

Abstract
The G‐protein‐coupled receptor kinase 2 (adrbk2/GRK2) has been implicated in vertebrate Hedgehog (Hh) signalling based on the effects of its transient knock‐down in mammalian cells and zebrafish embryos. Here, we show that the response to Hh signalling is effectively abolished in the absence of Grk2 activity. Zebrafish embryos lacking all Grk2 activity are refractory to both Sonic hedgehog (Shh) and oncogenic Smoothened (Smo) activity, but remain responsive to inhibition of cAMP‐dependent protein kinase (PKA) activity. Mutation of the kinase domain abrogates the rescuing activity of grk2 mRNA, suggesting that Grk2 acts in a kinase‐dependent manner to regulate the response to Hh. Previous studies have suggested that Grk2 potentiates Smo activity by phosphorylating its C‐terminal tail (CTT). In the zebrafish embryo, however, phosphomimetic Smo does not display constitutive activity, whereas phospho‐null mutants retain activity, implying phosphorylation is neither sufficient nor necessary for Smo function. Since Grk2 rescuing activity requires the integrity of domains essential for its interaction with GPCRs, we speculate that Grk2 may regulate Hh pathway activity by downregulation of a GPCR.
Keywords: Grk2, Hedgehog signalling, phosphorylation, PKA, Smo
Subject Categories: Development & Differentiation; Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
Introduction
The Hedgehog (Hh) signalling pathway plays crucial roles in the embryonic development of most animals as well as in tissue homeostasis, metabolism and physiological processes in juveniles and adults. Accordingly, aberrant Hh pathway activity has been implicated in many human disorders including birth defects and cancers 1. Hh ligands exert their functions principally through a signalling cascade that controls the balance between the activator and repressor forms of Gli family transcription factors. In the absence of Hh, these proteins are phosphorylated by cAMP‐dependent protein kinase (PKA), which promotes their partial cleavage by the proteasome, yielding C‐terminally truncated transcriptional repressors 2, 3, 4. The binding of Hh ligand to its receptor, Patched (Ptch), relieves the inhibition of Smoothened (Smo), an atypical member of the G‐protein‐coupled receptor (GPCR) superfamily, and promotes its translocation to the plasma membrane in Drosophila 5, or to the primary cilium (PC) in vertebrates 6. Activated Smo prevents Gli cleavage and promotes the activation of its full‐length form, which induces transcription of Hh pathway target genes 1. Exactly how Smo mediates the regulation of Gli activity is still not fully understood, but various studies highlight its interaction with the kinesin family member Kif7 7. In addition, recent analyses have implicated classical GPCR signalling pathways in modulating Gli activity 8, 9, 10.
G‐protein‐coupled receptor kinases (GRKs) play important roles in desensitisation of activated GPCRs by directly phosphorylating their C‐terminal tails (CTTs), thereby promoting the recruitment of β‐arrestin to mediate internalisation of the activated receptors 11. Several studies have implicated GRK2 as a positive regulator of the Hh pathway 12, 13, 14. The depletion of GRK2 by RNA interference in tissue culture cells leads to a downregulation of Hh activity 14, an effect recapitulated by morpholino‐mediated knock‐down of grk2 in zebrafish embryos 13. Further studies have suggested that GRK2 is essential for promoting Hh pathway activity by direct phosphorylation of Smo, which is also phosphorylated by casein kinase 1 (CK1) in vertebrates and by PKA in Drosophila 15, 16, 17. Such a role for Grk2 in potentiating the activity of Smo contrasts with its canonical function in GPCR desensitisation.
Here, we have re‐examined the role of Grk2 function in Hh signalling by using targeted mutagenesis to eliminate its activity completely from both zebrafish embryos and cultured mammalian cells. We show that Grk2 plays a critical role in Hh signal transduction upstream of the Gli transcription factors, but downstream of Smo both in mammalian cells and in the zebrafish embryo and that maternally supplied Grk2 is sufficient to support Hh signalling during the early stages of zebrafish development. We also show that localisation of Smo to the PC is independent of Grk2 in both zebrafish and mammalian cells. In addition, we present evidence that in zebrafish embryos, phosphorylation of putative GRK2 sites in the Smo CTT is neither necessary nor sufficient for Hh pathway activation. Our results suggest that Grk2 functions as a positive regulator of Hh signalling, not by direct phosphorylation of Smo but by regulation of a more distal step in the pathway.
Results
Generation of a mutant allele of zebrafish grk2 using zinc finger nucleases
Mammalian genomes contain two paralogous genes, Grk2 and Grk3 that encode closely related proteins. By contrast, BLAST searches of the zebrafish genome revealed a single gene, designated grk2, the predicted protein sequence of which is more similar to that of mammalian GRK3 (Fig 1A). Previous functional analyses of this gene have relied upon morpholino‐mediated transient gene knock‐down in zebrafish embryos 14, an approach that has well‐documented limitations 18. To investigate the effects of complete loss of Grk2 function, a null allele of the zebrafish grk2 gene was generated using zinc finger nuclease (ZFN)‐mediated targeted mutagenesis. The ZFN recognition site was designed to target the end of the first exon of the gene (Fig 1B). The mutagenesis rate was relatively low (< 5%), yielding only a single grk2 mutant allele, designated grk2 i283. This mutation is the result of a single base pair (C–G) deletion at coding sequence (CDS) 111, 111delC, causing a frame shift predicted to lead to a 72 residue C‐terminally truncated protein containing only 37 out of the 688 amino acids that comprise Grk2. As shown in Fig 1C, the truncated region includes most of the regulator of G‐protein signalling (RGS) domain, the entire kinase domain and the Pleckstrin homology (PH) domain; since these are all important functional domains of the protein, grk2 i283 is likely to be a null allele. Animals homozygous for grk2 i283 obtained by in‐crossing adult heterozygotes completed embryogenesis and showed no defects in specification of neuronal or muscle fibre cell types, both of which depend upon Hh pathway activity in zebrafish 19, 20. By 4dpf (days post‐fertilisation), however, the mutant larvae had failed to form a normal swim bladder (n = 16), an organ known to be Hh dependent 21. This phenotype is thus consistent with a late developmental requirement for Grk2 in Hh signalling; no homozygous animals survived beyond 20dpf (Fig 1D).
Figure 1. Generation of a zebrafish grk2 null mutation.
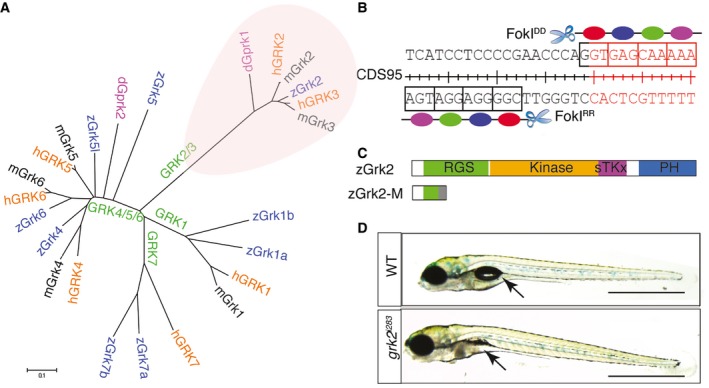
- Phylogenic tree showing the relationship of the various vertebrate Grk genes and the Drosophila gprk gene.
- Schematic representation of the exon 1 nucleotide sequence of zebrafish grk2 gene targeted by the ZFN.
- Schematic representation of the zebrafish Grk2 protein domains and the truncated peptide predicted to be encoded by the grk2 i283 mutant allele.
- Homozygous grk2 i283 mutant at 4dpf compared to wild type (WT) sibling (N = 80). Arrows indicate the position of swim bladder. Scale bar, 1 mm.
Grk2 activity is essential for the response to Hh signals
The lack of obvious early developmental defects indicative of a loss of Hh pathway activity in grk2 i283 homozygotes contrasts with the previously reported effects on muscle cell‐type specification caused by morpholino‐mediated knock‐down of grk2 (Fig EV1) 14. Translation‐blocking morpholinos can inhibit maternally as well as zygotically expressed mRNA and maternally derived grk2 has previously been shown to be present in newly fertilised eggs 22. To investigate the activity of maternally derived Grk2 protein, chimeric fish carrying grk2 homozygous mutant germ lines were generated using the established germ cell transplantation technique 23. Maternal‐zygotic (MZ) mutant embryos generated by crossing chimeric females to chimeric males lacked all Grk2 protein, as revealed by Western blot analysis (Fig 2A): strikingly, these embryos displayed a curved body axis, U‐shaped somites and severe cyclopia, a phenotype almost indistinguishable from that of smo homozygous mutants 24 (Fig 2B). At 24hpf (hours post‐fertilisation), the levels of mRNA encoded by Ptch2, a direct target of Hh pathway activity, were substantially reduced in both the mesoderm and the neural tube of MZgrk2 embryos, as judged by in situ hybridisation (Fig 2C). Similarly, nkx2.2a and olig2 transcripts, markers of V3 interneurons and motor neurons, respectively, were below the levels of detection in both the neural tube and brain (Fig 2C). Like smo mutants, the MZgrk2 embryos displayed a dramatic loss of Hh‐dependent muscle cell types at 30hpf (Figs 2D and EV1): the myotomes were devoid of all Eng‐expressing muscle pioneers (MPs) and medial fast‐twitch fibres (MFFs), while Prox1+ve superficial slow‐twitch fibres (SSFs) were absent from all but the most anterior somites, where, in contrast to smo mutants, a few SFFs (1 ± 1 per somite) were present (Fig EV1). Injection of mRNA encoding GFP‐tagged wild‐type Grk2 (grk2‐GFP) into MZgrk2 embryos efficiently suppressed the mutant phenotype (100%; N = 50) and fully recovered all Hh‐dependent gene expression and muscle cell types (Figs 2C and D, and EV1).
Figure EV1. MZgrk2 shows severe Hh defects.
