

Abstract
Key points
Regulation of autophagy in human muscle in many aspects differs from the majority of previous reports based on studies in cell systems and rodent muscle.
An acute bout of exercise and insulin stimulation reduce human muscle autophagosome content.
An acute bout of exercise regulates autophagy by a local contraction‐induced mechanism.
Exercise training increases the capacity for formation of autophagosomes in human muscle.
AMPK activation during exercise seems insufficient to regulate autophagosome content in muscle, while mTORC1 signalling via ULK1 probably mediates the autophagy‐inhibiting effect of insulin.
Abstract
Studies in rodent muscle suggest that autophagy is regulated by acute exercise, exercise training and insulin stimulation. However, little is known about the regulation of autophagy in human skeletal muscle. Here we investigate the autophagic response to acute one‐legged exercise, one‐legged exercise training and subsequent insulin stimulation in exercised and non‐exercised human muscle. Acute one‐legged exercise decreased (P<0.01) lipidation of microtubule‐associated protein 1A/1B‐light chain 3 (LC3) (∼50%) and the LC3‐II/LC3‐I ratio (∼60%) indicating that content of autophagosomes decreases with exercise in human muscle. The decrease in LC3‐II/LC3‐I ratio did not correlate with activation of 5′AMP activated protein kinase (AMPK) trimer complexes in human muscle. Consistently, pharmacological AMPK activation with 5‐aminoimidazole‐4‐carboxamide riboside (AICAR) in mouse muscle did not affect the LC3‐II/LC3‐I ratio. Four hours after exercise, insulin further reduced (P<0.01) the LC3‐II/LC3‐I ratio (∼80%) in muscle of the exercised and non‐exercised leg in humans. This coincided with increased Ser‐757 phosphorylation of Unc51 like kinase 1 (ULK1), which is suggested as a mammalian target of rapamycin complex 1 (mTORC1) target. Accordingly, inhibition of mTOR signalling in mouse muscle prevented the ability of insulin to reduce the LC3‐II/LC3‐I ratio. In response to 3 weeks of one‐legged exercise training, the LC3‐II/LC3‐I ratio decreased (P<0.05) in both trained and untrained muscle and this change was largely driven by an increase in LC3‐I content. Taken together, acute exercise and insulin stimulation reduce muscle autophagosome content, while exercise training may increase the capacity for formation of autophagosomes in muscle. Moreover, AMPK activation during exercise may not be sufficient to regulate autophagy in muscle, while mTORC1 signalling via ULK1 probably mediates the autophagy‐inhibiting effect of insulin.
Key points
Regulation of autophagy in human muscle in many aspects differs from the majority of previous reports based on studies in cell systems and rodent muscle.
An acute bout of exercise and insulin stimulation reduce human muscle autophagosome content.
An acute bout of exercise regulates autophagy by a local contraction‐induced mechanism.
Exercise training increases the capacity for formation of autophagosomes in human muscle.
AMPK activation during exercise seems insufficient to regulate autophagosome content in muscle, while mTORC1 signalling via ULK1 probably mediates the autophagy‐inhibiting effect of insulin.
Abbreviations
- ACC
acetyl‐CoA carboxylase
- AICAR
5‐aminoimidazole‐4‐carboxamide riboside
- AMPK, 5′AMP
activated protein kinase
- ATG
autophagy related gene
- BCA
bicinchoninic acid
- BMI
body mass index
- EDL
extensor digitorum longus
- GLUT4
glucose transporter 4
- IP
immunoprecipitation
- KD
kinase‐dead
- KRH
Krebs–Ringer–Henseleit
- LC3
microtubule‐associated protein 1A/1B‐light chain 3
- mKO
muscle‐specific knock‐out
- mTOR
mammalian target of rapamycin
- mTORC1
mTOR complex 1
- mTORC2
mTOR complex 2
- PMSF
phenylmethylsulphonyl fluoride
- PVDF
polyvinylidene difluoride
- PWL
peak work load
- ULK1
Unc51 like kinase 1
maximal oxygen uptake
- WT
wild‐type
Introduction
Macroautophagy, hereafter referred to as autophagy, is the major intracellular degradation system by which cytoplasmic materials are engulfed by autophagosomes and delivered to the lysosomes for degradation. This is part of a dynamic recycling system that provides substrates for cellular renovation and energy homeostasis (Mizushima & Komatsu, 2011). Autophagy is essential for cell differentiation, development and survival (Levine & Kroemer, 2008), and in skeletal muscle knockout of various proteins linked to autophagy leads to dysregulation of muscle metabolism and function (Raben et al. 2008; Masiero et al. 2009; Grumati et al. 2010; He et al. 2012). Microtubule‐associated protein 1A/1B‐light chain 3 (LC3) immunoblotting is the most commonly used biochemical method to evaluate autophagy in intact muscle tissue, and the autophagosome‐bound LC3‐II content and the LC3‐II/LC3‐I ratio are recognized as valuable markers of autophagosome content in tissues (Barth et al. 2010; Klionsky et al. 2012). In contrast, they should only cautiously be used as direct markers of autophagic flux (Mizushima & Yoshimori, 2007; Rubinsztein et al. 2009).
Evaluated by LC3 immunoblotting, acute exercise has been found to increase autophagosome content in mouse muscle in some (Grumati et al. 2011; He et al. 2012; Jamart et al. 2013; Pagano et al. 2014; Vainshtein et al. 2015) but not all studies (Kim et al. 2012). Furthermore, exercise training increases the basal content of autophagosomes in mouse muscle (Kim et al. 2013; Lira et al. 2013; Jiang et al. 2014). In human skeletal muscle, autophagosome content was increased after 18 h of ultra‐endurance running (Jamart et al. 2012), but remained unchanged (Masschelein et al. 2014) or even decreased in response to less strenuous cycling exercise protocols (Moller et al. 2015; Schwalm et al. 2015). The regulation of autophagy in response to endurance exercise training in humans has not been investigated.
In rodents, autophagy is critical for whole body glucose tolerance (He et al. 2012) as well as insulin action in liver (Yang et al. 2010) and skeletal muscle cells (Shi et al. 2015). Recently it was shown that insulin markedly inhibits markers of autophagosome content in muscle of healthy humans (Vendelbo et al. 2014; Kruse et al. 2015), but not in patients with type 2 diabetes when studied during euglycaemia (Kruse et al. 2015). This suggests that autophagy may also be vital for regulation of glucose homeostasis in humans. We and others have repeatedly shown that both acute exercise (Richter et al. 1982; Cartee et al. 1989; Wojtaszewski et al. 1997; Frosig et al. 2007 b) and exercise training (James et al. 1985; Dela et al. 1995; Frosig et al. 2007 a) potently improve insulin sensitivity for glucose uptake in human skeletal muscle. Whether this adaptation to exercise is related to altered autophagic capacity or regulation is currently unknown.
Several candidate signalling molecules have been suggested to regulate autophagy. In human skeletal muscle, 5′AMP activated protein kinase (AMPK) exists as three distinct heterotrimeric complexes (α2/β2/γ3, α2/β2/γ1 and α1/β2/γ1) that are differentially activated during exercise (Winder & Hardie, 1996; Wojtaszewski et al. 2000 b; Birk & Wojtaszewski, 2006). In cells, AMPK induces autophagy (Meley et al. 2006; Liang et al. 2007; Herrero‐Martin et al. 2009; Lee et al. 2010; Vingtdeux et al. 2011; Sanchez et al. 2012) by phosphorylation of Unc‐51 like kinase‐1 (ULK1) on Ser‐555 (Egan et al. 2011; Ost et al. 2014; Bujak et al. 2015). This signalling axis to autophagy has recently been supported by correlative evidence in human skeletal muscle (Moller et al. 2015). In contrast, the mammalian target of rapamycin (mTOR) complex 1 (mTORC1), a well‐described insulin signalling molecule (Manning & Cantley, 2007; Zoncu et al. 2011; Dan et al. 2014), suppresses autophagy via Ser‐757 phosphorylation of ULK1 (Jung et al. 2010; Kim et al. 2011; Castets et al. 2013).
