

Abstract
Retrotransposons, the ancestors of retroviruses, have the potential for gene disruption and genomic takeover if not kept in check. Paradoxically, although host cells repress these elements by multiple mechanisms, they are transcribed and are even activated under stress conditions. Here, we describe a new mechanism of retrotransposon regulation through transcription start site (TSS) selection by altered nucleosome occupancy. We show that Fun30 chromatin remodelers cooperate to maintain a high level of nucleosome occupancy at retrotransposon‐flanking long terminal repeat (LTR) elements. This enforces the use of a downstream TSS and the production of a truncated RNA incapable of reverse transcription and retrotransposition. However, in stressed cells, nucleosome occupancy at LTR elements is reduced, and the TSS shifts to allow for productive transcription. We propose that controlled retrotransposon transcription from a nonproductive TSS allows for rapid stress‐induced activation, while preventing uncontrolled transposon activity in the genome.
Keywords: chromatin remodeling, retrotransposable elements, transcriptional regulation
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Transcription
Introduction
Eukaryotic genomes are packaged in a complex DNA–protein structure called chromatin. In addition to compacting and protecting the genome, chromatin serves as a versatile regulatory platform. Nucleosomes, which consist of 147 base pairs of DNA wrapped around a histone protein octamer, are the fundamental repeating units of chromatin. Nucleosome positioning and posttranslational modifications to histones alter DNA accessibility and, consequently, transcription levels. In addition to the effects of nucleosome position and composition, chromatin modifications can attract or repel effector proteins for further impact on transcription and other processes (reviewed in 1).
Retrotransposable elements are genomic parasites that are both key drivers of evolution and the ancestors of retroviruses 2, 3, 4, 5. These elements can stably integrate into the host cell genome and replicate via an RNA intermediate. In addition to exogenous retroviral invaders, the human genome is host to several families of endogenous retroviruses (ERVs). Novel insertion events can abrogate gene function and cause disease. In addition, since supporting a high parasitic load is energetically demanding, cellular control over transposition is imperative. Host cells have developed numerous silencing mechanisms to repress these elements, including DNA methylation, RNA interference (RNAi), repressive histone modifications, and ATP‐dependent chromatin remodeling 6, 7, 8. For example, in budding yeast the mobility of the Ty1 retrotransposon is restricted by the activity of many host factor (RTT) genes, including several chromatin and transcription factors (reviewed in 9), and in plants, small RNA play key roles in silencing of retrotransposons (reviewed in 10). However, evidence from yeast to mammals shows that stressed cells permit transposition 11, 12, 13, 14, 15, 16, even targeting insertion to coding regions 17. For example, the budding yeast Ty5 retrotransposons normally target heterochromatin for insertion 18, 19. However, stressed host cells eliminate this targeting, allowing Ty5 to act as a powerful mutagen 17. It is also becoming clear that retrotransposons may be involved in plasticity 20, 21 and tissue‐specific gene regulation in mammals, often serving as promoters to coding genes and functional ncRNA 22, 23, 24. Thus, it is clear that regulation of retroelements demands a more sophisticated regulatory program than the simple repression that was once widely assumed.
The common laboratory strain of the fission yeast Schizosaccharomyces pombe is host to one gypsy‐class retrotransposable element, Tf2 25. Fission yeast does not methylate DNA, but represses Tf2 transcription with a combination of histone deacetylation, RNAi, and physical gene clustering mediated by the fission yeast CENP‐B homologs 26, 27, 28, 29. Like retroviruses, Tf2 retrotransposons spread via an RNA intermediate and are flanked by long terminal repeats (LTRs), which serve as both promoter and essential untranslated region (UTR) 30. Although the exact mechanism of retrotransposition varies between groups of LTR retrotransposons, reverse transcription is primed from a primer binding site (PBS) located just downstream of the LTR sequence. In the case of Tf2, the primer itself also comes from the LTR, though other retroelements prime reverse transcription with host nucleic acids, for example, tRNA molecules 30, 31. Thus, the entire transcript, including UTRs, is essential to LTR retroelement propagation.
ATP‐dependent chromatin remodelers are important transcriptional regulators, altering the positioning and composition of nucleosomes and, thus, the accessibility of DNA. Chromatin remodelers are integral to all processes that require access to the DNA molecule, including replication, repair, recombination, and transcription. Helicase‐like DNA translocases of the Snf2 family catalyze the remodeling reaction and are known to function both alone and in complexes. A diverse toolkit of Snf2 remodelers appears to have emerged early in eukaryotic evolution, with the structure and function of remodeler subfamilies being well conserved from yeasts to metazoans 32. The Fun30/SMARCAD1 chromatin remodelers have been implicated in centromeric function 33, 34, DNA double‐strand break repair 35, 36, 37, transcriptional repression 38, maintaining heterochromatin through DNA replication 39, and maintaining chromatin boundaries 40. Fission yeast has three Fun30 subfamily remodelers, Fft1, Fft2, and Fft3, of which only Fft3 is characterized 40, 41.
Here, we describe a novel mechanism of retrotransposon regulation via transcription start site selection by modulation of nucleosome occupancy. It is known from studies of the Ty1 retrotransposon in budding yeast that a shorter transcript, giving rise to a truncated Gag protein is involved in copy number control at the level of virus‐like particle formation 42. Here, we show that, as in the case of integrated HIV sequences, chromatin remodelers in fission yeast are crucial to blocking the transcription of full‐length retrotransposon transcripts. Rather than simply blocking retroelement transcription with a well‐positioned nucleosome, TSS control is part of a versatile strategy to harness LTR‐promoted transcription. This allows retroelements to play a role in the host cell stress response and may have implications for LTR and chromatin involvement in multicellular differentiation and plasticity.
Results
Fun30 chromatin remodelers regulate retrotransposon transcript abundance and length
In our investigation of the Fft2 remodeler, we noticed that Tf2 retrotransposons are the most dramatically upregulated class of genes in an fft2Δ strain (Fig 1A). A ura4+ reporter gene inserted 3′ of Tf2‐11 is also clearly derepressed in an fft2Δ fft3Δ double deletion mutant leading to reduced growth on counterselective FOA plates (Fig EV1). Intrigued, we decided to study the role of this previously uncharacterized chromatin remodeler in retrotransposon regulation. To verify the increased transcription, we performed a Northern blot with a sense strand‐specific riboprobe against the Tf2 elements (Fig 1B). The Northern blot confirmed a strong Tf2 upregulation in fft2Δ and showed an even more dramatic derepression in the double fft2Δ fft3Δ mutant. Tf2 element derepression in fft3Δ is also clear, though weaker than in fft2Δ. A size shift was also suggested by the blot, with the Tf2 mRNA gel mobility being slightly decreased in the single and double mutants (Fig 1B). Next, we performed reverse transcription and qPCR for different regions of the Tf2 mRNA 26 (Fig 1C). In this assay, the different regions of the RNA are detected in different ratios in mutant vs. wt samples. In wt, RNA molecules that contain amplicon 2 are relatively abundant, while amplicon 1 is rare. In fft mutants, however, amplicon 1 is upregulated, leading to a strong ratio increase. This assay thus validated the size shift and suggested that the additional sequence in the mutant samples is at the 5′ end of the transcript (Fig 1C and D). The full 5′UTR of fission yeast Tf1 and Tf2 elements is needed for the reverse transcription stage of the retrotransposon life cycle 30. Interestingly, RNA‐sequencing shows that the full 5′UTR is not transcribed for any of the 13 copies of Tf2 in wild‐type (WT) cells (43; shown in Fig 2B). Our results confirm that in WT cells, mRNA molecules with this extended 5′UTR are relatively rare from the population (Fig 1D). In contrast, transcripts with the full 5′UTR are abundant in Fft2 and Fft3 remodeler mutants, with the double mutant showing over an 80‐fold increase relative to WT. Elevation of Tf2 protein coding RNA relative to wt was more modest: four‐ to sixfold in the single mutants and 11‐fold in the double mutant. 5′ RACE (rapid amplification of cDNA ends) analysis reveals a clear difference between the lengths of the primary RNA species in WT and double fft2Δ fft3Δ mutant cells (Fig 1E). Sequencing the RACE products shows a WT transcript originating from a transcription start site just upstream of the translation start codon. In contrast, the longer mRNA species that appears in the single mutants and dominates in the double is initiated at the beginning of the R sequence of the LTR (Fig 1F). This means that although retrotransposons are transcribed at moderate levels in WT cells, these transcripts are unable to support reverse transcription. In the absence of Fft2 and Fft3, however, transcripts able to prime reverse transcription are abundant.
Figure 1. Tf2 retrotransposons are upregulated in Fft mutants.
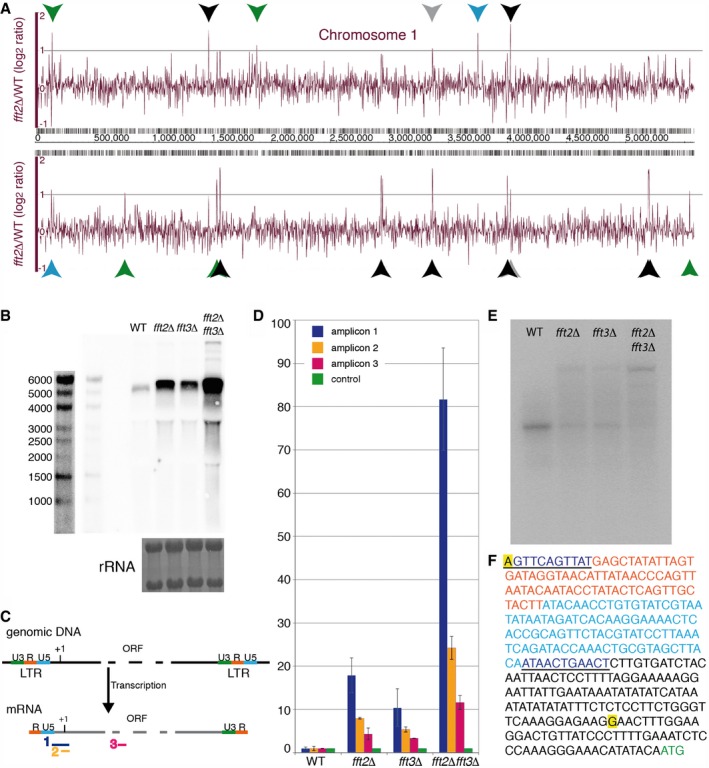
- Transcription in fft2Δ relative to WT over chromosome 1. The top and bottom panels represent fold change in tiling array signal from the forward and reverse strands, respectively. Coordinates are shown in the middle panel; genes more than twofold upregulated are marked: Tf2 retrotransposons (black); antisense to Tf2 elements (gray); coding genes (green); noncoding RNAs (blue).
- Northern blot for the Tf2 ORF; rRNA staining of the same membrane by methylene blue is shown below. Two different exposures of the molecular size marker are shown (left).
