

Abstract
DNA damage activates the ATM and ATR kinases that coordinate checkpoint and DNA repair pathways. An essential step in homology‐directed repair (HDR) of DNA breaks is the formation of RAD51 nucleofilaments mediated by PALB2–BRCA2; however, roles of ATM and ATR in this critical step of HDR are poorly understood. Here, we show that PALB2 is markedly phosphorylated in response to genotoxic stresses such as ionizing radiation and hydroxyurea. This response is mediated by the ATM and ATR kinases through three N‐terminal S/Q‐sites in PALB2, the consensus target sites for ATM and ATR. Importantly, a phospho‐deficient PALB2 mutant is unable to support proper RAD51 foci formation, a key PALB2 regulated repair event, whereas a phospho‐mimicking PALB2 version supports RAD51 foci formation. Moreover, phospho‐deficient PALB2 is less potent in HDR than wild‐type PALB2. Further, this mutation reveals a separation in PALB2 function, as the PALB2‐dependent checkpoint response is normal in cells expressing the phospho‐deficient PALB2 mutant. Collectively, our findings highlight a critical importance of PALB2 phosphorylation as a novel regulatory step in genome maintenance after genotoxic stress.
Keywords: ATM, ATR, DNA damage response, PALB2 phosphorylation, RAD51
Subject Categories: DNA Replication, Repair & Recombination; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Upon genotoxic stresses, cells activate the multi‐faceted DNA damage response (DDR), which includes signaling pathways that control cell cycle progression as well as DNA repair pathways 1, 2. The DDR is mainly coordinated by the kinases ATM (ataxia‐telangiectasia mutated) and ATR (ATM and Rad3‐related). These kinases are activated by DNA lesions and direct the DDR pathways through phosphorylation of downstream targets such as CHK1, CHK2, CtIP, and p53 3, 4.
The DDR maintains genome stability, thereby supporting cellular fitness, suppressing tumorigenesis, and has major impact on cancer therapy responses 1, 5, 6. A particularly relevant setting is hereditary breast cancer, where several DDR genes are germ line mutated leading to a familial cancer predisposition. Among the mutated genes are the HDR factors BRCA2 and PALB2 that are found mutated in familial breast, pancreatic, and ovarian cancers 7, 8, 9, 10. PALB2 and BRCA2 interact together, and PALB2 supports BRCA2 localization and stability to promote RAD51 loading and HDR 11, 12, 13, 14. Recently, ATM has also emerged as a breast cancer tumor suppressor, suggesting a link between ATM and the PALB2–BRCA2 pathway 10.
ATM and ATR are important cell cycle checkpoint regulators in the DDR, and intriguingly, BRCA2 and PALB2 were recently identified as checkpoint factors that block cell cycle progression in G2 phase following DNA damage by ionizing radiation 15, 16. As part of the DDR, the G2 checkpoint blocks cell proliferation by preventing mitotic entry after DNA damage, thereby prohibiting transmission of damaged DNA to daughter cells 17, 18. Notably, BRCA2 and PALB2 appear to function in checkpoint maintenance as they are not involved in the immediate activation of ATM and ATR 16. It remains to be understood how the function of BRCA2 and PALB2 in checkpoint control is integrated with DDR signaling and DNA repair by HDR.
Homology‐directed repair constitutes a key DNA double‐strand break (DSB) repair pathway together with non‐homologous end joining (NHEJ) 19. NHEJ is a fast process with the ability to repair DSBs throughout the cell cycle. HDR is an essentially error free multi‐step repair process, which is restricted to S and G2 phases due to the presence of an intact sister chromatid 19, 20. HDR requires processing of DNA ends by nucleolytic enzymes, a mechanism called DNA end resection, which is initiated by the collaborative action of MRE11 and CtIP in an ATM‐ and ATR‐dependent manner 21, 22, 23, 24. The exonucleases EXO1 and DNA2 then resect the 5′‐end to generate long stretches of single‐stranded DNA (ssDNA) immediately coated by the trimeric DNA‐binding protein complex RPA 24. RPA is exchanged with RAD51 to form the RAD51 nucleofilament through the action of the PALB2–BRCA2 complex 25, 26, 27, 28, 29, 30. Finally, the nucleofilament mediates the homology search and strand invasion on the sister chromatid, which is followed by post‐synapsis where RAD51 dissociates from the DNA ends to allow final steps including DNA synthesis 19.
The roles of ATM and ATR in HDR following DNA end resection are poorly understood. Here, we have investigated the interplay between ATM/ATR, PALB2, and RAD51 in checkpoint control and HDR. We show that ATM and ATR promote RAD51 function by mediating PALB2 phosphorylation, which is required for RAD51 foci formation. However, these phosphorylation events are dispensable for PALB2‐dependent checkpoint control. Our data provide evidence for a regulatory link between the tumor suppressor functions of ATM/ATR and PALB2–BRCA2 in maintaining genome stability.
Results and Discussion
DNA damage induces ATM/ATR‐dependent phosphorylation of PALB2
To further investigate the role of PALB2 in the DNA damage response, we explored the occurrence of potential post‐translational modification of PALB2. We treated U2OS cells with ionizing radiation (IR) and collected samples at different recovery time points. Western blot analysis revealed that 2 h after IR treatment PALB2 could be detected as a diffuse band with retarded mobility compared to control sample. The shifted appearance became more prominent at later recovery time points up to 24 h post‐IR, indicating increased post‐translational modification of PALB2 (Fig 1A). To test whether the retarded mobility is due to phosphorylation of PALB2, we treated lysate from the 24 h time point with phosphatase and monitored the PALB2 signal (Fig 1A). Phosphatase treatment completely reverted PALB2 to the lower migrating band, implying that PALB2 becomes phosphorylated after IR.
Figure 1. PALB2 phosphorylation is induced upon DNA damage in an ATM/ATR‐dependent manner.

- U2OS cells were harvested at the indicated time points after treatment with ionizing radiation (15 Gy) followed by analysis by SDS–PAGE and immunoblotting with antibodies recognizing PALB2 and vinculin (NT, no treatment). The lysate from the 24 h time point was treated with phosphatase (Ppase).
- U2OS cells were left untreated or treated with ATM (KU 55933, 10 μM), ATR (ETP‐464, 1 μM), or DNA‐PK (NU7441, 1 μM) inhibitors 30 min prior to exposure to IR (15 Gy, 2 h recovery). Immunoblots were performed with the indicated antibodies.
- U2OS cells were left untreated (NT) or exposed to HU (2 mM) for the indicated times. The lysate from the 24 h time point was treated with phosphatase (Ppase). Immunoblots were performed with the indicated antibodies.