The present study evaluated markers of autophagy during and in recovery from acute one‐legged exercise as well as in response to one‐legged exercise training in human skeletal muscle. Furthermore, we investigated if improved insulin sensitivity after acute exercise and training was associated with altered regulation of autophagy in muscle. Finally, by correlative evaluation in humans and studies in rodents we sought to elucidate the molecular signals regulating autophagy in response to exercise and insulin stimulation in muscle.
Methods
Ethical approval
All participants gave their informed consent before participating in the studies. The studies were approved by the local ethics committee (nos. KF1277313, 01‐180/01, KF 01‐070/96 and KF 01 261127) and performed in accordance with the Declaration of Helsinki. Protocols for animal studies were approved by the Danish Animal Experimental Inspectorate and complied with the European Convention for the protection of Vertebrate Animals used for Experiments and Other Scientific Purposes.
Acute exercise study
Samples from the vastus lateralis muscle from seven healthy moderately trained male subjects [age 27 ± 2 years, weight 84 ± 3 kg, and body mass index (BMI) 24.8 ± 0.6 kg m−2, maximal oxygen uptake () 52 ± 2 ml O2 min−1 kg−1] were obtained as previously described (Treebak et al. 2014). Samples from this study have been used for another publication (Treebak et al. 2014). The subjects initially came to the laboratory on two separate days to complete an incremental cycle ergometer test to determine peak oxygen uptake, and an incremental one‐legged knee extensor exercise test on a modified Krogh cycle ergometer (Andersen et al. 1985) to determine the peak work load (PWL) of the knee extensors. Three of the subjects exercised with their non‐dominant leg and the other four with their dominant leg. Two to 3 weeks after the initial baseline testing, subjects arrived overnight fasted at the laboratory on two separate occasions after having refrained from physical activity for 2 days. After 30–40 min of rest in the supine position, one muscle biopsy was obtained from the vastus lateralis muscle of each leg using a 5 mm Bergstrom needle with suction. Subjects rested for another 20 min and then performed a single 60 min bout of one‐legged knee extensor exercise at ∼80% PWL with two 5 min intervals at ∼100% PWL to ensure activation of the majority of muscle fibres in the thigh region. The other leg served as a rested control. Immediately after termination of the exercise bout, muscle biopsies were obtained from vastus lateralis muscles of both legs. In the overnight fasted situation, biopsies were also obtained in both legs 4 h after termination of exercise. All biopsies were obtained from different incisions and separated by at least 5 cm. Muscle biopsies were obtained under local anaesthesia [∼2–3 ml xylocaine (20 mg ml−1 lidocaine), Astra, Stockholm, Sweden] and were immediately frozen in liquid nitrogen and stored at −80°C for further analysis.
Recovery from exercise +/– insulin study
Samples from the vastus lateralis muscle from 12 young, healthy men (age 24 ± 1 years; weight 80 ± 2 kg; BMI 23 ± 0.5 kg m−2; 4.6 ± 0.1 l min−1) were obtained as previously described (Frosig et al. 2007 b). Samples from this study have been used for other publications (Frosig et al. 2007 b, 2009; Treebak et al. 2009). The subjects underwent the same preliminary incremental cycle ergometer test and incremental one‐legged knee extensor exercise test prior to the test day as described in the acute exercise study. On the morning following 4 days of standardized diet [65 energy (E)% carbohydrates, 20 E% fat and 15 E% protein] the subjects arrived at the laboratory after an overnight fast. After a light, carbohydrate‐rich (∼80 E%) breakfast (∼5% daily energy intake) the subjects performed 60 min of dynamic one‐legged knee extensor exercise (1 kick s−1) at a work intensity averaging ∼80% of the PWL of the knee extensors. Two 5 min intervals of work at ∼100% PWL in the exercise bout were included. After exercise, the subjects rested in the supine position. Four hours after exercise the subjects underwent a 100 min euglycaemic–hyperinsulinaemic clamp (1.5 mU min−1 kg−1) initiated with a bolus injection of insulin (9 mU kg−1) (Actrapid, Novo Nordisk, Bagsværd, Denmark) given over 1 min. Needle biopsies were obtained as described in the acute exercise study immediately before initiation and at termination of the clamp in both the rested and the previously exercised leg. As reported, 4 h after acute exercise, a ∼80% increase in insulin‐stimulated glucose uptake was observed when compared to the rested control leg (Frosig et al. 2007 b).
Exercise training +/– insulin study
Samples from the vastus lateralis muscle from eight healthy young men (aged 25 ± 1 years; weight 81 ± 2 kg; BMI 24.6 ± 0.5 kg m−2; and 49 ± 1 ml O2 min−1 kg−1) were obtained as previously described (Frosig et al. 2007 a). Samples from this study have been used for other publications (Frosig et al. 2004, 2007 a; Rose et al. 2007; Brandauer et al. 2013). Subjects underwent the same preliminary incremental cycle ergometer test and incremental one‐legged knee extensor exercise test before the test day as described in the acute exercise study. After 2 days of a controlled diet (55.5 ± 0.4% carbohydrate, 29.4 ± 0.4% fat, 15.1 ± 0.2% protein), the subjects arrived at the laboratory after an overnight fast. After 30 min of supine rest, needle biopsies were obtained as in the acute exercise study. Next, the subjects underwent a one‐legged knee extensor exercise training regimen consisting of four sessions during the 1st week, five sessions during the 2nd week and six sessions during the 3rd week. The duration gradually increased from 1 to 2 h per session. Training intensity varied between 70 and 85% PWL and was progressively increased in accordance with an expected increase in PWL during this type of training (Richter et al. 1998). Included in each training session was work for 5–7 min at 100% PWL to ensure recruitment of the majority of muscle fibres in the knee extensor region (Gollnick et al. 1974). After the training intervention, muscle biopsies were obtained 15 h after the last exercise bout. Subsequently, the subjects underwent a 120 min hyperinsulinaemic‐euglycaemic clamp as in the recovery from exercise +/– insulin study. Needle biopsies were obtained after 10 and 120 min of insulin infusion. As reported, training resulted in a ∼20% increase in PWL of the knee extensors, ∼35% increases in maximal activities of citrate synthase and 3‐hydroxyacylCoA dehydrogenase as well as a ∼60% increase in insulin‐stimulated glucose uptake (Frosig et al. 2004).
Swimming exercise in rats
Male Wistar rats (150–200 g) were maintained on a 12:12 h light–dark cycle and received standard rodent diet (altromin, cat. no. 1324; Brogaarden, Lynge, Denmark) and water ad libitum. Rats swam pair‐wise in containers with water temperature kept at 35 ± 1°C. Rats completed in total 4 h of swimming divided in eight bouts of 30 min. Each exercise bout was separated by 5 min of rest, where the rats were towel dried and placed in individual cages. Exercised rats were either killed immediately at exercise stop (Exercise) or allowed to recover for 2 h with ad libitum access to water (Recovery). The resting control rats were food deprived for a period corresponding to the exercise period in the Exercise and Recovery groups before being killed. From all animals, tibialis anterior muscle was removed and frozen in liquid nitrogen and stored at –80°C for further analysis.