- qPCR scheme. Amplicon 1 spans the end of the LTR and into the ORF. Amplicon 2 does not include the LTR but covers the translation start site. Amplicon 3 is entirely within the ORF.
- Bar graph representing RNA levels for the amplicons depicted in (C), relative to a control locus (SPAC1F8.07c) and to WT levels, as measured by qPCR. Error bars represent standard deviation of duplicate reverse transcriptions of biological triplicates.
- 5′ RACE products run on a 1.5% agarose gel.
- The sequence of a Tf2 retrotransposon transcript from the ‘R’ sequence of the upstream LTR to the start codon of the coding region. The long and short RACE products (E) were sequenced, and the transcript start sites are highlighted (yellow). Orange font, R. Light blue font, U5. Dark blue font, self‐primer and primer binding site. Green font, start codon.
Figure EV1. Slow growth phenotype and Tf2 element derepression in fft2Δ fft3Δ cells.
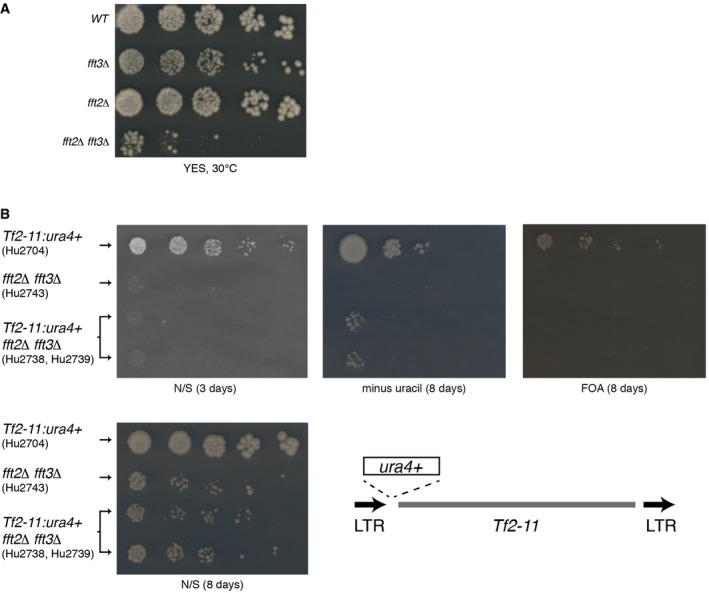
- WT (Hu0029), fft2Δ (Hu1955), fft3Δ (Hu1867), and fft2Δfft3Δ (Hu2000) cells were grown to log phase, spotted on YES media in fivefold serial dilutions, and grown at 30°C.
- Fivefold dilutions of cells grown to log phase were spotted on nonselective (N/S), selective (minus uracil), or counterselective (FOA) media. Plates were incubated at 30°C for 3 days (N/S) or 8 days (minus uracil, FOA). Schematic shows the location of the ura4+ insert, upstream of the Tf2‐11 translation start codon.
Figure 2. Transcription initiation occurs at alternative TSS in the U3 LTR region in Fft mutants.
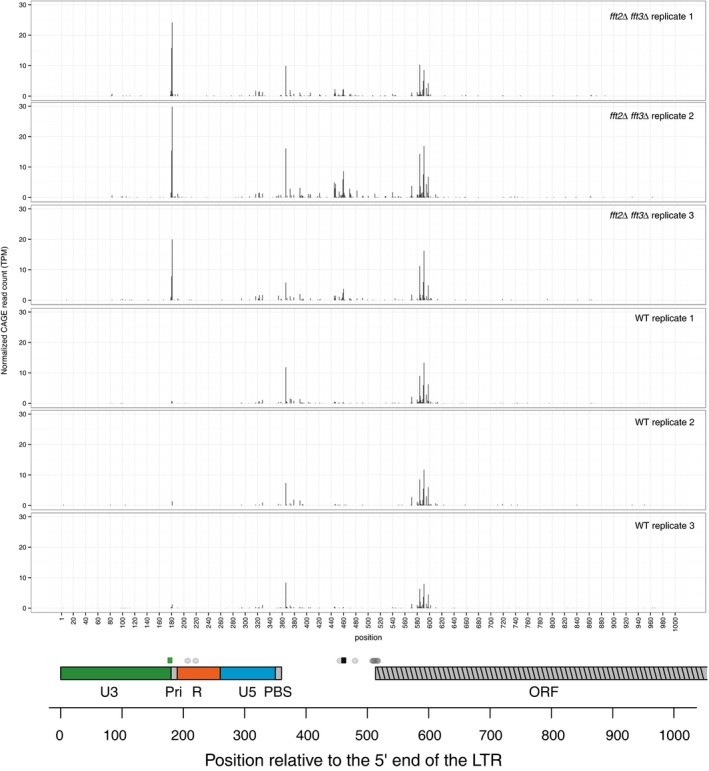
Top panel: Y‐axis shows the CAGE signal intensity (tag per million normalized number of mapped CAGE tags on plus strand). X‐axis shows LTR and 5′ of Tf2 ORF. Three biological replicates are shown for each strain: fft2Δfft3Δ and WT. Bottom: LTR and tf2 ORF are colored: U3 (green), R (red), U5 (blue), self‐primer and primer binding sequence (PBS; gray), beginning of ORF (gray striped). TSS of all Tf2 transcripts from Rhind et al 43 are shown as gray dots above the sequence elements. The black square indicates the 5′ end of the Tf2 mRNA in WT, and the green square indicates the 5′ end in fft2Δ fft3Δ detected by 5′ RACE.
To verify this observation, we sequenced cap‐selected 5′ ends of RNAs (CAGE tags 44) from WT and fft2Δ fft3Δ cells and mapped these tags to the LTR and 5′ of the Tf2 ORF. CAGE tag counts at given TSS are a measure of capped RNA production from that location. Indeed, a strong upstream CAGE peak is only present in fft2Δ fft3Δ cells, and this peak coincides precisely with the RACE‐defined TSS (Fig 2). To validate that the shorter RNA produced in wt cells is capped, we used 5′‐cap‐sensitive RNA ligase‐mediated RACE (RLM‐RACE). A total of 21 wt RLM‐RACE cDNA clones were sequenced. They verified the CAGE results with capped mRNA, with most sequence reads starting in the region around 600 bp relative to the 5′ end of the LTR coinciding with wt CAGE signals (Table EV1; Fig 2). The CAGE signals were highly consistent over three replicates, and since RLM‐RACE uses a different chemistry than CAGE, this confirms that the shorter forms of RNA that dominate in wt are produced from a downstream TSS. There are multiple downstream TSS sites in a region around 600 bp from the 5′ end of the LTR (Table EV1).
Fft2 and Fft3 position a nucleosome to block full‐length retrotransposon transcription
Next, we wondered whether the effect of Fft2 and Fft3 on Tf2 transcript length is direct, as we had previously observed that Fft3 binds LTRs 40 (Fig 3A, bottom). We performed chromatin immunoprecipitation coupled with DNA microarray (ChIP‐chip) analysis of endogenous C‐terminal epitope‐tagged Fft2 and found that Fft2 is localized to both LTR elements and Tf2 coding regions (Fig 3A, top). We were also curious about the complementary roles of the paralogs, as most species only have one Fun30 subfamily remodeler. To dissect this relationship further, we performed ChIP‐chip of Fft2‐myc in an fft3Δ background and of Fft3‐myc in an fft2Δ background. The LTR and Tf2 binding of Fft2 increases in the absence of Fft3 (Fig 3A, top), which in combination with the transcriptional phenotype suggests that these Fft remodelers are able to substitute for one another to some extent.
Figure 3. Fft remodelers bind to and affect nucleosome occupancy at Tf2 LTRs.
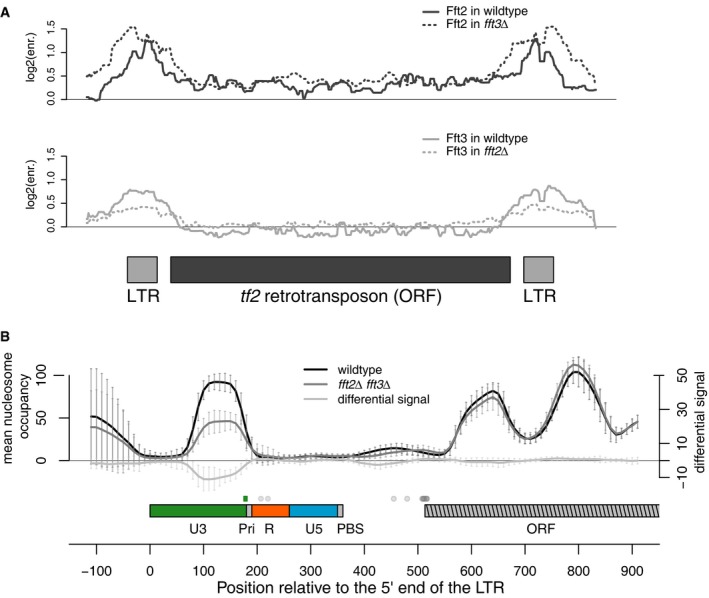
- Fft2 and Fft3 are both enriched at LTRs, while Fft2 is also enriched at the tf2 coding region. Fft2 LTR enrichment increases in fft3Δ cells, while Fft3 LTR enrichment decreases slightly in fft2Δ cells. Top: Fft2‐myc enrichment in WT (solid line) and in fft3Δ (dashed line). Bottom: Fft3‐myc enrichment in WT (solid line) and in fft2Δ (dashed line). ChIP‐chip signal is normalized against no‐epitope control arrays.
- Nucleosome occupancy over the LTR U3 is significantly reduced (average reduction ˜50%; average P‐values at each position < 0.001, Poisson test) in fft2Δ fft3Δ cells. MNase‐seq data aligned at the start of tf2 5′LTRs is shown for WT (black line), and fft2Δ fft3Δ (dark gray line). Light gray line indicates significance of difference between WT and mutant signals: Amplitude reflects significance (P‐value) and sign reflects the sign of occupancy change in mutant vs. WT cells. Error bars show standard deviation of nucleosome occupancy averaged over 13 tf2 elements. LTR and tf2 ORF are colored: U3 (green), R (red), U5 (blue), self‐primer and primer binding sequence (PBS; gray), beginning of ORF (gray striped). TSS of all tf2 transcripts from Rhind et al 43 are shown as gray dots above the sequence elements, while the green dot indicates the TSS in fft2Δ fft3Δ.
To determine whether Fft2 and Fft3 regulate the Tf2 TSS by altering nucleosome occupancy or positioning, we sequenced mononucleosomal DNA fragments generated by micrococcal nuclease (MNase) digestion. We observed an approximately 50% decrease in occupancy of the nucleosome in the LTR U3 region in the fft2Δ fft3Δ double mutant (Fig 3B). This nucleosome is positioned directly upstream of and slightly overlapping the TSS of the longer 5′UTR and presumably prevents transcription initiation at this site in WT cells. In contrast, nucleosome occupancy at this position was substantially reduced, but to a lesser degree in fft3Δ MNase‐seq samples that were prepared in parallel 41. We hypothesize that, as observed at the HIV LTR 8, the LTR U3 sequence appears so refractory to nucleosome formation that this nucleosome is lost in the absence of Fft2/3 remodeling. We also observed an increase in the transcription‐associated histone H3K9 acetylation 5′ of the Tf2 ORF in fft mutants (Fig EV2).
