

Abstract
The spleen plays critical roles in immunity and also provides a permissive microenvironment for hematopoiesis. Previous studies have reported that the TALE‐class homeodomain transcription factor Pbx1 is essential in hematopoietic stem and progenitor cells (HSPCs) for stem cell maintenance and progenitor expansion. However, the role of Pbx1 in the hematopoietic niche has not been investigated. Here we explored the effects that genetic perturbation of the splenic mesenchymal niche has on hematopoiesis upon loss of members of the Pbx family of homeoproteins. Splenic mesenchyme‐specific inactivation of Pbx1 (SKO) on a Pbx2‐ or Pbx3‐deficient genetic background (DKO) resulted in abnormal development of the spleen, which is dysmorphic and severely hypoplastic. This phenotype, in turn, affected the number of HSPCs in the fetal and adult spleen at steady state, as well as markedly impairing the kinetics of hematopoietic regeneration in adult mice after sub‐lethal and lethal myelosuppressive irradiation. Spleens of mice with compound Pyx deficiency 8 days following sublethal irradiation displayed significant downregulation of multiple cytokine‐encoding genes, including KitL/SCF, Cxcl12/SDF‐1,IL‐3,IL‐4,GM‐CSF/Csf2 IL‐10, and Igf‐1, compared with controls. KitL/SCF and Cxcl12/SDF‐1 were recently shown to play key roles in the splenic niche in response to various haematopoietic stresses such as myeloablation, blood loss, or pregnancy. Our results demonstrate that, in addition to their intrinsic roles in HSPCs, non‐cell autonomous functions of Pbx factors within the splenic niche contribute to the regulation of hematopoiesis, at least in part via the control of KitL/SCF and Cxcl12/SDF‐1. Furthermore, our study establishes that abnormal spleen development and hypoplasia have deleterious effects on the efficiency of hematopoietic recovery after bone marrow injury.
Keywords: Cxcl12/SDF‐1, extramedullary hematopoiesis, GM‐CSF, hematopoietic stem cells, Nkx2‐5+ spleen niche KitL/SCF, Pbx transcription factors, spleen hypoplasia
Introduction
The adult mammalian spleen is a secondary lymphoid organ with prominent roles in immune responses (Bronte & Pittet, 2013). During mouse development, at gestational day (E) 9.5, the first recognizable sign of the developing spleen is an apparent condensation of round, undifferentiated cells of splanchnic/visceral mesodermal derivation (Brendolan et al. 2007). This early condensation comprises mesenchymal cells surrounded by columns of epithelial‐like cells (Funayama et al. 1999; Hecksher‐Sorensen et al. 2004). The latter are transient structures, initially found on both sides of the midline gut endoderm. By E10, these columns of epithelial‐like cells persist only on the left‐hand side, lateral to the developing stomach, within a structure named dorsal mesogastrium (Dm), the future greater omentum (Hecksher‐Sorensen et al. 2004; Burn et al. 2008; reviewed by Brendolan et al. 2007). The murine spleen anlage proper is detectable at approximately E10.5–11, as mesenchymal progenitor cells condense on the left side within the Dm, adjacent to the stomach and dorsal pancreas. By E10.5, splenic mesenchymal cells proliferate considerably and begin the expression of splenic markers including Hox11/Tlx1, Bapx1/Nkx3‐2, Wt‐1, Tcf21, Nkx2‐5, Pbx1 and Sox11. Mouse embryos with loss‐of‐function for each of the above proteins develop spleen agenesis or hypoplasia (reviewed by Brendolan et al. 2007). Given the close association of spleen and dorsal pancreas during the early development of the spleen, it is difficult to distinguish these two tissues morphologically before E12.5. Thus, only by using spleen and pancreatic molecular markers can we can delineate their anatomical domains in the early mouse embryo (reviewed by Brendolan et al. 2007). Through morphogenesis and growth of the mesenchyme, as well as interaction of the mesenchymal cells with invading endothelial and hematopoietic cells, the spleen develops as an asymmetrical organ in the upper left part of the abdomen (reviewed by Brendolan et al. 2007).
Histologically, the adult spleen is organized into red and white pulp (RP and WP, respectively). Monocytes and red blood cells (RBCs) are enriched in the RP, and the WP harbors B and T lymphocytes and macrophages, critical for B cell maturation (Brendolan et al. 2007; den Haan et al. 2012; Tarlinton & Good‐Jacobson, 2013). While it has critical roles in immunity, the spleen also provides a permissive microenvironment for hematopoiesis, in addition to the fetal liver and the bone marrow (BM), which are the preponderant hematopoietic sites during embryogenesis and postnatal life, respectively (Wolber et al. 2002; Xue et al. 2011; Bronte & Pittet, 2013). Various studies report hematopoietic activity and the presence of hematopoietic stem cells (HSCs) in the fetal spleen as early as E13 (Potocnik et al. 2000; Kiel et al. 2005a; Mikkola & Orkin, 2006; Cao et al. 2013). Notably, clinical procedures such as G‐CSF administration and BM transplantation, which mobilize hematopoietic stem and progenitor cells (HSPCs) into circulation, as well as myeloablative treatments such as chemotherapy, irradiation and other BM injuries, elicit marked hematopoietic activity within the adult spleen, particularly in the RP (Kiel et al. 2005b; Griseri et al. 2012; Leuschner et al. 2012).
Among the earliest molecular markers of the murine spleen primordium is the TALE‐class homeodomain transcription factor Pbx1 (Moens & Selleri, 2006; Longobardi et al. 2014). We have previously shown that Pbx1 is expressed in the lateral plate mesoderm (LPM) of the mouse embryo as early as E8.5, in the dorsal mesogastrium by E9.5, and in the columnar epithelial‐like cells, as well as the underlying splenopancreatic mesenchyme, by E10–11. By E11.5, all mesenchymal cells of the spleen anlage express Pbx1. Pbx1 is also detectable in the prospective spleen capsule and at low levels also in cells associated with sinusoidal vessels (Brendolan et al. 2005; Koss et al. 2012). Furthermore, HSCs, lymphoid and erythroid progenitors, which colonize the developing spleen starting at approximately E13–15, also express Pbx1 (DiMartino et al. 2001; Stanley et al. 2002; Brendolan et al. 2007; Ficara et al. 2008). In addition, the related family member Pbx2 is also abundant in E13.5 spleen mesenchymal cells. Spleen mesenchymal Pbx1 expression persists to P0‐P3, although starting at E16.5, protein levels are considerably lower (Koss et al. 2012). Lastly, lineage tracing has shown that Pbx1‐positive progenitors give rise to the stromal cells of the WP and RP in the adult spleen (Castagnaro et al. 2013). In summary, Pbx1 is expressed in the mesenchyme giving rise to the spleen anlage and ubiquitously in the spleen proper throughout spleen morphogenesis during embryonic development.
Transcription factors of the Pbx family (Pbx1, Pbx2, Pbx3) are master regulators of morphogenesis and organogenesis. They govern the development of multiple organs in the mammalian embryo including the craniofacial complex, axial and appendicular skeleton, heart, pancreas, kidney, and spleen (Selleri et al. 2001; Kim et al. 2002; Brendolan et al. 2005; Capellini et al. 2006, 2008, 2010, 2011; Stankunas et al. 2008; Ferretti et al. 2011; Vitobello et al. 2011; Koss et al. 2012; Hurtado et al. 2015). Mouse embryos with constitutive inactivation of Pbx1 show asplenia due to loss or decreased expression of genes essential for early spleen induction or morphogenesis, such as Hox11, Wt1 and Nkx2‐5 (Brendolan et al. 2005). On the other hand, mesenchymal‐specific inactivation of Pbx1 causes abnormal spleen growth and morphogenesis that result in splenic hypoplasia with fragmentation of the primordium (Koss et al. 2012). This phenotype is caused by a marked proliferation defect of the splenic mesenchyme and is exacerbated by compound loss of one allele of Pbx2 or Pbx3 (Koss et al. 2012). Thus, mice with mesenchymal loss of Pbx1 can be used as a model to dissect the consequences of genetic perturbation of the spleen mesenchyme on hematopoietic colonization, development, and function in vivo.
During embryogenesis, mouse embryos with constitutive homozygous deletion of Pbx1 (Pbx1 –/–) die at E15.5, exhibiting severe fetal anemia with reduced hematocrit and loss of common myeloid progenitors (CMPs) in the fetal liver (DiMartino et al. 2001; Selleri et al. 2001). Furthermore, in chimeras generated by injecting Pbx1 –/– embryonic stem cells into immunodeficient Rag1‐null blastocysts, Pbx1 –/– cells cannot compensate for the lack of common lymphoid progenitors (CLPs), B cells, and natural killer cells (Sanyal et al. 2007). Accordingly, the hematopoietic defects observed in Pbx1 –/– embryos have been attributed to cell‐autonomous functions of Pbx1 in fetal hematopoietic progenitors. Subsequently, it has been shown that in adult murine hematopoiesis, Pbx1 is essential for maintaining quiescence, self‐renewing potential, and multipotency of long‐term repopulating HSCs (LT‐HSCs), while promoting the expansion of hematopoietic progenitors (Ficara et al. 2008). Conditional inactivation of Pbx1 in the hematopoietic system leads to perturbation of cell cycle and transforming growth factor (TGF)‐β pathway‐associated gene expression in LT‐HSCs, which likely drives their loss of self‐renewal potential and impairs long‐term reconstitution (Ficara et al. 2008). Furthermore, it has been reported that Pbx1 loss is associated with a striking reduction of the total number of B cell progenitors, common lymphoid progenitors (CLPs), and common myeloid progenitors (CMPs) in the BM (Ficara et al. 2008, 2013). Taken together, these studies have established that Pbx1 is an essential intrinsic regulator of hematopoiesis. Whether loss of Pbx1 activity in non‐hematopoietic cells contributes to the observed hematopoietic defects in a non‐cell autonomous manner needs further investigation.