Incubation of intact mouse skeletal muscle in vitro
C57Bl/6 female mice overexpressing a muscle‐specific, kinase‐dead AMPKα2 construct (AMPK KD) and corresponding wild type (WT) littermates (Mu et al. 2001) were used in the 5‐aminoimidazole‐4‐carboxamide riboside (AICAR) experiments, and female C57BL/6JBomTac (Taconic, Lille Skensved, Denmark) mice were used in the mTOR kinase inhibitor experiments. All mice were 10–16 weeks old at the time of experimentation. Mice were kept on a 12:12 h light–dark cycle and received standard rodent diet (altromin, cat. no. 1324; Brogaarden) and water ad libitum. Soleus muscle in the AICAR experiments and both soleus and extensor digitorum longus (EDL) muscles in the mTOR kinase inhibitor experiments from both legs were carefully removed from 1 h fasted and anaesthetized mice (6 mg pentobarbital and 0.24 mg lidocaine per 100 g body weight). Mice were killed by cervical dislocation after muscles had been removed. Muscles were gently stretched to resting tension (4–5 mN) in incubation chambers with continuous force recording (Multi Myograph system; Danish Myo‐Technology, Aarhus, Denmark). These chambers contained 4 ml of heated (30°C) constantly 95% O2/5% CO2‐bubbled Krebs–Ringer–Henseleit (KRH) buffer supplemented with 2 mm pyruvate and 8 mm mannitol, as described previously (Jensen et al. 2007). When relevant, muscles were incubated for 40 min with 2 mm AICAR (Toronto Research Chemicals, Toronto, Canada) in the AICAR experiments, and with either 0.1% DMSO or 640 nm AZD8055 for 60 min in the mTOR kinase inhibitor experiments as described (Kleinert et al. 2014). When appropriate, insulin (Actrapid, Novo Nordisk) was added during the last 30 min of inhibitor and DMSO presence, resulting in an insulin concentration of 60 nm in the mTOR kinase inhibitor experiments. Muscles were washed in ice‐cold KRH buffer, blotted dry and snap‐frozen in liquid nitrogen before being stored at −80°C for subsequent analyses.
Cultured human myotubes
After consuming a controlled isocaloric diet (60 E% carbohydrate, 15 E% protein and 25 E% fat) for 6–8 days, five young, healthy, moderately trained male subjects who had fasted overnight arrived in the laboratory. After 30 min of supine rest, a needle biopsy was obtained as in the acute exercise study. The biopsies were collected in cold PBS supplemented with 1% PeSt (100 U ml–1 penicillin and 100 μg ml–1 streptomycin). Satellite cells were isolated and cultured as described (Al‐Khalili et al. 2003). Myotubes were serum starved overnight, washed once in sterile PBS and incubated with DMEM (5 mm 1 g glucose–1 L–1) in the presence or absence of AICAR (1 mm) with an incubation time of 30 min.
RNA isolation, reverse transcription and real time PCR
In remaining muscle samples (n = 2–6) from the acute exercise study, total RNA was isolated from ∼15 mg pulverized wet muscle tissue by a guanidinium thiocyanate‐phenol‐chloroform extraction method (Chomczynski & Sacchi, 1987) with modifications (Pilegaard et al. 2000). Superscript II RNase H−‐system and Oligo dT (Invitrogen, Carlsbad, CA, USA) were used to reverse transcribe the mRNA to cDNA as previously described (Pilegaard et al. 2000). Real‐time PCR was performed with an ABI 7900 sequence‐detection system (Applied Biosystems, Foster City, CA, USA). Primers and TaqMan probes for cyclophilin, SQMST1 and MAP1LC3 were from Applied Biosystems (Copenhagen, Denmark). The obtained cycle threshold (C T) values reflecting the initial content of the transcript in the samples were converted to an arbitrary amount by using standard curves obtained from a serial dilution of a representative pooled sample. For each sample, the amount of a given target cDNA was normalized to cyclophilin mRNA content, which did not differ with intervention and between legs.
Western blotting
All materials were from Sigma‐Aldrich (St Louis, MO, USA) unless stated otherwise. Freeze‐dried and dissected muscle tissue and cell lysates were homogenized in ice‐cold buffer [50 mm Hepes, 150 mm NaCl, 20 mm Na4P2O7, 20 mm β‐glycerophosphate, 10 mm NaF, 2 mm Na3VO4, 2 mm EDTA, 1% Nonidet P‐40, 10% glycerol, 2 mm phenylmethylsulphonyl fluoride (PMSF), 1 mm MgCl2, 1 mm CaCl2, 10 μg ml−1 leupeptin, 10 μg ml−1 aprotinin and 3 mm benzamidine], and the homogenate was rotated end‐over‐end at 4°C for 1 h before being centrifuged for 30 min [16,000 g (acute exercise study & mouse experiments) or 17,500 g (acute exercise and insulin study & training study)]. The supernatant (lysate) was harvested and total protein content was determined using the bicinchoninic acid method (BCA no. 23225, Pierce, Rockford, IL, USA). Next, lysates were diluted in MilliQ ultrapure water and 6× Laemlii buffer (340 mm Tris base, pH 6.8, 11% SDS, 20% glycerol, 0.05% bromophenol blue and 225 mm freshly added dithiothrietol) at the same protein concentration in all samples.
Total protein and phosphorylation were measured by standard immunoblotting techniques as described previously (Frosig et al. 2007 b). For a given measurement, equal volumes and protein amounts of all samples were separated by SDS‐PAGE (PROTEAN Tetra, BioRad, Hercules, USA) using self‐cast 15‐well minigels (7–15% acrylamide). Next, regions of interest were identified based on the molecular weight markers, cut from the gels and transferred semi‐dry (TE‐77, Amersham Biosciences, Piscataway, NJ, USA) onto the same polyvinylidene difluoride (PVDF) membrane (Immobilon‐P, 0.45 μm, Millipore, Billerica, MA, USA) for subsequent blocking in TBS with 0.05% Tween 20 and 2% skimmed milk protein or BSA for 60 min at room temperature and antibody probing. Primary antibodies used were anti‐phospho‐acetyl coA carboxylase (ACC)1 Ser‐79 (also detects equivalent site on mouse ACC2 at Ser‐212) (Upstate Biotechnology, Lake Placid, NY, USA, cat. no. 07‐303), anti‐p62 (BD Biosciences, San Jose, CA, USA, cat. no. 610832), anti‐microtubule‐associated protein 1A/1B‐light chain 3 (LC3) (Novus Bio., Littleton, Canada, cat. no. NB100‐2220), anti‐β‐actin (cat. no. 4967), anti‐phospho‐ULK1‐Ser‐555 (cat. no. 5869S) and anti‐phospho‐ULK1‐Ser‐757 (cat. no. 6888) (Cell Signaling Technology, Danvers, MA, USA) and anti‐ULK1 (Sigma‐Aldrich, Denmark, cat. no. A7481). Membranes were incubated with primary antibodies overnight at 4°C, followed by incubation with HRP‐conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 60 min at room temperature. Bands were visualized in a Bio‐Rad ChemiDoc™ MP Imaging System after incubation with enhanced chemiluminescence (ECL+; Amersham Biosciences). Band densitometry was performed using Image Lab version 4.0 (BioRad). Loading was optimized for linearity for all antibodies prior to use. On each gel two internal controls were loaded in each side of the gel, and data were related to the average of these on each gel to minimize variation. Coomassie staining after development was performed by submersion of PVDF membranes for ∼10 min in 0.5% Coomassie Blue G‐250 in 50% ethanol/10% acetic acid, removal of excess Coomassie stain with distilled water followed by destaining in 50% ethanol/10% acetic acid until bands were clearly visible.