Figure EV2. Transcription‐associated histone mark elevated in fft2 and fft3 mutants.
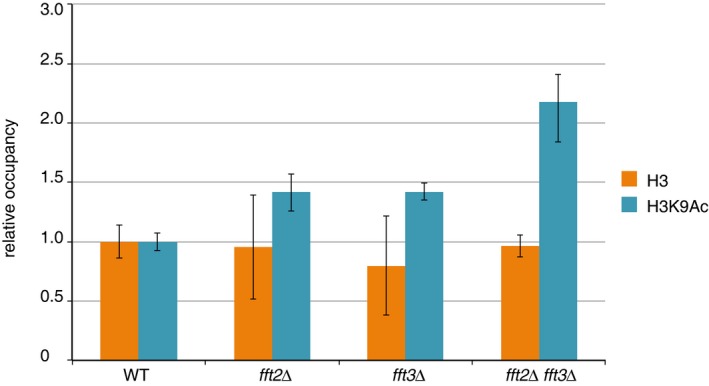
Acetylation of H3K9 is elevated at Tf2 elements in fft2Δ, fft3Δ, and fft2Δ fft3Δ. Error bars represent the standard deviation of biological triplicate samples. Occupancy is relative to a control gene (SPAC1F8.07c) promoter and to WT.
Fft2 and Fft3 position nucleosomes over solo LTRs, reducing transcription of adjacent genes
Next, we asked whether Fft2/3 remodeling at the 261 solo LTR elements scattered throughout the genome could affect the transcription of adjacent genes. Genes with a 5′ LTR are transcribed at the same level as genes without a 5′ LTR (Fig EV3), suggesting that a 5′ LTR is not generally active. While the sequence of many of these LTRs is deteriorated, we nevertheless find a nucleosome occupancy similar to what we observe at Tf2 5′ LTRs. The occupancy of this nucleosome is reduced in fft2Δ fft3Δ cells (Fig 4A). We observe that genes with a 5′ LTR are significantly upregulated in fft2Δ fft3Δ cells (Fig 4B), likely due to increased exposure of the U3 LTR promoter. This is true for both coding and noncoding genes.
Figure EV3. Genes with an upstream LTR are not repressed in WT cells.
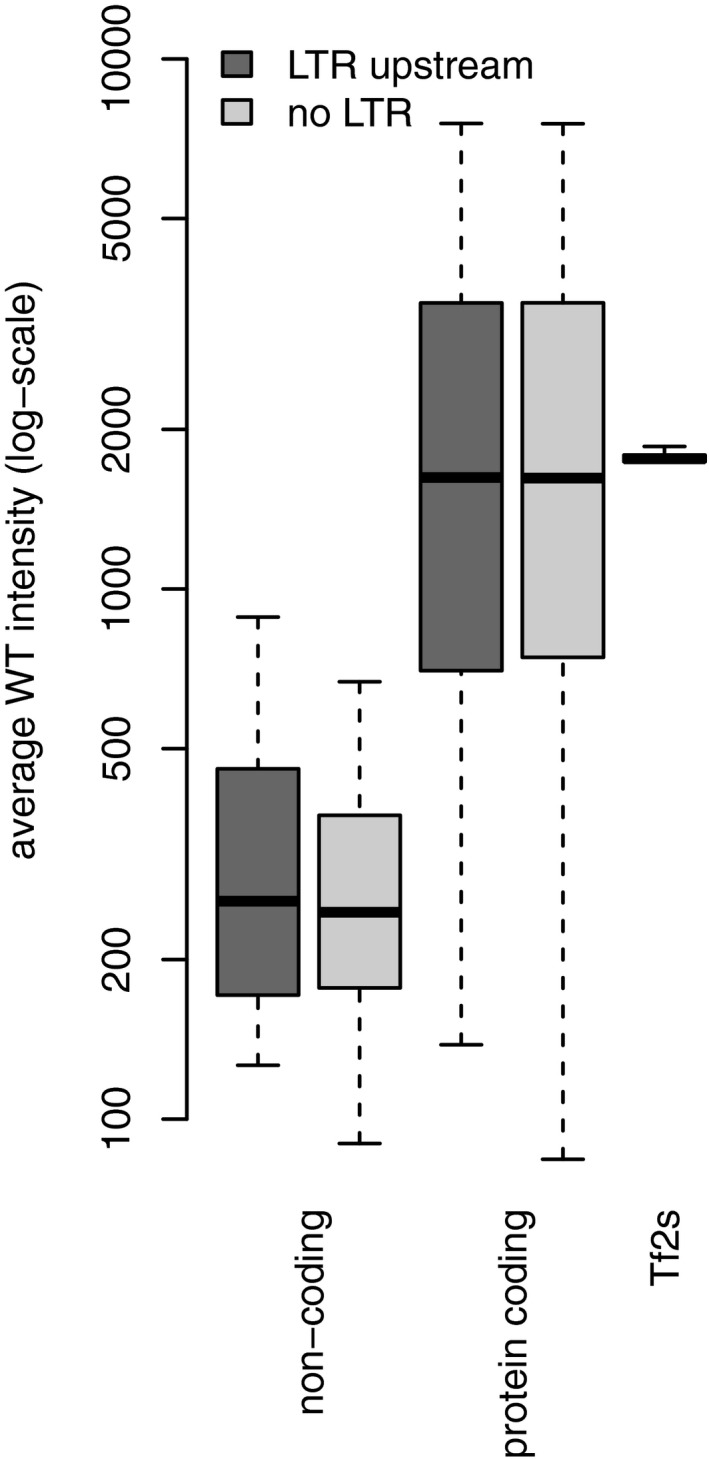
Transcripts with an upstream LTR are neither more nor less abundant than transcripts without an upstream LTR. Average signal intensity across genes of wild‐type RNA hybridized to tiling array shown as box plot for each of the following categories: noncoding with LTR (n = 69)/without LTR (n = 1,665); protein coding with LTR (n = 246)/without LTR (n = 4,887); Tf2s (n = 13).
Figure 4. LTR‐proximal genes are upregulated in fft2Δ fft3Δ.
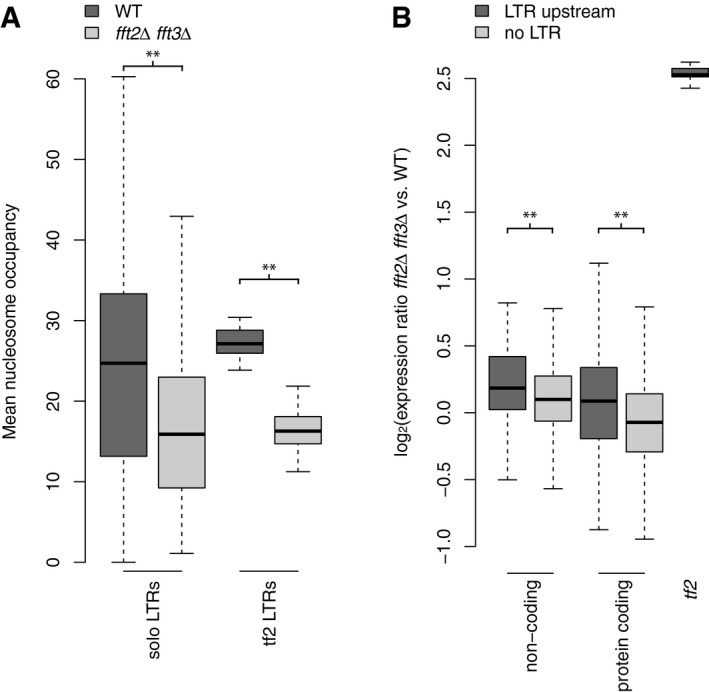
- Mean nucleosome occupancy over solo LTRs (n = 236) and tf2‐associated LTRs (n = 25), in WT and in fft2Δ fft3Δ.
- Mean ratio of expression (fft2Δ fft3Δ/WT) for each of the following categories: noncoding with LTR (n = 69)/without LTR (n = 1,665) (**P = 0.008); protein coding with LTR (n = 246)/without LTR (n = 4,887) (**P < 0.001); Tf2s (n = 13).
Data information: Data shown as box plots, significance of difference between categories was assessed by Wilcoxon–Mann–Whitney test (**P < 0.001).
Retrotransposon TSS and the cellular stress response
Interestingly, both fft2 and fft3 are downregulated at the mRNA level upon stress treatment 15. Given the evidence that transposon activation is a widespread stress response in eukaryotes 11, 14, 15, we hypothesized that permitting the low level transcription of a truncated retrotransposon transcript allows for rapid activation as a part of the stress response. To see whether the same TSS shift is indeed part of the natural cellular response to stress, we exposed cells to transient heat (39°C) or oxidative stress (0.5 mM H2O2). RT–qPCR revealed an increase in longer transcripts, as well as an overall increase in transcription, following heat and oxidative stress treatments (Fig 5A). This indicates that cells switch between Tf2 retrotransposon transcription initiation sites in response to environmental conditions.
Figure 5. Tf2 elements undergo an Fft remodeler‐dependent TSS shift when stressed.
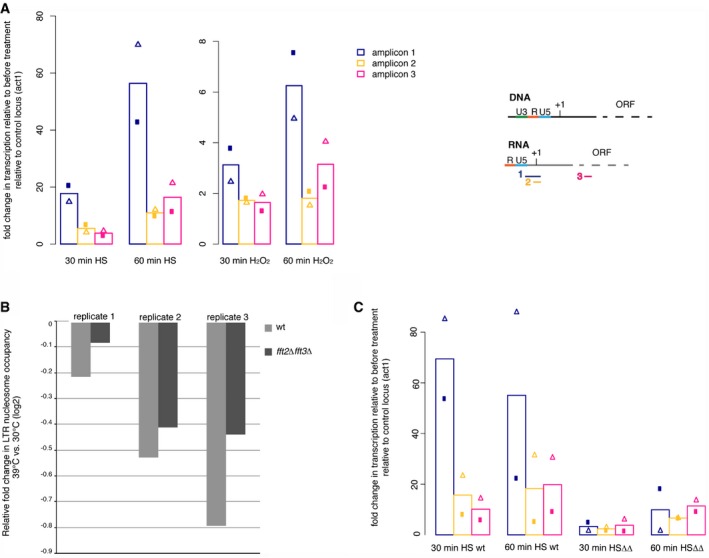
- Heat and oxidative stress cause a Tf2 TSS shift in WT cells, resulting in the production of reverse transcription competent Tf2 transcripts. Transcription fold change as in Fig 1D, but relative to act1 transcription and to transcription in the same culture immediately prior to stress, as measured by qPCR.