- Untreated U2OS cells or cells exposed to either HU (2 mM, 24 h) or IR (15 Gy, 2 h recovery) alone or together with ATM inhibitor (KU55933, 10 μM) or inhibitors to ATR (ATR#1: ETP‐464, 1 μM; ATR#2: AZ‐20, 3 μM) were harvested and lysates were prepared. The inhibitors were added 30 min prior to HU or IR treatment. Immunoblots were performed with the indicated antibodies.
- ATR kinase assay was performed with N‐terminal PALB2 (aa 1‐560; GST–PALB2–N (Fig 2A)) as a substrate and HA‐tagged ATR as kinase and visualized by Fujifilm Phosphroimager. 293T cells transfected with empty vector (EV), GST, and ATR inhibition (AZ‐20, 30 μM) are controls. HA‐ATR and GST–PALB2–N were controlled by immunoblotting with HA and GST antibodies.
The primary phosphorylation cascades after DNA damage are carried out by the ATM/ATR kinases 3, 4. To explore whether ATM or ATR is responsible for phosphorylating PALB2, we treated cells with ATM or ATR inhibitors prior to exposure to IR (Fig 1B) 31, 32. Subjecting cells to either ATM or ATR inhibitor decreased PALB2 phosphorylation to levels observed in non‐irradiated cells. In contrast, inhibiting the activity of DNA‐PK, an ATM‐related kinase active in NHEJ did not markedly affect the IR‐induced phosphorylation of PALB2. These results suggest that ATM and ATR mediate phosphorylation of PALB2 in response to DNA damage. This finding is in line with a recent report by Guo et al (published while this manuscript was under review)33, showing ATM/ATR‐dependent phosphorylation of PALB2 upon IR as well as a previous proteome‐wide analysis identifying 3 potential ATM/ATR target sites in the N‐terminus of PALB2 by mass spectrometry 34.
While ATM is mainly activated by double‐strand breaks caused by DNA‐damaging agents such as IR, ATR is activated in response to single‐stranded DNA containing lesions 3. Such lesions are very prominent following DNA replication stress, which can be induced by agents such as hydroxyurea (HU). HU depletes the cellular nucleotide pool 35, 36, and this leads to replication fork stalling and eventually to DNA breaks 3. To test whether PALB2 phosphorylation is induced by replication stress, we treated cells with HU for increasing time and analyzed PALB2 mobility on Western blot. The phosphorylation of PALB2 was induced after 8 h of HU being sustained during treatment until 24 h (Fig 1C). Unlike IR‐induced PALB2 phosphorylation, the HU‐induced phosphorylation was less sensitive to ATM inhibition while retained sensitivity to ATR inhibition (Fig 1D). This result suggests that following replication stress PALB2 is predominantly phosphorylated in an ATR‐dependent manner, which is further supported by sustained phosphorylation of PALB2 in cells depleted of ATM by siRNA compared to ATR siRNA (Fig EV1A). Furthermore, purified N‐terminal version of PALB2 (aa 1‐560) was phosphorylated by ATR in vitro (Fig 1E). Altogether, our results indicate that in response to DNA perturbation PALB2 phosphorylation is mediated by the checkpoint kinases ATM and ATR. Furthermore, both IR and HU‐induced phosphorylation of PALB2 could be detected in the human colorectal carcinoma cell line HCT116 and human breast epithelial cell line MCF10a implying that PALB2 phosphorylation is part of a general genotoxic stress response (Fig EV1B).
Figure EV1. Analysis of ATM/ATR‐dependent PALB2 phosphorylation in U2OS, HCT116 and MCF10a cells.

- U2OS cells were transfected with UNC (negative control), ATM or ATR siRNA and 48 h later left untreated or treated with HU (2 mM, 24 h, left panel) or IR (15 Gy, 2 h recovery, right panel). The cell lysates were analyzed by SDS–PAGE and Western blotting with PALB2, ATM, ATR, and vinculin antibodies.
- HCT116 and MCF10a cells were left untreated (NT) or treated with IR (15 Gy, 2 h recovery) or HU (2 mM for 7 h). The lysates were subsequently treated with phosphatase and analyzed by SDS–PAGE. Immunoblotting was performed with PALB2 and vinculin antibodies.
- Immunoprecipitation of the PALB2 cell lines with pS/Q antibody was performed as in Fig 2D. Cells were either left untreated (NT) or exposed to IR (15 Gy, 2 h recovery) or treated with ATM (KU55933, 1 μM) and ATR (AZ‐20, 3 μM) inhibitors 30 min before exposure to IR (15 Gy, 2 h recovery). The values under the IP blot show relative band intensities in the IP samples normalized to the expression levels of the input samples. The non‐treated WT sample was arbitrarily set to one, and the densitometry was performed using ImageJ/Fiji.
ATM/ATR mediates phosphorylation of serine residues in the PALB2 N‐terminus
PALB2 interacts with a number of proteins that are essential for the HDR pathway, such as BRCA1, BRCA2, and RAD51 37. The N‐terminal PALB2 contains coiled‐coil motifs that interact with BRCA1 whereas the C‐terminus forms a WD40‐type β‐propeller that mediates the interaction with BRCA2 and RAD51 (Fig 2A) 12, 13, 14. Additionally, there is an interaction site with RAD51 in the N‐terminus of PALB2 25, 26. The human PALB2 sequence contains seven serine residues with the ATM/ATR‐specific S/Q motif. Guo et al 33 recently found two sites, S157 and S376, to be targeted by ATM/ATR in response to DNA damage. However, the mass spectrometry analysis performed by Matsuoka et al 34 identified three N‐terminal S/Q motifs phosphorylated upon IR. We mutated these serine residues, S59, S157, and S376, to alanine to obtain phosphorylation impaired mutant (triple mutant S‐A, TMA‐PALB2) or to aspartic acid for a phosphorylation mimicking mutant (triple mutant S‐D, TMD‐PALB2). These constructs were used to generate doxycycline‐inducible cell lines expressing siRNA‐resistant FLAG/HA‐tagged wild‐type (WT) PALB2 or the respective phosphorylation mutants (Fig 2B). Similar to WT PALB2, both the TMA and the TMD mutants formed foci upon IR treatment of these cell lines (Fig 2C). To test whether PALB2 phosphorylation is dependent on the three S/Q sites, we performed the in vitro ATR kinase assay with purified N‐terminal (aa 1‐560) WT and TMA‐PALB2. TMA‐PALB2 was poorly phosphorylated, implying that the three N‐terminal S/Q sites are targeted by ATR (Fig 2D). Moreover, we investigated PALB2 phosphorylation in the WT and TMA cell lines after IR. Cell lysates were immunoprecipitated with phospho‐S/Q antibody recognizing phosphorylated S/Q residues on ATM/ATR substrates. Detection of exogenous PALB2 with the HA antibody revealed IR‐induced phosphorylation of S/Q sites on WT PALB2 (Fig 2E, compare lanes 3 and 4), which was decreased in the presence of ATM and ATR inhibitors (Fig EV1C). Notably, TMA‐PALB2 was less phosphorylated than WT PALB2 (Fig 2E, compare lanes 4 and 5). Collectively, our results indicate that ATM/ATR‐mediated PALB2 phosphorylation largely, although not completely, depends on the three N‐terminal S/Q sites.