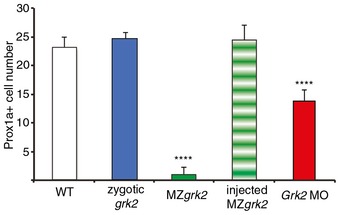
Numbers of Prox1a+ve cells in wild‐type (WT), zygotic grk2 mutants, MZgrk2, MZgrk2 injected with Grk2‐GFP and Grk2 ATG morphant embryos at 30hpf (n = 8). The error bars indicate SD. Asterisks indicate statistically significant differences based on unpaired Student's t‐test. ****P < 0.0001.
Figure 2. Loss of Grk2 impairs Hh signal transduction in zebrafish.
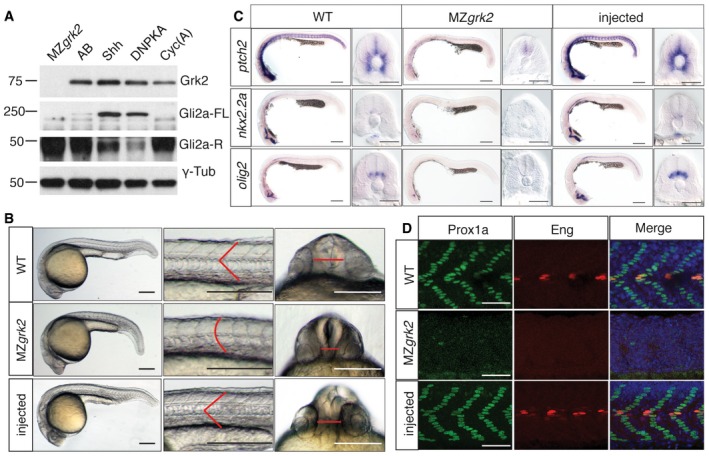
- Western blot analysis of the different forms of Gli2a in MZgrk2 embryos compared to wild‐type (AB), shh mRNA‐ and dnPKA mRNA‐injected wild‐type and cyclopamine (Cyc(A))‐treated wild‐type 20hpf embryos. Gli2a‐FL levels are low relative to Gli2aR levels in wild‐type embryos, but are elevated in response to pathway activation (shh and dnPKA mRNA injected). Gli2aR levels are increased while Gli2a‐FL is undetectable in CycA‐treated and MZgrk2 mutant embryos. Probing the same blot with rabbit anti‐GRK3 (which recognises the zebrafish Grk2 protein) reveals a complete loss of full‐length Grk2 in MZgrk2 embryos. Probing with anti‐γ‐tubulin was performed as a loading control. Three biological replicates of this analysis were performed.
- Phenotype of wild‐type (WT), MZgrk2 and grk2‐GFP mRNA‐injected MZgrk2 embryos at 24hpf (n = 20 for each sample). The red lines indicate the shape of the somites (middle panels) and the separation of the eyes (right hand panels). Scale bar, 200 μm.
- In situ hybridisation of ptch2, nkx2.2a and olig2 transcripts in wild‐type, MZgrk2‐and grk2‐GFP‐injected MZgrk2 24hpf embryos. Each panel shows a full view of the embryo on the left and a cross‐sectional view of a somite on the right (n = 30 for each sample). Scale bars, 200 μm (whole mounts), 50 μm (sections).
- Expression of Prox1a and Engrailed (Eng) proteins in somites of wild‐type, MZgrk2‐ and grk2‐GFP mRNA‐injected MZgrk2 embryos at 30hpf. Each panel shows Prox1a in green, Eng in red and the merged images with DAPI staining in blue (n = 10 for each sample). Scale bar, 50 μm.
Hh pathway activity modulates the PKA‐dependent processing of the Gli2 and Gli3 transcription factors: in the zebrafish, ubiquitous activation of the pathway by ectopic Shh expression or inhibition of PKA activity leads to accumulation of the full‐length activator form of Gli2a, whilst Smo inhibition by cyclopamine (CycA) promotes production of its truncated repressor form at the expense of the full‐length activator form 25, 26 (Fig 2A). Consistent with the phenotypic similarities to smo, Western blot analysis revealed a loss of full‐length Gli2a and accumulation of its truncated repressor form in MZgrk2 embryos (Fig 2A).
Grk2 modulates Hh pathway upstream of Gli processing
To investigate at which point in the transduction of the Hh signal Grk2 is required, the pathway was activated at three different levels by injection of mRNA encoding either: (i) the Shh ligand; (ii) an oncogenic mutant form of Smo, SmoA1 27; or (iii) a dominant negative form of the PKA regulatory subunit, dnPKA. Injection of each of these mRNAs into wild‐type embryos has similar effects, causing the transformation of most of the myotome into slow‐twitch muscle fibres and the ectopic expression of Eng 28, 29, 30 (Fig 3A). Injection of dnPKA mRNA into MZgrk2 mutant embryos similarly led to the widespread induction of slow‐twitch fibres and Eng expression (Fig 3A). MZgrk2 embryos injected with shh or mSmoA1 mRNA, by contrast, showed strongly attenuated responses; only a few additional slow‐twitch fibres (3 ± 2 and 7 ± 4 per somite, n = 8) were detected scattered throughout the length of the trunk, in addition to those restricted to the anterior somites of uninjected controls (Fig 3A). Consistent with these phenotypic effects, overexpressing Shh or mSmoA1 in MZgrk2 failed to block production of the truncated repressor form of Gli2a (Fig 3B).
Figure 3. Grk2 acts upstream of Gli processing.
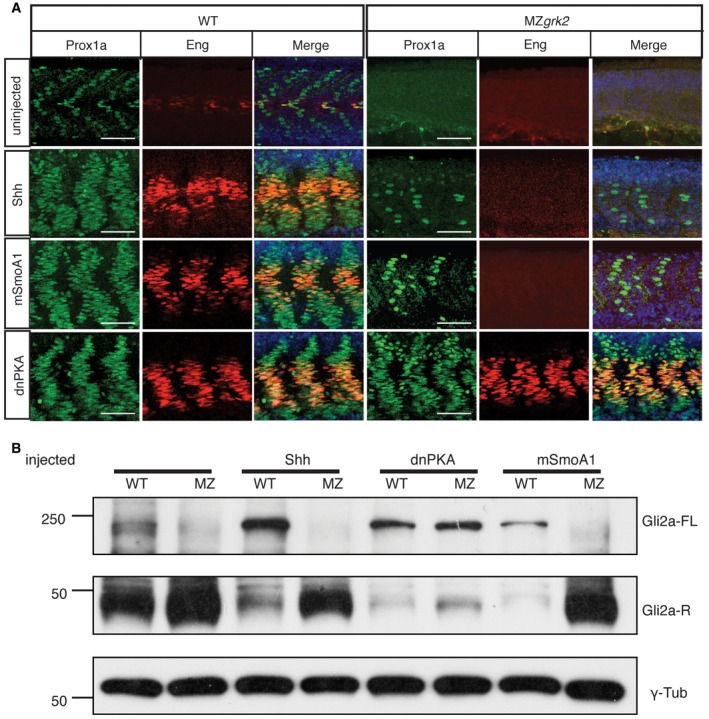
- Prox1a and Eng expression in the myotome of 30hpf wild‐type (WT) and MZgrk2 embryos injected with mRNAs encoding Shh, dnPKA or mSmoA1‐GFP. Each panel shows Prox1a in green, Eng in red and the merged images with DAPI staining in blue. The co‐labelling for Prox1a and Eng (orange) is indicative of MP differentiation (n = 30 for each sample). Scale bar, 50 μm.
- Western blot showing levels of Gli2a‐FL and Gli2a‐R forms in 20hpf wild‐type (WT) and MZgrk2 (MZ) embryos injected with mRNA encoding Shh, dnPKA or mSmoA1‐GFP, respectively. γ‐tubulin was used as a loading control. Three biological replicates of this analysis were performed.
To extend the zebrafish analysis to mammalian cells, we used the lentiCRISPR‐Cas9 mutagenesis method to generate several clonal Flp‐In‐3T3 fibroblast cell lines lacking Grk2 (Grk2 −/− cells). In wild‐type cells, SAG treatment caused an increase in Gli1 and Ptch1 protein and a decrease in Gli3 repressor levels; by contrast, the Grk2 −/− cells did not respond to Shh or SAG (Fig 4A and C). The defect in Hh responsiveness correlated with the amount of residual Grk2 protein in these cell lines (Fig 4A). Overexpression of PKI, an inhibitor of PKA, in both wild‐type and Grk2 −/− cells caused constitutively active pathway activity even without Hh ligand (Fig 4B); however, as in zebrafish embryos, the constitutively active mutant mSmoA1 did not rescue pathway activation in Grk2 −/− cells (Fig 4C).
Figure 4. Mammalian Grk2 −/− cells exhibit strong loss of Hh signalling upstream of Gli processing.
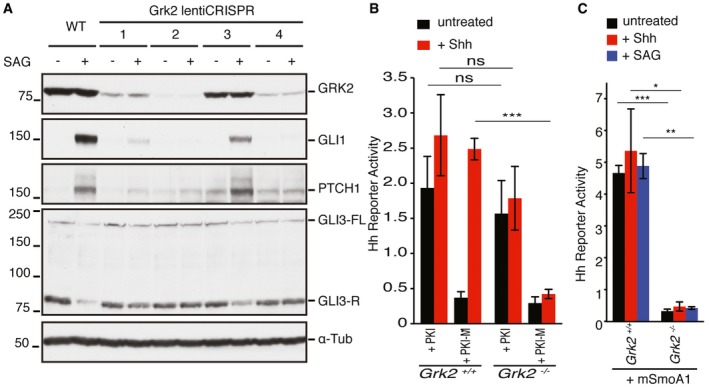
-
AWestern blot analysis of GRK2, GLI1, PTCH1 and GLI3 protein levels in Flp‐In‐3T3 cells with lentiCRISPR of four different guide RNAs in the presence or absence of SAG. α‐tubulin was used as a loading control. Guide RNA 2 was found to remove Grk2 efficiently and used for subsequent experiments. GLI3 panel shows both full‐length (GLI3FL) and repressor form of GLI3 (GLI3R) (n = 3).