- Heat shock‐associated TSS shift is accompanied by a reduction in LTR nucleosome occupancy. The degree of nucleosome loss is greater in WT cells than in fft2Δ fft3Δ cells, where nucleosome occupancy is already very low (Figs 2B and EV3). LTR nucleosome occupancy shown relative to occupancy of the Tf2 ORF +1 nucleosome, measured by MNase‐qPCR.
- Exposure to stress conditions does not increase Tf2 transcript levels further in fft2Δ fft3Δ cells. Transcription fold change as in Fig 1D, but relative to act1 transcription and to transcription in the same culture immediately prior to stress, as measured by qPCR.
Data information: Bars show mean of two biological replicates, with triangles and circles showing the values of the individual replicates.
To see whether chromatin remodeling directly enables this stress‐induced TSS switch, we compared LTR nucleosome occupancy in WT and fft2Δ fft3Δ cells before and after heat shock. Using MNase‐qPCR to measure LTR nucleosome occupancy, we observed that in WT, the occupancy of the nucleosome in the U3 region of the LTR is clearly reduced by stress (Fig 5B). Reduction in LTR nucleosome occupancy in fft2Δ fft3Δ, from its much lower starting point before heat shock, was milder (15% reduction on average) (Figs 5B and EV4). Accordingly, exposure to stress conditions does not lead to a further increase in Tf2 transcription levels compared to unstressed cells (Fig 5C). This indicates that the LTR chromatin state in fft2Δ fft3Δ cells mirrors that of stressed cells.
Figure EV4. LTR nucleosome occupancy is reduced upon heat shock.
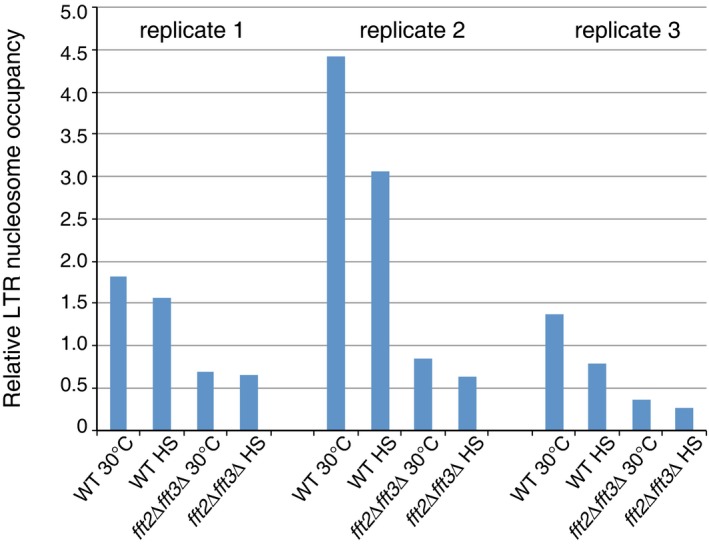
This reduction is more pronounced in WT cells than in fft2Δ fft3Δ cells. LTR nucleosome occupancy shown relative to occupancy of the Tf2 ORF + 1 nucleosome, measured by qPCR.
LTR nucleosome positioning is dependent on the catalytic function of Fft2 and Fft3
We wondered whether the effect of the two Fun30 chromatin remodelers on LTR nucleosome occupancy was due to active remodeling or to some other function, perhaps recruitment of another factor. We had previously generated a strain in which Fft3 ATPase function is abrogated by a point mutation 41, and decided to generate an equivalent point mutation (K581R) in fft2 (Fig 6A). Neither the level of Fft2 protein expression (Fig EV5A) nor Fft2 targeting to chromatin (Fig EV5B) appears to be altered in fft2‐K581R‐myc as compared with fft2‐myc.
Figure 6. ATP‐dependent remodeling function is essential to retrotransposon regulation by Fft2 and Fft3.
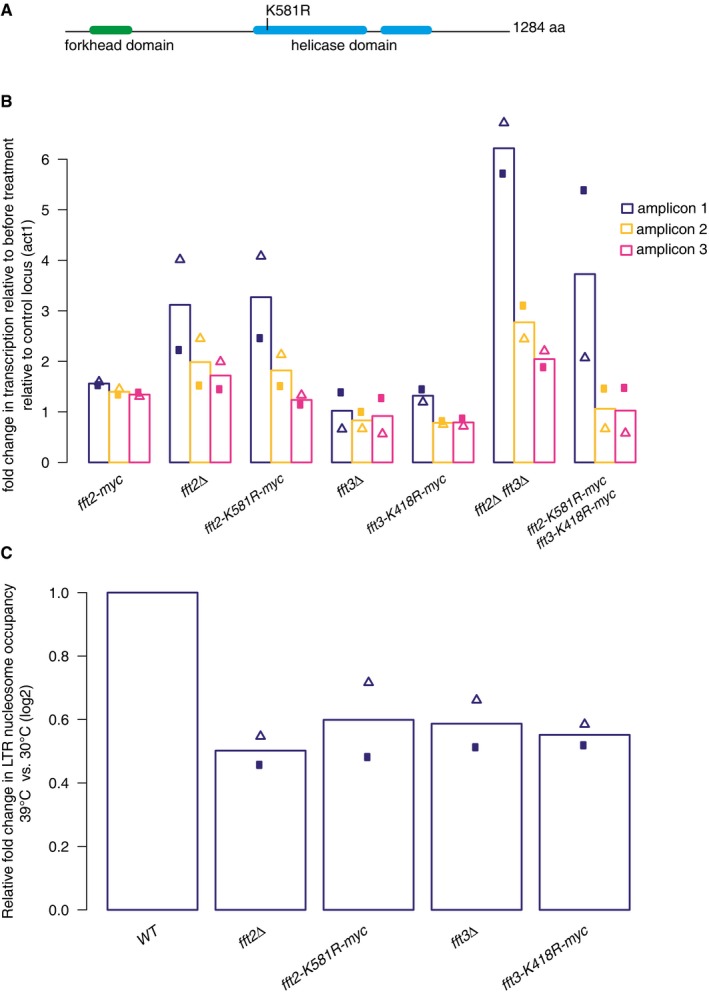
- Schematic of the Fft2 protein showing the location of the catalytic function‐abrogating point mutation K581R.
- Bar graph representing RNA levels as in Fig 1D, relative to a control locus (act1) and to WT levels, as measured by qPCR.
- TSS shift in Fft deletion and catalytic mutants is accompanied by reduced LTR nucleosome occupancy, measured by MNase‐qPCR as in Fig 5B.
Data information: Bars show mean of two biological replicates, with triangles and circles showing the values of the individual replicates.
Figure EV5. Fft2‐K581R‐myc is expressed and binds to chromatin at WT levels.
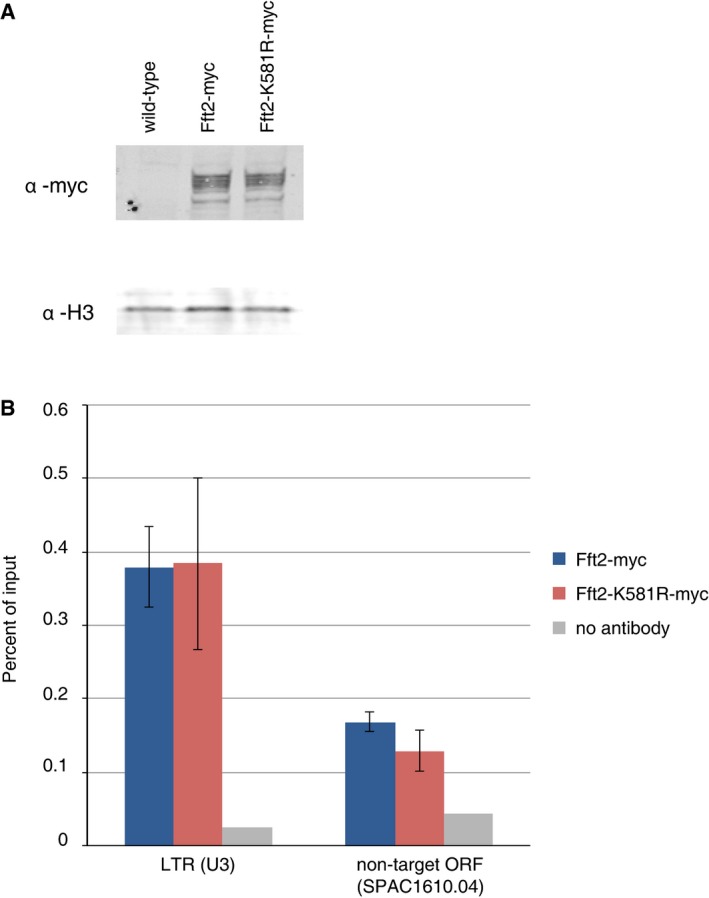
- Whole‐cell lysates, including an epitope‐free WT control, were immunoblotted with α‐myc, to determine Fft2 expression level, and with α‐H3, to confirm equal loading.
- Chromatin immunoprecipitation indicates that Fft2‐K581R‐myc is recruited to LTR elements to the same degree as functional Fft2‐myc. Results are shown as percent input, and a no‐antibody control indicates the level of nonspecific protein A binding. Error bars reflect technical duplicates of biological duplicates.
First, we compared retrotransposon TSS usage between Fun30 remodeler catalytic and deletion mutants. Using reverse transcription followed by qPCR, as in Fig 1C and D, we observed comparable increases in long transcript abundance in single catalytic (fft2‐K581R and fft3‐K418R) and deletion (fft2Δ and fft3Δ) mutants (Fig 6B). This indicates that the chromatin remodelers’ catalytic function is essential for preventing the production of full‐length Tf2 transcripts. In contrast, the increase in full‐length transcript abundance in a double catalytic mutant (fft2‐K581R fft3‐K418R) is significantly less than the increase in a double deletion mutant (fft2Δ fft3Δ), pointing to an additional noncatalytic role for the remodelers. Indeed, the abundance of full‐length Tf2 transcripts in fft2‐K581R fft3‐K418R is similar to that in the single mutants. Because remodeler targeting to LTRs is not compromised by the catalytic point mutation, it is possible that the catalytically dead remodeler blocks the access of the functioning remodeler. This could cause an almost complete loss of nucleosome remodeling, even in the single catalytic mutants.
Finally, we measured nucleosome occupancy in the single catalytic and deletion remodeler mutants by MNase‐qPCR. In all four strains (fft2Δ, fft2‐K581R, fft3Δ, and fft3‐K418R), nucleosome occupancy was reduced to 50–60% of WT occupancy (Fig 6C). This further supports a direct role for chromatin remodeling in regulating the TSS of retrotransposons.