Figure 2. Extensive PALB2 phosphorylation after DNA damage requires three N‐terminal ATM/ATR consensus target sites.
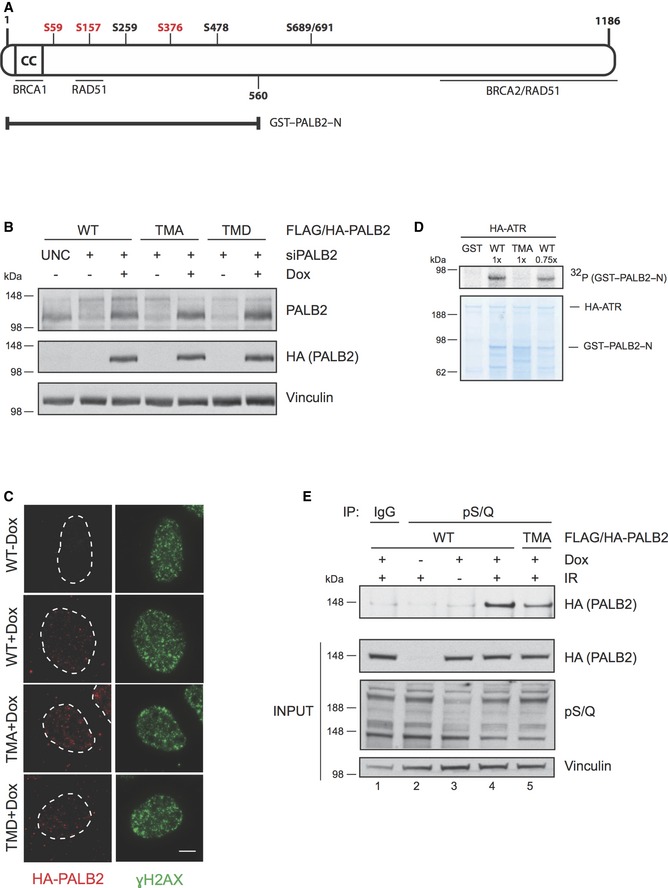
- Schematic figure of the human PALB2 protein. The N‐terminus of PALB2 contains a coiled‐coil region (CC) that mediates the interaction between PALB2 and BRCA1. PALB2 interacts with BRCA2 via the C‐terminal WD40 domain, which also harbors interaction sites for RAD51. An additional region for RAD51 interaction is located in the N‐terminus at amino acid residues 101–184. PALB2 has seven potential S/Q phosphorylation sites of which serines S59, S157, and S376 (depicted in red) were mutated either to alanine (triple mutant S to A, TMA) to create a phospho‐deficient mutant, or aspartic acid (triple mutant S to D, TMD) for a phospho‐mimicking mutant. GST–PALB2–N (amino acids 1‐560) contains the N‐terminal serines.
- Characterization of the doxycycline‐inducible cell lines expressing siRNA‐resistant wild‐type (WT) or mutant (TMA or TMD) FLAG/HA‐tagged PALB2. The cell lines were transfected with UNC (negative control) or PALB2 siRNA for 24 h prior to induction with doxycycline. Immunoblots were performed with the indicated antibodies.
- Cell line characterization of FLAG/HA‐tagged PALB2 localization 2 h following exposure to 15 Gy of IR. Z‐stack max intensity image projection and background subtraction was done using ImageJ/Fiji. Scale bar = 5 μm.
- Kinase assay was performed as described in Fig 1E except that the SDS–PAGE gel was Coomassie stained following electrophoresis. WT was used at two different concentrations (1× and 0.75×).
- Doxycycline‐inducible stable U2OS cell lines were left untreated or treated with doxycycline and/or IR (15 Gy, 2 h recovery). Following protein extraction, the lysates were immunoprecipitated (IP) with IgG or antibody recognizing phosphorylated S/Q sites (pS/Q) and analyzed by SDS–PAGE. Immunoblots were performed with the indicated antibodies.
Phosphorylation of PALB2 supports RAD51 foci formation
Homology‐directed repair is utilized to mend DNA DSBs that appear in S and G2 phases of the cell cycle. PALB2 is critical for the recruitment of both BRCA2 and RAD51 to the sites of damage during HDR 37. To address the functional requirement of PALB2 phosphorylation in HDR, we examined the extent of RAD51 foci formation in the PALB2 cell lines following DNA damage. Two hours post‐IR, the formation of RAD51 foci was significantly impaired in cells expressing the TMA construct of PALB2 compared to cells expressing the wild‐type PALB2 (Fig 3A). Further, similar data on the TMA mutant were obtained on RAD51 foci formation in response to HU‐induced DNA lesions (Fig 3B). Importantly, the phospho‐mimetic version of PALB2, TMD, was largely competent in RAD51 foci formation. Furthermore, expression of a single site S59A mutant or a double site S157/376A mutant supported formation of RAD51 foci (Fig EV2A). However, abrogating the phosphorylation of all three sites reduced RAD51 foci. This result is in line with the study by Guo et al 33, where the RAD51 foci formation was found to be unaffected in the presence of the S157/376 mutant. Finally, we ensured that the observed effect on RAD51 foci formation did not reflect a clonal difference between the cell lines (Fig EV2B).
Figure 3. PALB2 phosphorylation promotes RAD51 foci formation.

- PALB2 cell lines transfected with PALB2 siRNA followed by inducible expression of siRNA‐resistant PALB2 versions were exposed to 15 Gy of IR and fixed for IF 2 h later. Z‐stack images were acquired with a 60× oil objective (Deltavision). Z‐stack max intensity projection, background subtraction, and foci count were done using ImageJ/Fiji (*P < 0.0001, unpaired Student's t‐test). Scale bar = 5 μm.
- PALB2 cell lines were transfected and the exogenous PALB2 induced as in Fig 3A before they were pulsed with 10 μM EdU for 20 min prior to addition of 2 mM HU. Twenty‐four hours later, the cells were fixed and processed for IF as in (A). Cells in S phase (EdU+) at the time of HU treatment were Click‐IT labeled with an Alexa Fluor 647 azide and RAD51 foci in EdU‐positive cells were enumerated using ImageJ/Fiji (*P < 0.0001, unpaired Student's t‐test). Scale bar = 5 μm.