Extensive studies of the BM microenvironment have identified multiple extrinsic signals that regulate the biology of HSCs and control the generation of all mature blood cells (Mendelson & Frenette, 2014). Genetic experiments have demonstrated that diverse cell types are key components of the BM stem cell niche, including arterioles, sinusoidal endothelium, Prx‐1+ or Osx+ osteoprogenitors, Col‐2.3+ osteoblasts, macrophages, megakaryocytes, and Leptin receptor+ perivascular cells (Calvi et al. 2003; Omatsu et al. 2010; Ding et al. 2012; Sugimura et al. 2012; Ding & Morrison, 2013; Greenbaum et al. 2013; Kunisaki et al. 2013; Mizoguchi et al. 2014). All of these cell types constitute a tightly controlled microenvironment by providing regulatory signals in the form of bound or secreted molecules. Stem cell factor (SCF)/KitL, Cxcl12, interleukin (IL)‐6, thrombopoietin, osteopontin, pleiotrophin, and E‐selectin, as well as components of the Notch and Wnt signaling pathways, are among the critical cytokines and angiocrine factors that regulate the balance between HSC dormancy and their expansion and differentiation in homeostatic or regenerative conditions (Avecilla et al. 2004; Stier et al. 2005; Trompouki et al. 2011; Varnum‐Finney et al. 2011; Himburg et al. 2012; Winkler et al. 2012; Poulos et al. 2013; Lento et al. 2014; Morrison & Scadden, 2014; Zhao et al. 2014). Unlike for the BM, the cellular and molecular characterization of the spleen hematopoietic niche has been very limited. However, similar to the BM, the spleen contains stromal cells and vascular niches that can support the maintenance of spleen‐resident myeloid progenitors, as well as the proliferation and differentiation of HSPCs during stress conditions (Kiel et al. 2005b; Bertrand et al. 2006; Mueller & Germain, 2009; Castagnaro et al. 2013; Dutta et al. 2015; Inra et al. 2015). Furthermore, cytokines such as Cxcl12 and IL‐6, which promote HSC and progenitor maintenance and proliferation in the BM, are present also in the murine spleen (den Haan et al. 2012). In this study, we utilized a mouse model with spleen mesenchyme‐specific loss of Pbx1, on a Pbx2‐ or Pbx3‐deficient genetic background, to identify potential non‐cell autonomous functions of Pbx homeoproteins in hematopoiesis.
Material and methods
Mice
The mouse lines used in these studies have been previously described by our and other laboratories: Pbx2 –/– constitutive mutants (Selleri et al. 2004); Pbx3 –/– constitutive mutants (Rhee et al. 2004); Pbx1 conditional mutants (Pbx1 flox/flox) (Koss et al. 2012); Nkx2‐5 IRESCre deleters (Stanley et al. 2002); and R26‐stop‐eYFP reporters (Srinivas et al. 2001). Mice carrying compound engineered alleles were obtained by interbreeding, as appropriate for the present studies. Genotypes of all mice were confirmed by PCR of tail DNA using gene‐specific primers as reported in the studies listed above. For all experiments, control and mutant adult mice (3–5 months old) were age‐matched.
Histology
Embryonic and adult mouse spleens were excised and fixed in 4% paraformaldehyde (PFA). For BM histology, 4% PFA‐fixed femurs were decalcified using Decalcifying Solution (Richard‐Allan Scientific) and then processed for embedding in paraffin. Paraffin sections were dewaxed, rehydrated and stained with hematoxylin and eosin (Sigma) following the manufacturer's instructions.
Flow cytometry
Samples were run on FACSAriaII‐SORP (Becton Dickinson) at Weill Cornell Flow Cytometry Core. Data were analyzed using BD facsdiva Software. BM cell suspensions were obtained by flushing femurs in phosphate‐buffered saline (PBS). Single cell suspensions of spleens were generated by digestion in Liver Digest Medium (Invitrogen) for 30 min at 37 °C. BM and spleen cells were strained through 70‐μm nylon mesh filters and washed in PBS. Red blood cells were lysed in BD Pharm Lyse and washed in PBS. Staining of cells for FACS analysis was performed in DMEM containing 1% fetal bovine serum (FBS), 2 mm EDTA, and 25 mm HEPES staining buffer. For blocking, we used 5% normal rat serum or anti‐mouse CD16/32. The following conjugated monoclonal antibodies were used: c‐Kit (2B8), Sca‐1 (D7), CD16/32 (93), and CD71 (RI7217) from Biolegend, as well as CD34 (RAM34) from BD Biosciences. Mouse Lineage antibody cocktail with isotype control was obtained from BD Biosciences (BD Horizon V450‐conjugated), which included CD3e (500A2), CD11b (M1/70), B220 (RA3‐6B2), Ter‐119, and Gr‐1 (RB6‐8C5).
Irradiation and peripheral blood analysis
Age‐matched control and mutant mice of 3–5 months received a sub‐lethal dose irradiation of 650 Rads. Retro‐orbital peripheral blood was collected using microhematocrit capillary tubes (Fisher Scientific) and was diluted 1 : 4 in PBS/EDTA solution. white blood cell (WBC) counts with differential, hemoglobin, and platelets were obtained using Bayer Advia 120 Hematology Analyzer (Bayer Healthcare). For transplantation assays, CD45.1 Control, SKO and DKO recipient mice were lethally irradiated (950 Rads) and transplanted with 105 CD45.2 whole bone marrow cells. Spleens were isolated at day 8 post‐transplantation.
Colony‐forming unit assays
For Spleen Colony Assays, spleens were dissected from sub‐lethally irradiated mice 12 days following irradiation and fixed overnight in Bouin's solutions. Macroscopic nodules visible without magnification were counted. For methylcellulose colony assays, fetal spleens were isolated from E16.5 or E17.5 embryos. Control spleens (Pbx1 f/f , Pbx1 f/f ;Pbx2 +/−, Pbx1 f/f ;Pbx3 +/− or Pbx1 f/+ ;Nkx2‐5 Cre/+ ) were segregated from mutant spleens (Pbx1 f/f ; Nkx2‐5 Cre/+) based on size and morphology and pooled separately. Subsequently, the genotype of each spleen/embryo was confirmed by PCR. The pools of control and mutant spleens were dissociated in parallel to single cell suspensions in Liver Digest Medium (Invitrogen) for 30 min at 37 °C. BM and spleen cells were strained through 70‐μm nylon mesh filters and washed in PBS. Red blood cells were lysed in BD Pharm Lyse and washed in PBS. Cell pellets were then resuspended in IMDM medium supplemented with 2% FBS and counted. In all, 10 500 fetal spleen cells, 100 000 adult spleen cells or 35 000 BM cells of each genotype were mixed with methylcellulose‐containing medium (Stem Cell Technologies Methocult M3434, which contains SCF, IL‐6, IL‐3 and Epo), and seeded in six‐well cell culture plates in duplicates. CFU‐Es were counted on day 3 of culture, and CFU‐GM, BFU‐E, and CFU‐GEMM colonies were counted after 8–9 days of culture under a microscope.
Real‐time quantitative PCR
RNA was isolated using the RNeasy kit (Qiagen). RNA was treated with RNase‐free DNase (Qiagen). cDNA was prepared from equal amounts of mRNA of control and DKO spleens, using Superscript III First‐Strand Synthesis System (Invitrogen). Quantitative RT‐PCR was performed on cDNA using Power SYBR® Green PCR Master Mix (Life Technologies) on TaqMan 7900HT real‐time PCR machine (Applied Biosystems). Transcript levels that were altered more than 2‐fold in DKO spleens compared with controls were initially identified by performing RT‐qPCR on a 384‐well PrimePCR assay platform. The panel included predesigned mouse primers for the top 88 genes known to be involved in hematopoiesis pathways from BioRad (Hemapoiesis Tier 1 M384; Catalogue #10040355). A full primer list will be provided upon request.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Statistical significance of differences was assessed by comparing datasets using unpaired two‐tailed Student's t‐tests. A P‐value ≤ 0.05 was considered significant.
Results
Hematopoietic defects in embryonic spleen with mesenchyme‐specific loss of Pbx1
To characterize hematopoietic development in mice with mesenchymal inactivation of Pbx1, we utilized the Nkx2‐5‐Cre deleter strain (Stanley et al. 2002). As we previously reported (Koss et al. 2012), embryos with conditional loss of Pbx1 in Nkx2‐5‐positive cells of the splenic anlage (Pbx1 f/f ;Nkx2‐5 Cre/+) (hereafter referred to as SKO) and embryos with conditional loss of Pbx1 on a Pbx2‐ or Pbx3‐deficient background (Pbx1 f/f ;Pbx2 +/− ;Nkx2‐5 Cre/+ or Pbx1 f/f ;Pbx3 +/− ;Nkx2‐5 Cre/+; DKO) display spleen hypoplasia and fragmentation in late gestation (Fig. 1A). In addition, mutant spleens are strikingly pale, suggesting a potential defect in erythropoiesis. We previously described that during development, spleen hypoplasia in Pbx conditional mutant embryos at E14.5 is not associated with increased apoptosis, or with gross defects in erythroid cell invasion and vasculogenesis (Koss et al. 2012). Here we asked whether erythropoiesis or development of other hematopoietic lineages was affected in this model later in development or in the adult. Accordingly, we set out to assess whether this phenotype arose solely from lack of hematopoietic progenitor colonization in mutant spleens, due to niche dysfunction, or concomitantly from proliferation and/or differentiation defects in hematopoietic progenitors, secondary to Pbx1 deletion in the splenic stroma.
Figure 1.