AMPK activity assay
AMPK activity was measured on muscle lysates by immunoprecipitation (IP) of the γ3 subunit (isolating the α2/β2/γ3 complex from lysates) followed by IP of the α2 (isolating the α2/β2/γ1 complex from lysates) and lastly the α1 subunit (isolating the α1/β2/γ1 complex from lysates). Assay conditions for performing activity measurements on the different IPs have been described previously (Birk & Wojtaszewski, 2006).
Statistics
Data are reported as means ± SEM. All statistical analyses were performed in SigmaPlot 12.5 (Systat Software, San Jose, CA, USA) using paired t‐test in Fig. 4, one‐way ANOVA in Fig. 1 E, two‐way ANOVA in Figs 3 and 6, and two‐way repeated‐measures ANOVA in Figs 1 A–D, 5 and 7. In the case of significant interaction effects, the Tukey test was used post hoc. Correlation analyses were performed by calculating Pearson's product moment correlation coefficient. Statistical significance was set at P < 0.05.
Figure 4. Effect of acute AICAR treatment on LC3 lipidation in human myotubes .
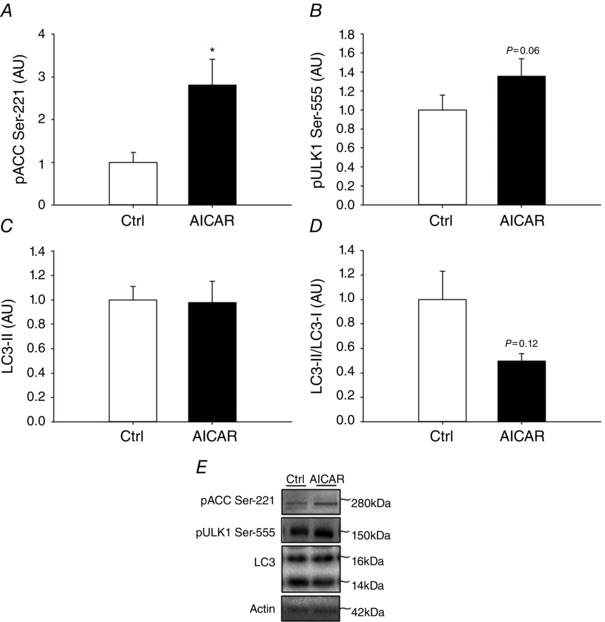
A–D, phosphorylation of ACC at Ser‐221 (A), ULK1 at Ser‐555 (B), protein content of LC3‐II (C) and LC3‐II/LC3‐I ratio (D) in human myotubes incubated either with or without AICAR for 30 min. E, representative Western blots; n = 5. Data are presented as means ± SEM. *Significantly different (P<0.05) from Ctrl. AICAR, 5‐aminoimidazole‐4‐carboxamide riboside; AU, arbitrary units; Ctrl, control.
Figure 1. Effect of acute exercise on autophagy markers in human and rat skeletal muscle .
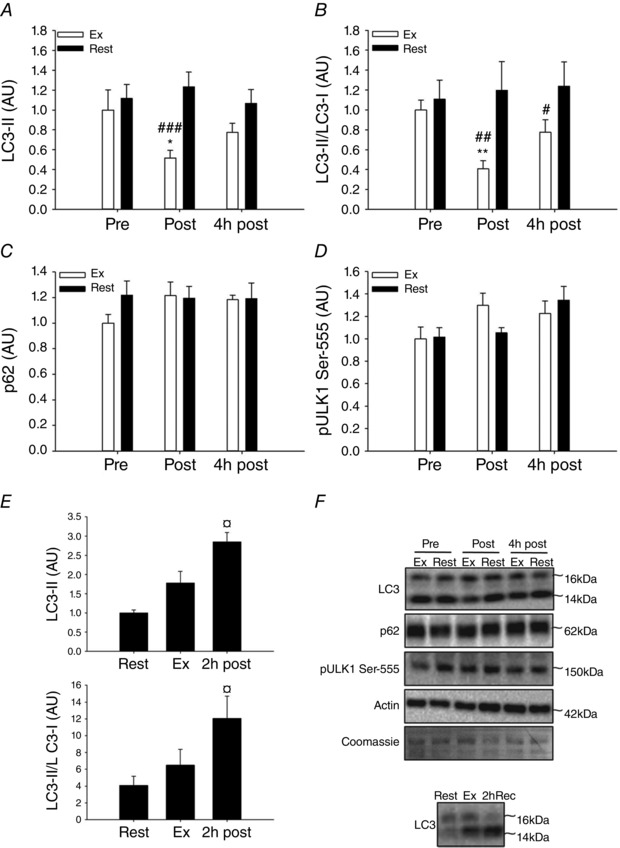
A–D, LC3‐II protein content (A), LC3‐II/LC3‐I ratio (B), p62 protein content (C) and ULK1 Ser‐555 phosphorylation (D) in human skeletal muscle at rest (Pre), immediately after one‐legged knee extensor exercise (Post) and 4 h after exercise (4 h post) in the exercised and the rested leg. E, LC3‐II protein content and LC3‐II/LC3‐I ratio in rat skeletal muscle at rest, immediately after 4 h of swimming exercise and after 2 h of recovery from exercise termination. F, representative Western blots. Data are presented as means ± SEM; n = 6–7 in A–D and n = 8 in E. *,**Significantly different (P<0.05, P<0.01) from pre within exercised leg; #,##,###significantly different (P<0.05, P<0.01, P<0.001) from rested leg within trial; ¤significantly different (P<0.05) from rest. AU, arbitrary units; Ex, exercised leg (A–D), post exercise (E); Pre, pre exercise; Rest, rested leg (A–D), rest (E); Post, post exercise; 2 h post, 2 h post exercise; 4 h post, 4 h post exercise.
Figure 3. Effect of acute AICAR treatment on LC3 lipidation in mouse skeletal muscle of WT and AMPK KD mice .
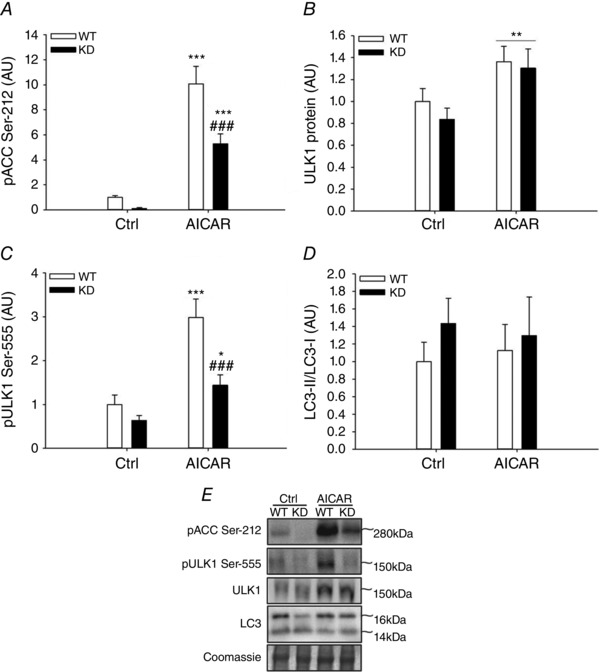
A–D, LC3‐II/LC3‐I ratio (A), phosphorylation of ACC at Ser‐212 (B) and ULK1 protein content (C), and phosphorylation of ULK1 at Ser‐555 (D) in soleus muscle from AMPK KD mice and WT littermates incubated with either saline (Ctrl) or AICAR for 40 min. E, representative Western blots; n = 6. Data are presented as means ± SEM. *,*** significantly different (P<0.05, P<0.001) from Ctrl within genotype; **main effect (P<0.01) of AICAR; ###significantly different (P<0.001) from WT within AICAR. AICAR, 5‐aminoimidazole‐4‐carboxamide riboside; AU, arbitrary units; Ctrl, control; KD, kinase‐dead; WT, wild type.