-
B, CHh reporter activity assay for wild‐type and Grk2 −/− cells. Cells were treated with PKA peptide inhibitor (PKI) and mutant form of PKA peptide inhibitor (PKI‐M), respectively, in the presence or absence of SHH (B). Cells were transfected with mSmoA1‐GFP‐expressing constructs in the presence of SHH or SAG (C). Data represent the mean and ± SD (n = 3). Unpaired Student's t‐test was used for analysis. ***P < 0.001; **P < 0.01; *P < 0.05 and n.s. (not significant).
Phosphorylation of the carboxyl terminal tail is not essential for Smoothened activation
That loss of Grk2 suppresses ectopic pathway activation caused by the SmoA1 mutation, is consistent with the view that Grk2‐dependent phosphorylation of the CTT of Smo is required for its activity. Indeed, previous studies using mammalian cell culture assays have reported that the constitutive activity of SmoA1 can be abolished by mutation of the putative CK1 and GRK2 phosphorylation sites in its CTT 16. We found, however, that this mutant form, mSmoA1SA, retained constitutive activity in the zebrafish embryo, as evidenced by the increased numbers of Prox1+ve and Eng+ve fibres in the myotome (Fig 5A and B). This effect cannot simply be accounted for by an increased stability of the mutated protein, as levels of mSmoA1SA in injected embryos were significantly lower than those of wild‐type mSmo expressed from injected mRNA (Fig EV2A). Notably, the increase in Prox1+ve cells was less than that induced by mSmoA1 expression (Fig 5A and B) whilst the ectopic Eng expression was restricted to fast‐twitch muscle fibres (Fig 5A), indicating that mutation of the phosphorylation sites attenuates, but does not abolish, Smo activity. Consistent with this, quantitative PCR analysis revealed a less robust upregulation of ptch2 transcription by mSmoA1SA compared to mSmoA1 (Fig EV2B).
Figure 5. Phosphorylation of the CTT is not essential for Smo function.
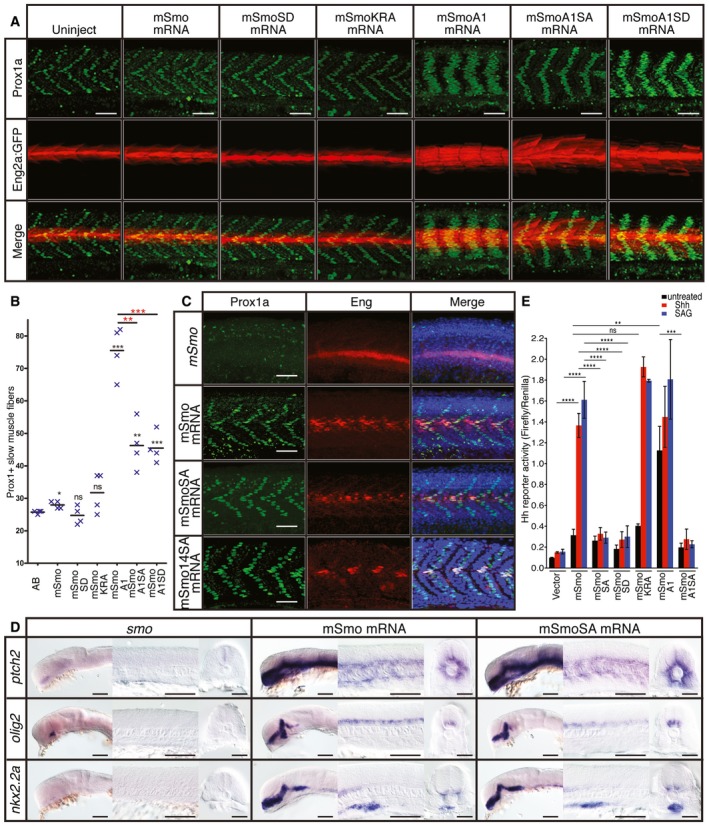
- Expression of Prox1a (green) and the eng2a:GFP reporter (red) in otherwise wild‐type 30hpf embryos injected with mRNA encoding wild‐type and mutant forms of mouse Smo (mSmo). Ectopic MPs are indicated by fibres co‐labelled with Proxa1 and GFP; ectopic MFFs are labelled only with GFP. Images are representative of embryos in the following proportions of each sample: 28/28 (mSmo; mSmoA1; mSmoSD; mSmoKRA); 9/28 (mSmoA1SA); and 12/28 (mSmoA1SD). Scale bar, 50 μm.
- Average number of Prox1a+ve slow fibres in wild‐type embryos injected with mRNA encoding different forms of mSmo. Prox1‐positive cells were quantified in four somites in each of four embryos for each sample. The error bars indicate SD. Unpaired Student's t‐test was used to determine the statistical significance between uninjected embryos and the various Smo mutants (black asterisk) and between mSmoA1 and mSmoA1SA or mSmoA1SD (red asterisk). ***P < 0.001; **P < 0.01; *P < 0.05 and n.s. (not significant).
- Prox1a and Eng expression in smo hi1640 mutant embryos injected with mRNA encoding mSmo (n = 10), mSmoSA (n = 9) and mSmo14SA (n = 6). Note the full recovery of SSFs and MPs compared to the uninjected controls. Scale bar, 50 μm.
- In situ hybridisation for transcripts of ptch2, olig2 and nkx2.2 in 24hpf smo hi1640 mutant embryos injected with mRNA encoding mSmo and mSmoSA (n = 6 for each sample). Scale bars, 100 μm (lateral view), 50 μm (sections).
- Hh reporter assay of the activity of wild‐type and mutant forms of mSmo in Smo −/− MEFs in response to Shh or SAG stimulation. Note that mSmoA1 shows constitutive activity in the absence of either Shh or SAG, whereas mSmoKRA does not; all mutants affecting phosphorylation failed to restore the response to Shh or SAG. Data represent the mean and ± SD (n = 3). Unpaired Student's t‐test was used for analysis. ****P < 0.0001; ***P < 0.001; **P < 0.01; and n.s. (not significant).
Figure EV2. Phosphorylation and dimerisation of Smo does not impair or enhance Hh activity.
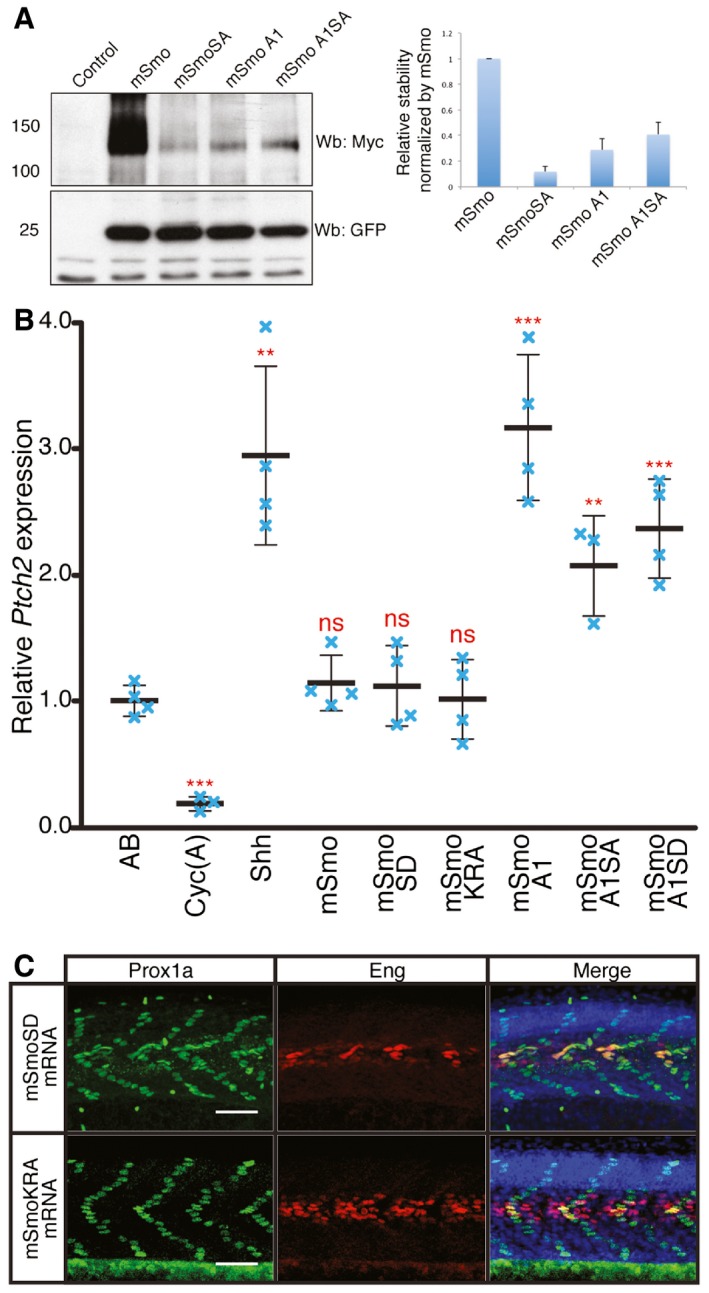
- Western blot analysis of wild‐type and mutant forms of Smo expressed from bicistronic myc‐tagged mSmo and GFP constructs injected into wild‐type zebrafish embryos. The histogram shows the quantification of Myc‐tagged mSmo vs. GFP (n = 3). The error bars indicate SD.
- Normalised expression of ptch2 in 18hpf wild‐type (AB), Cyc(A)‐treated embryos and embryos injected with mRNA encoding Shh or various Smo mutants. Individual data points (blue crosses) are shown; mean is indicated by thick solid line, and SD by thin bars. Unpaired Student's t‐test was used to determine the statistical significance between control (AB) and experimental embryos. ***P < 0.001; **P < 0.01; and n.s. (not significant).