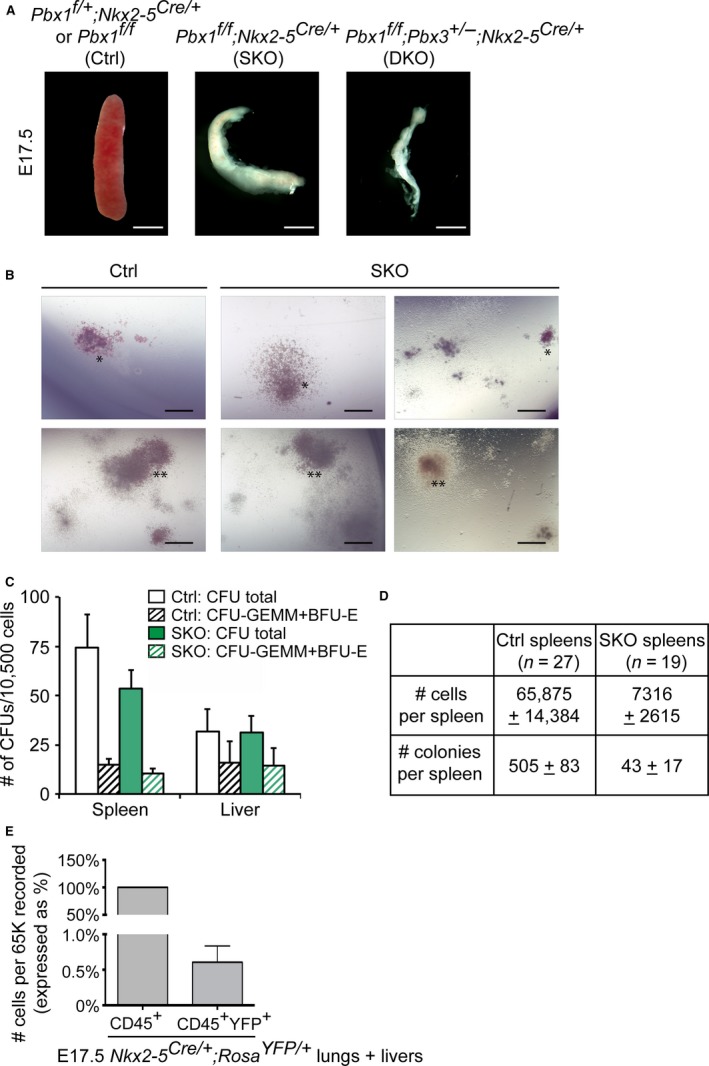
Spleen mesenchyme specific inactivation of Pbx1 results in hypoplastic embryonic spleens with low hematopoietic progenitor cell content but does not affect in vitro hematopoietic progenitor function. (A) Gross appearance of E17.5 spleens from control and embryos with loss of Pbx1 alone (SKO) or loss of two Pbx genes (DKO) in Nkx2‐5‐positive splenic mesenchyme. Scale bars: 1 mm. (B,C) Methylcellulose colony assays performed with E16.5–17.5 spleen cells. (B) Representative images of BFU‐Es (*) and CFU‐GEMMs (**). Scale bars: 150 μm. (C) Colony counts at day 8 of culture. Histogram shows average data from three independent experiments. (D) Table summarizing total cell numbers (P = 0.016) and numbers of hematopoietic progenitors (P = 0.005) observed in a typical control and SKO spleen. (E) Lineage tracing analysis by flow cytometry in Nkx2‐5 Cre/+ ;Rosa YFP /+ embryonic tissues confirms the absence of ectopic Nkx2‐5‐Cre activity in leukocytes. Data are mean percentage + SEM of CD45+ YFP + cells of total CD45+ cells collected from six embryos.
By methylcellulose colony forming unit (CFU) assays, we evaluated whether the potential of mutant splenic hematopoietic progenitors to differentiate in vitro into myelomonocytic and erythroid lineages was affected in our mouse model. Splenocytes and liver cells were derived from littermate control and SKO embryos at E16.5 or E17.5. Both the size of individual colonies and the types of colonies formed were not different in control and mutant spleens (Fig. 1B). Also, the number of CFUs formed when equal numbers of control and SKO dissociated spleen cells were plated were similar at both developmental stages tested (Fig. 1C). Liver cells, which exhibit low Nkx2‐5‐Cre activity, were plated in parallel as technical controls (Fig. 1C). These results indicated that hematopoietic progenitors in SKO spleens have no intrinsic proliferation or differentiation defects once they are provided the necessary factors and cytokines in vitro. However, due to the substantial reduction of SKO spleen size, the average number of cells recovered from each dissociated SKO spleen 9‐fold lower than that obtained from control spleen. Taking into account that higher numbers of SKO spleens had to be pooled to obtain equal numbers of dissociated cells as in control spleens, the absolute number of hematopoietic progenitors with in vitro clonogenic potential was significantly lower in SKO spleens (Fig. 1D) (P = 0.005). However, the frequency of progenitors, i.e. the percentage of clonogenic progenitors per spleen, was not strikingly different in control and SKO spleens (0.79 and 0.58% of total spleen cells, respectively; P = 0.077). As adequate cell numbers from pooled and dissociated DKO spleens could not be obtained due to the striking hypoplasia, even when pooling up to five spleens with genotype Pbx1 f/f ;Pbx2 +/− ;Nkx2‐5 Cre/+ or Pbx1 f/f ;Pbx3 +/− ;Nkx2‐5 Cre/+, clonogenic activity of spleens with Pbx compound gene deletion could not be assayed. Thus, the present study did not investigate whether resident embryonic DKO spleen hematopoietic progenitors might have functional defects.
Furthermore, we assessed whether Pbx1 inactivation by the Nkx2‐5‐Cre deleter strain could cause potential alterations of Pbx1 expression within hematopoietic cells, due to Nkx2‐5 Cre activity in these cells, thus contributing to the severity of the phenotype. To this end, we generated Nkx2‐5 Cre/+ ;Rosa YFP/YFP embryos and performed lineage tracing of Nkx2‐5‐Cre+ cells. By FACS analysis, we determined that only a negligible percentage of CD45+ leukocytes were YFP+, demonstrating that Nkx2‐5 Cre is not active in hematopoietic cells (Fig. 1E). These experiments firmly established that the observed phenotype is not exacerbated by Pbx1 loss in hematopoietic cells.
Loss of Pbx homeoproteins in spleen mesenchyme compromises hematopoietic recovery in myelosuppressed adult mice
To analyze the effects of spleen hypoplasia on homeostatic hematopoiesis in adult mice, we performed complete blood counts of SKO and DKO mice. This revealed a significant increase of white blood cells (WBCs) and red blood cells (RBCs) in DKO mice vs. controls at steady‐state (Fig. 2A,B). Increase in circulating lymphocytes in DKO mice accounted for the increase of total WBCs (Supporting Information Fig. S1A). In contrast, the number of other WBC lineages, including neutrophils and eosinophils, was similar in controls and mutant mice (Fig. S1B). Furthermore, to investigate the impact of spleen hypoplasia/dysplasia on hematopoietic regeneration after stress, we elicited active extramedullary hematopoiesis. Accordingly, adult mice were sub‐lethally irradiated, which causes BM vascular damage and severe loss of cycling hematopoietic progenitors (Dainiak, 2002). Due to strong positive feedback loops, as yet poorly understood, those resistant and surviving dormant HSCs, or other hematopoietic progenitors, exit their niches and move to the spleen via circulation. Once they reach the spleen, they undergo self‐renewing divisions, and expand to generate new progenitor populations, which also proceed to proliferate and differentiate to replenish mature blood cell populations (Harandi et al. 2010; Peslak et al. 2011). In light of this knowledge, we compared the radioprotective capacity of spleens from control and Pbx mutant adult mice. Peripheral blood was analyzed over 25 days post‐irradiation to monitor progressive reconstitution of the hematopoietic system. Irradiated control mice recovered circulatory RBCs and platelets significantly more rapidly than SKO and DKO mice. In irradiated DKO mice RBC and platelet counts never reached their steady‐state levels over the course of the experiment and were 1.4‐ and 1.8‐fold lower, respectively, than in controls (Fig. 2A–C).
Figure 2.
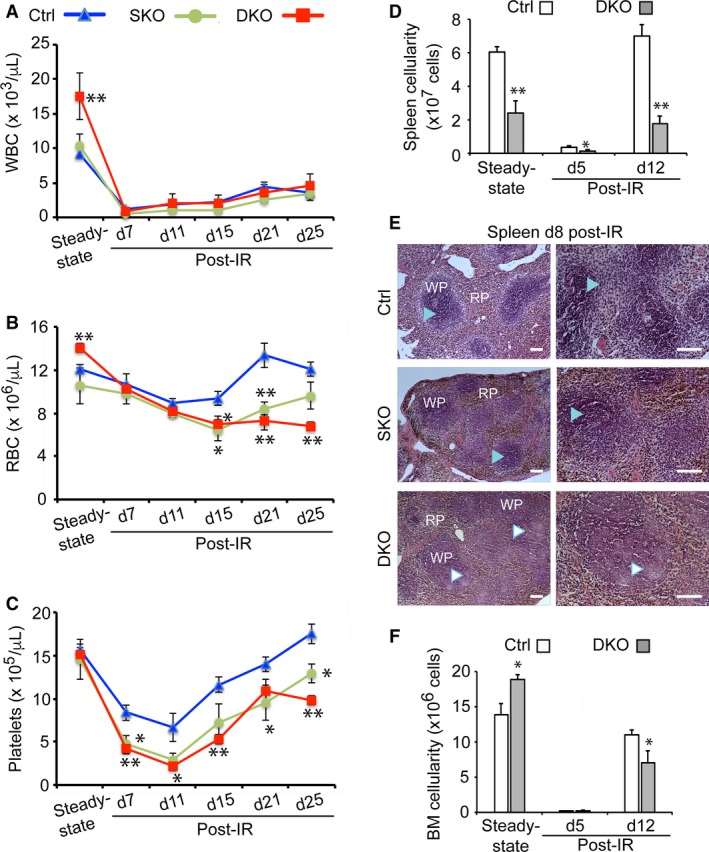
Loss of Pbx1 in spleen mesenchyme results in delayed hematopoietic recovery after acute radiation‐induced stress. (A‐C) Periodic sampling of peripheral blood to monitor multilineage hematologic recovery kinetics after increasing times (measured in days). (D) Following sublethal irradiation (IR) for age‐matched control (Ctrl; n = 10), SKO (n = 8), DKO (n = 10) adult mice. Each data point represents mean ± SEM. * P ≤ 0.05; ** P ≤ 0.01. (D) Histogram showing the average + SEM of control (n = 5) and DKO (n = 4) spleens before and 5 or 12 days following irradiation. P < 0.002. (E) H&E staining of spleen sections from 3‐month‐old mice at day 8 post‐irradiation reveals hypocellular WP in DKO spleens (indicated by white arrowheads) as opposed to normal WP histology in control and SKO (light blue arrowheads). Scale bars: 200 μm. (F) Histogram illustrating average bone marrow (BM) cell counts (as total nucleated blood cells) + SEM. Analysis conducted in two femurs per mouse (number of mice analyzed = 5), at the indicated time points. P = 0.04 at steady‐state; P = 0.05 at day (d) 12 post‐irradiation.