Figure 6. Effect of acute mTOR inhibition for insulin‐stimulated LC3 lipidation in mouse skeletal muscle .
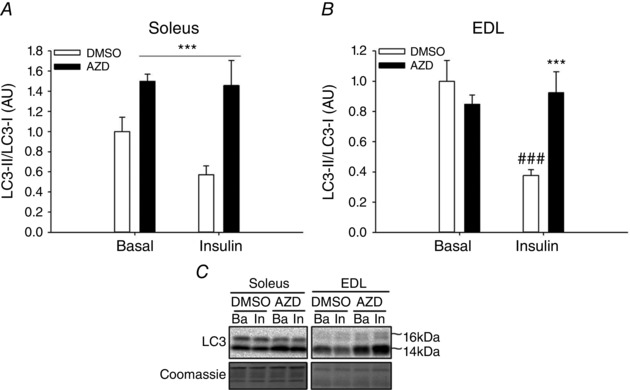
A and B, LC3‐II/LC3‐I ratio in soleus (A) and EDL (B) muscle incubated with either DMSO as basal control or AZD8055 and subsequently incubated with either saline (basal) or insulin. C, representative Western blots; n = 6–8. Data are presented as means ± SEM. A: ***main effect (P<0.001) of AZD. B: ***significantly different (P<0.001) from DMSO within insulin; ###significantly different (P<0.001) from basal within DMSO. AU, arbitrary units; AZD, AZD8055; Ba, basal; EDL, extensor digitorum longus; In, insulin.
Results
Acute exercise
In response to high intensity acute one‐legged exercise we observed a decreased (P<0.01) LC3‐II content (Fig. 1 A) resulting in a decreased LC3‐II/LC3‐I ratio (Fig. 1 B) in the exercised compared with the contralateral rested muscle. This indicates that exercise reduces the autophagosome content in human muscle due to a local contraction‐induced mechanism. In contrast to the marked changes in LC3, the content of the autophagosomal cargo adapter protein, p62, did not change with exercise (Fig. 1 C). A decrease in LC3‐II and LC3‐II/LC3‐I ratio in human muscle after exercise is in contrast to most rodent studies that report increased LC3‐II/LC3‐I ratio in response to exercise (Grumati et al. 2011; He et al. 2012; Pagano et al. 2014; Liu et al. 2015). To positively control the dynamics of LC3 immunoblotting and to confirm this apparent species difference in regulation of autophagy, rats were studied before and after 4 h of swimming exercise as well as 2 h into recovery from exercise. We found that exercise in rats increased the LC3‐II content and the LC3‐II/LC3‐I ratio and the response was significantly different (P<0.05) from resting values after 2 h of recovery (Fig. 1 E). Although the exercise protocols are not comparable with our human studies, this observation seems consistent with our notion of a species difference in exercise‐induced regulation of autophagy.
AMPK has been proposed as an important component in exercise‐induced regulation of autophagy. To evaluate this in human muscle, we correlated changes in AMPK activity with the observed changes in LC3‐II/LC3‐I ratio. However, activity of none of the three identified AMPK trimer complexes in human muscle was associated with regulation of LC3 (Fig. 2 A–C). The AMPK activities used for the correlation analyses have previously been published (Treebak et al. 2014). Briefly, α2β2γ3 and α2β2γ1 AMPK complex activity increased with exercise ∼25 and ∼2‐fold, respectively, while changes in α1β2γ1 AMPK complex activity with exercise were not significantly different from rested values (Treebak et al. 2014). Ser‐555 on ULK1 is a proposed AMPK target (Egan et al. 2011; Ost et al. 2014; Bujak et al. 2015) and notably changes (after exercise minus before exercise) in phosphorylation on this site in exercised muscle correlated (P<0.01) with the change in activity of AMPK α1β2γ1 complexes (Fig. 2 F). Hence in human skeletal muscle, activation of AMPKα1‐containing complexes during exercise may regulate ULK1, but based on the absence of a correlation between AMPK activity and the LC3‐II/LC3‐I ratio, AMPK activation does not appear to be closely linked to regulation of autophagy in muscle. To test this interpretation, we incubated WT and AMPK KD muscle with the AMPK agonist AICAR. As expected, AICAR robustly activated AMPK based on ACC Ser‐212 phosphorylation (P<0.001), a widely used endogenous AMPK activity read‐out, and this response was markedly blunted (P<0.001) in AMPK KD muscle (Fig. 3 A). Furthermore, AICAR stimulation increased (P<0.01) the content of ULK1 (Fig. 3 B) as well as ULK‐1 Ser‐555 phosphorylation (Fig. 3 C) in both WT and AMPK KD muscle, but the change in phosphorylation was markedly reduced (P<0.001) in AMPK KD muscle (Fig. 3 C). Of particular note and consistent with the correlation data, we did not observe any change in LC3‐II/LC3‐I ratio with AICAR stimulation in either WT or AMPK KD muscle (Fig. 3 D). To verify this in human muscle cells, myotubes isolated and cultured from young male donors were incubated with or without AICAR for 30 min. As expected, AICAR robustly activated AMPK based on ACC Ser‐221 phosphorylation (P<0.05; Fig. 4 A), a widely used endogenous AMPK activity read‐out. Furthermore, AICAR stimulation tended to increase strongly (P = 0.06) the phosphorylation of ULK‐1 at Ser‐555 (Fig. 4 B). Consistent with the data in mouse muscle, we did not observe any significant change in LC3‐II protein content in human myotubes (Fig. 4 C). This supports AMPK as an upstream kinase of ULK1, but AMPK activation may not be sufficient to increase autophagosome content in skeletal muscle. Curiously, LC3‐I tended to increase with AICAR, suggesting that our AICAR stimulation protocol led to adaptations at the level of the autophagic gene transcriptional program and control of autophagic capacity in cells.
Figure 2. Associations between changes with exercise in AMPK complex activity and LC3 lipidation as well as ULK1 phosphorylation in human skeletal muscle .
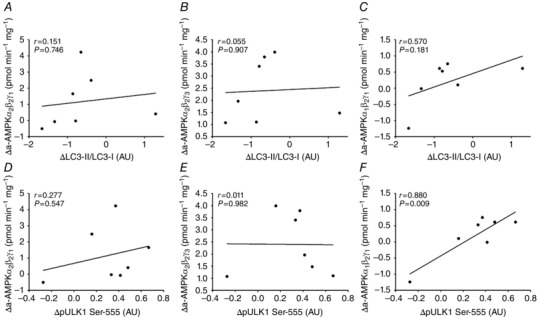
Associations between acute changes with exercise (post values – pre values) in the exercised leg in LC3‐II/LC3‐I ratio and AMPK α2β2γ3 (A), AMPK α2β2γ1 (B) and AMPK α1β2γ1 (C) complex activity as well as phosphorylation of ULK1 at Ser‐555 and AMPK α2β2γ3 (D), AMPK α2β2γ1 (E) and AMPK α1β2γ1 (F) complex activity in human skeletal muscle; n = 7. Data are presented as means ± SEM. AU, arbitrary units.