Tf body disruption
In WT cells, Tf2 elements cluster within the nucleus in subnuclear structures called tf bodies 28. This clustering is lost upon stress treatment and in CENP‐B mutant strains 28. We therefore asked whether the disrupted chromatin structure and TSS regulation we had observed in fft2/3 mutants would also perturb tf body integrity. We performed fluorescence in situ hybridization (FISH) against the Tf2 elements in WT, fft2Δ, fft3Δ, and fft2Δ fft3Δ cells and observed a significant (P < 0.001, χ2‐test) declustering effect in the double fft2Δ fft3Δ mutant (Fig 7A and B). This suggests that transcription and chromatin structure may be epistatic to CENP‐B‐mediated Tf2 regulation by clustering.
Figure 7. Fft2 and Fft3 are required for Tf2 element clustering in tf bodies.
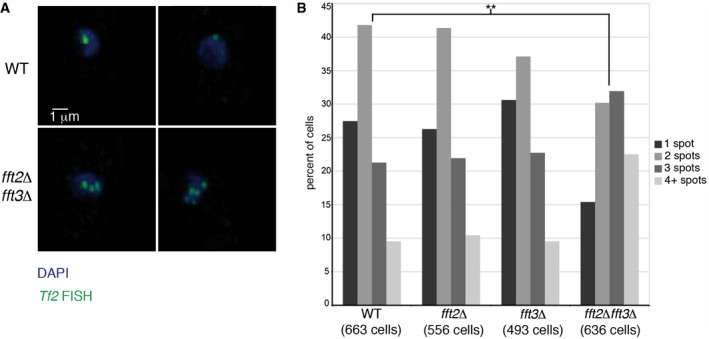
- Fluorescence in situ hybridization with a probe against tf2 elements (green), and DAPI staining (blue) in WT and fft2Δ fft3Δ cells.
- The number of tf2 element signal clusters is significantly elevated (**P = 9.8 × 10−18, χ2‐test) in fft2Δ fft3Δ cells, indicating a loss of retrotransposon clustering. Tf2 clustering was counted in WT cells (n = 663), fft2∆ (n = 556), fft3∆ (n = 493), fft2∆ fft3∆ (n = 636). Significance of difference between strains was assessed by chi‐square test.
Increased mobility of the Tf2 retrotransposon in Fun30 mutants
Our above results provide strong support for a repressive role of Fft2 and Fft3 at Tf2 elements. To directly test the retromobility of Tf2 elements, we used the synthetic Tf2‐12‐neoAI construct with the neomycin marker gene containing an artificial intron inserted in an opposite orientation downstream of the Tf2 ORF 16. This construct can only give rise to G418‐resistant colonies by splicing out the artificial intron and production of cDNA, which is mobilized and inserted into the genome. Using this assay, the fft3∆ and fft2∆fft3∆ mutant strains showed > 100‐fold increase in median Tf2 transposition frequency and about a 40‐fold increase in transposition rates per cell generation compared to wild type (Fig 8A and B). Three independent G418‐resistant derivatives and the fft3∆ and fft2∆fft3∆ tf2‐neoAI strains were subjected to Southern blotting using a probe for the neoA gene, and new genomic Tf2 insertion sites were clearly detected (Fig 8C). Thus, not only do Fft2 and Fft3 repress the expression of full‐length Tf2 transcripts, but at least Fft3 also restricts the mobility of the Tf2 element.
Figure 8. Tf2 retromobility assay.
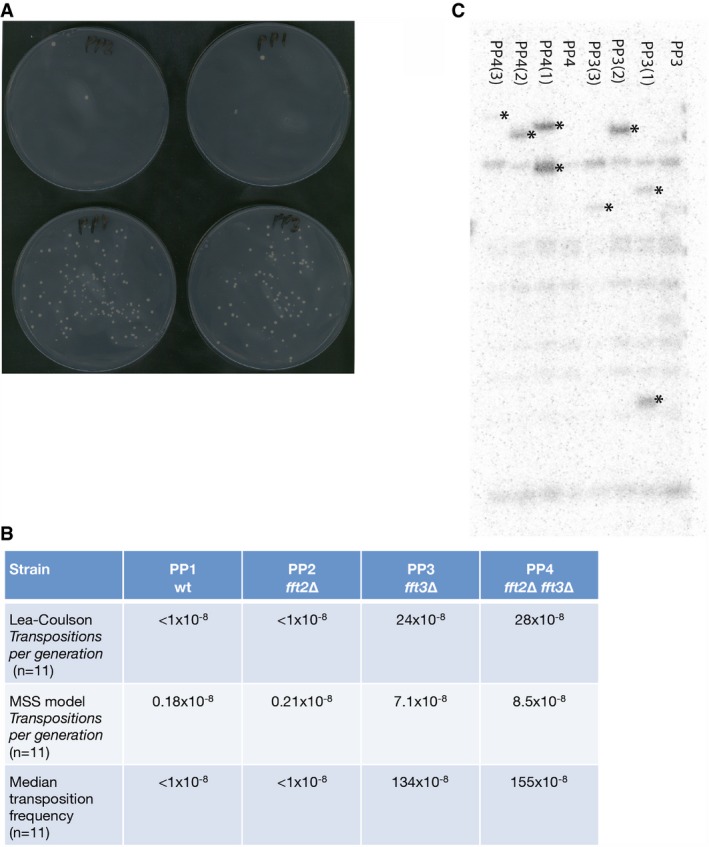
- Pictures of YES plates with G418 showing growth of G418‐resistant subclones in fft3Δ (PP3) and fft2Δfft3Δ (PP4) mutant strains with new integrations of Tf2‐12‐neoAI. Pictures were taken after 3 days of growth at 32°C.
- A table showing the Tf2 transposition frequencies and rates calculated with fluctuation tests using FALCOR.
- Southern blot probed for neoA in fft3Δ (PP3) and fft2Δfft3Δ mutant strains (PP4) and derived G418‐resistant subclones. The bands marked by asterisks represent new unique Tf2 insertions.
Discussion
Here, we provide a mechanistic understanding of the relationship between cellular stress and retroelement activation (Fig 9). Taken together, our findings reveal TSS control as an important mechanism of retroelement regulation. We provide evidence of a direct role for nucleosome occupancy and ATP‐dependent chromatin remodeling in this regulation and demonstrate coordination with the cellular response to stress. Fft2 and Fft3 cooperate to position a nucleosome over the LTR U3 sequence, blocking transcription initiation. This leads to the exclusive generation of truncated, reverse transcription‐incompetent Tf2 transcripts in WT cells while maintaining a transcriptionally permissive chromatin environment around the retrotransposon. We also provide evidence for Fft3 restricting the in vivo mobility of Tf2 elements using a retromobility assay. When cells are exposed to environmental stress, the fft2 and fft3 genes are downregulated. This results in the loss of the LTR nucleosome from the energetically unfavorable U3 sequence, allowing a shift in TSS and production of full‐length transcripts capable of retrotransposition.
Figure 9. A model of transcription at Tf2 retrotransposons.
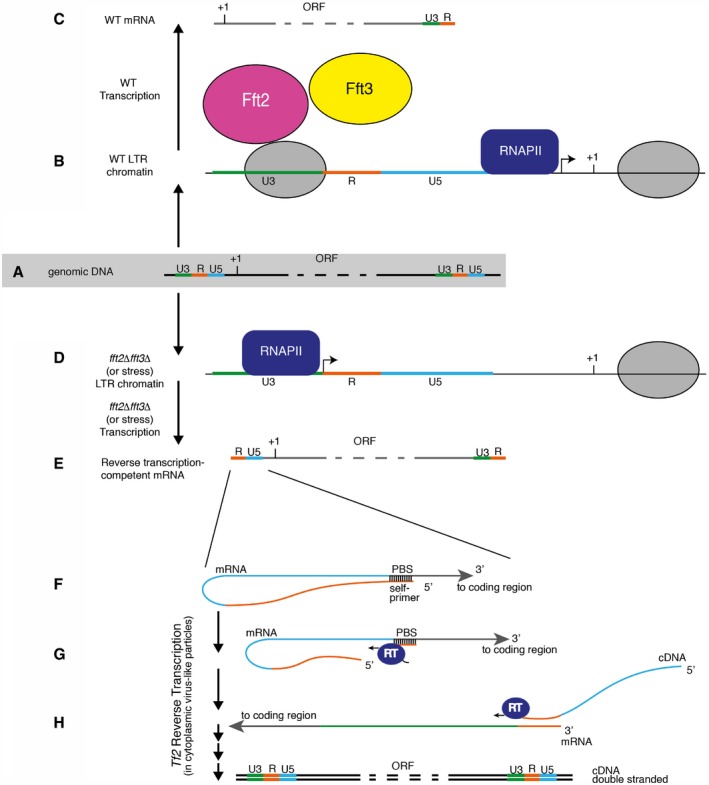
- Genomic DNA: Tf2 retrotransposons are flanked by LTRs, which are composed of U3, R, and U5 sequences.
- In WT cells, Fft2 and/or Fft3 position a nucleosome over the U3 and U3/R border. As a result, transcription initiates downstream of the LTR, just upstream of the protein coding region.
- The mRNA produced in WT cells is unable to support reverse transcription, an essential step in the retrotransposon life cycle.
- In cells lacking or downregulating Fft2, Fft3, or both, a promoter and transcription start site in the LTR are exposed.
- The resultant mutant mRNA is capable of supporting reverse transcription.
- A self‐primer in the R region hybridizes to the primer binding site (PBS) in the U5.
- The tf2‐encoding reverse transcriptase (RT) cleaves the self‐primer from the rest of the mRNA and generates cDNA complementary to the U5 and R sequences while digesting the mRNA template.
- The RT and short cDNA transcript are able to hybridize at the 3′ end of the mRNA, continuing reverse transcription and eventually producing a double‐stranded cDNA capable of reintegration into the genome.
- A putative role of Fft3 in restricting Tf2 integration is depicted. Gray ovals represent nucleosomes; RNAPII, RNA polymerase II. Steps (F–H) are based on the mechanism elucidated by Levin 30.
Although Tf2 is an LTR retrotransposon that spreads via a self‐priming mechanism, the method of regulation described here (Fig 9) has relevance for all LTR retrotransposons, endogenous retroviruses (ERVs), and retroviruses. For any of these retroelements, an incomplete transcript will be incapable of generating cDNA and integrating into the host genome. Our model is consistent with observations that reduced histone production results in Tf2 upregulation 45 and that deletion of the fission yeast HIRA histone chaperone hip1 leads to Tf2 upregulation 46. We had previously observed an extended Tf2 5′UTR in hst4Δ cells, which we at the time attributed to a retrotransposon processing defect 26. We now suspect that Hst4, a class III histone deacetylase, is involved in stabilizing the U3 nucleosome, perhaps via its role in deacetylating H3K56 47, 48. H3K56ac is known to be important for nucleosome “breathing” 49, and deacetylation of this residue could be particularly important for nucleosome stability at energetically unfavorable sequences like the LTR U3. Unlike the exposed histone tail residues, K56 is in the nucleosome core and is not readily accessible. Hst4 may cooperate with an ATP‐dependent chromatin remodeler, in this case Fft2/3, for efficient access to its target lysine residue. Such a cooperation would be similar to that between HDAC1/2 and CHD3/4 in the NuRD complex, where in vitro histone deacetylase activity is enhanced twofold by the presence of ATP 50. An alternative explanation could be that Fft2 and Fft3 are sensitive to the histone acetylation state of target nucleosomes, as is the case for the Swr1 remodeler complex SWR‐C. When H3K56 is acetylated, the dimer exchange function of SWR‐C becomes deregulated, allowing it to insert either H2A.Z/H2B or H2A/H2B histone dimers 51.