Adult DKO spleens, like their embryonic counterparts, exhibited dramatically reduced size and cellularity relative to controls. Nonetheless, they showed significant recovery of cellularity between days 5 and 12 post‐irradiation, similar to controls (Fig. 2D). Notably, unlike control and SKO, DKO spleen sections displayed histological alterations; specifically, absence of clear demarcation between WP and RP, as well as mild hypocellularity, as assessed at 8 days post‐irradiation (Fig. 2E). In addition, mild to moderate persistent lymphocytolysis was observed in the WP of DKO, unlike in control and SKO spleen sections, which likely reflects delayed tissue repair (Fig. 2E). However, WP and RP organization appeared normal in DKO spleens under steady‐state conditions (Fig. S1C). In addition, DKO mice exhibited significantly increased BM cellularity in homeostatic conditions, but displayed a delayed recovery in total BM cell numbers at day 12 after irradiation (Fig. 2F). Overall, hematopoietic tissue repair in spleen, and consequently BM, and reconstitution of mature circulating blood cells, are delayed in compound Pbx‐mutant spleens after acute radiation‐induced stress.
Loss of Pbx homeoproteins in splenic mesenchyme results in limited expansion of HSPCs in mutant spleens in vivo and in vitro
As regeneration of peripheral blood erythrocytes and platelets in DKO mice was significantly defective, we examined the ability of DKO spleens to support proliferation of hematopoietic progenitors. This was assessed by counting spleen colony forming units at day 12 (CFU‐S12) following sub‐lethal irradiation. Day 12 colonies are formed due to the proliferation and differentiation activity of short‐term HSCs (ST‐HSCs) and multipotent progenitors (MPPs) into hematopoietic cells. Thus, the ability to form CFU‐S12 is a reflection of the presence of progenitors with hematopoietic reconstitution capacity (Yang et al. 2005). Whereas all control spleens and two of three SKO spleens displayed multiple, macroscopic surface colonies, none of the DKO spleens examined showed comparable colonies (Fig. 3A). Interestingly, the average number of colonies formed by each SKO spleen was also slightly decreased. Histological analysis of sectioned spleens, however, revealed that DKO spleens do form colonies of proliferative primitive progenitors, although these colonies fail to enlarge in size compared with control and SKO spleens (Fig. 3B). The present studies do not rule out that the severity of the phenotype might be exacerbated by organ hypoplasia, which could physically restrict HSPC proliferation.
Figure 3.
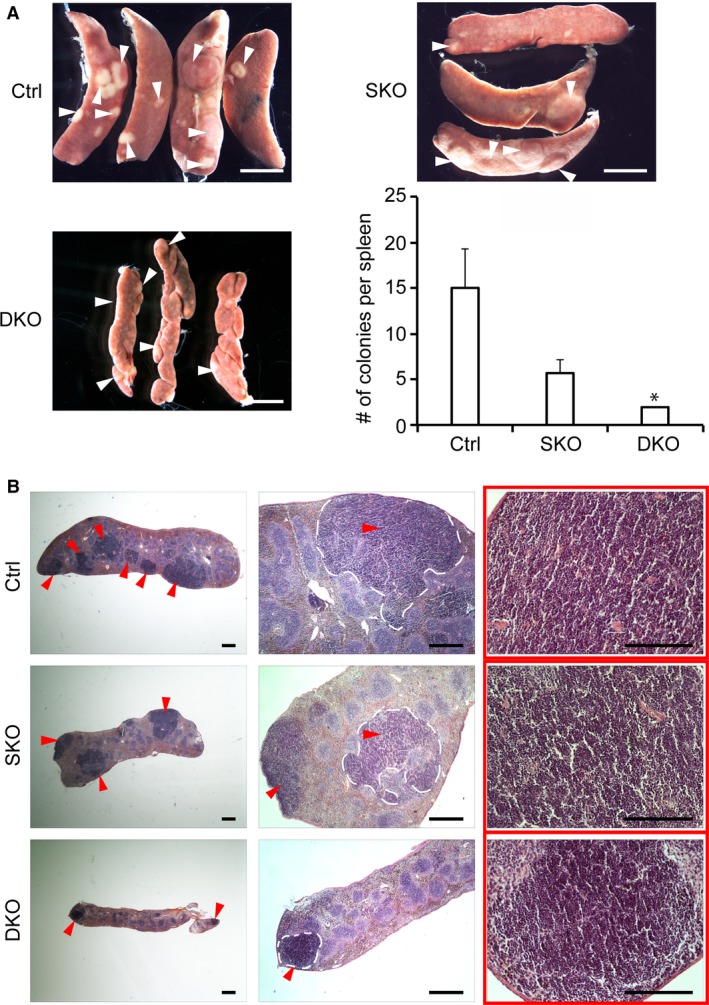
DKO spleens form sparse and minute in vivo colony‐forming units (CFU‐S). (A) Light micrographs of CFU‐S colonies at day 12 (CFU‐S12) (highlighted by white arrowheads) in sub‐lethally irradiated control (Ctrl), SKO, and DKO mice. The number of CFU‐S12 colonies was counted after fixation of the spleen and represented by a histogram. The size and number of colonies were significantly reduced in DKO spleens (P = 0.03). Scale bars: 2 mm. (B) H&E stained histological sections of the spleens shown in (A) demonstrate robust stress‐induced HSPC expansion in control and SKO spleens but not in DKOs. Red arrowheads point to CFU‐S12, some of which are delimited by dashed white lines. Scale bars: 500 μm.
Given that, overall, the hematopoietic phenotypes we observed were more pronounced in the DKO model, we performed the subsequent experiments using controls and DKO mice in sub‐lethal irradiation experiments. Accordingly, we further assessed the in vitro potential of hematopoietic progenitors derived from DKO spleens to proliferate and differentiate in methylcellulose culture assays (Fig. 4A‐C). At steady state, DKO mice exhibited a 4‐fold reduction in the number of spleen granulocyte‐progenitors (CFU‐GMs) and a significant increase in BM multipotent progenitors (CFU‐GEMMs) (Fig. 4B,C). By day 5 following myeloablation due to sub‐lethal irradiation, control and DKO spleens became depleted of myeloid and multipotent progenitors to similar extents. In contrast, numbers of colonies of rapidly proliferating mature erythroid progenitors (CFU‐E), which contribute to maintenance of peripheral RBC counts, were 3‐ to 4‐fold lower in DKO spleens than controls at both day 5 and day 10 after sub‐lethal irradiation (Fig. 4A). In addition, in DKO splenocyte cultures, no BFU‐Es were detectable at day 10 after irradiation, and the number of CFU‐GEMMs, CFU‐G, CFU‐M, and CFU‐GMs were reduced by 6‐ to 17‐fold relative to those formed by control splenocytes (Fig. 4B). Similarly, the number of all three types of myeloid progenitor colonies formed by DKO BM cells, isolated at day 10 post‐irradiation, was reduced 2‐ to 3‐fold compared with control BM cells (Fig. 4C).
Figure 4.
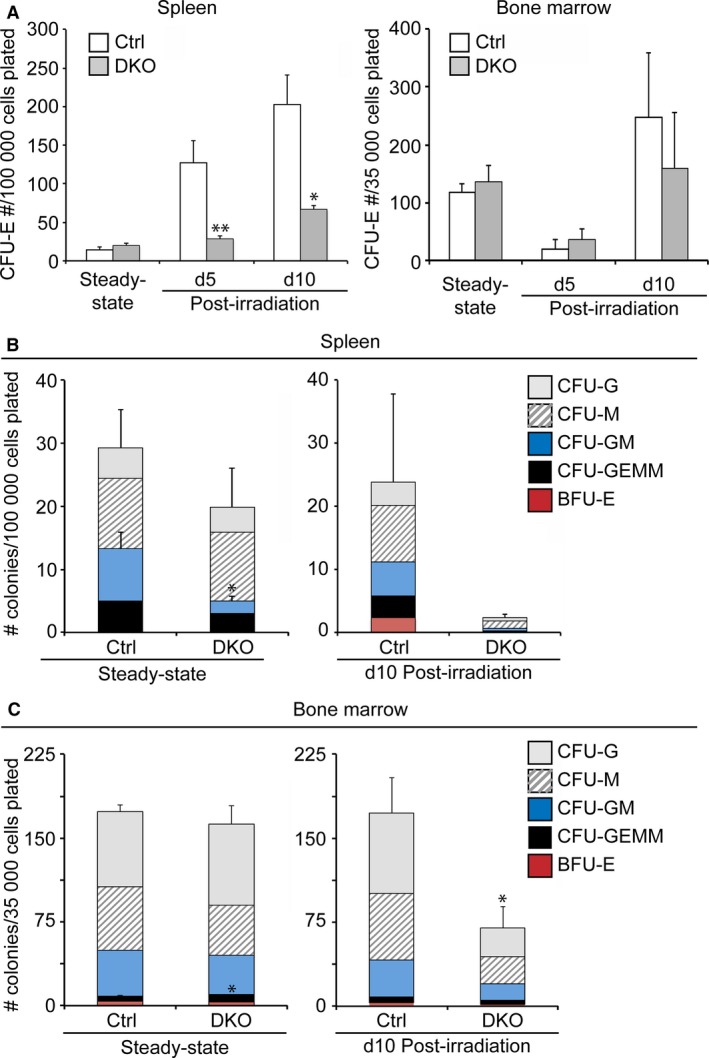
Impaired in vitro hematopoietic progenitor function in Pbx1 f/f ;Pbx2 +/− ;Nkx2‐5 Cre/+ (DKO) mutant spleens compared with controls. (A‐C) Control (Ctrl) and DKO spleens and bone marrow were harvested at the indicated time points (day; d) before or after irradiation. Subsequently, single cell suspensions were prepared and plated on Methocult™ media containing SCF, IL‐3, IL‐6, EPO, insulin and transferrin. The number of colonies of erythroid (CFU‐E, BFU‐E), myeloid (CFU‐GM, CFU‐M, CFU‐G), and multipotent progenitors (CFU‐GEMM) was counted after 3 or 9 days of culture. n = 3 or 4 per group; error bars indicate mean + SEM. *P < 0.05; **P < 0.01.