- HeLa DR‐GFP cell line was used to analyze homologous recombination activity. Cells were transfected with PALB2 siRNA followed by transfection of I‐SceI together with empty vector (EV), WT, TMA‐, or TMD‐PALB2 the next day. 72 h later cells were fixed and analyzed by FACS. The percentage of cells with GFP signal from the empty vector sample was set to one (dotted line). The graph shows a representative image from three biological repeats (error bars = SEM).
- Phosphorylation of PALB2 supports RAD51 foci. Cells were transfected with siPALB2 overnight followed by forward transfection of siFBH1/siBLM for 6 h then induced for expression of PALB2 (WT and TMA). 24 h following induction cells were pulsed with EdU and exposed to HU as in (B). Cells in S phase (EdU+) at the time of HU treatment were Click‐IT labeled with an Alexa Fluor 647 azide and RAD51 foci in EdU‐positive cells were enumerated using ImageJ/Fiji (*P < 0.0001, unpaired Student's t‐test). Representative images displaying RAD51 (green) and HA‐PALB2 (red) localization in HU‐treated EdU‐positive (not shown) cells. Scale bar = 5 μm.
Figure EV2. Analysis of the role of PALB2 phosphorylation in RAD51 foci formation.

- Influence of PALB2 phosphorylation mutants on RAD51 foci formation following exposure to IR. U2OS cells were siRNA transfected to knock down endogenous PALB2, then transfected with FLAG‐HA WT, S59A, S157/376A, or TMA–PALB2. Twenty‐four hours following construct transfection cells were irradiated with 15 Gy of IR and fixed and processed for IF 2 h later. Transfected cells were identified by HA antibody reactivity and scored for RAD51 foci (WT n = 53, S59A n = 54, S157/376A n = 52, TMA n = 51) (P‐values: *P = 0.0257, **P = 0.0029, ***P = 0.0003, unpaired Student's t‐test). Transient expression of the PALB2 constructs was confirmed by Western blot probing for the HA tag. Scale bar = 5 μm.
- Proficiency of clonal PALB2 cell lines to form RAD51 foci in response to DNA damage. Cell lines were assessed for RAD51 foci formation without siRNA transfection and transgene induction. Cells were exposed to IR (15 Gy) or HU (2 mM) and processed for RAD51 foci as in Fig 3A and B, respectively. Representative images are shown. Scale bar = 5 μm.
PALB2 promotes the localization of RAD51 to sites of HR both directly and through interaction with BRCA2. As such, the need for PALB2 phosphorylation in RAD51 foci formation may reflect altered interaction with BRCA2 and/or its direct interaction with RAD51. To this end, we observed reduced interaction between RAD51 and the TMA‐PALB2 following exposure to IR (Fig EV3A), while the interaction of the TMA mutant with BRCA2 was maintained (Fig EV3B). Further, the TMD‐PALB2 mutant retained interaction with RAD51 comparable to WT PALB2. These results indicate that following DNA damage, the ATM/ATR‐mediated phosphorylation of PALB2 promotes its interaction with RAD51, and this could at least in part account for the stimulation of RAD51 repair foci. In addition, the TMA mutant was not able to stimulate the HDR activity to the same extent as WT PALB2 in a DR‐GFP reporter assay, suggesting that PALB2 phosphorylation promotes HDR (Fig 3C). Moreover, the phosphorylation mutants interacted with BRCA1 to the same degree as WT PALB2, indicating that PALB2 phosphorylation does not markedly affect the formation of the BRCA1–PALB2–BRCA2 complex (Fig EV3C).
Figure EV3. Analysis of interactions between PALB2 versions and key HDR proteins, as well as immunoblots upon siRNA depletion of BLM and FBH1.
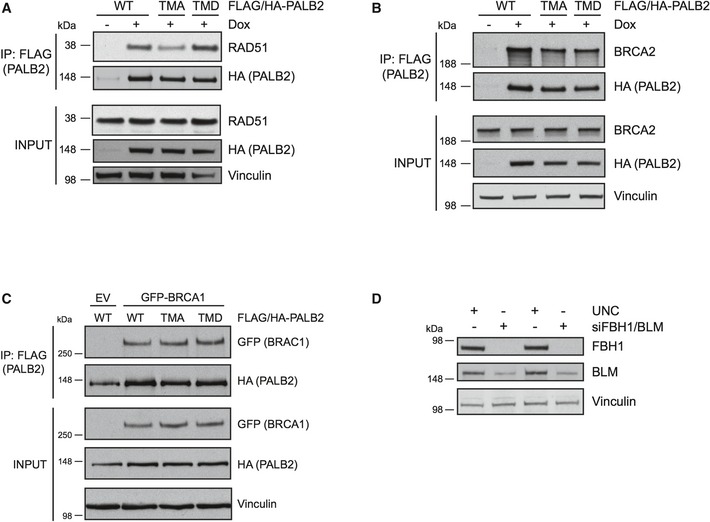
- The PALB2 cell lines were untreated or treated with doxycycline followed by exposure to IR treatment (15 Gy, 2 h recovery). The exogenous FLAG/HA‐tagged PALB2 was immunoprecipitated with FLAG‐agarose and the IP and input samples were run on SDS–PAGE. The membranes were blotted with RAD51 and HA antibodies and vinculin was used as a loading control.
- The cells were treated and analyzed as in (A). Immunoblotting was performed with BRCA2, HA, and vinculin antibodies.
- 293T cells were transfected with empty vector (EV) or GFP‐BRCA1 together with FLAG‐/HA‐tagged WT, TMA, or TMD PALB2. Immunoprecipitation was performed using FLAG‐agarose, and the IP and input samples were probed with GFP and HA antibodies. Vinculin was used as a loading control.
- Representative immunoblot for Fig 3D showing depletion of FBH1 and BLM. The cell lysates were analyzed by Western blotting and probed with FBH1, BLM, and vinculin antibodies.
Although loading of RAD51 could be affected by PALB2 phosphorylation, we hypothesized that unstable RAD51 nucleoprotein filaments might underlie the TMA‐PALB2 phenotype. To explore this further, we took advantage of the fact that RAD51 filaments can be disrupted by helicases such as the F‐box helicase 1 (FBH1) 38. In addition, helicases as Bloom syndrome protein (BLM) play a major role in resolving HDR intermediates after HU 39. Thus, co‐depleting FBH1 and BLM allow estimation of the ability of TMA‐PALB2 cells to form RAD51 foci after exposure to HU (Figs 3D and EV3D). While the lack of FBH1 and BLM only marginally affected RAD51 foci formation in WT PALB2 cells, the RAD51 foci were fully restored in the TMA cells. This result indicates that PALB2 phosphorylation plays a role in maintaining the RAD51 filaments. Similar to WT PALB2, the TMA mutant is localized to the RAD51 foci formed after depletion of FBH1 and BLM (Fig 3D, right panel).