We observe that Fft2 and Fft3 enzymes both contribute to repression of full‐length Tf2 mRNA expression. However, only Fft3 was found to have a role in repressing Tf2 mobility in vivo. We speculate that Fft3 has an additional role in restricting transposition during the integration step, and therefore, loss of Fft2 alone is not sufficient to boost the mobility of the retrotransposon (Fig 9). It is possible that Fft3 affects the chromatin structure of target regions or interferes with the function of the DNA‐binding protein Sap1, recently shown to interact with the Tf1 integrase and target integration to RNA pol II promoters 52, 53.
Activating retrotransposons, and even targeting them to coding regions, in response to stress has been observed in numerous species 11, including fission yeast 15, 16. It has long been postulated that the resulting novel regulatory patterns could be an adaptive response to environmental challenges 12. Indeed, newly inserted Tf1 retrotransposons have been shown to target RNA polymerase II promoters 52, 54, 55 and to affect the transcription levels of their new neighbor genes 56, 57. The mechanism described here would allow the cell to retain any novel regulatory benefits after the stress response ends, because LTRs could be allowed to stimulate transcription of neighboring genes without the risk of genomic takeover by repetitive elements. If retrotransposon integration targets the promoters of the genes activated by the current stressor, this could lead to a stably upregulated transcriptional program. The result would be cells with a survival advantage in an altered, for example, warmer or nutrient‐depleted environment.
The need for retrotransposon TSS control extends beyond the yeast genome. Our findings provide an exciting mechanistic explanation for the rather perplexing recent findings that widespread LTR and retrotransposon‐directed transcription is crucial for mammalian pluripotency and cell fate determination. Both human and mouse development exhibit close ties to transposable elements, with MuERV‐L and HERV‐H activation being suggested to mark pluripotency or trigger and regulate embryonic development, respectively 23, 58, 59. Indeed, up to 30% of mouse and human TSS are in transposable elements. These transcripts demonstrate cell type‐specific regulation 60 and have, in some cases, been shown to correlate with pluripotency 24. A copy of HERV is highly transcribed in a human myeloid leukemia cell line 61, suggesting differential regulation of transposable elements in disease. Host cells have coopted LTR sequences as important cis‐regulatory elements, with stem cell‐specific transcription factors targeting LTR sequences 62, 63 and stem cell‐specific LTR‐derived transcripts associating with enhancers 24. Furthermore, it has long been known that global demethylation during mammalian gametogenesis and early embryonic development results in the derepression of ERVs and LTR retrotransposons 64. All of this would seem to expose a cell to intolerable levels of retrotransposition, but would be safe in the context of a strictly regulated TSS. In contrast, when the retrotransposon sequence falls at the 3′ end of a transcript, it appears to be targeted for repression 22. Such transcripts pose a higher risk to genomic integrity, as they may contain all LTR sequences required for successful retrotransposition.
The mechanism of retroelement control we describe here is of particular interest in the context of integrated exogenous retroviruses. The human Swi/Snf subfamily chromatin remodeler BAF may repress HIV sequences that have integrated into the genome by positioning a nucleosome over the TSS of the HIV sequence, in the LTR 8. A similar remodeling complex, PBAF, has also been found to support HIV proviral transcription 65. Based on our findings in fission yeast, it would be interesting to see whether truncated HIV transcripts are initiated downstream of the nucleosome that BAF positions. One of the major challenges in curing HIV is the persistence of latent, integrated infection (reviewed in 66). A better understanding of the role that chromatin plays in latency may suggest new drug targets. It would also be very interesting to see whether BAF or other chromatin remodelers position nucleosomes to steer TSS choice for the many human transcripts that are promoted by LTR sequences.
From an evolutionary perspective, it is interesting to note that the expansion of the Fun30 remodeler subfamily in the fission yeasts occurred in parallel with other major evolutionary changes, including the elimination of most transposon families and a transition to nontransposon centromeres 43. Given the involvement of the S. pombe Fun30 remodelers in both centromeric function and retrotransposon regulation, it is tempting to speculate that the diversification of this subfamily either allowed for or was favored by these changes. Finally, the ability of fission yeast Fun30 remodelers to bind and alter the chromatin structure at LTR elements 40, 41 may be yet another example of the generalization of a mechanism originally selected for its function in controlling transposable elements. The ability of LTR elements to function as chromatin boundaries may stem from their previously acquired ability to attract chromatin remodelers like those of the Fun30 subfamily.
Here, we show an exciting mechanism that underpins host–retroelement symbiosis in fission yeast. Strict TSS control, regulated by chromatin remodelers and nucleosome positioning, provides context for the interesting possibility that ERVs may activate mammalian development. It will be important to determine how phylogenetically widespread such mechanisms are, and to see whether they can be manipulated to improve treatment of retroviral infections.
Materials and Methods
Construction of strains
Standard genetic techniques were used to construct strains 67. Primers used in this study are described in Table EV2. Primers 1–2, used to tag Fft2 by homologous recombination, were generated with the PPPP tool 68. The catalytic mutant fft2‐K581R mutation was generated by homologous recombination with the primer 25–26 amplicon and screened by sequencing the primer 27–28 amplicon. Strains used in this study are described in Table EV3.
Growth conditions
Cells were grown according to standard methods 67. Duplicate liquid cultures were grown in YES media at 30°C and 180 rpm shaking. For all experiments, cells were harvested in mid‐log growth (5 × 106 cells/ml). Heat stress was achieved by transferring log‐phase cultures to 39°C for the indicated length of time (30 or 60 min). Oxidative stress was achieved by adding H2O2 to cultures to a concentration of 0.5 mM and harvesting after 30 or 60 min. For spotting growth assays, cells were grown in liquid culture to mid‐log phase, diluted in PBS, and spotted on solid media in fivefold serial dilutions. Nonselective medium (N/S) is PMG with 100 mg/l uracil; selective medium (−ura) is PMG without added uracil; counterselective medium (FOA) is PMG with 100 mg/l uracil and 1 g/l 5‐fluoroorotic acid (US Biologicals). Assays were performed in duplicate, and plates were incubated at 30°C.
Tf2 retromobility assay
Precultures of strains PP1, PP2, PP3, and PP4 carrying the Tf2‐12‐neoAI allele were grown in YES medium and used to inoculate 11 different 20 ml YES cultures for each strain at a starting concentration of 2 × 103 cells/ml. The cultures were grown for approximately 9 generations, and cells were plated at high densities (10 × 107 cells per plate) onto YES plates containing 200 μg/ml G418 at 32°C. After 3 days, the number of G418‐resistant colonies were scored and mobility rates were determined using fluctuation tests with Luria–Delbruck and MSS (maximum likelihood) models using the FALCOR (Fluctuation AnaLysis CalculatOR) tool according to 69. New Tf2 integration sites in G418‐resistant subclones of PP3 and PP4 strains were confirmed by Southern blot as described in 16.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as described in 70 and in 71. Chromatin extracts were prepared from biological triplicate cultures. Duplicate or triplicate ChIP was then performed on each biological replicate using the following antibody quantities per ChIP: 2 μg mouse 9E10 α‐myc (Sigma M4439); 3 μg rabbit α‐H3 (Ab1791); 2 μl α‐H3K9Ac (Millipore 07‐352). Fragmentation, labeling, and hybridization to the GeneChip S. pombe Tiling 1.0FR Arrays (Affymetrix) were performed by the Affymetrix core facility at Karolinska Institutet (BEA) according to standard protocols (http://www.affymetrix.com). In all cases, two or three biological replicates were hybridized separately.
RNA extraction and reverse transcription
Total RNA was extracted from mid‐log‐phase triplicate cultures of Hu303, Hu0029, Hu1955, Hu1867, Hu2000. In brief, cells were harvested by 5 min of centrifugation at 3,000 rpm. Pellets were resuspended in TES (10 mM Tris–HCl pH 7.5, 10 mM EDTA, and 0.5% SDS) and transferred to 65°C preheated acid phenol. After 45 min of shaking (1,400 rpm) incubation at 65°C, RNA was extracted with phenol–chloroform. Reverse transcription for quantification by qPCR was performed using Superscript™ II Reverse Transcriptase and random hexamers (Invitrogen) according to kit instructions. Levels of different species of Tf2 mRNA were quantified using primers 3–8 as described in 26. Prior to reverse transcription, RNA was treated with Turbo DNase I (Ambion #1907) according to the manufacturer instructions.
For microarray analysis, RNA was treated according to the Affymetrix total RNA labeling protocol (http://www.affymetrix.com) and hybridized to GeneChip S. pombe Tiling 1.0FR Arrays (Affymetrix) by the Affymetrix core facility at Karolinska Institutet (BEA). In all cases, two or three biological replicates were hybridized separately.
CAGE analysis
RNA concentration was determined using the NanoDrop ND‐1000 (Thermo Fisher Scientific, Waltham, MA, United States), and the RNA quality was determined using the Bioanalyzer RNA Pico kit (Agilent Technologies, Santa Clara, CA, USA). CAGE libraries were prepared as described 44 with an input of 5,000 ng total RNA. Samples were run individually, and four CAGE libraries with different barcodes were pooled just prior to sequencing and applied to the same sequencing lane. Libraries were sequenced using Illumina Hi‐Seq 2000. Sequenced reads were trimmed to remove linker sequences and subsequently filtered for a minimum sequencing quality of 30 in 50% of the bases. Trimmed and filtered reads were mapped both to the Schizosaccharomyces pombe assembly ASM294v2 and to a 1,000‐nt sequence from the start of the U3 LTR region using Bowtie 72 (version 0.12.7), and uniquely mapped reads with up to two mismatches were extracted. 5′ ends of mapped CAGE tags (reads) to the LTR region were aggregated in CAGE tag start sites (CTSSs) and normalized to tags per million mapped reads (TPM) according to the total number of mapped reads to ASM294v2 per CAGE library.
Microarray data normalization and analysis
Raw microarray data files (.cel format) were normalized using either R or Affymetrix Tiling Analysis Software (TAS). TAS was used to generate log2 ratios of Fft2‐myc or Fft3‐myc to a control no‐epitope myc‐ChIP (Hu303) using two‐sample analysis quantile normalization together and a bandwidth of 100. Probe signals were assigned to S. pombe genome coordinates (Sanger 2007). TAS was also used to generate log2 ratios of mutant to WT expression arrays using the same normalization. Data analysis was performed in R (http://www.r-project.org) using the Bioconductor (http://www.bioconductor.org) packages “affy”, “affxparser”, and “preprocessCore” with standard parameters. CEL‐files were imported and quantile normalized as described in 73.