- Prox1a and Eng expression in smo hi1640 mutant embryos injected with mRNA encoding mSmoSD (n = 7) and mSmoKRA (n = 7). These mutant embryos show full recovery of SSFs and MPs. Embryos shown are at 30hpf. Scale bar, 50 μm.
By contrast to the constitutive activity of mSmoA1SA, a form of Smo containing phosphomimetic mutations of the predicted CTT phosphorylation sites, mSmoSD, showed no evidence of constitutive activity (Figs 5A and B, and EV2B); this contrasts with the reported constitutive activity of this mutant form in cultured cells 16. On the other hand, introduction of these phosphomimetic residues into mSmoA1 attenuated its activity to a similar degree as the S‐A mutations (Figs 5A and B, and EV2B). To explore the potential significance of phosphorylation of the CTT further, we used a more stringent smo rescue assay to test the function of the mutant forms of mSmo 31. In line with previous studies, we found that injection of mRNA encoding GFP‐tagged forms of mSmo completely restored the specification of Hh‐dependent cell types in the myotome of zebrafish smo mutants at 30hpf (Fig 5C). Mutation of all the predicted phosphorylation sites in the mSmo CTT (mSmoSA) had no effect on the efficiency of rescue, as assayed by Prox1a and Eng expression (Fig 5C) as well as the expression pattern of ptch2, nkx2.2a and olig2 transcripts (Fig 5D). We considered the possibility that other previously unidentified Grk2 sites might be sufficient to mediate Smo activation; based on the consensus Grk2 phosphorylation sites32, we identified two further putative sites within the mSmo CTT; mutagenesis of these additional sites in mSmoSA (mSmo14SA) did not affect the rescue efficiency (Fig 5C).
Phosphorylation of the Smo CTT has been postulated to neutralise the positive charges caused by a cluster of lysine/arginine residues, thereby promoting dimerisation that promotes activation of pathway components downstream of Smo, a view supported by FRET‐based analysis of Smo conformation 16. In line with this, replacement of the lysine and arginine cluster by alanines in mSmoKRA was reported to render the mutant protein constitutively active in tissue culture assays 16. Expression of mSmoKRA in wild‐type zebrafish embryos, however, showed no evidence of constitutive pathway activity, although it could fully rescue the smo mutant phenotype (Figs 5A and B, and EV2B and C).
In the light of these findings, we re‐examined the activity of these Smo mutant forms in mammalian tissue culture cells. In our hands, neither mSmoSD nor mSmoKRA showed any evidence of constitutive pathway activity (Fig 5E). However, in contrast to the findings in zebrafish embryos, mutant forms refractory to phosphorylation, viz. mSmoSA and mSmoA1SA, failed to restore the response to Hh in Smo −/− MEFs (Fig 5E).
Phosphorylation of Smo has been implicated in promoting its accumulation within the PC; in the absence of phosphorylation, Smo reportedly fails to localise to the PC of cultured mammalian cells 16. We found that GFP‐tagged mSmo localised to the PC of wild‐type zebrafish embryos and that co‐injection of Shh with the mSmo mRNA potentiated this localisation (Fig 6A). As expected, the constitutively active mSmoA1 also localised to the PC, but so did mSmoA1SA, mSmoSA, mSmoSD and mSmoKRA (Fig 6B). These findings suggest that phosphorylation of the putative GRK2 sites is not required for localisation of Smo to the PC in zebrafish embryos. Moreover, we found that wild‐type mSmo localised normally to the PC in MZgrk2 embryos, as did the mSmoSA mutant form, indicating ciliary localisation of Smo is also independent of Grk2 activity (Fig 6C). In line with this, mSmo also translocated to the PC in response to Shh or SAG in Grk2 −/− cells (Fig EV3).
Figure 6. Cilia localisation of wild‐type and mutant Smo.
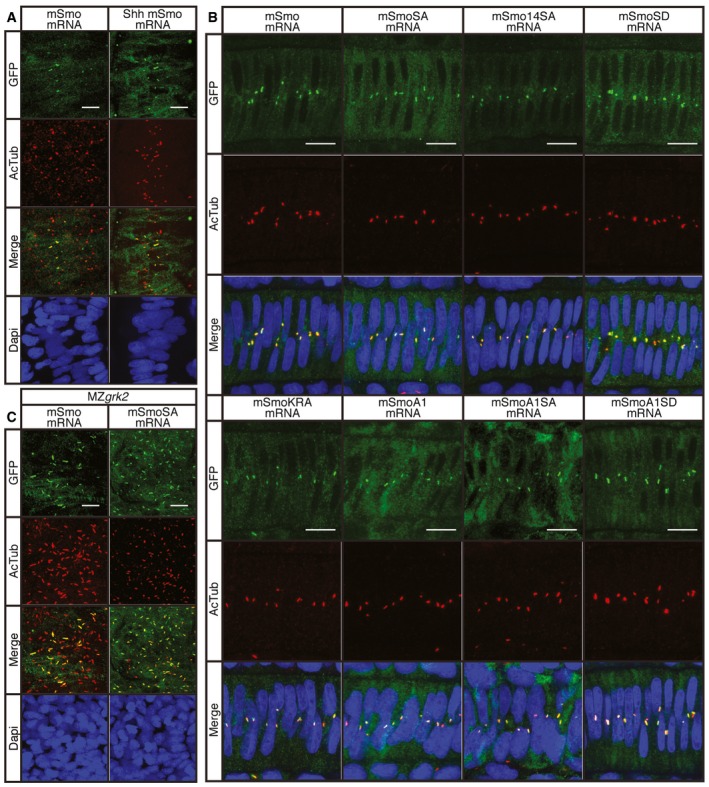
- Wild‐type 18hpf embryos injected with mRNA encoding GFP‐tagged mSmo (green) showing localisation to the PC of myotomal cells labelled with anti‐acetylated tubulin (AcTub; red), stimulated in response to Shh injection (n = 4). Differences in PC distribution are due to morphological changes in the myotome induced by ectopic expression of Shh as revealed by the distribution of nuclei (DAPI stained; blue). Scale bar, 10 μm.
- Notochord cells of wild‐type embryos expressing GFP‐tagged wild‐type and mutant forms of mSmo. Note the localisation to the PC (labelled with anti‐AcTub; red) in each case (n = 4 for each sample). Scale bar, 10 μm.
- MZgrk2 18hpf embryos injected with mRNA encoding GFP‐tagged wild‐type mSmo or mSmoSA (green) showing localisation to the PC (labelled with anti‐AcTub; red) in myotomal cells. More Smo is localised to the PC in MZgrk2 mutants compared to wild type (panel A). Scale bar, 10 μm.
Figure EV3. Smo translocates to the primary cilium independently of Grk2.
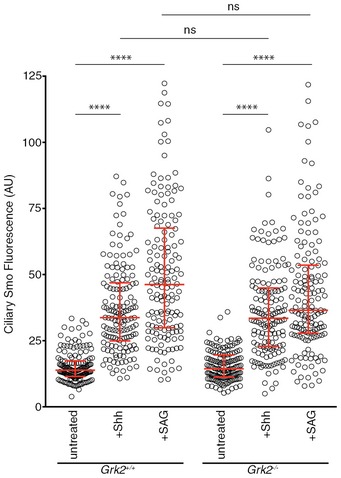
Accumulation of endogenous Smo in the PC of wild‐type and Grk2 −/− Flp‐In‐3T3 cells after 4 h of Shh‐N and SAG treatment. The medians with interquartile range of the fluorescence intensities of endogenous Smo are shown, quantified from multiple cilia (n = 150). Data were analysed using Kruskal–Wallis non‐parametric ANOVA. ****P < 0.001 and n.s. (not significant).
Kinase activity is required for Grk2 function in Hh pathway
Grk2 has been reported to play both kinase‐dependent and kinase‐independent roles in different processes, including Hh signalling 15. Given that phosphorylation of the Smo CTT appears not to be essential for Smo activity in the zebrafish embryo, we next asked whether the kinase activity of Grk2 is dispensable for its function in Hh signalling. Injection of mRNA encoding the archetypal kinase dead form, Grk2K220R, was previously reported not to rescue the effects of morpholino‐mediated knock‐down of Grk2 in zebrafish embryos 13. We found, however, that injection of this mRNA efficiently rescued both the myotomal expression of Prox1a and Eng and the neural tube expression of ptch2 and nkx2.2a (Figs 7B and EV4A). Injection of mRNA encoding two other kinase dead forms of Grk2 (Grk2K220M and Grk2D335N) 17, 33 by contrast failed to rescue fully the expression of any of these markers; however, some Prox1a+ve SSFs were restored by Grk2K220M (10 ± 3, n = 8) (Fig 7B).
Figure 7. Domains of Grk2 required for Hh pathway activity.