We further tested the expansion of short‐term radio‐protective progenitors following bone marrow transplantation in mutant mice. Using methylcellulose colony assays, we evaluated the ability of mutant spleens to provide a niche for the development of erythroid cells in the immediate post‐transplant period after lethal irradiation. A total of 105 donor CD45.2 BM cells were transplanted into lethally irradiated control, SKO, and DKO (CD45.1) recipients. Spleens were subsequently isolated at day 8. Unlike in controls, CFU‐Es and BFU‐Es were absent not only in DKO but also in SKO spleens (Supporting Information Fig. S2). These results suggest that the mutant phenotype can be elicited by the absence of Pbx1 alone, albeit with different degrees of severity and varying under different types of induced stress, as demonstrated by our assays. These additional findings further strengthen the conclusion that Pbx‐mutant splenic mesenchyme has a reduced capacity to support stress‐induced spleen erythropoiesis. Overall, two different scenarios could be envisaged to explain the strikingly impaired expansion of HSPCs in DKO spleens: (1) dysfunction of the splenic niche, involving insufficient production of growth factors; and (2) acquisition of intrinsic proliferation and differentiation defects by the hematopoietic progenitors upon colonization of Pbx‐deficient splenic mesenchyme.
Loss of Pbx transcription factors in spleen mesenchyme causes decrease in numbers of hematopoietic progenitors in vivo
To evaluate whether spleens and BM of DKO mice contained abnormal numbers of hematopoietic progenitors before in vitro culture, we quantified HSPCs by FACS analysis using various cell surface markers (Fig. 5A‐D). FACS analysis revealed that steady‐state DKO spleens exhibited marked reduction of lineage‐negative, Sca‐1+, cKit+ (LSK) HSCs, specifically LT‐HSCs, as well as early erythroid progenitors that normally form BFU‐E colonies in semisolid culture (Fig. 5E). During hematopoietic regeneration at day 12 post‐irradiation, spleens of DKO mice harbored significantly lower numbers of ST‐HSCs and CMPs compared with control spleens (Fig. 5E). Notably, ST‐HSCs are a potent radio‐protective stem cell population with the ability to rapidly regenerate hematopoietic cells (Yang et al. 2005). Thus, the marked decrease in the number of ST‐HSCs in DKO spleens (Fig. 5E) and BM (Fig. 5F) is likely linked to the slower recovery kinetics of erythrocyte and platelet numbers in mutants compared with controls (as shown in Fig. 2). Although not statistically significant, a marked decrease in the numbers of megakaryocyte‐erythrocyte progenitors (MEPs) and more mature erythroid progenitors (CFU‐E and proerythroblasts) was also observed in DKO spleens. The reduced frequency of MEPs and CMPs in mutant spleens is of particular note because these progenitors have been shown to be required and sufficient for radio‐protection from bone marrow failure in lethally irradiated mice, as well as for the initial phase of spleen and peripheral blood repopulation (Nakorn et al. 2002). We further assessed erythropoietic recovery at an earlier timepoint (day 8) post‐irradiation by performing flow cytometry for Ter119 and CD71 markers. This experiment demonstrated the lack of Lineage–Ter119−CD71+ erythroid precursors in SKO and DKO spleens compared with controls (Fig. 5G). Overall, these results (Figs 4 and 5) indicate that spleen mesenchymal defects perturb both steady‐state and stress‐induced hematopoiesis in a non‐lineage‐specific manner, starting from LT‐HSCs, the most primitive progenitor cell population. Our findings also highlight that impaired stem cell maintenance is observed in DKO spleens prior to the appearance of defects in the BM compartment, where a significant reduction of ST‐HSCs and BFU‐Es was observed after irradiation. Thus, an imbalance in spleen hematopoietic progenitor populations can affect the maintenance of select classes of BM HSPCs during stress hematopoiesis. Taken together, our observations suggest that the decrease of CFUs detected when DKO spleen cells are cultured (shown in Fig. 4), rather than resulting from a proliferation and/or differentiation defect in resident progenitors, reflects the reduction in progenitor cell numbers in mutant spleens at the time of their isolation. This defect is underpinned by loss of Pbx factors in the mesenchyme, with consequent impairment of the splenic niche.
Figure 5.
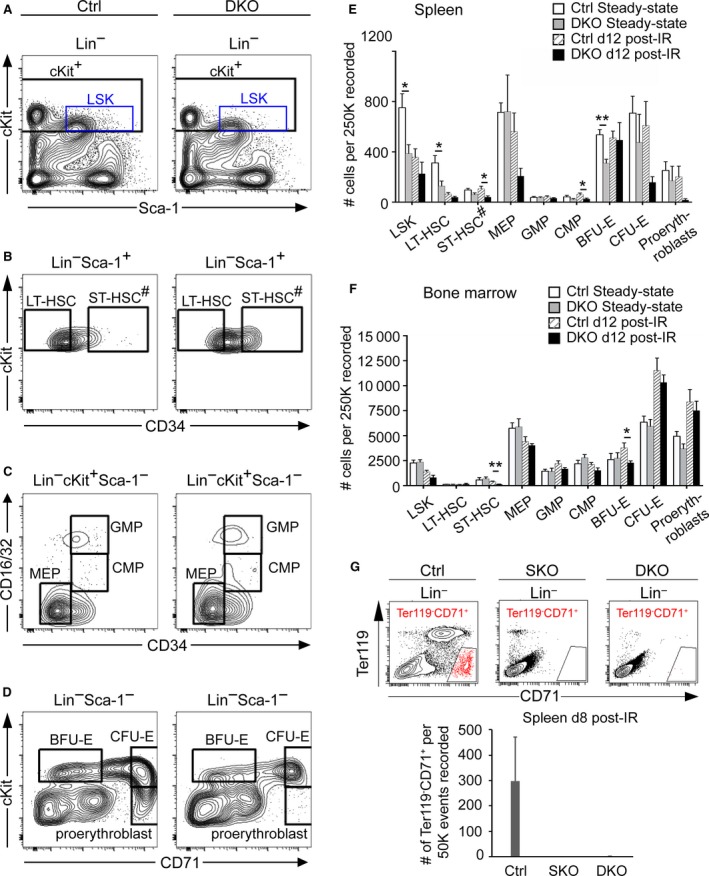
Decreased numbers of hematopoietic stem and progenitor cells in Pbx‐deficient splenic mesenchyme. (A–D) Representative FACS analyses are shown for spleen cells from control (Ctrl) and DKO age‐matched, non‐irradiated mice. HSCs (LSK cells) are defined as Lin– cKit+Sca1+; LT‐HSCs as Lin− cKit+Sca1+ CD34−; ST‐HSCs# as Lin− cKit+Sca1+ CD34+ [note: this phenotyping also includes spleen‐specific stress erythroid progenitors (Harandi et al. 2010)]; MEPs as Lin− cKit+Sca1− CD34−FcγRII/III −; GMPs as Lin− cKit+Sca1− CD34+FcγRII/III +; CMPs as Lin− cKit+Sca1− CD34+FcγRII/III lo; BFU‐Es as Lin− cKit+Sca1− CD71lo; CFU‐Es as Lin− cKit+Sca1− CD71+; proerythroblasts as Lin− cKitloSca1− CD71+. (E,F) Flow cytometric quantitation of indicated populations in spleen and BM. Ctrl n = 5, DKO n = 4 per time point. (G) Flow cytometry for spleen erythroid progenitors (Lin−Ter119− CD71+) at day 8 following irradiation. Top panel: representative contour plots for CD71 vs. Ter119 after gating on lineage negative population. Bottom panel: bar charts displaying the average numbers of erythroid progenitors in Ctrl, SKO, DKO spleens (n = 3 per cohort). Error bars indicate mean + SEM; *P ≤ 0.05; **P < 0.01.
Dysregulation of cytokines and angiocrine factors in Pbx mutant spleens after irradiation
To investigate the mechanisms underlying the decreased numbers of progenitors in mice with loss of Pbx homeoproteins in the spleen mesenchyme, we performed gene expression analysis on control and DKO spleens in conditions of stress‐induced extramedullary hematopoiesis. Using a predesigned real‐time (RT) PCR assay panel (BioRad), we assessed the expression of 88 top‐ranked genes involved in hematopoiesis. Subsequently, by RT qPCR we validated the 36 genes that showed differential expression between control and DKO spleens in the assay panel. Also by RT qPCR, we examined differential expression levels of additional genes that have been implicated in hematopoiesis but were not present in the RT PCR assay panel, thus analyzing the expression levels of 96 genes in total. We found that 19 genes coding for secreted cytokines and cell surface receptors that engage in paracrine signaling were statistically significantly down‐regulated in the spleens of irradiated Pbx‐mutant mice compared to controls (Fig. 6A,B). Notably, cytokines are important hematopoietic growth factors and elicit diverse cellular responses (Robb, 2007). mRNA levels for all of the 19 cytokine and angiocrine factor that we found altered in DKO spleens were previously shown to be pivotal for self‐renewal of HSCs and maintenance of their numbers in the BM and/or spleen, such as KitL and Cxcl12 (Ding et al. 2012; Ding & Morrison, 2013; Inra et al. 2015). In addition, other factors including TNF‐α (Mizrahi & Askenasy, 2014), VEGFR2 (Chute et al. 2007; Hooper et al. 2009), IL‐10 (Kang et al. 2007), and Notch pathway components (Notch1, Dll1) have been reported to enhance HSC repopulating activity and to mediate multilineage reconstitution of the hematopoietic system in response to stress/transplantation assays (Butler et al. 2010; Delaney et al. 2010; Mendelson & Frenette, 2014). Other molecules, such as IL‐3, IL‐11, and Csf2/GM‐CSF, which we also found down‐regulated in DKO spleens, support the expansion of HSCs in vitro (Miller et al. 2002; Shelton et al. 2004; Bayne et al. 2012; Broxmeyer, 2013; Wohrer et al. 2014). In contrast, additional cytokines that we found perturbed in DKO spleens promote differentiation along specific lineages, e.g. lymphoid, such as IL‐4, IL‐15, IL‐7, CD40L, Flt3L; or myeloid, such as Csf3/G‐CSF and interferon (IFN)‐γ (Solanilla et al. 2000; Sitnicka et al. 2002; Ranson et al. 2003; Robb, 2007; Jack et al. 2009; Schurch et al. 2014). Thus, concomitant down‐regulation of these factors in DKO spleens likely accounts for the profoundly decreased numbers of HSCs, myeloid, and erythroid progenitors after radiation injury, as well as underlying the delay in peripheral blood reconstitution in DKO mice.