PALB2 phosphorylation maintains genome stability
In addition to its role in HR, PALB2 maintains the G2 checkpoint following DNA damage 15, 16. However, it is unclear whether the checkpoint function of PALB2 is related to its function in HDR. To address the potential requirement for PALB2 phosphorylation in the G2 checkpoint, we examined the ability of the PALB2 mutants to restore the checkpoint. Expression of either the TMA‐ or TMD‐PALB2 could restore checkpoint control following IR as efficiently as WT PALB2 (Fig 4A, left panel). This result indicates that N‐terminal S/Q phosphorylation of PALB2 is dispensable for G2 checkpoint control. Moreover, this finding reveals a potential separation of function for PALB2 between checkpoint control and the RAD51 foci formation required for HR.
Figure 4. PALB2 phosphorylation promotes genome maintenance.

- U2OS and PALB2 cell lines were transfected with UNC (negative control) or PALB2 siRNA and 24 h later doxycycline was added to the PALB2 cell lines. Left panel, cells were treated the following day with IR (5 Gy) and 2 h later nocodazole was added for 6 h, or treated with only nocodazole for 6 h. Right panel, cells were treated with IR (5 Gy) and left to recover for 16 h. Cells were fixed and stained with the pMPM2 antibody to detect the mitotic cells by flow cytometry. The percentages of mitotic cells in the IR + nocodazole samples were normalized to the nocodazole samples (left panel, n = 3, error bars = SEM). For the samples with the 16‐h recovery after IR, the percentage of mitotic cells in the WT PALB2‐expressing cells was set to one (right panel) (n = 3, **P < 0.005, *P < 0.05, unpaired Student's t‐test, error bars = SEM).
- The relative intensity of phosphorylated H2AX (pS139) was examined in the total population of WT and TMA‐PALB2‐expressing cell lines 18 h post‐exposure to IR (5 Gy). Cell lines were prepared as above, fixed at the indicated time point, and stained for pH2AX. Cells were imaged with a 20× air objective on a Scan^R workstation (Olympus); mean relative pH2AX intensity was calculated from background subtracted images using the Scan^R analysis software (*P < 0.0001, unpaired Student's t‐test, error bars = SEM).
- Number of 53BP1 nuclear bodies in TMA‐PALB2‐expressing G1 daughter cells. Cell lines were prepared as above and left untreated (NT) or exposed to IR (3 Gy), and 16 h later cytochalasin B was added for 8 h to block cytokinesis. The irradiated binucleated cells were scored for 53BP1 nuclear bodies. Scale bar = 10 μm.
Once the genome is repaired, cells reenter the cell cycle via a process termed checkpoint recovery 17. The need for PALB2 N–terminal S/Q phosphorylation to promote RAD51 repair foci suggests that cells with disturbed PALB2 phosphorylation may encounter difficulties in completing DNA repair in a timely manner. To this end, we noted impaired checkpoint recovery following IR in cells expressing TMA‐PALB2 compared to WT or TMD‐PALB2 (Fig 4A, right panel). This corresponded to elevated levels of the DNA damage marker phosphorylated H2AX in TMA‐PALB2 expressing cells compared to WT cells within this recovery period (Fig 4B). The elevated levels of phosphorylated H2AX may reflect increased number of unrepaired DNA breaks that delay checkpoint recovery. Because cells with impaired DNA repair can move through the G2 checkpoint under a threshold of DNA breaks, we investigated genome stability in TMA‐PALB2 cells that recovered from the G2 checkpoint. To distinguish the G1 daughter cells that originated from recovered G2 cells, we blocked cytokinesis in recovering IR‐treated cells, trapping the daughter cells in a binuclear state. Examining the extent of 53BP1 nuclear bodies, a DNA damage marker 40, in these daughter cells revealed that TMA‐PALB2 expressing cells contained higher numbers of 53BP1 nuclear bodies compared to WT PALB2 cells (Figs 4C and EV4). This result suggests that TMA‐PALB2 cells escaping the G2 checkpoint can proceed through mitosis with unrepaired DNA breaks. These breaks are transferred to the daughter cells, indicating that PALB2 need to be appropriately phosphorylated for genome maintenance.
Figure EV4. Identification of binuclear cells with actin as marker.

Actin staining of binuclear cells presented in Fig 4C. Actin and 53BP1 were visualized by immunostaining of samples prepared as described in Fig 4C. Scale bar = 10 μm.
Collectively, our results indicate that phosphorylation of the N‐terminal S/Q sites of PALB2 is required to maintain genome stability by efficient repair of DNA damage. The phosphorylation of these residues (S59, S157, and S376) is mediated by ATM and ATR upon exposure to genotoxic insults. Phosphorylation‐deficient PALB2 is inefficient in promoting RAD51 foci, leading to impaired DNA repair and genome instability. Notably, ATM is an emerging breast cancer tumor suppressor, and based on our data, we now propose that ATM can suppress tumor development by promoting HDR through PALB2‐mediated phosphorylation.
Materials and Methods
Cells and reagents
The human osteosarcoma cell line (U2OS), human embryonic kidney 293T cells, and HeLa cell line harboring DR‐GFP were grown in Dulbecco's modified Eagle's medium with 10% FBS. The human colorectal carcinoma cell line HCT116 was cultured in McCoy's 5A media containing 10% FBS, and the human breast epithelial MCF10a cells were grown in Dulbecco's modified Eagle's medium with F12 nutrient mixture and supplemented with 5% horse serum, EGF (100 mg/ml), hydrocortisone (1 mg/ml), cholera toxin (1 mg/ml), and insulin (10 mg/ml). All media also contained 1% PenStrep antibiotics.