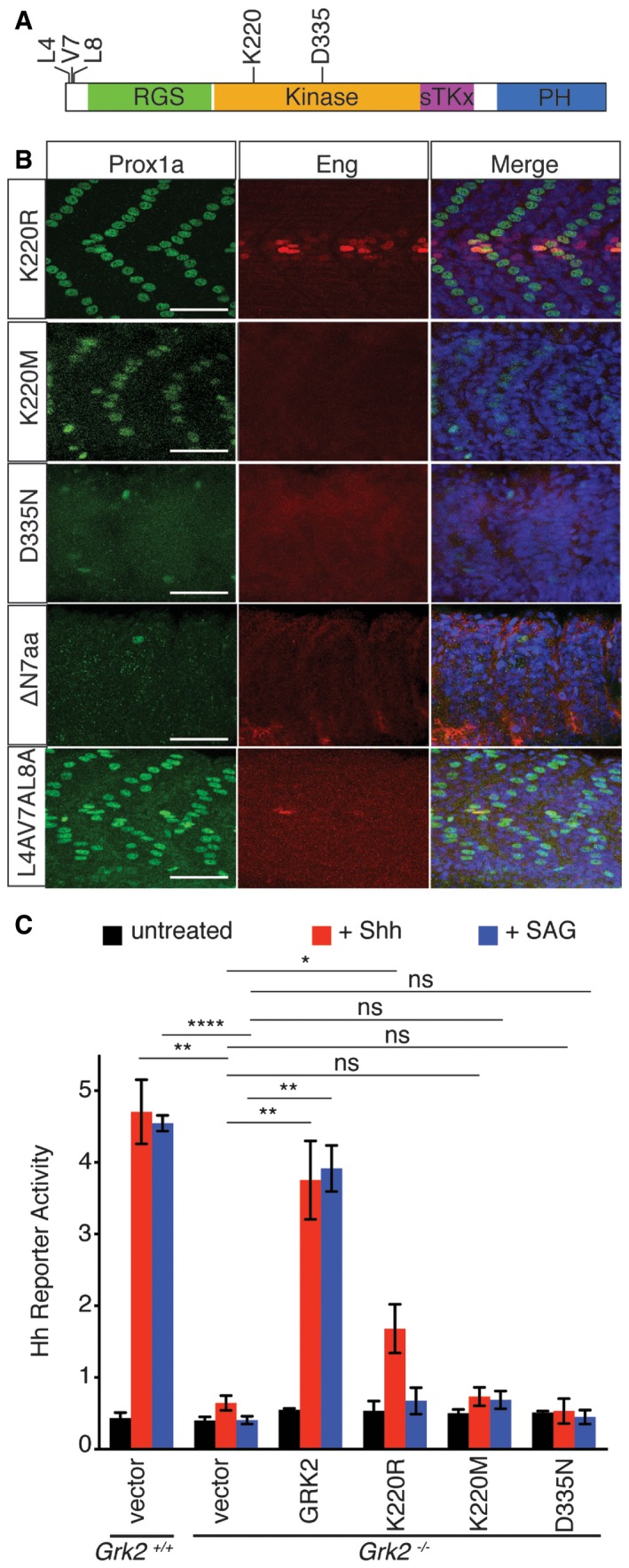
- Schematic representation of the zebrafish Grk2 protein showing the location of residues assayed for the effects of their mutation on rescuing activity.
- Prox1a (green) and Eng (red) expression in the myotome of 30hpf MZgrk2 embryos that were injected with mRNA encoding: Grk2K220R‐GFP; Grk2K220M‐GFP; Grk2D335N‐GFP; Grk2ΔN7aa‐GFP; and mGrk2L4AV7AL8A‐GFP. Merged panels show nuclear staining with DAPI in blue (n = 20 for each sample). Scale bar, 50 μm.
- Hh reporter activity assay in wild‐type and Grk2 −/− 3T3 cells transfected with the equivalent wild‐type and mutant kinase domain forms of mouse Grk2, Grk2K220R, Grk2K220M and Grk2D335N after 24‐h Shh‐N and SAG treatment. Data represent the mean and ± SD (n = 3). Unpaired Student's t‐test was used for analysis. ****P < 0.0001; **P < 0.01; *P < 0.05; and n.s. (not significant).
To explore the disparity between the kinase mutants further, we tested their activity in the Grk2 −/− cells. The lack of Hh responsiveness of the cells was restored by re‐introducing wild‐type mouse GRK2. The kinase mutants GRK2K220R, GRK2K220M and GRK2D335N failed to restore full Hh responsiveness as indicated by absence of luciferase activity compared to wild‐type cells, although GRK2K220R‐transfected cells showed an attenuated response to Shh (~40% compared to wild type), but not to SAG, consistent with GRK2K220R retaining some kinase activity (Fig 7C).
The Grk2 domains have been well characterised by structural and functional analyses 34; injection of mRNA encoding zebrafish Grk2∆RGS, Grk2∆kinase and Grk2∆PH showed that none of these mutant forms of Grk2 can rescue the Hh defects of MZgrk2 mutants, indicating that each of these domains is essential for Grk2 function in Hh signalling (Fig EV4B). The N‐terminal and C‐terminal domains of Grk2 have been shown to be important for interaction with GPCRs and Gβγ, respectively. Injection of mRNA encoding Grk2∆CMargin completely rescued the Prox1a and Eng expression in the myotome, whereas mRNA encoding Grk2∆NMargin had no activity (Fig EV4B). Grk2 has been proposed to interact with GPCRs via an alpha helix in the N‐terminal region of the molecule 35; mRNA encoding a smaller deletion of the first seven residues, Grk2∆N7aa, or mutations of key amino acids involved with GPCR interaction, Grk2L4AV7AL8A were tested for MZgrk2 rescue. Grk2∆N7aa failed to rescue the MZgrk2 phenotype, whereas Grk2L4AV7AL8A partially rescued (Fig 7B).
Figure EV4. MZgrk2 is fully rescued by mRNA encoding Grk2K220R and Grk2 domain swapping assay.
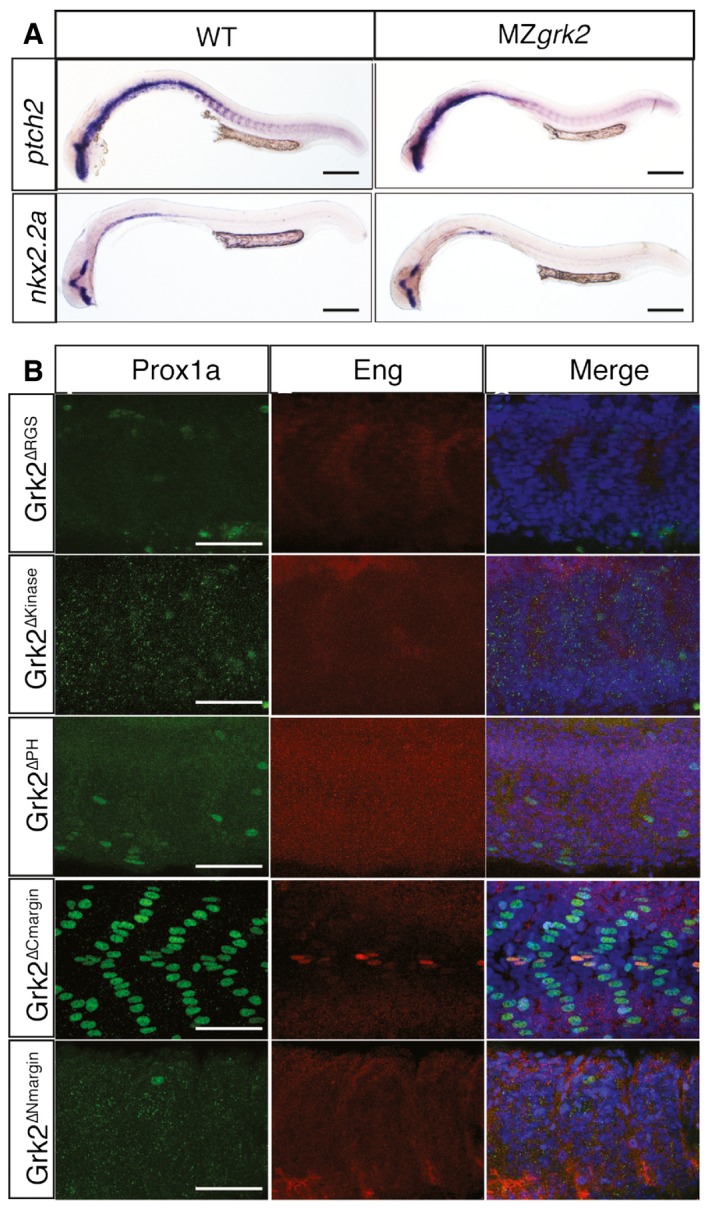
- In situ hybridisation of ptch2 and nkx2.2a in 24hpf wild‐type (left column) and MZgrk2 embryos expressing Grk2K220R‐GFP (right column) (n = 30 for each sample). Scale bar, 200 μm.
- Prox1a (green) and Eng (red) expression in the myotome of 30hpf MZgrk2 embryos injected with mRNA encoding Grk2ΔRGS‐GFP, Grk2Δkinase‐GFP, Grk2ΔPH‐GFP, Grk2ΔCmargin‐GFP and Grk2ΔNmargin‐GFP. Merge images show nuclei labelled with DAPI (blue) (n = 20). Scale bar, 50 μm.
Discussion
A role for GRK2 in Hh signalling was first suggested by siRNA‐mediated knock‐down experiments in cultured mammalian cells and validated in vivo by morpholino‐mediated knock‐down experiments in zebrafish 12, 14. The generation and analysis of a zebrafish grk2 null allele presented here has confirmed an essential requirement for Grk2 in the response of cells to Hh signalling; zebrafish embryos lacking all Grk2 function are devoid of almost all response to Hh activity and resemble embryos homozygous for smo null mutations 24. In contrast to smo, however, the maternal expression of grk2 is sufficient to support normal embryonic development up to 4dpf. Our findings indicate a more critical requirement for Grk2 activity than has hitherto been appreciated. The mild morphant phenotype in zebrafish embryos together with the moderate reduction in Shh responsiveness of cells elicited by Grk2 siRNA led to the conclusion that GRK2 acts only to potentiate the response of cells to Hh 13, 14. However, our analysis of both grk2 embryos and mutant cells reveals that the response to Hh is effectively abolished in the absence of Grk2. Notably, neither zygotic grk2 nor MZgrk2 zebrafish embryos show the developmental retardation reported to occur in Grk2 morphants 22, suggesting this to be an off‐target effect of the morpholino. This underlines the need for caution in interpreting effects associated with morpholino antisense oligonucleotide injection and the importance of analysing stable transmissible null mutations when characterising gene function. The dramatic loss of Hh signalling observed in the zebrafish MZgrk2 mutants stands in contrast to the rather mild phenotype of the mouse Grk2 mutation 14, which might be explained by partial redundancy between Grk2 and the paralogous Grk3 gene. Notably, our Grk2 −/− cells, which show a strong loss of Shh responsiveness, do not express Grk3 (Fig EV5).