Data was visualized using Podbat 33, R, and the Integrated Genome Browser (IGB, Affymetrix). R was used to generate average gene occupancy graphs. Box‐and‐whisker plots were created in R using the “boxplot” function with standard parameters. Significance tests between data subsets were performed using the Wilcoxon–Mann–Whitney test function “wilcox.test” with standard parameters.
Micrococcal nuclease (MNase) digestion
Mononucleosomal DNA fragments were prepared and purified as described in 74. Fragments were amplified and labeled with the NEBNext® ChIP‐Seq Library Prep Master Mix Set for Illumina® (NEB #E6240) and sequenced on an Illumina Miseq v.3. Paired‐end reads were mapped to the S. pombe genome using Bowtie2 75 with standard parameters. DANPOS 76 was then used to remove clonal reads, normalize by quantile normalization, calculate nucleosome occupancy, and compute differential signals. Averaging of occupancy data and visualization was carried out using R.
Primers 21–24 were used for quantification by qPCR. We normalized LTR nucleosome occupancy against the Tf2 ORF +1 nucleosome because occupancy of this nucleosome appeared very stable in our MNase‐seq data.
Northern blots
Ten micrograms of hot phenol isolated total RNA was electrophoresed in a MOPS–acetate–formaldehyde agarose gel and transferred to Hybond N+ (GE Healthcare) in 20× SSC. The RNA was UV crosslinked (Auto Crosslink 1,200 mJ, Stratalinker, Stratagene). A 656‐bp fragment from the Tf2 retrotransposon coding region was cloned into pBluescript (Stratagene), and the resulting plasmids were used as templates for probe synthesis. 32P‐labeled riboprobes were prepared using MaxiScript in vitro Transcription Kit (Ambion) according to the manufacturer's directions. Probes were hybridized in 5× Denhardt's solution, 6× SSC, 10 mM EDTA, 0.5% SDS, 0.1 mg/ml salmon sperm DNA (Invitrogen) at 68°C for 16 h. The blots were washed and exposed to phosphoimager screens (Fuji). Screens were scanned using Molecular Imager FX (BioRad). Blot was performed with biological duplicates of Hu0029, Hu1955, Hu1867, and Hu2000 RNA.
5′ RACE
Classical RACE (Fig 1) was performed with Turbo DNase I (Ambion #1907)‐treated RNA from Hu303, Hu1955, Hu1867, and Hu2000 using the 5′ RACE System for Rapid Amplification of cDNA Ends, version 2.0 (Invitrogen), and oligonucleotides 9 (GSP1) and 10 (GSP2). 5′‐cap‐sensitive RNA ligase‐mediated RACE (RLM‐RACE) was performed to validate TSS in RNA from Hu0029 wt cells (Table EV1). A First‐Choice RLM‐RACE kit (Ambion #1700) was used, and RNA was treated with calf intestine alkaline phosphatase (CIP) to eliminate fragmented and uncapped mRNA. RACE and RLM‐RACE products were cloned into pGEM‐T (Promega) and electroporated into E. coli cells for amplification and sequencing.
Fluorescence in situ hybridization (FISH)
Cells from Hu0029, Hu1955, Hu1867, and Hu2000 were harvested from mid‐log YES cultures grown at 30°C and prepared for FISH as described in 77. Briefly, cells were fixed by adding paraformaldehyde to 1.75% for 45 min. Chromosomal DNA was denatured by 15‐min incubations with 2× SSC, 2× SSC + 10% formamide, 2× SSC + 20% formamide, and 2× SSC + 40% formamide, successively, at 37°C. The probe was prepared from a PCR product amplifying Tf2 element DNA (primers 14 and 15 as described in 28). The PCR product was digested to an average length of 200 bp using DNase I and labeled with Digoxigenin‐11‐dUTP (Roche) using Pol I (Roche). Images were taken using a Nikon A1+ laser scanning microscope with a 60× Lambda S oil‐immersion objective (NA 1.4). Imaging was set up fulfilling the Nyquist criterion in xy and z, with a minimum zoom of 2. For each cell, z‐stacks were acquired with 0.2‐μm spacing and subjected to a blind 3D deconvolution algorithm using the NIS Elements Advanced Research software version 4.12.
Western blot
Protein extracts from log‐phase cultures were prepared using a FastPrep‐24 machine (MP Biomedicals) and separated by 4–12% SDS–PAGE. Immunoblot analysis was carried out using anti‐myc (9E10, Sigma) and anti‐H3 (ab1791, Abcam) primary antibodies. A secondary infrared imaging system (Li‐Cor) was used for detection, with Odyssey goat anti‐mouse IRDYE 800CW and anti‐rabbit IRDYE 680RD (both Li‐Cor) as secondary antibodies.
Data accession
Raw and processed data are available for download at http://www.ncbi.nlm.nih.gov/geo/ under the accession number GSE57069.
Author contributions
JP and KE planned the experiments. JP, BS, AS, MB, JB, CS, and ON performed the experiments. KE, JP, RA, AS, MA, BA, and BS analyzed and interpreted the data. JP, BS, RA, AS, and KE made the figures. JP and KE wrote and edited the text with input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Review Process File
Acknowledgements
We thank the Bioinformatics and Expression Analysis (BEA) core facility at Karolinska Institutet for their assistance. We thank Peter Espenshade for kindly providing the Tf2‐12‐neoAI strain. This study was in part performed at the Live Cell Imaging Unit, Department of Biosciences and Nutrition, Karolinska Institutet, Huddinge, Sweden, supported by grants from the Knut and Alice Wallenberg Foundation, the Swedish Research Council, the Centre for Biosciences, and the Jonasson donation to the School of Technology and Health, Kungliga Tekniska Högskolan, Huddinge, Sweden. Work in the KE laboratory is supported by grants from the Swedish Cancer Society and the Swedish Research Council (VR). KE received a Wenner‐Gren stipend for work as a visiting researcher at Institut Pasteur, France. B.A. was supported the grant ANR‐06‐BLAN‐0271. Work in the AS laboratory was supported by the Novo Nordisk Foundation and Lundbeck Foundation. R.A. was supported by ERC grant 638273. CAGE sequencing was done at the National High‐throughput DNA sequencing center, University of Copenhagen.
EMBO Reports (2016) 17: 753–768
References
- 1. Struhl K, Segal E (2013) Determinants of nucleosome positioning. Nat Struct Mol Biol 20: 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Malik HS, Henikoff S, Eickbush TH (2000) Poised for contagion: evolutionary origins of the infectious abilities of invertebrate retroviruses. Genome Res 10: 1307–1318 [DOI] [PubMed] [Google Scholar]
- 3. Fedoroff NV (2012) Presidential address. Transposable elements, epigenetics, and genome evolution. Science 338: 758–767 [DOI] [PubMed] [Google Scholar]
- 4. Kidwell MG, Lisch DR (2000) Transposable elements and host genome evolution. Trends Ecol Evol 15: 95–99 [DOI] [PubMed] [Google Scholar]
- 5. Cordaux R, Batzer MA (2009) The impact of retrotransposons on human genome evolution. Nat Rev Genet 10: 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thayer RE, Singer MF, Fanning TG (1993) Undermethylation of specific LINE‐1 sequences in human cells producing a LINE‐1‐encoded protein. Gene 133: 273–277 [DOI] [PubMed] [Google Scholar]
- 7. Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11: 204–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rafati H, Parra M, Hakre S, Moshkin Y, Verdin E, Mahmoudi T (2011) Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol 9: e1001206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Curcio MJ, Lutz S, Lesage P (2015) The Ty1 LTR‐retrotransposon of budding yeast. Microbiol Spectr 3: 1–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ito H (2013) Small RNAs and regulation of transposons in plants. Genes Genet Syst 88: 3–7 [DOI] [PubMed] [Google Scholar]
- 11. Levin HL, Moran JV (2011) Dynamic interactions between transposable elements and their hosts. Nat Rev Genet 12: 615–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McClintock B (1984) The significance of responses of the genome to challenge. Science 226: 792–801 [DOI] [PubMed] [Google Scholar]
- 13. Coufal NG, Garcia‐Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O'Shea KS, Moran JV, Gage FH (2009) L1 retrotransposition in human neural progenitor cells. Nature 460: 1127–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Todeschini AL, Morillon A, Springer M, Lesage P (2005) Severe adenine starvation activates Ty1 transcription and retrotransposition in Saccharomyces cerevisiae . Mol Cell Biol 25: 7459–7472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen D, Toone WM, Mata J, Lyne R, Burns G, Kivinen K, Brazma A, Jones N, Bahler J (2003) Global transcriptional responses of fission yeast to environmental stress. Mol Biol Cell 14: 214–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sehgal A, Lee CY, Espenshade PJ (2007) SREBP controls oxygen‐dependent mobilization of retrotransposons in fission yeast. PLoS Genet 3: e131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dai J, Xie W, Brady TL, Gao J, Voytas DF (2007) Phosphorylation regulates integration of the yeast Ty5 retrotransposon into heterochromatin. Mol Cell 27: 289–299 [DOI] [PubMed] [Google Scholar]
- 18. Gai X, Voytas DF (1998) A single amino acid change in the yeast retrotransposon Ty5 abolishes targeting to silent chromatin. Mol Cell 1: 1051–1055 [DOI] [PubMed] [Google Scholar]
- 19. Xie W, Gai X, Zhu Y, Zappulla DC, Sternglanz R, Voytas DF (2001) Targeting of the yeast Ty5 retrotransposon to silent chromatin is mediated by interactions between integrase and Sir4p. Mol Cell Biol 21: 6606–6614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singer T, McConnell MJ, Marchetto MC, Coufal NG, Gage FH (2010) LINE‐1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends Neurosci 33: 345–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Brennan PM, Rizzu P, Smith S, Fell M et al (2011) Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479: 534–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Faulkner GJ, Carninci P (2009) Altruistic functions for selfish DNA. Cell Cycle 8: 2895–2900 [DOI] [PubMed] [Google Scholar]
- 23. Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, Firth A, Singer O, Trono D, Pfaff SL (2012) Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fort A, Hashimoto K, Yamada D, Salimullah M, Keya CA, Saxena A, Bonetti A, Voineagu I, Bertin N, Kratz A et al (2014) Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat Genet 46: 558–566 [DOI] [PubMed] [Google Scholar]
- 25. Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S et al (2002) The genome sequence of Schizosaccharomyces pombe . Nature 415: 871–880 [DOI] [PubMed] [Google Scholar]
- 26. Durand‐Dubief M, Sinha I, Fagerstrom‐Billai F, Bonilla C, Wright A, Grunstein M, Ekwall K (2007) Specific functions for the fission yeast Sirtuins Hst2 and Hst4 in gene regulation and retrotransposon silencing. EMBO J 26: 2477–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yamanaka S, Mehta S, Reyes‐Turcu FE, Zhuang F, Fuchs RT, Rong Y, Robb GB, Grewal SI (2013) RNAi triggered by specialized machinery silences developmental genes and retrotransposons. Nature 493: 557–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cam HP, Noma K, Ebina H, Levin HL, Grewal SI (2008) Host genome surveillance for retrotransposons by transposon‐derived proteins. Nature 451: 431–436 [DOI] [PubMed] [Google Scholar]