Figure 6.
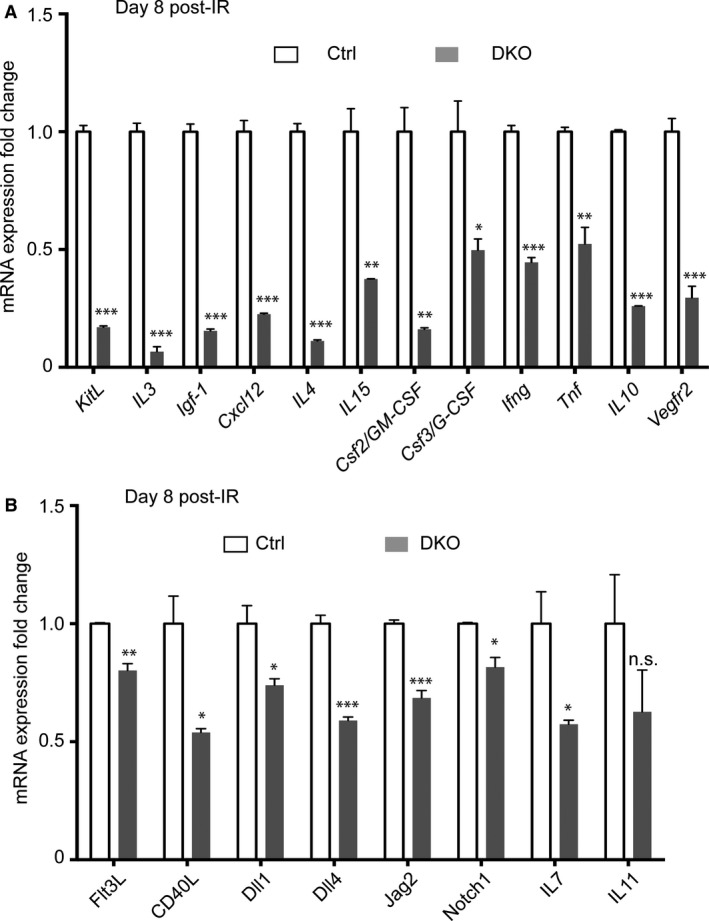
Impaired expression of cytokine‐encoding genes in DKO spleens at steady‐state and during post‐injury hematopoiesis. (A,B) Control (Ctrl; n = 3) and DKO (n = 3) spleens were enzymatically digested to generate single cell suspensions and erythrocytes were lysed, following which RNAs were prepared from the cell pellets. Graphs show differentially expressed genes that code for secreted factors and receptors with known functions in facilitating hematopoietic regeneration. Notably, KitL (also known as SCF) and Cxcl12 are markedly down‐regulated in Pbx mutant spleens. The housekeeping transcript Hprt was used to normalize RT‐qPCR. Error bars indicate mean + SEM; *P < 0.05; **P < 0.01; ***P < 0.001; n.s., non‐significant. (A) Transcripts for 12 factors are downregulated 2‐ to 15‐fold in irradiated Pbx DKO vs. control spleens. (B) Transcripts of seven additional cytokines are downregulated < 2‐fold in irradiated DKO spleens vs. controls.
Lastly, we assessed whether any of the cytokine‐encoding genes discussed above were induced by irradiation, with subsequent hematopoietic damage, in wild‐type mice. We found that in wild‐type animals the transcripts for the 19 cytokines and angiocrine factors that were down‐regulated in Pbx‐mutant spleens were significantly induced, except for Igf‐1, at day 8 following irradiation compared with steady state levels (Supporting Information Fig. S3A). Unlike in wild‐type, irradiation did not induce the expression of KitL, IL3, Cxcl12, IL4, Csf2, Csf3, IL10, Vegfr2 or Dll4 in spleens of DKO mice. The latter factors were instead downregulated, or remained unchanged following myeloablation in Pbx DKO mice (Supporting Information Fig. S3B). Thus, defects in both basal and stress‐induced hematopoiesis in Pbx compound mutant mice with Pbx1 spleen‐specific loss is accompanied by marked abnormalities in the expression of hematopoietic growth factors.
Discussion
In humans, asplenia increases the risk of fatal infections, including sepsis mediated by encapsulated bacteria, underscoring the essential role of the spleen in controlling immune responses to pathogens (Bolze et al. 2013; Bronte & Pittet, 2013). Here, we focused on elucidating the role of the spleen in fetal and adult murine hematopoiesis. The generation of all mature blood and immune cells from HSCs through the progressive differentiation of lineage‐restricted, phenotypically defined progenitors is a tightly controlled process. Hematopoiesis mainly occurs in fetal liver and adult bone marrow (Morrison & Scadden, 2014). In this study, we use a mouse model with spleen mesenchyme‐specific deletion of Pbx1 on a Pbx2‐ or Pbx3‐deficient genetic background to reach a deeper understanding of the contribution of the splenic mesenchyme to systemic hematopoiesis. Our experiments demonstrate that a genetically intact spleen mesenchyme is necessary to support normal HSPC content in embryonic and adult spleens. This, in turn, influences multiple processes that occur after acute radiation‐induced stress and myelosuppression, including: (1) the kinetics of tissue repair of the spleen and BM; (2) the reconstitution of circulatory red blood cells and platelets; and (3) the regeneration of hematopoietic progenitors with clonogenic activity.
The main culprit for the decreased content of HSPCs in compound Pbx1/2 and Pbx1/3 mutant spleens likely resides in splenic mesenchyme hypoplasia concomitant with structural defects such as disorganization of stromal cells, the latter requiring further exploration. Genetic rescue of organ size by inactivation of p15Ink4b, a cell cycle inhibitor, partially restores spleen mesenchyme proliferation and organ growth, as we reported (Koss et al. 2012). In future experiments, this approach could be attempted to test whether hypoplasia is the primary defect underlying the hematopoietic phenotype in mice with compound loss of Pbx transcription factors in the spleen. However, there is no guarantee that these experiments would unequivocally answer the question, given that only a partial rescue of splenic size can be obtained by eliminating p15Ink4b function in Pbx mutant embryos (Koss et al. 2012). Therefore, unambiguous dissection of the causative scenario underlying the impaired splenic hematopoiesis in Pbx mutants will require additional long‐term experimental approaches. For example, it remains to be elucidated whether partial splenectomy in wild‐type mice would mimic, at least in part, the stress‐induced hematopoiesis defects we observed in irradiated DKO mice. Notably, our CFU‐S assays show that hematopoietic cells after sub‐lethal irradiation are still able to migrate and home into the mutant spleens, despite their smaller size. However, these experiments are not quantitative and thus do not unequivocally resolve the possibility that organ hypoplasia might impair, at least in part, the ability of the spleen to home hematopoietic cells.
In DKO mice we found a significant increase in the numbers of peripheral WBCs (mostly lymphocytes and neutrophils) and RBCs in homeostasis conditions. This phenotype is likely due to the inability of the DKO spleen to sequester lymphocytes, monocytes and neutrophils, as well as to inefficient clearing of aged RBCs, as a result of space limitation or functional deficiency of the splenic stromal component. DKO spleens also displayed lower numbers of LSK HSCs and LT‐HSCs, which could result at least in part from dysfunction of the niche, including the inability to maintain mobilized HSCs within the spleen mesenchyme.
Our study highlights an important role of the adult spleen in efficient and speedy regeneration of RBCs and platelets after myelosuppression. Indeed, poor hematopoietic reconstitution in our mutant mouse model with hyposplenia was associated with a marked depletion of ST‐HSCs, MEPs, CMPs, CFU‐Es and proerythroblasts and diminished proliferation of primitive progenitors in vivo. The reduction of progenitors in DKO spleens of irradiated mice can be attributed at least in part to the profound down‐regulation of multiple genes encoding hematopoietic growth factor and angiocrine factors, including KitL (also known as SCF) and Cxcl12, as we revealed by quantitative RT‐PCR (Fig. 7). Notably, it was recently reported that in response to various haematopoietic stresses such as myeloablation, blood loss, or pregnancy, all of which mobilize HSCs from the bone marrow to the spleen and induce extramedullary haematopoiesis, KitL/SCF and Cxcl12 play a key role in the splenic niche (Inra et al. 2015). In particular, endothelial and stromal splenic cell expression of these two factors, which we show here are controlled by Pbx homeodomain proteins, is required to sustain all the new blood‐forming HSCs migrating into the spleen (Inra et al. 2015).
Figure 7.
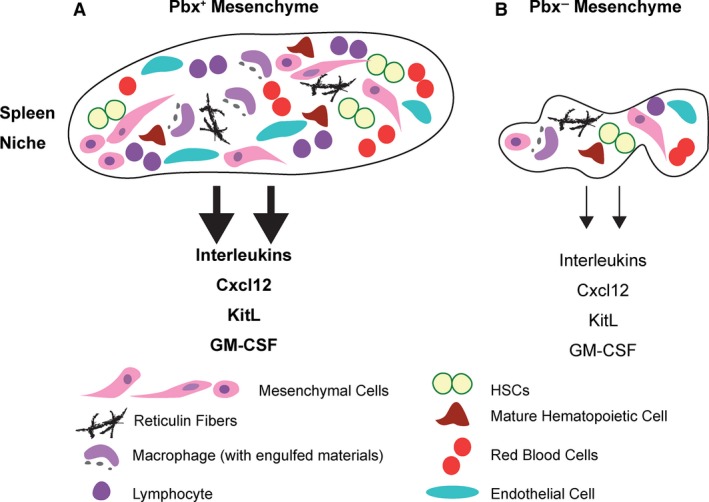
Pbx‐dependent spleen hypoplasia and morphogenesis defects cause impaired production of cytokines and growth factors in the splenic niche. (A) Normal splenic niche function requires developmental expression of Pbx genes in the Nkx2–5+ mesenchyme (Pbx+ Mesenchyme). (B) Splenic mesenchyme‐specific inactivation of Pbx1 on Pbx2‐ or Pbx3‐deficient background (Pbx− mesenchyme) results in spleen stromal hypoplasia and morphogenesis defects that impact the niche. The cellular components of the splenic niche are illustrated at the bottom of the cartoon. Our study establishes that genetic perturbation of splenic stromal development due to loss of Pbx factors is associated with impaired expansion and maintenance of HSCs, as well as myeloid and erythroid progenitors, during injury‐induced hematopoietic regeneration. This Pbx‐dependent hematopoietic phenotype results at least in part from the downregulation of genes encoding interleukins, colony‐stimulating factors, KitL and Cxcl12, all of which are essential growth factors for hematopoietic reconstitution after BM damage.