Figure EV5. Grk3 is not expressed in NIH3T3 cells.
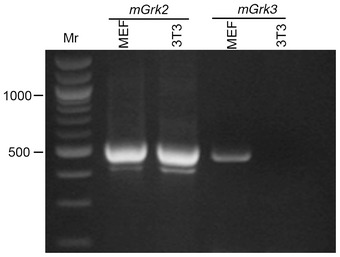
RT–PCR assay of Grk2 and Grk3 expression in MEF and NIH 3T3 cells.
Several analyses have led to the conclusion that the principal role of Grk2 in Hh signalling is to potentiate activity of Smo by phosphorylating its CTT 15, 16, 17. In Drosophila, although PKA is primarily responsible for Smo phosphorylation, CK1 and Gprk2 have also been implicated in this process 15, 17, 36. Drosophila and vertebrate Smo have divergent CTTs, and the PKA phosphorylation sites are not found in vertebrate Smo. The phosphorylation is postulated to neutralise the positive charge of the lysine/arginine cluster in the CTT, causing a conformational change that promotes dimerisation and ciliary localisation, activating Smo 16. Our in vivo data challenge this model: first, SmoSA as well as Smo14SA, which lacks all the predicted phosphorylation sites, fully rescued the smo mutant phenotype. Second, mutation of the phosphorylation sites in the SmoA1 gain of function mutant attenuated but did not abolish its constitutive activity. Third, the phosphomimetic SmoSD mutant, while remaining responsive to Hh, lacked constitutive activity in zebrafish embryos. Finally, the SmoKRA mutant, in which the CTT lysine/arginine residues are replaced with alanine, showed no evidence of constitutive activity. In addition, we found that wild‐type Smo and SmoSA localised to the PC both in wild‐type and in grk2 mutant embryos. Based on these findings, we conclude that phosphorylation of the CTT, at least at the predicted GRK2/CK1 sites, is dispensable for Smo localisation and neither necessary nor sufficient for Smo activation. Our findings that the SmoSD and SmoKRA mutants lack constitutive activity in MEFs, together with the ability of Smo to localise to the PC of Grk2 −/− cells, support this conclusion. On the other hand, the finding that SmoSA mutant lacked activity in the MEF assay is in line with previous reports 16, and consistent with phosphorylation of the GRK2/CK1 sites being necessary, if not sufficient, for Smo activity, at least in these mammalian cells. Why there should be a difference in the requirement for these sites between zebrafish and mammals is unclear, though such differences in Hh pathway activity between zebrafish and mammals are not unprecedented; the serine–threonine kinase, STK36, for instance, has been shown to be required for efficient Hh signal transduction in zebrafish yet is dispensable for the response to Hh in mouse 37, 38. Direct analysis of the phosphorylation status of Smo in wild‐type and grk2 mutant zebrafish and mouse embryos will be required to shed further light on this issue.
Previous studies in Drosophila have concluded that Gprk2 plays both kinase‐dependent and kinase‐independent roles in the response of cells to Hh activity, a conclusion based on the ability of the K220M kinase domain mutant to effect a partial rescue of the gprk2 mutant phenotype in the wing imaginal disc 15. In line with this, we found that the same mutant form of Grk2 could partially rescue the MZgrk2 mutant phenotype in zebrafish. More surprisingly, the archetypal K220R “kinase dead” mutant effected a complete rescue of the MZgrk2, contrary to the previous report that it could not rescue the grk2 morphant phenotype. This latter finding could reflect the conservative nature of this mutation, the positively charged lysine residue, essential for interaction with the alpha/beta phosphate of ATP 39, 40, being replaced by a similarly positively charge arginine residue, which might thereby result in the mutant form retaining significant catalytic activity. On the other hand, we found that the Grk2D335N mutant form completely failed to rescue the MZgrk2 mutant; while this implies that the kinase domain is essential for all responses to Hh activity, we cannot exclude the possibility that this substitution also disrupts some other aspect of Grk2 function.
If Grk2 does not function by activating Smo, what then is its target? Our findings are consistent with a role for Grk2 in controlling the trafficking of a G‐protein‐coupled receptor (GPCR): first, the kinase activity is required for Grk2 function in Hh pathway; second, loss of the N‐terminal region of Grk2, involved in interaction with GPCR, abolishes its rescuing activity. Recent studies have implicated several orphan GPCRs in modulation of the Hh pathway via classical G‐protein signalling mechanisms 8, 9: in the neural tube, for instance, GPR161 suppresses Hh pathway activity promoting cAMP production via the Gαs–adenylate cyclase pathway; this in turn leads to activation of PKA, thereby promoting the production of the repressor forms of Gli. Hh pathway activation counters this effect by promoting internalisation of GPR161, leading to a decrease in cellular cAMP levels and hence a reduction in PKA activity 8. It is tempting to speculate that this GPCR could be a target of Grk2. Testing this hypothesis will require the generation of mutations in the two zebrafish GPR161 orthologues.
Materials and Methods
Zebrafish strains and husbandry
Adult fish were maintained on a 14‐h light/10‐h dark cycle at 28°C in the AVA (Singapore)‐certificated IMCB Zebrafish Facility. Zebrafish strains used were grk2 i283 (this study); smo hi1640 31; Tg(Eng2a:eGFP) i233 41.
Generation, selection and genotyping of zebrafish mutant alleles
Plasmids encoding zinc finger nuclease (ZFN) were synthesised by ToolGen, Korea. Capped mRNA was produced using mMessage mMachine kit according to the manufacturer's protocol and injected into 1‐cell stage embryos at a dose of 100 pg per embryo. G0 adults derived from embryos injected with ZFN mRNA were in‐crossed and their progenies were individually genotyped by PCR using the forward primer (5′‐CTC TCT CGC GCA TCA ACA TCA TCA TCT‐3′) and the reverse primer (5′‐GGT GAA CTA GCT CTT TAT TAC TGA ACA‐3′) followed by Sanger sequencing using primer (5′‐CTG GCT GGA CTC GGT GCT GGT GT‐3′). Founders transmitting a single allele (grk2 i283, 111delC) were selected and used to establish mutant line.
The smo hi1640 mutant allele was genotyped by PCR using the forward primer (5′‐CTA CTT TGT TGC GTC TCC AAG ATG TC‐3′) and the reverse primer (5′‐GAG GGT CTC CTC TGA GTG ATT GAC TAC‐3′). Homozygous mutants were identified by the presence of smo hi1640 mutant allele and the absence of the wild‐type allele using the same forward primer as before and a reverse primer (5′‐CCA GAC CAC ATG GCC AAT TTC TCG‐3′).
Generation of MZgrk2 by germ cell replacement
Germ cell replacement was performed as described 23. In brief, donor embryos from a grk2 i283/+ × grk2 i283/+ in‐cross were labelled by injecting GFP‐nanos‐3′UTR RNA 42. Wild‐type host embryos were injected with morpholino antisense oligonucleotide against dead end (5′‐GCT GGG CAT CCA TGT CTC CGA CCA T‐3′) to block germ cell development 43. Cells were transplanted from donors (dome stage) into hosts at the same stage. Donor embryos were genotyped by PCR using the primers described above. Transplanted host embryos were screened at 24hpf for successful transfer of donor germ cells indicated by GFP expression 23.
Cell lines
Flp‐In‐3T3 and 293FT cells (Life Technologies) were cultured in Dulbecco's modified Eagle's medium (DMEM) containing high glucose (Thermo Scientific) and supplemented with 10% foetal bovine serum (FBS) (Atlanta Biologicals), 1 mM sodium pyruvate, 2 mM l‐glutamine, 1× MEM non‐essential amino acids solution, penicillin (40 U/ml) and streptomycin (40 μg/ml), in a humidified atmosphere containing 5% CO2 at 37°C. Experiments in Smo −/− MEFs were performed as previously described 44. To induce ciliation, cells were grown to confluence in medium containing 10% FBS and then switched to medium containing 0.5% FBS with Hh pathway agonists (SHH‐N and 200 nM SAG) for 24 h (for reporter assays and Western blotting) and 4 h (for Smo immunofluorescence staining). SHH‐N‐conditioned media was generated as previously described 45.
Generation of Grk2 −/− cells
Flp‐In‐3T3 Grk2 −/− cells were generated by CRISPR‐Cas9 gene editing technology as previously described 46. Briefly, guide RNA sequences targeting Grk2 (guide‐1: 5′‐AAT GCT ACG GAG CAT GTC C‐3′; guide‐2: 5′‐CTG GAA CAC GTC CCC TCG G‐3′; guide‐3: 5′‐TCA GTG TGC ATC GAA TCA T‐3′; guide‐4: 5′‐TGC ATC GAA TCA TCG GGC GT‐3′) were cloned into lentiCRISPR v2 plasmid (Addgene#52961). To produce lentivirus, 293FT (3 × 106 cells) seeded in 10‐cm plates were transfected with 8 μg lentiCRISPR v2 plasmid, 4 μg pCMV‐VSV‐G (Addgene#8454), 4 μg psPAX2 (Addgene#12260) and 48 μl of 1 mg/ml polyethylenimine (Polysciences). Lentivirus was collected 60 h posttransfection and filtered using a 0.45‐μm low‐protein binding membrane (Pall Corporation). Flp‐In‐3T3 cells were transduced twice with the lentivirus for 48 h and later selected in puromycin‐containing DMEM (2 μg/ml) for 10 days.
Hh reporter assays
Flp‐In‐3T3 wild‐type and Grk2 −/− (lentiCRISPR guide‐2) cells were seeded in 96‐well plates and transfected using X‐tremeGENE 9 (Roche) following manufacturer's instructions. The transfection mix consisted of a 4:1 w/w ratio of a firefly luciferase reporter driven by an 8xGli‐responsive promoter and a Renilla luciferase reporter driven by a constitutive TK promoter (Promega) along with a vector control and GRK2, mSmoA1 and YF‐PKI/PKI‐M. Cells were grown to confluence and then serum‐starved for 24 h with SHH‐N (used at 1:4 dilution of the conditioned media) and SAG (200 nM). SHH‐N‐conditioned media was produced using HEK293 cells 45. Reporter activity was measured using the Dual‐Luciferase Reporter kit (Promega) and read on a Synergy H1 Hybrid Multi‐Mode Microplate Reader (BioTek). The GLI luciferase to Renilla luciferase ratio is reported as “Hh reporter activity”.