Recovery from exercise +/– insulin stimulation
To study the effect of insulin on regulation of autophagy either alone or in recovery from exercise, muscle biopsies were obtained in both legs 4 h after one‐legged exercise. The LC3‐II content was still decreased (P<0.01) in the previously exercised muscle (Fig. 5 A, basal state), although this was neither reflected in the LC3‐II/LC3‐1 ratio (Fig. 5 B) nor in the content of p62 protein (Fig. 5 C). Two hours of insulin stimulation at this time point resulted in a substantial (∼80%) further reduction (P<0.01) in the LC3‐II/LC3‐I ratio independent of previous exercise (Fig. 5 B). This illustrates that insulin reduces the content of autophagosomes in human muscle. While insulin did not change Ser‐555 phosphorylation of ULK1 (Fig. 5 D), phosphorylation of ULK1 at Ser‐757 was increased (P<0.01) 4 h after exercise and increased (P<0.01) even further with insulin stimulation (Fig. 5 E). As ULK1 phosphorylation at Ser‐757 is a proposed autophagy‐inhibiting mTORC1 target, this led us to further examine the role of mTOR in mediating insulin‐stimulated suppression of autophagy. Interestingly, inhibition of mTOR signalling in mouse muscle with the global mTOR (mTORC1 and mTORC2) inhibitor AZD8055, previously shown to block insulin‐stimulated p70S6K1 Thr‐389 phosphorylation (Kleinert et al. 2014), completely prevented the insulin‐stimulated reduction in the LC3‐II/LC3‐I ratio (Fig. 6 A, B), firmly establishing this signalling link in mouse muscle.
Figure 5. Effect of insulin after acute exercise on autophagy markers in human skeletal muscle .
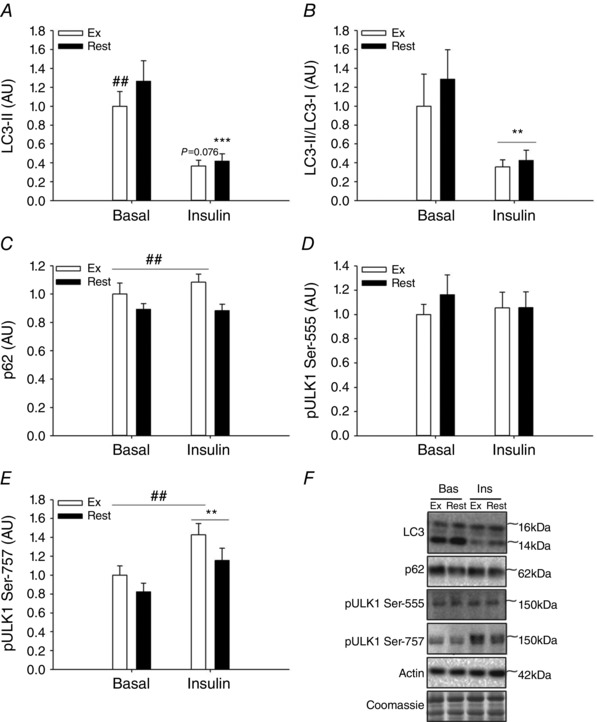
A–F, LC3‐II protein content (A), LC3‐II/LC3‐I ratio (B), p62 protein content (C), and phosphorylation of ULK1 at Ser‐555 (D) and Ser‐757 (E) in human skeletal muscle 4 h after one‐legged knee extensor exercise before (Basal) and after (Insulin) a euglycaemic–hyperinsulinaemic clamp in the previously exercised and the rested leg. E, representative Western blots; n = 10–12 due to lack of sample in two subjects. ***Significantly different (P<0.001) from basal within leg; **main effect (P<0.01) of insulin independent of leg; ##significantly different (P<0.01) from rested leg within basal in A and main effect (P<0.01) of leg independent of insulin in C and E. AU, arbitrary units; Bas, basal; Ex, exercised leg; Ins, Insulin; Rest, rested leg.
Exercise training +/– insulin stimulation
To study the effect of exercise training on regulation of autophagy, muscle biopsies were obtained in both legs before and after 3 weeks of one‐legged knee extensor endurance training. Interestingly, training decreased (P<0.05) the LC3‐II/LC3‐I ratio (Fig. 7 B) and increased (P<0.05, Pre vs. Ins10) the protein content of p62 (Fig. 7 C) in both legs, suggesting that the adaptation was not induced locally by contractions but rather induced by changes in circulating systemic factors. Notably, 15 h after the last exercise bout, 120 min (but not 10 min) of insulin stimulation resulted in a marked further reduction (P<0.001) in LC3‐II/LC3‐I ratio that was similar in trained and untrained muscle (Fig. 7 B). In contrast to the acute response to exercise, the reduced LC3‐II/LC3‐I ratio after training was due to increased protein content of LC3‐I (P<0.01; ∼75%) and unchanged LC3‐II levels (Fig. 7 A), suggesting that the capacity for autophagosome formation is increased with exercise training. To understand the mechanism for the increase in LC3‐I and p62 protein content after the training in both the trained and the untrained leg, mRNA levels in response to acute exercise were studied in a subset of samples (n = 2–6) from the acute exercise study. Here, mRNA levels of LC3 (MAP1LC3) tended (P = 0.087) to increase (Fig. 3 E) and p62 (SQMST) mRNA significantly increased (P<0.05) 4 h after acute one‐legged knee extensor exercise in both the previously exercised and the rested leg (Fig. 7 F). This shows that exercise activates the autophagic gene transcriptional apparatus in the recovery period after contractions and provides a possible explanation for increased content of autophagic proteins in both legs measured after training.
Figure 7. Effect of insulin on autophagy markers before and after exercise training and mRNA levels of autophagy genes in response to acute exercise in human skeletal muscle .