Intriguingly, in Pbx‐mutant spleens we found unchanged levels of transcripts for critical cytokines including: (1) Bmp4 (Harandi et al. 2010; Paulson et al. 2011) and Epo (Peslak et al. 2012), which are required for regeneration of the erythroid lineage after acute stress; (2) Fgf2, which is involved in the recovery of HSPCs (Itkin et al. 2012; Zhao et al. 2012); and (3) IL6 (Zhao et al. 2014), which is essential for rapid myeloid cell restoration during neutropenia (Supporting Information Fig. S3C). Interestingly, at day 8 following sub‐lethal irradiation, unlike in the spleen Epo expression in DKO kidneys was significantly upregulated (by 6‐folds) compared to controls (Supporting Information Fig. S3D), probably reflecting persistent anemia in DKO mice. The high levels of Epo transcript were not accompanied by enhanced erythrocyte recovery in these mutant mice, probably caused by the inefficient replenishing of circulating RBCs by Pbx‐mutant spleens. The mRNA levels of other factors with demonstrated roles in hematopoiesis, such as FasL, Csf1 (M‐CSF), and Jag1 (Mossadegh‐Keller et al. 2013; Poulos et al. 2013; Choi et al. 2014), were also found to be similar in irradiated control and Pbx‐mutant spleens (Supporting Information Fig. S3C), suggesting the presence of complex downstream regulatory mechanisms that will require additional investigations. Lastly, among the cytokine‐encoding genes that we profiled at the mRNA level, most were induced by irradiation in wild‐type spleens, but failed to be induced in DKO spleens. These results underscore the notion that the hematopoietic defects observed in Pbx DKO mice are underpinned by profound dysregulation of cytokines and angiocrine factors in the splenic niche. It is worthy of mention that the genes coding for IFN‐γ, Flt3L, CD40L, Jag2, Notch1 and IL‐7 were induced in response to irradiation in both control and DKO spleens alike (Supporting Information Fig. S3A‐B). In light of these findings, it could be envisaged that the two classes of factors discussed above are controlled through different upstream mechanisms, either cell‐autonomous within the spleen niche, or non‐cell autonomous in the splenic hematopoietic compartment, which will require future investigations.
Concluding remarks
In this study we explored the mechanisms by which a Pbx‐deficient spleen mesenchymal niche is not competent to support stress hematopoiesis. Our work elucidates a previously unknown non‐cell autonomous role for Pbx homeodomain proteins, in part through the control of key factors such as KitL/SCF and Cxcl12, in the expansion and maintenance of HSPCs in the splenic niche. Overall, this research broadens our knowledge of the functional contributions of the Pbx family of transcription factors to hematopoiesis beyond previous studies.
Conflict of interests
The authors declare there are no conflicts of interest.
Supporting information
Fig. S1. Differential WBC counts, spleen histology, and bone marrow cellularity in control and Pbx mutant mice at steady‐state or at steady‐state and during recovery from sub‐lethal irradiation (cf. Fig. 2).
Fig. S2. BFU‐E and CFU‐E analysis in SKOs and DKOs vs. controls (Ctrl) at day 8 post lethal irradiation (cf. Fig. 4).
Fig. S3. Cytokine gene expression analysis after irradiation‐induced injury in control and Pbx DKO spleens, as well as in control and DKO kidneys (for Epo) (cf. Fig. 6).
Acknowledgements
We thank Shahin Rafii and Bi‐Sen Ding for many invaluable discussions and support; Michael Cleary for critical suggestions; Richard Harvey for the Nkx2‐5 Cre deleter strain; Jason McCormick at WCMC's Flow Cytometry Core for FACS analysis; and Robert Aho for artwork. All research was supported by NIH grant R01 HD061403 to L. Selleri.
References
- Avecilla ST, Hattori K, Heissig B, et al. (2004) Chemokine‐mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med 10, 64–71. [DOI] [PubMed] [Google Scholar]
- Bayne LJ, Beatty GL, Jhala N, et al. (2012) Tumor‐derived granulocyte‐macrophage colony‐stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand JY, Desanti GE, Lo‐Man R, et al. (2006) Fetal spleen stroma drives macrophage commitment. Development 133, 3619–3628. [DOI] [PubMed] [Google Scholar]
- Bolze A, Mahlaoui N, Byun M, et al. (2013) Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science 340, 976–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendolan A, Ferretti E, Salsi V, et al. (2005) A Pbx1‐dependent genetic and transcriptional network regulates spleen ontogeny. Development 132, 3113–3126. [DOI] [PubMed] [Google Scholar]
- Brendolan A, Rosado MM, Carsetti R, et al. (2007) Development and function of the mammalian spleen. BioEssays 29, 166–177. [DOI] [PubMed] [Google Scholar]
- Bronte V, Pittet MJ (2013) The spleen in local and systemic regulation of immunity. Immunity 39, 806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broxmeyer HE (2013) Erythropoietin: multiple targets, actions, and modifying influences for biological and clinical consideration. J Exp Med 210, 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burn SF, Boot MJ, de Angelis C, et al. (2008) The dynamics of spleen morphogenesis. Dev Biol 318, 303–311. [DOI] [PubMed] [Google Scholar]
- Butler JM, Nolan DJ, Vertes EL, et al. (2010) Endothelial cells are essential for the self‐renewal and repopulation of Notch‐dependent hematopoietic stem cells. Cell Stem Cell 6, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvi L, Adams G, Weibrecht K, et al. (2003) Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846. [DOI] [PubMed] [Google Scholar]
- Cao H, Oteiza A, Nilsson SK (2013) Understanding the role of the microenvironment during definitive hemopoietic development. Exp Hematol 41, 761–768. [DOI] [PubMed] [Google Scholar]
- Capellini TD, Di Giacomo G, Salsi V, et al. (2006) Pbx1/Pbx2 requirement for distal limb patterning is mediated by the hierarchical control of Hox gene spatial distribution and Shh expression. Development 133, 2263–2273. [DOI] [PubMed] [Google Scholar]
- Capellini TD, Zewdu R, Di Giacomo G, et al. (2008) Pbx1/Pbx2 govern axial skeletal development by controlling Polycomb and Hox in mesoderm and Pax1/Pax9 in sclerotome. Dev Biol 321, 500–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capellini TD, Vaccari G, Ferretti E, et al. (2010) Scapula development is governed by genetic interactions of Pbx1 with its family members and with Emx2 via their cooperative control of Alx1. Development 137, 2559–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capellini TD, Zappavigna V, Selleri L (2011) Pbx homeodomain proteins: TALEnted regulators of limb patterning and outgrowth. Dev Dyn 240, 1063–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagnaro L, Lenti E, Maruzzelli S, et al. (2013) Nk2–5+ Islet1+ mesenchymal precursors generate distinct spleen stromal cell subsets and participate in restoring stromal network integrity. Immunity 38, 782–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Saez B, Anders L, et al. (2014) D‐cyclins repress apoptosis in hematopoietic cells by controlling death receptor Fas and its ligand FasL. Dev Cell 30, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chute JP, Muramoto GG, Salter AB, et al. (2007) Transplantation of vascular endothelial cells mediates the hematopoietic recovery and survival of lethally irradiated mice. Blood 109, 2365–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dainiak N (2002) Hematologic consequences of exposure to ionizing radiation. Exp Hematol 30, 513–528. [DOI] [PubMed] [Google Scholar]
- Delaney C, Heimfeld S, Brashem‐Stein C, et al. (2010) Notch‐mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med 16, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMartino JF, Selleri L, Traver D, et al. (2001) The Hox cofactor and proto‐oncogene Pbx1 is required for maintenance of definitive hematopoiesis in the fetal liver. Blood 98, 618–626. [DOI] [PubMed] [Google Scholar]
- Ding L, Morrison SJ (2013) Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Saunders TL, Enikolopov G, et al. (2012) Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta P, Hoyer FF, Grigoryeva LS, et al. (2015) Macrophages retain hematopoietic stem cells in the spleen via VCAM‐1. J Exp Med 212, 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti E, Li B, Zewdu R, et al. (2011) A conserved Pbx‐Wnt‐p63‐Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev Cell 21, 627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficara F, Murphy MJ, Lin M, et al. (2008) Pbx1 regulates self‐renewal of long‐term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell 2, 484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficara F, Crisafulli L, Lin C, et al. (2013) Pbx1 restrains myeloid maturation while preserving lymphoid potential in hematopoietic progenitors. J Cell Sci 126, 3181–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama N, Sato Y, Matsumoto K, et al. (1999) Coelom formation: binary decision of the lateral plate mesoderm is controlled by the ectoderm. Development 126, 4129–4138. [DOI] [PubMed] [Google Scholar]
- Greenbaum A, Hsu YMS, Day RB, et al. (2013) CXCL12 in early mesenchymal progenitors is required for haematopoietic stem‐cell maintenance. Nature 495, 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griseri T, McKenzie BS, Schiering C, et al. (2012) Dysregulated hematopoietic stem and progenitor cell activity promotes interleukin‐23‐driven chronic intestinal inflammation. Immunity 37, 1116–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Haan JM, Mebius RE, Kraal G (2012) Stromal cells of the mouse spleen. Front Immunol 3, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harandi OF, Hedge S, Wu DC, et al. (2010) Murine erythroid short‐term radioprotection requires a BMP4‐dependent, self‐renewing population of stress erythroid progenitors. J Clin Invest 120, 4507–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecksher‐Sorensen J, Watson RP, Lettice LA, et al. (2004) The splanchnic mesodermal plate directs spleen and pancreatic laterality, and is regulated by Bapx1/Nkx3.2. Development 131, 4665–4675. [DOI] [PubMed] [Google Scholar]