In situ hybridisation and immunofluorescence
Standard in situ hybridisation was performed with anti‐Dig alkaline phosphatase and chromogenic substrate NBT/BCIP as previously described 47. RNA probes were prepared from templates as previously described: ptch2 (formerly ptc1) 48, nkx2.2a 49 and olig2 50.
Whole‐mount antibody staining was performed as previously described at the following dilutions: mAb4D9 (anti‐Engrailed, DSHB) at 1:50 26; rabbit anti‐Prox1a at 1:5,000 (Millipore); mouse anti‐acetylated α‐tubulin at 1:500 (Sigma); chicken anti‐GFP at 1:500 (Abcam) 51. The secondary antibodies were as follows: Alexa488‐conjugated donkey anti‐mouse, anti‐rabbit and anti‐chicken, Alexa546‐conjugated donkey anti‐mouse and anti‐rabbit, Alexa647‐conjugated donkey anti‐mouse and anti‐rabbit secondary antibodies (1:1,000, Invitrogen). Rabbit polyclonal antibodies against mouse Smo were used for the detection of ciliary localisation of Smo in Flp‐In‐3T3 cells as previously described 6. Ciliary Smo quantifications were done as previously described 52. Prox1a+ve cells were counted in 3–5 somites from 4 to 10 embryos rostral to the end of yolk extension.
Protein analysis
Standard Western blotting on fish embryo lysates was performed as previously described 53. Primary antibodies used were as follows: rabbit anti‐zebrafish Gli2a (1:5,000) 41; mouse anti‐γ‐tubulin at 1:5,000 (Sigma); mouse anti‐myc 1:2,000 (9E10, Santa Cruz Biotechnology, Inc); rabbit anti‐GRK3 1:2,500 (sc‐563, raised against C‐terminal of bovine Grk3; Santa Cruz Biotechnology, Inc) 14; mouse anti‐GFP 1:3,000 (632569, Clontech); and mouse anti‐Myc 1:3,000 (Santa Cruz Biotechnology, Inc). The secondary antibodies were HRP‐conjugated goat anti‐mouse and anti‐rabbit at 1:20,000. Western blotting on cell lines was performed as previously described 52, using the following antibodies: mouse anti‐Grk2 antibody (sc‐13143, raised against 468–689 of human GRK2; Santa Cruz Biotechnology); mouse anti‐GLI1 antibody (2643, Cell Signaling); goat anti‐GLI3 antibody (AF3690, R&D); mouse anti‐α‐tubulin antibody (T6199, Sigma); and rabbit anti‐PTCH1 6.
To test the stability of Smo, 600 pg mRNA of the bicistronic Smo P2A construct was injected. Approximately 100 embryos were harvested at 18hpf and deyolked as previously described 53. About 0.5 μl of PBS with proteinase inhibitor was added per embryo. Vigorous pipetting was used to lyse the cells. Lysates were incubated on ice for 30 min with occasional vigorous pipetting. Lysates were spun down at 16,200 g for 15 min, and the supernatant was removed for GFP Western blot analysis. The pellet‐containing insoluble Smo was washed once with PBS and solubilised with 50 mM Tris pH 6.8, 1% Triton X‐100 and 8 M urea. Insoluble material was removed by centrifuging at 16,200 g for 15 min, and the supernatant was used for Myc Western blot analysis. Extract from approximately 40 embryos was loaded in each well. The intensity of Myc and GFP signal was quantified using ImageJ. Mutant Smo stability was assessed by normalising the Myc/GFP intensity ratio against wild‐type Smo.
DNA constructs, synthetic RNA synthesis, morpholinos and microinjection
Wild‐type and all mutant forms of zebrafish grk2 and mouse Smo were tagged with GFP at their C‐termini and cloned into pCS2+ vector. To test Smo stability, a bicistronic Smo was Myc‐tagged at its C‐terminus followed by a self‐cleaving P2A peptide sequence and GFP. Capped mRNA was synthesised using the SP6 mMessage mMachine Kit (Ambion), using plasmids linearised by NotI. shh and dnPKA mRNA for injection were synthesised as described 48. For smo, 500 pg of mRNA was injected. For transient knock‐down of zGrk2, 1 nl of the 0.1 mM ATG antisense morpholino: 5′‐AGG TCC GCC ATC TTC GCC CTC TGG G‐3′ (synthesised by Gene Tools, Philomath, OR), was injected into embryos at the 1‐cell stage as previously described 14. To test the activity of wild‐type and mutant forms of GRK2 in cell lines, mouse Grk2 was tagged with a C‐terminal GFP and cloned into pEF5/FRT/V5‐DEST (Life Technologies). YF‐PKI and YF‐PKI‐M were kindly provided by Dr. Takanari Inoue (Johns Hopkins University, USA). To test the activity of mutant forms of Smo in Smo −/− MEFs, the same constructs used for generating mRNA were used.
Quantitative RT–PCR and RT–PCR
RNA was isolated from 30 embryos at the 18 somite stage (ss) using Trizol (Invitrogen). Two milligrams of RNA was then used for generating cDNA using SuperScript III reverse transcriptase (Invitrogen) and oligodT primers (Fermentas). About 0.2 μl of cDNA was used for the qPCR (Kapa Sybr Fast Universal qPCR kit, Kapa Biosystem). Normalisation was done against β‐actin, and primers used for the quantification of β‐actin and Patched2 are as follows: actin F: 5′‐CTC TTC CAG CCT TCC TTC CT‐3′; actin R: 5′‐CAC CGA TCC AGA CGG AGT AT‐3′; ptch2 F: 5′‐CCT GGT GTG TGC CAT CCT CCT G‐3′; and ptch2 R: 5′‐TCC ATA GCA ACG GCT GAA CGA G‐3′. RNA of tissue culture and cDNA was prepared as described above. One microlitre of cDNA was used for RT–PCR. The mGrk2 primers are F: 5′‐GCT CAG GAG GTA AAA GAA AGT CC‐3′ R: 5′‐CGT TCC TTG ATC TGT GTC TCT TC‐3′; and the mGrk3 primers are F: 5′‐GGC AGC ATA AAA CCA AAG ACA AG‐3′ R: 5′‐TAG CGT AAT CTT CCT CTT GAC CA‐3′.
Author contributions
PWI, RTHL and ZZ conceived the study; PWI, RTHL, GVP, RR and ZZ designed the experiments; AI, RTHL, GVP and ZZ performed the experiments; PWI, RTHL, GVP, RR and ZZ analysed the data; PWI, RTHL, GVP and ZZ drafted the manuscript. All authors reviewed and provided feedback on the final draft of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
This study was supported by the Toh Kian Chui Foundation and the Agency for Science Technology and Research (A*STAR) (to PWI), National Institutes of Health grants GM102261 and DP2GM10544801 (to RR) and a fellowship award from the American Heart Association (to GVP). We thank Marc G. Caron for the zebrafish grk2 plasmid and Niah Weixin for support in maintaining zebrafish lines. The mAb 4D9, developed by the Goodman laboratory, was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242.