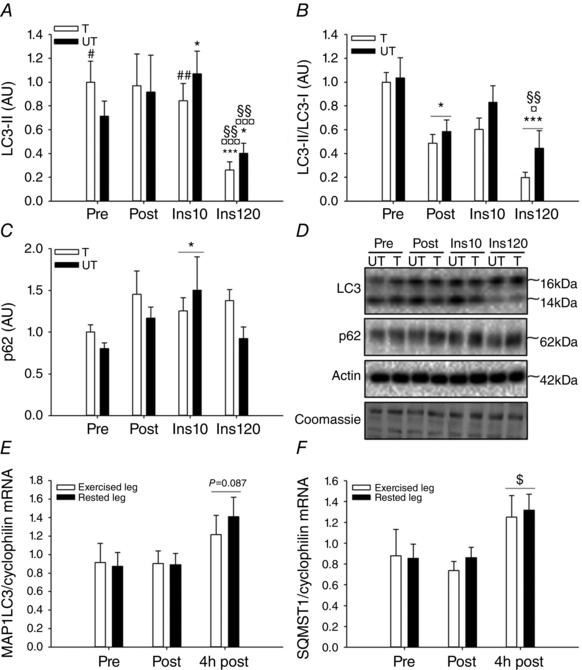
A–C, LC3‐II protein content (A), LC3‐II/LC3‐I ratio (B) and p62 protein content (C) in human skeletal muscle before and after 3 weeks of one‐legged knee extensor exercise training and after 10 and 90 min of a euglycaemic–hyperinsulinaemic clamp after the training intervention. D, representative Western blots; n = 6–8 due to lack of sample in two subjects in A–C. E and F, MAP1LC3 (LC3 gene) (E) and SQMST1(p62 gene) (F) mRNA levels in human skeletal muscle at rest (Pre), immediately after one‐legged knee extensor exercise (Post) and 4 h after exercise (4 h post) in the exercised and the rested leg. Data are presented as means ± SEM. E and F: n = 2 in pre ex leg, n = 5 in pre rest leg, n = 4 in post ex leg, n = 4 in post rest leg, n = 4 in 4 h post ex leg, n = 6 in 4 h post rest leg due to lack of sample. *,**,***/¤,¤¤,¤¤¤/§,§§,§§§Significantly different (P<0.05, P<0.01, P<0.001) from pre/post/insulin 10; #,##significantly different (P<0.05, P<0.01) from untrained leg within intervention; $significant different (P<0.05) from pre independent of leg. AU, arbitrary units; Ex, exercised leg; Ins10, 10 min into the euglycaemic–hyperinsulinaemic clamp after the training intervention; Ins90, 90 min into the euglycaemic–hyperinsulinaemic clamp after the training intervention; Pre, pre‐training intervention/ pre‐exercise; Post, post‐training intervention/exercise; Rest, rested leg; T, trained leg; UT, untrained leg; 4 h post, 4 hours post‐exercise.
Discussion
This study indicates that regulation of autophagy in human muscle in many aspects differs from the majority of cell and rodent reports. Notably, in human muscle, acute exercise, exercise training and insulin stimulation leads to a reduction in the LC3‐II/LC3‐I ratio, a commonly used marker of autophagy. Our data further suggest that exercise‐induced activation of AMPK leads to regulation of ULK1, but AMPK activation alone is not sufficient to regulate autophagy. In contrast, mTOR signalling via ULK1 is a probable signalling axis regulating autophagy in response to insulin stimulation.
Autophagy is critical for muscle function and metabolic homeostasis. Consequently, understanding how exercise and insulin stimulation regulates autophagy in human muscle is currently of major interest. During autophagy cytoplasmic materials are engulfed by autophagosomes in a process where LC3‐I is lipidated to form the autophagosome‐resident anchoring protein LC3‐II. This targets the material as well as LC3‐II for lysosomal degradation or recycling. As reviewed, the content of LC3‐II is considered a valid indicator of the content of autophagosomes under most conditions (Tanida et al. 2005; Klionsky et al. 2012). In contrast, using either the content of LC3‐II or the ratio between LC3‐II and LC3‐I as a direct marker of autophagic flux should be done with caution, as the content of autophagosomes depend on both the rate of formation and the rate of lysosomal degradation (Mizushima & Yoshimori, 2007; Rubinsztein et al. 2009). To illustrate this, autophagosomes may accumulate at a given time point both as a consequence of accelerated formation (increased autophagic flux) or suppressed degradation (reduced autophagic flux).
Here we show that acute one‐legged knee extensor exercise substantially reduces LC3 lipidation and subsequently the LC3‐II/LC3‐I ratio in skeletal muscle and that this effect lasts at least 4 h into recovery from exercise. This adaptation to exercise has recently been observed in response to both high and low intensity whole body cycling exercise (Moller et al. 2015; Schwalm et al. 2015), supporting that exercise is a stimulus that consistently reduces the content of autophagosomes in human muscle. This is in direct contrast to the majority of rodent studies showing that the LC3‐II/LC3‐I ratio increases in response to muscle contraction (Grumati et al. 2011; He et al. 2012; Pagano et al. 2014; Liu et al. 2015). It can be speculated that macroautophagy in human muscle is less well suited for rapid generation of energy substrates compared to rodents, and thus suppression of this autophagic pathway in humans may be a mechanism to conserve energy during exercise. In this context, it is possible that other degradative pathways such as proteasomal degradation or other types of autophagy (e.g. chaperone‐assisted selective autophagy) are more important for maintenance of human muscle function as suggested in response to acute resistance exercise (Ulbricht et al. 2015). Alternatively, exercise‐induced stimulation of autophagy in humans may be associated with a rapid acceleration of the autophagosome degradation process in excess of the rate of formation, resulting in a lower overall autophagosome content. Some studies have used p62 as a surrogate marker of lysosomal degradation of autophagosomes (Bjorkoy et al. 2005; Pankiv et al. 2007; Klionsky et al. 2012), while other studies argue that p62 protein content is insufficient in this context (Mizushima & Yoshimori, 2007; Rubinsztein et al. 2009; Barth et al. 2010; Ju et al. 2010). Here we observe that the reduction in the LC3‐II/LC3‐I ratio induced by either exercise or insulin stimulation occurred independent of changes in p62 protein content. Similar observations have recently been found in response to cycling exercise at 55% of for 2 h (Schwalm et al. 2015), cycling exercise at 50% for 60 min (Moller et al. 2015) as well as in response to insulin stimulation in human skeletal muscle despite a significant decrease in LC3‐II/LC3‐I ratio (Vendelbo et al. 2014; Kruse et al. 2015). Collectively, this suggests that p62 protein content is not dynamically regulated in response to changes in autophagy and hence may not be a suitable marker of lysosomal degradation of autophagosomes in human skeletal muscle.
In the present study, exercise was restricted to the knee extensors of one leg. Hence we demonstrate for the first time that exercise‐induced regulation of autophagy in humans (exercised vs. rested muscle) should at least in part be ascribed to a local contraction‐induced mechanism. Based on studies in cell cultures, the energy sensing kinase AMPK induces autophagy (Meley et al. 2006; Liang et al. 2007; Herrero‐Martin et al. 2009; Lee et al. 2010; Vingtdeux et al. 2011; Sanchez et al. 2012) via Ser‐555 phosphorylation of ULK1 (Egan et al. 2011; Ost et al. 2014; Bujak et al. 2015). Consistently, knock‐out of the catalytic α2 AMPK subunit prevents exercise‐induced LC3 lipidation as well as increased LC3‐II/LC3‐I ratio in mouse skeletal muscle (Liu et al. 2015). Very recently, a link between exercise‐induced total AMPK activation (AMPKα Thr‐172 phosphorylation), ULK1 phosphorylation and a reduction in the LC3‐II/LC3‐I ratio was suggested in human muscle (Moller et al. 2015; Schwalm et al. 2015) indicating that the exercise signalling axis regulating autophagy is conserved between species. Here we show that exercise‐induced changes in α1β2γ1 AMPK activity correlate with the changes in ULK1 Ser‐555 phosphorylation. This supports the link between AMPK and ULK1 and suggests that it is primarily α1β2γ1 AMPK complexes that mediate signalling to ULK1 in humans. This is consistent with our previous observation that PT‐1, a pharmacological activator specific to the α1β2γ1 and α2β2γ1, but not α2β2γ3 AMPK complexes, induces ULK1 Ser‐555 phosphorylation in mouse muscle (Jensen et al. 2015). However, in contrast to previous suggestions (Moller et al. 2015; Schwalm et al. 2015), exercise‐induced changes in AMPK activities or ULK1 Ser‐555 phosphorylation did not correlate with changes in the LC3‐II/LC3‐I ratio in the present study. To further elucidate the role of AMPK to regulate autophagy in muscle, we observed that pharmacological activation of AMPK in incubated primary human myotubes and mouse muscle leads to ULK1 Ser‐555 phosphorylation and furthermore that the latter response depends on intact AMPK activity in mouse muscle. This firmly establishes the link between AMPK and ULK1. Of particular note, AMPK activation did not result in changes in content of LC3‐II in both mouse muscle and human myotubes. Hence our data support a signalling link between AMPK and ULK1 in both rodent and human muscle as well as in human muscle cells, but AMPK‐mediated Ser‐555 phosphorylation of ULK1 alone may not be a sufficient stimulus to induce changes in autophagosome content. Of note, this does not rule out that the AMPK–ULK1 signalling axis is contributing to regulation of autophagy during exercise. Clearly more research is needed to explain the apparent species differences in regulation of autophagy during exercise as observed using LC3 immunoblotting. In this context, more sensitive methods to track the different phases of autophagic flux in human muscle preparation are needed. Moreover, a more detailed time resolution based on biopsy sampling both during and after exercise might prove useful.