- 29. Lorenz DR, Mikheyeva IV, Johansen P, Meyer L, Berg A, Grewal SI, Cam HP (2012) CENP‐B cooperates with Set1 in bidirectional transcriptional silencing and genome organization of retrotransposons. Mol Cell Biol 32: 4215–4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Levin HL (1995) A novel mechanism of self‐primed reverse transcription defines a new family of retroelements. Mol Cell Biol 15: 3310–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin JH, Levin HL (1997) A complex structure in the mRNA of Tf1 is recognized and cleaved to generate the primer of reverse transcription. Genes Dev 11: 270–285 [DOI] [PubMed] [Google Scholar]
- 32. Flaus A, Martin DM, Barton GJ, Owen‐Hughes T (2006) Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res 34: 2887–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sadeghi L, Bonilla C, Stralfors A, Ekwall K, Svensson JP (2011) Podbat: a novel genomic tool reveals Swr1‐independent H2A.Z incorporation at gene coding sequences through epigenetic meta‐analysis. PLoS Comput Biol 7: e1002163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Durand‐Dubief M, Will WR, Petrini E, Theodorou D, Harris RR, Crawford MR, Paszkiewicz K, Krueger F, Correra RM, Vetter AT et al (2012) SWI/SNF‐like chromatin remodeling factor Fun30 supports point centromere function in S. cerevisiae . PLoS Genet 8: e1002974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry A, Burma S et al (2012) The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 489: 581–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen X, Cui D, Papusha A, Zhang X, Chu CD, Tang J, Chen K, Pan X, Ira G (2012) The Fun30 nucleosome remodeller promotes resection of DNA double‐strand break ends. Nature 489: 576–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eapen VV, Sugawara N, Tsabar M, Wu WH, Haber JE (2012) The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol Cell Biol 32: 4727–4740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Byeon B, Wang W, Barski A, Ranallo RT, Bao K, Schones DE, Zhao K, Wu C, Wu WH (2013) The ATP‐dependent chromatin remodeling enzyme Fun30 represses transcription by sliding promoter‐proximal nucleosomes. J Biol Chem 288: 23182–23193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rowbotham SP, Barki L, Neves‐Costa A, Santos F, Dean W, Hawkes N, Choudhary P, Will WR, Webster J, Oxley D et al (2011) Maintenance of silent chromatin through replication requires SWI/SNF‐like chromatin remodeler SMARCAD1. Mol Cell 42: 285–296 [DOI] [PubMed] [Google Scholar]
- 40. Stralfors A, Walfridsson J, Bhuiyan H, Ekwall K (2011) The FUN30 chromatin remodeler, Fft3, protects centromeric and subtelomeric domains from euchromatin formation. PLoS Genet 7: e1001334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Steglich B, Stralfors A, Khorosjutina O, Persson J, Smialowska A, Javerzat JP, Ekwall K (2015) The Fun30 chromatin remodeler Fft3 controls nuclear organization and chromatin structure of insulators and subtelomeres in fission yeast. PLoS Genet 11: e1005101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saha A, Mitchell JA, Nishida Y, Hildreth JE, Ariberre JA, Gilbert WV, Garfinkel DJ (2015) A trans‐dominant form of Gag restricts Ty1 retrotransposition and mediates copy number control. J Virol 89: 3922–3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rhind N, Chen Z, Yassour M, Thompson DA, Haas BJ, Habib N, Wapinski I, Roy S, Lin MF, Heiman DI et al (2011) Comparative functional genomics of the fission yeasts. Science 332: 930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takahashi H, Lassmann T, Murata M, Carninci P (2012) 5′ end‐centered expression profiling using cap‐analysis gene expression and next‐generation sequencing. Nat Protoc 7: 542–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou ZX, Zhang MJ, Peng X, Takayama Y, Xu XY, Huang LZ, Du LL (2013) Mapping genomic hotspots of DNA damage by a single‐strand‐DNA‐compatible and strand‐specific ChIP‐seq method. Genome Res 23: 705–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Greenall A, Williams ES, Martin KA, Palmer JM, Gray J, Liu C, Whitehall SK (2006) Hip3 interacts with the HIRA proteins Hip1 and Slm9 and is required for transcriptional silencing and accurate chromosome segregation. J Biol Chem 281: 8732–8739 [DOI] [PubMed] [Google Scholar]
- 47. Xhemalce B, Miller KM, Driscoll R, Masumoto H, Jackson SP, Kouzarides T, Verreault A, Arcangioli B (2007) Regulation of histone H3 lysine 56 acetylation in Schizosaccharomyces pombe . J Biol Chem 282: 15040–15047 [DOI] [PubMed] [Google Scholar]
- 48. Maas NL, Miller KM, DeFazio LG, Toczyski DP (2006) Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell 23: 109–119 [DOI] [PubMed] [Google Scholar]
- 49. Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen‐Hughes T, van Noort J, Rhodes D, Chin JW (2009) A method for genetically installing site‐specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol Cell 36: 153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL (1998) Chromatin deacetylation by an ATP‐dependent nucleosome remodelling complex. Nature 395: 917–921 [DOI] [PubMed] [Google Scholar]
- 51. Watanabe S, Radman‐Livaja M, Rando OJ, Peterson CL (2013) A histone acetylation switch regulates H2A.Z deposition by the SWR‐C remodeling enzyme. Science 340: 195–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hickey A, Esnault C, Majumdar A, Chatterjee AG, Iben JR, McQueen PG, Yang AX, Mizuguchi T, Grewal SI, Levin HL (2015) Single‐nucleotide‐specific targeting of the Tf1 retrotransposon promoted by the DNA‐binding protein Sap1 of Schizosaccharomyces pombe . Genetics 201: 905–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jacobs JZ, Rosado‐Lugo JD, Cranz‐Mileva S, Ciccaglione KM, Tournier V, Zaratiegui M (2015) Arrested replication forks guide retrotransposon integration. Science 349: 1549–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guo Y, Levin HL (2010) High‐throughput sequencing of retrotransposon integration provides a saturated profile of target activity in Schizosaccharomyces pombe . Genome Res 20: 239–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Majumdar A, Chatterjee AG, Ripmaster TL, Levin HL (2011) Determinants that specify the integration pattern of retrotransposon Tf1 in the fbp1 promoter of Schizosaccharomyces pombe . J Virol 85: 519–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feng G, Leem YE, Levin HL (2013) Transposon integration enhances expression of stress response genes. Nucleic Acids Res 41: 775–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Leem YE, Ripmaster TL, Kelly FD, Ebina H, Heincelman ME, Zhang K, Grewal SI, Hoffman CS, Levin HL (2008) Retrotransposon Tf1 is targeted to Pol II promoters by transcription activators. Mol Cell 30: 98–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Peaston AE, Evsikov AV, Graber JH, de Vries WN, Holbrook AE, Solter D, Knowles BB (2004) Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev Cell 7: 597–606 [DOI] [PubMed] [Google Scholar]
- 59. Santoni FA, Guerra J, Luban J (2012) HERV‐H RNA is abundant in human embryonic stem cells and a precise marker for pluripotency. Retrovirology 9: 111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T et al (2009) The regulated retrotransposon transcriptome of mammalian cells. Nat Genet 41: 563–571 [DOI] [PubMed] [Google Scholar]
- 61. Kanamori‐Katayama M, Itoh M, Kawaji H, Lassmann T, Katayama S, Kojima M, Bertin N, Kaiho A, Ninomiya N, Daub CO et al (2011) Unamplified cap analysis of gene expression on a single‐molecule sequencer. Genome Res 21: 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bourque G, Leong B, Vega VB, Chen X, Lee YL, Srinivasan KG, Chew JL, Ruan Y, Wei CL, Ng HH et al (2008) Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res 18: 1752–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kunarso G, Chia NY, Jeyakani J, Hwang C, Lu X, Chan YS, Ng HH, Bourque G (2010) Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet 42: 631–634 [DOI] [PubMed] [Google Scholar]
- 64. Maksakova IA, Mager DL, Reiss D (2008) Keeping active endogenous retroviral‐like elements in check: the epigenetic perspective. Cell Mol Life Sci 65: 3329–3347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Easley R, Carpio L, Dannenberg L, Choi S, Alani D, Van Duyne R, Guendel I, Klase Z, Agbottah E, Kehn‐Hall K et al (2010) Transcription through the HIV‐1 nucleosomes: effects of the PBAF complex in Tat activated transcription. Virology 405: 322–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lusic M, Giacca M (2015) Regulation of HIV‐1 latency by chromatin structure and nuclear architecture. J Mol Biol 427: 688–694 [DOI] [PubMed] [Google Scholar]
- 67. Moreno S, Klar A, Nurse P (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe . Methods Enzymol 194: 795–823 [DOI] [PubMed] [Google Scholar]
- 68. Penkett CJ, Birtle ZE, Bahler J (2006) Simplified primer design for PCR‐based gene targeting and microarray primer database: two web tools for fission yeast. Yeast 23: 921–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hall BM, Ma CX, Liang P, Singh KK (2009) Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria‐Delbruck fluctuation analysis. Bioinformatics 25: 1564–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Durand‐Dubief M, Ekwall K (2009) Chromatin immunoprecipitation using microarrays. Methods Mol Biol 529: 279–295 [DOI] [PubMed] [Google Scholar]
- 71. Norman‐Axelsson U, Durand‐Dubief M, Prasad P, Ekwall K (2013) DNA topoisomerase III localizes to centromeres and affects centromeric CENP‐A levels in fission yeast. PLoS Genet 9: e1003371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Steglich B, Filion GJ, van Steensel B, Ekwall K (2012) The inner nuclear membrane proteins Man1 and Ima1 link to two different types of chromatin at the nuclear periphery in S. pombe . Nucleus 3: 77–87 [DOI] [PubMed] [Google Scholar]
- 74. Lantermann A, Stralfors A, Fagerstrom‐Billai F, Korber P, Ekwall K (2009) Genome‐wide mapping of nucleosome positions in Schizosaccharomyces pombe . Methods 48: 218–225 [DOI] [PubMed] [Google Scholar]
- 75. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, Li W (2013) DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res 23: 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ekwall K, Nimmo ER, Javerzat JP, Borgstrom B, Egel R, Cranston G, Allshire R (1996) Mutations in the fission yeast silencing factors clr4+ and rik1+ disrupt the localisation of the chromo domain protein Swi6p and impair centromere function. J Cell Sci 109(Pt 11): 2637–2648 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Review Process File