The doxycycline‐inducible stable U2OS cell lines expressing the Flag/HA‐tagged siRNA‐resistant versions of wild‐type and mutant PALB2 were established by cloning PALB2 cDNA into pcDNA4/TO‐FLAG‐HA vector (Invitrogen). The phosphorylation‐deficient and mimicking versions of PALB2 were generated by site‐directed mutagenesis of the siRNA‐resistant WT PALB2 by mutating S59, S157, and S376 to alanine residues (TMA) or aspartic acid residues (TMD), respectively. The following primers were used for the TMA mutation: S59A FW 5′‐GAA GAA CAA GAT TGT TTG GCT CAG CAG GAT CTC TCA C‐3′ and RV 5′‐GTG AGA GAT CCT GCT GAG CCA AAC AAT CTT GTT CTT C‐3′, S157A FW 5′‐GCA GCA GAA GAG GAC ATT TAT TGC ACA GGA GAG AG‐3′ and RV 5′‐CTC TCT CCT GTG CAA TAA ATG TCC TCT TCT GCT GC‐3′, S376A FW 5′‐TGA AAA TCT TCA GGA AAG TGA GAT TCT AGC TCA ACC TAA GAG TCT TAG C‐3′ and RV 5′‐GCT AAG ACT CTT AGG TTG AGC TAG AAT CTC ACT TTC CTG AAG ATT TTC A‐3′. For the TMD mutation, the following primers were used: S59D FW 5′‐GTA GAA GAA CAA GAT TGT TTG GAT CAG CAG GAT CTC TCA CCG C‐3′ and RV 5′‐GCG GTG AGA GAT CCT GCT GAT CCA AAC AAT CTT GTT CTT CTA C‐3′, S159D FW 5′‐AGA AAG AAG CAG CAG AAG AGG ACA TTT ATT GAT CAG GAG AGA GAC TGT G‐3′ and RV 5′‐CAC AGT CTC TCT CCT GAT CAA TAA ATG TCC TCT TCT GCT GCT TCT TTC T‐3′, S376D FW 5′‐GGA AAG TGA GAT TCT AGA TCA ACC TAA GAG TCT TAG C‐3′ and RV 5′‐GCT AAG ACT CTT AGG TTG ATC TAG AAT CTC ACT TTC C‐3′. The PFU ultra‐high‐fidelity polymerase (Agilent) was used according to the manufacturer's protocol.
The generated PALB2 plasmids were then co‐transfected with pcDNA6‐TR (Invitrogen) expressing tetracycline repressor into U2OS cells using GenJet transfection reagent (SignaGen Laboratories) according to the manufacturer's protocol resulting in cell lines expressing FLAG/HA‐tagged siRNA‐resistant PALB2 WT, TMA, or TMD in a Tet‐on system. To induce expression of siRNA‐resistant PALB2, doxycycline (1 ng/ml) was added to the medium for approximately 24 h.
The GST‐tagged truncated versions of PALB2 harboring amino acids 1‐560 were generated by PCR using the following primers FW 5′‐ATG GAC GAG CCT CCC GGG A‐3′ and RV 5′‐TCA TTG AAT AAA TAA TTT TTC GTG CTG‐3′. The PCR product was inserted into the pGEX6 vector for recombinant protein production.
For siRNA transfections (48 h), Lipofectamine RNAiMAX (Invitrogen) was used according to the manufacturer's protocol. MISSION® siRNA universal negative control (UNC, Sigma) was used as a negative control, and the oligonucleotide sequences used for knockdown of PALB2 and BLM are 5′‐CUUAGAAGAGGACCUUAUU[dT][dT] and 5′‐GGAAGUUGUAUGCACUACC[dT][dT], respectively. A mix of two sequences 5′‐GGGAUGUUCUUUUGAUAAA[dT][dT] and 5′‐CCAUCCAACUUACACAUGA[dT][dT] was used for depleting FBH1.
To transfect 293T cells with expression plasmids, the calcium phosphate transfection method was used.
Hydroxyurea (Sigma) was used at a final concentration of 2 mM for the indicated time. The ATM inhibitor (KU55933, Tocris Bioscience) was used at final concentration of 10 μM. ATR (ETP‐464, Millipore) and DNA‐PK (NU7441, Tocris Bioscience) inhibitors were used at a final concentration of 1 μM, and ATR inhibitor (AZ‐20, Tocris Bioscience) was used at a final concentration of 3 μM. Furthermore, cytochalasin B was used at a concentration of 1 μg/ml. The indicated doses of ionizing radiation (IR) were given using a Faxitron X‐ray apparatus.
Immunoblotting and antibodies
Briefly, cells were snap‐frozen and lysed in cold EBC‐buffer (150 mM NaCl, 50 mM Tris pH 7.4, 1 mM EDTA, 0.5% NP‐40) containing protease inhibitors (1% vol/vol aprotinin, 5 μg/ml leupeptin, 1 mM PMSF), phosphatase inhibitors (1 mM NaF, 10 mM β‐glycerophosphate), and 1 mM DTT. The lysates were sonicated followed by centrifugation at 20,000 g for 10 min. The soluble proteins were then separated by SDS–PAGE and transferred to a nitrocellulose membrane. For detecting the phosphorylated PALB2, the samples were resolved on 8% polyacrylamide gels (Figs 1A–D, 2B and EV1A and B) where the unphosphorylated PALB2 runs at a size of ~100 kDa. Otherwise, samples were resolved on gradient 4–12% Bis‐Tris gels (Life Technologies) where FLAG/HA‐PALB2 runs at a size of ~150 kDa. The membranes were blocked in 5% milk, incubated with primary antibody diluted in 5% milk, washed, and incubated with secondary HRP‐conjugated antibody (1:10,000; Vector Laboratories) diluted in 5% milk. ECL‐mix (GE Healthcare) was used to visualize proteins on X‐ray film. The following primary antibodies were used for immunoblotting: phospho‐(Ser/Thr) ATM/ATR substrate (1:500; 4F7, 2909, Cell Signaling), BLM (1:5,000, 476, Abcam), BRCA2 (1:2,000; Ab‐1, Calbiochem), phospho‐CHK1‐S317 (1:1,500; 2344, Cell Signaling), CHK1 (1:10,000; DCS‐310; described by 41, phospho‐CHK2‐T68 (1:500; 2661, Cell Signaling), CHK2 (1:500; sc56296, Santa Cruz), FBH1 (FBXO18 1:100; sc81563, Santa Cruz), GST (1:2,000; sc138, Santa Cruz), GFP (1:1,000; 1181446000, Roche), HA (1:2,000; MMS‐101P, Covance), PALB2 (1:1,000; A301‐246A, Bethyl Laboratories), vinculin (1:50,000; V9131, Sigma).
Immunoprecipitation
Extracts for immunoprecipitation were prepared using immunoprecipitation buffer (50 mM Hepes, pH 7.5, 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 10% glycerol, 0.1% Tween) with protease inhibitors. Following preclearing with IgG‐coupled protein G beads (GE Healthcare), the lysates were incubated with protein G beads coupled to phospho‐S/Q antibody (1:20; 2851, Cell Signaling) or M2 FLAG‐agarose (Sigma) for 2 h at 4°C on a rotator. The beads were washed five times followed by elution of bound proteins in Laemmli sample buffer.
