulation of proximal T cell receptor signaling and tolerance induction by deubiquitinase Usp9X. J Exp Med 211: 1947–1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao Y, Majid MC, Soll JM, Brickner JR, Dango S, Mosammaparast N (2015) Noncanonical regulation of alkylation damage resistance by the OTUD4 deubiquitinase. EMBO J 34: 1687–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weber A, Paschen SA, Heger K, Wilfling F, Frankenberg T, Bauerschmitt H, Seiffert BM, Kirschnek S, Wagner H, Hacker G (2007) BimS‐induced apoptosis requires mitochondrial localization but not interaction with anti‐apoptotic Bcl‐2 proteins. J Cell Biol 177: 625–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M et al (2007) IAP antagonists target cIAP1 to induce TNFalpha‐dependent apoptosis. Cell 131: 682–693 [DOI] [PubMed] [Google Scholar]
- 52. Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weber A, Kirejczyk Z, Besch R, Potthoff S, Leverkus M, Hacker G (2010) Proapoptotic signalling through Toll‐like receptor‐3 involves TRIF‐dependent activation of caspase‐8 and is under the control of inhibitor of apoptosis proteins in melanoma cells. Cell Death Differ 17: 942–951 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File