EMBO Reports (2016) 17: 739–752
Reference
- 1. Ingham PW, Nakano Y, Seger C (2011) Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet 12: 393–406 [DOI] [PubMed] [Google Scholar]
- 2. Price MA, Kalderon D (1999) Proteolysis of cubitus interruptus in Drosophila requires phosphorylation by protein kinase A. Development 126: 4331–4339 [DOI] [PubMed] [Google Scholar]
- 3. Wang G, Wang B, Jiang J (1999) Protein kinase A antagonizes Hedgehog signaling by regulating both the activator and repressor forms of Cubitus interruptus. Genes Dev 13: 2828–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pan Y, Wang C, Wang B (2009) Phosphorylation of Gli2 by protein kinase A is required for Gli2 processing and degradation and the Sonic Hedgehog‐regulated mouse development. Dev Biol 326: 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Denef N, Neubüser D, Perez L, Cohen SM (2000) Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 102: 521–531 [DOI] [PubMed] [Google Scholar]
- 6. Rohatgi R, Milenkovic L, Scott MP (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science 317: 372–376 [DOI] [PubMed] [Google Scholar]
- 7. Endoh‐Yamagami S, Evangelista M, Wilson D, Wen X, Theunissen J‐W, Phamluong K, Davis M, Scales SJ, Solloway MJ, de Sauvage FJ et al (2009) The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr Biol 19: 1320–1326 [DOI] [PubMed] [Google Scholar]
- 8. Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, Jackson PK (2013) The ciliary G‐protein‐coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 152: 210–223 [DOI] [PubMed] [Google Scholar]
- 9. Singh J, Wen X, Scales SJ (2015) The Orphan G Protein‐coupled Receptor Gpr175 (Tpra40) Enhances Hedgehog Signaling by Modulating cAMP Levels. J Biol Chem 290: 29663–29675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bassilana F, Carlson A, DaSilva JA, Grosshans B, Vidal S, Beck V, Wilmeringwetter B, Llamas LA, Showalter TB, Rigollier P et al (2014) Target identification for a Hedgehog pathway inhibitor reveals the receptor GPR39. Nat Chem Biol 10: 343–349 [DOI] [PubMed] [Google Scholar]
- 11. Premont RT, Gainetdinov RR (2007) Physiological roles of G protein‐coupled receptor kinases and arrestins. Annu Rev Physiol 69: 511–534 [DOI] [PubMed] [Google Scholar]
- 12. Chen W, Ren X‐R, Nelson CD, Barak LS, Chen JK, Beachy PA, de Sauvage F, Lefkowitz RJ (2004) Activity‐dependent internalization of smoothened mediated by beta‐arrestin 2 and GRK2. Science 306: 2257–2260 [DOI] [PubMed] [Google Scholar]
- 13. Meloni AR, Fralish GB, Kelly P, Salahpour A, Chen JK, Wechsler‐Reya RJ, Lefkowitz RJ, Caron MG (2006) Smoothened signal transduction is promoted by G protein‐coupled receptor kinase 2. Mol Cell Biol 26: 7550–7560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Philipp M, Fralish GB, Meloni AR, Chen W, MacInnes AW, Barak LS, Caron MG (2008) Smoothened signaling in vertebrates is facilitated by a G protein‐coupled receptor kinase. Mol Biol Cell 19: 5478–5489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen Y, Li S, Tong C, Zhao Y, Wang B, Liu Y, Jia J, Jiang J (2010) G protein‐coupled receptor kinase 2 promotes high‐level Hedgehog signaling by regulating the active state of Smo through kinase‐dependent and kinase‐independent mechanisms in Drosophila . Genes Dev 24: 2054–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen Y, Sasai N, Ma G, Yue T, Jia J, Briscoe J, Jiang J (2011) Sonic Hedgehog dependent phosphorylation by CK1α and GRK2 is required for ciliary accumulation and activation of Smoothened. PLoS Biol 9: e1001083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maier D, Cheng S, Faubert D, Hipfner DR (2014) A broadly conserved g‐protein‐coupled receptor kinase phosphorylation mechanism controls Drosophila smoothened activity. PLoS Genet 10: e1004399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kok FO, Shin M, Ni C‐W, Gupta A, Grosse AS, van Impel A, Kirchmaier BC, Peterson‐Maduro J, Kourkoulis G, Male I et al (2015) Reverse genetic screening reveals poor correlation between morpholino‐induced and mutant phenotypes in zebrafish. Dev Cell 32: 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lewis KE, Eisen JS (2001) Hedgehog signaling is required for primary motoneuron induction in zebrafish. Development 128: 3485–3495 [DOI] [PubMed] [Google Scholar]
- 20. Ingham PW, McMahon AP (2001) Hedgehog signaling in animal development: paradigms and principles. Genes Dev 15: 3059–3087 [DOI] [PubMed] [Google Scholar]
- 21. Winata CL, Korzh S, Kondrychyn I, Zheng W, Korzh V, Gong Z (2009) Development of zebrafish swimbladder: the requirement of Hedgehog signaling in specification and organization of the three tissue layers. Dev Biol 331: 222–236 [DOI] [PubMed] [Google Scholar]
- 22. Jiang X, Yang P, Ma L (2009) Kinase activity‐independent regulation of cyclin pathway by GRK2 is essential for zebrafish early development. Proc Natl Acad Sci USA 106: 10183–10188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ciruna B, Weidinger G, Knaut H, Thisse B, Thisse C, Raz E, Schier AF (2002) Production of maternal‐zygotic mutant zebrafish by germ‐line replacement. Proc Natl Acad Sci USA 99: 14919–14924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen W, Burgess S, Hopkins N (2001) Analysis of the zebrafish smoothened mutant reveals conserved and divergent functions of hedgehog activity. Development 128: 2385–2396 [DOI] [PubMed] [Google Scholar]
- 25. Ben J, Elworthy S, Ng ASM, van Eeden F, Ingham PW (2011) Targeted mutation of the talpid3 gene in zebrafish reveals its conserved requirement for ciliogenesis and Hedgehog signalling across the vertebrates. Development 138: 4969–4978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maurya AK, Ben J, Zhao Z, Lee RTH, Niah W, Ng ASM, Iyu A, Yu W, Elworthy S, van Eeden FJM et al (2013) Positive and negative regulation of Gli activity by Kif7 in the zebrafish embryo. PLoS Genet 9: e1003955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA (2000) Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 406: 1005–1009 [DOI] [PubMed] [Google Scholar]
- 28. Blagden CS, Currie PD, Ingham PW, Hughes SM (1997) Notochord induction of zebrafish slow muscle mediated by Sonic hedgehog. Genes Dev 11: 2163–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Currie PD, Ingham PW (1996) Induction of a specific muscle cell type by a hedgehog‐like protein in zebrafish. Nature 382: 452–455 [DOI] [PubMed] [Google Scholar]
- 30. Hammerschmidt M, Bitgood MJ, McMahon AP (1996) Protein kinase A is a common negative regulator of Hedgehog signaling in the vertebrate embryo. Genes Dev 10: 647–658 [DOI] [PubMed] [Google Scholar]
- 31. Varga ZM, Amores A, Lewis KE, Yan YL, Postlethwait JH, Eisen JS, Westerfield M (2001) Zebrafish smoothened functions in ventral neural tube specification and axon tract formation. Development 128: 3497–3509 [DOI] [PubMed] [Google Scholar]
- 32. Asai D, Toita R, Murata M, Katayama Y, Nakashima H, Kang J‐H (2014) Peptide substrates for G protein‐coupled receptor kinase 2. FEBS Lett 588: 2129–2132 [DOI] [PubMed] [Google Scholar]
- 33. Ferguson SS, Ménard L, Barak LS, Koch WJ, Colapietro AM, Caron MG (1995) Role of phosphorylation in agonist‐promoted beta 2‐adrenergic receptor sequestration. Rescue of a sequestration‐defective mutant receptor by beta ARK1. J Biol Chem 270: 24782–24789 [DOI] [PubMed] [Google Scholar]
- 34. Penela P, Murga C, Ribas C, Lafarga V, Mayor F (2010) The complex G protein‐coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol 160: 821–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beautrait A, Michalski KR, Lopez TS, Mannix KM, McDonald DJ, Cutter AR, Medina CB, Hebert AM, Francis CJ, Bouvier M et al (2014) Mapping the putative G protein‐coupled receptor (GPCR) docking site on GPCR kinase 2: insights from intact cell phosphorylation and recruitment assays. J Biol Chem 289: 25262–25275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jia J, Tong C, Wang B, Luo L, Jiang J (2004) Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 432: 1045–1050 [DOI] [PubMed] [Google Scholar]
- 37. Wolff C, Roy S, Ingham PW (2003) Multiple muscle cell identities induced by distinct levels and timing of hedgehog activity in the zebrafish embryo. Curr Biol 13: 1169–1181 [DOI] [PubMed] [Google Scholar]
- 38. Chen M‐H, Gao N, Kawakami T, Chuang P‐T (2005) Mice deficient in the fused homolog do not exhibit phenotypes indicative of perturbed hedgehog signaling during embryonic development. Mol Cell Biol 25: 7042–7053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Singh P, Wang B, Maeda T, Palczewski K, Tesmer JJG (2008) Structures of rhodopsin kinase in different ligand states reveal key elements involved in G protein‐coupled receptor kinase activation. J Biol Chem 283: 14053–14062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hanks SK, Hunter T (1995) Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J 9: 576–596 [PubMed] [Google Scholar]
- 41. Maurya AK, Tan H, Souren M, Wang X, Wittbrodt J, Ingham PW (2011) Integration of Hedgehog and BMP signalling by the engrailed2a gene in the zebrafish myotome. Development 138: 755–765 [DOI] [PubMed] [Google Scholar]
- 42. Köprunner M, Thisse C, Thisse B, Raz E (2001) A zebrafish nanos‐related gene is essential for the development of primordial germ cells. Genes Dev 15: 2877–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weidinger G, Stebler J, Slanchev K, Dumstrei K, Wise C, Lovell‐Badge R, Thisse C, Thisse B, Raz E (2003) dead end, a novel vertebrate germ plasm component, is required for zebrafish primordial germ cell migration and survival. Curr Biol 13: 1429–1434 [DOI] [PubMed] [Google Scholar]
- 44. Nachtergaele S, Whalen DM, Mydock LK, Zhao Z, Malinauskas T, Krishnan K, Ingham PW, Covey DF, Siebold C, Rohatgi R (2013) Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. eLife 2: e01340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen JK, Taipale J, Young KE, Maiti T, Beachy PA (2002) Small molecule modulation of Smoothened activity. Proc Natl Acad Sci USA 99: 14071–14076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG et al (2014) Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Oxtoby E, Jowett T (1993) Cloning of the zebrafish krox‐20 gene (krx‐20) and its expression during hindbrain development. Nucleic Acids Res 21: 1087–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Concordet JP, Lewis KE, Moore JW, Goodrich LV, Johnson RL, Scott MP, Ingham PW (1996) Spatial regulation of a zebrafish patched homologue reflects the roles of sonic hedgehog and protein kinase A in neural tube and somite patterning. Development 122: 2835–2846 [DOI] [PubMed] [Google Scholar]
- 49. Barth KA, Wilson SW (1995) Expression of zebrafish nk2.2 is influenced by sonic hedgehog/vertebrate hedgehog‐1 and demarcates a zone of neuronal differentiation in the embryonic forebrain. Development 121: 1755–1768 [DOI] [PubMed] [Google Scholar]
- 50. Park H‐C, Mehta A, Richardson JS, Appel B (2002) olig2 is required for zebrafish primary motor neuron and oligodendrocyte development. Dev Biol 248: 356–368 [DOI] [PubMed] [Google Scholar]
- 51. Lee RTH, Nagai H, Nakaya Y, Sheng G, Trainor PA, Weston JA, Thiery JP (2013) Cell delamination in the mesencephalic neural fold and its implication for the origin of ectomesenchyme. Development 140: 4890–4902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pusapati GV, Hughes CE, Dorn KV, Zhang D, Sugianto P, Aravind L, Rohatgi R (2014) EFCAB7 and IQCE regulate hedgehog signaling by tethering the EVC‐EVC2 complex to the base of primary cilia. Dev Cell 28: 483–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang X, Zhao Z, Muller J, Iyu A, Khng AJ, Guccione E, Ruan Y, Ingham PW (2013) Targeted inactivation and identification of targets of the Gli2a transcription factor in the zebrafish. Biol Open 2: 1203–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File