Immunofluorescence
Cells grown on coverslips were treated as indicated and prepared for immunofluorescence as previously described 42. Primary antibodies used were actin (1:1,000; MAB1501, Millipore), phospho‐H2AX S139 (1:500; 2577, Cell Signaling), 53BP1 (1:1,000; sc22760, Santa Cruz), HA (1:500; MMS‐101P, Covance), and RAD51 (1:1,000, 70‐001, BioAcademia Jpn). Anti‐mouse Alexa Fluor 488, anti‐mouse Alexa Fluor 594, anti‐rabbit Alexa Fluor 488, anti‐rabbit Alexa Fluor 594 (1:2,000, A21202, A21203, R37118, and A21206, Life Technologies) were used as secondary antibodies. For RAD51, immunofluorescence cells were pre‐extracted twice for 3 min in CSK buffer (0.5% Triton X‐100, 20 mM Hepes pH 7.4, 100 mM NaCl, 3 mM MgCl2, and 300 mM sucrose) followed by fixation in 4% formaldehyde. Cells were permeablized in 0.5% Triton X‐100 followed by incubation in blocking buffer (1% BSA, 0.15% glycine, 0.1% Triton X‐100 in PBS++ wash buffer (1× PBS 0.1% Tween‐20, 1 mM CaCl2, 0.5 mM MgCl2)). Primary antibody was incubated for 1 h at room temperature in blocking buffer, followed by three washes with PBS++ wash buffer. Secondary antibody was incubated for an additional hour, washed 3× with PBS++ wash buffer, and nuclei counterstained with DAPI. EdU staining was done per manufacturer's instructions (Life Technologies). Z‐stack images were acquired on a Deltavision workstation, and images were processed and foci enumerated using Fiji (ImageJ). For phospho‐H2AX intensity analysis, stained coverslips were imaged and analyzed on the Olympus Scan^R workstation.
Flow cytometry
To prepare cells for flow cytometry, cells were fixed in 70% ethanol. The cells were stained with phospho‐MPM2 antibody (1:200; 05‐368, Millipore) for 1 h followed by 1‐h incubation with anti‐mouse Alexa Fluor 647 (1:1,000; A21236, Life Technologies) secondary antibody. DNA was stained with 0.1 mg/ml propidium iodide (PI) containing RNase (20 μg/ml) for 30 min at 37°C. Flow cytometry was performed on a FACS Calibur (BD Biosciences) using CellQuest Pro software (Becton Dickinson).
GST purification of recombinant proteins
GST–PALB2–N (WT and TMA) and GST were expressed in E. coli Rosetta (DE3) using 0.2 mM IPTG for induction at 37°C for 3 h. Harvested cells were lysed in GST buffer (20 mM Tris pH 7.5, 200 mM NaCl, 0.5% NP‐40) with protease inhibitors and sonicated. Following centrifugation at 20,000 g for 20 min., the cleared lysates were incubated with glutathione Sepharose beads (GE Healthcare), washed three times with 50 column volumes of GST buffer and eluted with 5 column volumes 50 mM Tris pH 7.5 containing 1 mM reduced glutathione. Finally, the samples were dialyzed to remove the glutathione and stored at −80°C.
Kinase assay
For the kinase assay, 293T cells transfected with empty vector (EV) or HA‐ATR were lysed in TGN buffer (150 mM NaCl, 50 mM Tris pH 7.4, 10% glycerol, 1% Tween‐20) containing protease inhibitors and phosphatase inhibitors (1 mM NaF, 50 mM β‐glycerophosphate, 1 mM sodium vanadate) and 1 mM DTT. The lysates were sonicated followed by centrifugation at 20,000 g for 10 min. Immunoprecipitation of HA‐ATR was performed by incubating the lysates with HA‐agarose (Sigma) for 2 h at 4°C on a rotator. The beads were washed three times with TGN buffer and two times with TGN buffer supplemented with 500 mM LiCl and finally two times with kinase buffer (10 mM Hepes pH 7.5, 50 mM NaCl, 50 mM β‐glycerophosphate, 10 mM MgCl2, 1 mM DTT). The HA‐ATR binding beads were incubated with GST‐PALB2‐N in a reaction mix containing kinase buffer, 1 mM DTT, 50 mM β‐glycerophosphate, 150 μM ATP and 10 mM MnCl2. 10 μCi [γ‐32P]‐ATP was added to the mix and the reaction was incubated at 30°C for 40 min with gentle shaking. Laemmli sample buffer was added, and the samples were resolved with SDS–PAGE. The gel was either stained with InstantBlue Coomassie stain (Expedeon) and dried, or the resolved proteins were transferred to a nitrocellulose membrane. The radioactive signal was detected using Fujifilm Phosphorimager.
Homologous recombination assay
The homologous recombination assay was performed essentially as described previously 43. HeLa cells expressing the integrated homologous recombination reporter DR‐GFP were transfected with PALB2 siRNA followed by transfection of an I‐SceI expression vector (pCBA‐I‐SceI) together with empty vector (EV) or siRNA‐resistant WT, TMA, or TMD PALB2 24 h later. After 72 h, cells were collected and fixed in 0.5% formaldehyde. The percentage of GFP‐positive cells was analyzed by flow cytometry using CellQuest Pro software (Becton Dickinson). The HeLa DR‐GFP cell line and the I‐SceI expression vector were kind gifts from Douglas NP Oliveira.
Author contributions
CSS and JKA conceived the project. JKA, BDL, and KA performed experiments and acquired data. All authors analyzed, interpreted, and concluded on results. The manuscript was written by CSS and JKA with contributions from BDL.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Markus Löbrich for critical comments to the manuscript, Valdemaras Petrosius for purifying PALB2, and Peter Haahr for advice on ATR assay. This work was supported by The Novo Nordisk Foundation (CSS), The Danish Cancer Society (CSS), The Lundbeck Foundation (CSS), the Danish Medical Research Council (CSS), and Sigrid Juselius Foundation (JKA).