- Himburg HA, Harris JR, Ito T, et al. (2012) Pleiotrophin regulates the retention and self‐renewal of hematopoietic stem cells in the bone marrow vascular niche. Cell Rep 2, 964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper AT, Butler JM, Nolan DJ, et al. (2009) Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2‐mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell 4, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado R, Zewdu R, Mtui J, et al. (2015) Pbx1‐dependent control of VMC differentiation kinetics underlies gross renal vascular patterning. Development 142, 2653–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inra CN, Zhou BO, Acar M, et al. (2015) A perisinusoidal niche for extramedullary haematopoiesis in the spleen. Nature 527, 466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itkin T, Ludin A, Gradus B, et al. (2012) FGF‐2 expands murine hematopoietic stem and progenitor cells via proliferation of stromal cells, c‐Kit activation, and CXCL12 down‐regulation. Blood 120, 1843–1855. [DOI] [PubMed] [Google Scholar]
- Jack GD, Zhang L, Friedman AD (2009) M‐CSF elevates c‐Fos and phospho‐C/EBPa (S21) via ERK whereas G‐CSF stimulates SHP2 phosphorylation in marrow progenitors to contribute to myeloid lineage specification. Blood 114, 2172–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ, Yang SJ, Park G, et al. (2007) A novel function of interleukin‐10 promoting self‐renewal of hematopoietic stem cells. Stem Cells 25, 1814–1822. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, Iwashita T, Yilmaz OH, et al. (2005a) Spatial differences in hematopoiesis but not in stem cells indicate a lack of regional patterning in definitive hematopoietic stem cells. Dev Biol 283, 29–39. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, et al. (2005b) SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121. [DOI] [PubMed] [Google Scholar]
- Kim SK, Selleri L, Lee JS, et al. (2002) Pbx1 inactivation disrupts pancreas development and in Ipf1‐deficient mice promotes diabetes mellitus. Nat Genet 30, 430–435. [DOI] [PubMed] [Google Scholar]
- Koss M, Bolze A, Brendolan A, et al. (2012) Congenital asplenia in mice and humans with mutations in a Pbx/Nk2–5/p15 module. Dev Cell 22, 913–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, et al. (2013) Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lento W, Ito T, Zhao C, et al. (2014) Loss of Beta‐catenin triggers oxidative stress and impairs hematopoietic regeneration. Genes Dev 28, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuschner F, Rauch PJ, Ueno T, et al. (2012) Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med 209, 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longobardi E, Penkov D, Mateos D, et al. (2014) Biochemistry of the tale transcription factors PREP, MEIS, and PBX in vertebrates. Dev Dyn 243, 59–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson A, Frenette PS (2014) Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med 20, 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkola HK, Orkin SH (2006) The journey of developing hematopoietic stem cells. Development 133, 3733–3744. [DOI] [PubMed] [Google Scholar]
- Miller C, Audet J, Eaves C (2002) Ex vivo expansion of human and murine hematopoietic stem cells. Methods Mol Med 63, 189. [DOI] [PubMed] [Google Scholar]
- Mizoguchi T, Pinho S, Ahmed J, et al. (2014) Osterix marks distinct waves of primitive and definitive stromal progenitors during bone marrow development. Dev Cell 29, 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrahi K, Askenasy N (2014) Physiological functions of TNF family receptor/ligand interactions in hematopoiesis and transplantation. Blood 124, 176–183. [DOI] [PubMed] [Google Scholar]
- Moens CB, Selleri L (2006) Hox cofactors in vertebrate development. Dev Biol 291, 193–206. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Scadden DT (2014) The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossadegh‐Keller N, Sarrazin S, Kandalla PK, et al. (2013) M‐CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature 497, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SN, Germain RN (2009) Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol 9, 618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakorn TN, Traver D, Weissman IL, et al. (2002) Myeloerythroid‐restricted progenitors are sufficient to confer radioprotection and provide the majority of day 8 CFU‐S. J Clin Invest 109, 1579–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omatsu Y, Sugiyama T, Kohara H, et al. (2010) The essential functions of adipo‐osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 33, 387–399. [DOI] [PubMed] [Google Scholar]
- Paulson RF, Shi L, Wu DC (2011) Stress erythropoiesis: new signals and new stress progenitor cells. Curr Opin Hematol 18, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peslak SA, Wenger J, Bemis JC, et al. (2011) Sublethal radiation injury uncovers a functional transition during erythroid maturation. Exp Hematol 39, 434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peslak SA, Wenger J, Bemis JC, et al. (2012) EPO‐mediated expansion of late‐stage erythroid progenitors in the bone marrow initiates recovery from sublethal radiation stress. Blood 120, 2501–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potocnik AJ, Brakebusch C, Fassler R (2000) Fetal and adult hematopoietic stem cells require Beta1‐Integrin function for colonizing fetal liver, spleen, and bone marrow. Immunity 12, 653–663. [DOI] [PubMed] [Google Scholar]
- Poulos MG, Guo P, Kofler NM, et al. (2013) Endothelial Jagged‐1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep 4, 1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranson T, Vosshenrich CA, Corcuff E, et al. (2003) IL‐15 is an essential mediator of peripheral NK‐cell homeostasis. Blood 101, 4887–4893. [DOI] [PubMed] [Google Scholar]
- Rhee JW, Arata A, Selleri L, et al. (2004) Pbx3 deficiency results in central hypoventilation. Am J Pathol 165, 1343–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb L (2007) Cytokine receptors and hematopoietic differentiation. Oncogene 26, 6715–6723. [DOI] [PubMed] [Google Scholar]
- Sanyal M, Tung JW, Karsunky H, et al. (2007) B‐cell development fails in the absence of the Pbx1 proto‐oncogene. Blood 109, 4191–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurch CM, Riether C, Ochsenbein AF (2014) Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell 14, 460–472. [DOI] [PubMed] [Google Scholar]
- Selleri L, Depew MJ, Jacobs Y, et al. (2001) Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development 128, 3543–3557. [DOI] [PubMed] [Google Scholar]
- Selleri L, DiMartino J, van Deursen J, et al. (2004) The TALE homeodomain protein Pbx2 is not essential for development and long‐term survival. Mol Cell Biol 24, 5324–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton JG, Steelman LS, White ER, et al. (2004) Synergy between PI3K/Akt and Raf/MEK/ERK pathways in IGF‐1R mediated cell cycle progression and prevention of apoptosis in hematopoietic cells. Cell Cycle 3, 370–377. [PubMed] [Google Scholar]
- Sitnicka E, Bryder D, Theilgaard‐Monch K, et al. (2002) Key role of Flt3 ligand in regulation of the common lymphoid progenitor but not in maintenance of the hematopoietic stem cell pool. Immunity 17, 463–472. [DOI] [PubMed] [Google Scholar]
- Solanilla A, Dechanet J, El Andaloussi A, et al. (2000) CD40‐ligand stimulates myelopoiesis by regulating flt3‐ligand and thrombopoietin production in bone marrow stromal cells. Blood 95, 3758–3764. [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, et al. (2001) Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankunas K, Shang C, Twu KY, et al. (2008) Pbx/Meis deficiencies demonstrate multigenetic origins of congenital heart disease. Circ Res 103, 702–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley EG, Biben C, Elefanty A, et al. (2002) Efficient Cre‐mediated deletion in cardiac progenitor cells conferred by a 3′UTR‐ires‐Cre allele of the homeobox gene Nk2–5. Int J Dev Biol 46, 431–439. [PubMed] [Google Scholar]
- Stier S, Ko Y, Forkert R, et al. (2005) Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med 201, 1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura R, He XC, Venkatraman A, et al. (2012) Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell 150, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlinton D, Good‐Jacobson K (2013) Diversity among memory B cells: origin, consequences, and utility. Science 341, 1205–1211. [DOI] [PubMed] [Google Scholar]
- Trompouki E, Bowman TV, Lawton LN, et al. (2011) Lineage regulators direct BMP and Wnt pathways to cell‐specific programs during differentiation and regeneration. Cell 147, 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum‐Finney B, Halasz LM, Sun M, et al. (2011) Notch2 governs the rate of generation of mouse long‐and short‐term repopulating stem cells. J Clin Invest 121, 1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitobello A, Ferretti E, Lampe X, et al. (2011) Hox and Pbx factors control retinoic acid synthesis during hindbrain segmentation. Dev Cell 20, 469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler IG, Barbier V, Nowlan B, et al. (2012) Vascular niche E‐selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance . Nat Med 18, 1651–1657. [DOI] [PubMed] [Google Scholar]
- Wohrer S, Knapp DJ, Copley MR, et al. (2014) Distinct stromal cell factor combinations can separately control hematopoietic stem cell survival, proliferation, and self‐renewal. Cell Rep 7, 1956–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolber FM, Leonard E, Michael S, et al. (2002) Roles of spleen and liver in development of the murine hematopoietic system. Exp Hematol 30, 1010–1019. [DOI] [PubMed] [Google Scholar]
- Xue Y, Lim S, Yang Y, et al. (2011) PDGF‐BB modulates hematopoiesis and tumor angiogenesis by inducing erythropoietin production in stromal cells. Nat Med 18, 100–110. [DOI] [PubMed] [Google Scholar]
- Yang L, Bryder D, Adolfsson J, et al. (2005) Identification of Lin–Sca1+kit+CD34+Flt3– short‐term hematopoietic stem cells capable of rapidly reconstituting and rescuing myeloablated transplant recipients. Blood 105, 2717–2723. [DOI] [PubMed] [Google Scholar]
- Zhao M, Ross JT, Itkin T, et al. (2012) FGF signaling facilitates postinjury recovery of mouse hematopoietic system. Blood 120, 1831–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JL, Ma C, O'Connell RM, et al. (2014) Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress‐induced hematopoiesis. Cell Stem Cell 14, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Differential WBC counts, spleen histology, and bone marrow cellularity in control and Pbx mutant mice at steady‐state or at steady‐state and during recovery from sub‐lethal irradiation (cf. Fig. 2).
Fig. S2. BFU‐E and CFU‐E analysis in SKOs and DKOs vs. controls (Ctrl) at day 8 post lethal irradiation (cf. Fig. 4).
Fig. S3. Cytokine gene expression analysis after irradiation‐induced injury in control and Pbx DKO spleens, as well as in control and DKO kidneys (for Epo) (cf. Fig. 6).