EMBO Reports (2016) 17: 671–681
References
- 1. Jackson SP, Bartek J (2009) The DNA‐damage response in human biology and disease. Nature 461: 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thompson LH (2012) Recognition, signaling, and repair of DNA double‐strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat Res 751: 158–246 [DOI] [PubMed] [Google Scholar]
- 3. Marechal A, Zou L (2013) DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5: a012716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shiloh Y, Ziv Y (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14: 197–210 [PubMed] [Google Scholar]
- 5. Dobbelstein M, Sorensen CS (2015) Exploiting replicative stress to treat cancer. Nat Rev Drug Discov 14: 405–423 [DOI] [PubMed] [Google Scholar]
- 6. Khanna A (2015) DNA damage in cancer therapeutics: a boon or a curse? Cancer Res 75: 2133–2138 [DOI] [PubMed] [Google Scholar]
- 7. Erkko H, Xia B, Nikkila J, Schleutker J, Syrjakoski K, Mannermaa A, Kallioniemi A, Pylkas K, Karppinen SM, Rapakko K et al (2007) A recurrent mutation in PALB2 in Finnish cancer families. Nature 446: 316–319 [DOI] [PubMed] [Google Scholar]
- 8. Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T et al (2007) PALB2, which encodes a BRCA2‐interacting protein, is a breast cancer susceptibility gene. Nat Genet 39: 165–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Couch FJ, Nathanson KL, Offit K (2014) Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science 343: 1466–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shiovitz S, Korde LA (2015) Genetics of breast cancer: a topic in evolution. Ann Oncol 26: 1291–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM (2006) Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 22: 719–729 [DOI] [PubMed] [Google Scholar]
- 12. Sy SM, Huen MS, Chen J (2009) PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA 106: 7155–7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang F, Fan Q, Ren K, Andreassen PR (2009) PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res 7: 1110–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, Yu X (2009) PALB2 links BRCA1 and BRCA2 in the DNA‐damage response. Curr Biol 19: 524–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cotta‐Ramusino C, McDonald ER III, Hurov K, Sowa ME, Harper JW, Elledge SJ (2011) A DNA damage response screen identifies RHINO, a 9‐1‐1 and TopBP1 interacting protein required for ATR signaling. Science 332: 1313–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Menzel T, Nahse‐Kumpf V, Kousholt AN, Klein DK, Lund‐Andersen C, Lees M, Johansen JV, Syljuasen RG, Sorensen CS (2011) A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. EMBO Rep 12: 705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lobrich M, Jeggo PA (2007) The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer 7: 861–869 [DOI] [PubMed] [Google Scholar]
- 18. Medema RH, Macurek L (2012) Checkpoint control and cancer. Oncogene 31: 2601–2613 [DOI] [PubMed] [Google Scholar]
- 19. Moynahan ME, Jasin M (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 11: 196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Symington LS, Gautier J (2011) Double‐strand break end resection and repair pathway choice. Annu Rev Genet 45: 247–271 [DOI] [PubMed] [Google Scholar]
- 21. Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP (2007) Human CtIP promotes DNA end resection. Nature 450: 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takeda S, Nakamura K, Taniguchi Y, Paull TT (2007) Ctp1/CtIP and the MRN complex collaborate in the initial steps of homologous recombination. Mol Cell 28: 351–352 [DOI] [PubMed] [Google Scholar]
- 23. Peterson SE, Li Y, Wu‐Baer F, Chait BT, Baer R, Yan H, Gottesman ME, Gautier J (2013) Activation of DSB processing requires phosphorylation of CtIP by ATR. Mol Cell 49: 657–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Symington LS (2014) End resection at double‐strand breaks: mechanism and regulation. Cold Spring Harb Perspect Biol 6: a016436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buisson R, Dion‐Cote AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, Stasiak A, Xia B, Masson JY (2010) Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol 17: 1247–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dray E, Etchin J, Wiese C, Saro D, Williams GJ, Hammel M, Yu X, Galkin VE, Liu D, Tsai MS et al (2010) Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat Struct Mol Biol 17: 1255–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jensen RB, Carreira A, Kowalczykowski SC (2010) Purified human BRCA2 stimulates RAD51‐mediated recombination. Nature 467: 678–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu J, Doty T, Gibson B, Heyer WD (2010) Human BRCA2 protein promotes RAD51 filament formation on RPA‐covered single‐stranded DNA. Nat Struct Mol Biol 17: 1260–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thorslund T, McIlwraith MJ, Compton SA, Lekomtsev S, Petronczki M, Griffith JD, West SC (2010) The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single‐stranded DNA. Nat Struct Mol Biol 17: 1263–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shahid T, Soroka J, Kong EH, Malivert L, McIlwraith MJ, Pape T, West SC, Zhang X (2014) Structure and mechanism of action of the BRCA2 breast cancer tumor suppressor. Nat Struct Mol Biol 21: 962–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC (2004) Identification and characterization of a novel and specific inhibitor of the ataxia‐telangiectasia mutated kinase ATM. Cancer Res 64: 9152–9159 [DOI] [PubMed] [Google Scholar]
- 32. Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR, Fernandez‐Capetillo O (2011) A cell‐based screen identifies ATR inhibitors with synthetic lethal properties for cancer‐associated mutations. Nat Struct Mol Biol 18: 721–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guo Y, Feng W, Sy SM, Huen MS (2015) ATM‐dependent Phosphorylation of the Fanconi Anemia Protein PALB2 Promotes the DNA Damage Response. J Biol Chem 290: 27545–27556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y et al (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316: 1160–1166 [DOI] [PubMed] [Google Scholar]
- 35. Neuhard J (1967) Studies on the acid‐soluble nucleotide pool in Escherichia coli. IV. Effects of hydroxyurea. Biochim Biophys Acta 145: 1–6 [DOI] [PubMed] [Google Scholar]
- 36. Krakoff IH, Brown NC, Reichard P (1968) Inhibition of ribonucleoside diphosphate reductase by hydroxyurea. Cancer Res 28: 1559–1565 [PubMed] [Google Scholar]
- 37. Prakash R, Zhang Y, Feng W, Jasin M (2015) Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol 7: a016600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karpenshif Y, Bernstein KA (2012) From yeast to mammals: recent advances in genetic control of homologous recombination. DNA Repair (Amst) 11: 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Colavito S, Prakash R, Sung P (2010) Promotion and regulation of homologous recombination by DNA helicases. Methods 51: 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Panier S, Boulton SJ (2014) Double‐strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15: 7–18 [DOI] [PubMed] [Google Scholar]
- 41. Sørensen CS, Hansen LT, Dziegielewski J, Syljuåsen RG, Lundin C, Bartek J, Helleday T (2005) The cell‐cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 7: 195–201 [DOI] [PubMed] [Google Scholar]
- 42. Klein DK, Hoffmann S, Ahlskog JK, O'Hanlon K, Quaas M, Larsen BD, Rolland B, Rosner HI, Walter D, Kousholt AN et al (2015) Cyclin F suppresses B‐Myb activity to promote cell cycle checkpoint control. Nat Commun 6: 5800 [DOI] [PubMed] [Google Scholar]
- 43. Pierce AJ, Hu P, Han M, Ellis N, Jasin M (2001) Ku DNA end‐binding protein modulates homologous repair of double‐strand breaks in mammalian cells. Genes Dev 15: 3237–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File