The present study confirms that insulin is a potent inhibitor of autophagy in human skeletal muscle (Vendelbo et al. 2014). Four hours after acute exercise, we have previously observed improved insulin‐stimulated glucose uptake, activation of glycogen synthase and signalling to protein elongation in muscle (Richter et al. 1989; Wojtaszewski et al. 2000 a; Frosig et al. 2007 b). Here we show that insulin‐stimulated suppression of LC3 lipidation and the LC3‐II/LC3‐I ratio are similar in rested and exercised muscle, suggesting that improved insulin sensitivity after exercise is not related to altered regulation of autophagy in humans. Still, in the present study only one fairly high physiological plasma insulin concentration (∼100 μU ml–1) was used, and it cannot be excluded that improved insulin‐stimulated regulation of autophagy could be observed at lower insulin concentrations.
mTORC1 signalling is sufficient for inhibition of autophagy in cells and mouse muscle (Jung et al. 2010; Castets et al. 2013). Moreover, insulin‐stimulated mTORC1 activation (Ser‐2448 phosphorylation) coincides with decreased LC3 lipidation in human skeletal muscle in both fasted and fed subjects (Vendelbo et al. 2014). In accordance, in the present study, insulin stimulation led to increased phosphorylation of ULK1 at Ser‐757, a proposed mTORC1 target, concomitantly with reduction in LC3‐II/LC3‐I ratio. We therefore investigated whether mTORC1 is an upstream signalling component sufficient to lower the LC3‐II/LC3‐I ratio in response to insulin stimulation in muscle. By incubation of mouse muscle with a global mTOR inhibitor, we were able to prevent the insulin‐stimulated reduction in LC3‐II/LC3‐I ratio. This demonstrates a distinct role of mTOR (mTORC1 and/or mTORC2) signalling for insulin‐stimulated regulation of autophagy in mouse muscle.
In response to exercise training we observed a reduced LC3‐II/LC3‐I ratio and a trend towards increased p62 protein content in both the trained and the untrained muscle in contrast to some (Kim et al. 2013; Lira et al. 2013; Jiang et al. 2014) but not all studies in rodents (Lee et al. 2012; Luo et al. 2013; Tam et al. 2015). The decreased LC3‐II/LC3‐I ratio with training was primarily induced by an increased LC3‐I protein abundance. These observations are in accordance with activation of the autophagic transcriptional programme in recovery from exercise as recently demonstrated (Schwalm et al. 2015), and supported by the observed changes in gene expression of LC3 and p62 in both exercised and rested muscle 4 h after acute one‐legged exercise in the present study. As LC3‐I and p62 protein content increased in both legs after one‐legged exercise training, release of a systemic factor during high intensity, high volume one‐legged exercise is probably the underlying signal. Our results could imply that the capacity for autophagy is increased with training, although additional research is clearly needed to unravel the functional consequences of the observed adaptations to training as well as the underlying regulatory signals. Fifteen hours after the last exercise training bout, we have previously reported a marked (∼60%) increase in insulin sensitivity to stimulate glucose uptake in muscle (Frosig et al. 2007 a). However, in accordance with the observations after acute exercise, we do not find indications that improved insulin sensitivity is related to altered insulin action to suppress autophagy in muscle after exercise training.
Collectively, this study demonstrates that regulation of autophagy in human muscle in many aspects differs from the majority of previous reports based on studies in cell systems and rodent muscle. Insulin (known to suppress autophagy) and acute exercise (believed to stimulate autophagy) both reduce the LC3‐II/LC3‐I ratio in human muscle, suggesting that the content of autophagosomes is decreased by both stimuli. In contrast, exercise training reduces the LC3‐II/LC3‐I ratio, associated with improved capacity for formation of autophagosomes. Our results suggest that activation of α1β2γ1 AMPK complexes in response to exercise regulates ULK1 Ser‐555 phosphorylation, although the implications with regard to regulation of autophagy remain elusive. In contrast, mTORC1‐induced regulation of ULK1 Ser‐757 seems to be critical for insulin‐stimulated regulation of autophagy. Finally, the ability of insulin to suppress the LC3‐II/LC3‐I ratio is not affected by acute exercise or training at time points where insulin sensitivity for glucose uptake is improved.
Additional information
Competing interests
The authors have declared no competing interests.
Author contributions
CF, EAR, JW, JTT and BK designed and performed the human studies. MK and TEJ designed and performed the animal studies. AMF, AML and ABM collected and analysed the data. AMF and CF wrote the manuscript with all co‐authors revising it critically for important intellectual content. All authors approved the final version of the manuscript.
Funding
The study was supported by funding from an integrated project funded by the European Union (LSHMCT‐2004‐005272), the Novo Nordisk Foundation, the Lundbeck Foundation, the University of Copenhagen Excellence Program for Interdisciplinary Research (2016), and a 2015 Novo Nordisk Foundation Excellence grant. This work was furthermore carried out as part of the research programme of the UNIK: Food, Fitness and Pharma for Health and Disease. The UNIK project was supported by the Danish Ministry of Science, Technology and Innovation. PhD Scholarships to ABM and A‐ML was funded by The Danish Diabetes Academy supported by the Novo Nordisk Foundation.
Acknowledgements
We thank all of the volunteers who participated in the study. We are grateful to Professor Morris J. Birnbaum (Howard Hughes Medical Institute and University of Pennsylvania School of Medicine, USA) for providing the AMPK KD founder mice. We also thank the following for helpful discussions and advice: Rikke K. Henriksen, Jonas M. Kristensen and Professor Kurt Højlund (University of Southern Denmark) as well as Jens Frey Halling and Professor Henriette Pilegaard (University of Copenhagen). Finally, we generously acknowledge Irene Bech Nielsen, Betina Bolmgren, Jesper B. Birk, Peter Schjerling and Anja Jokipii for skilled technical assistance.
References
- Al‐Khalili L, Chibalin AV, Kannisto K, Zhang BB, Permert J, Holman GD, Ehrenborg E, Ding VD, Zierath JR & Krook A (2003). Insulin action in cultured human skeletal muscle cells during differentiation: assessment of cell surface GLUT4 and GLUT1 content. Cell Mol Life Sci 60, 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P, Adams RP, Sjogaard G, Thorboe A & Saltin B (1985). Dynamic knee extension as model for study of isolated exercising muscle in humans. J Appl Physiol (1985) 59, 1647–1653. [DOI] [PubMed] [Google Scholar]
- Barth S, Glick D & Macleod KF (2010). Autophagy: assays and artifacts. J Pathol 221, 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk JB & Wojtaszewski JF (2006). Predominant α2/β2/γ3 AMPK activation during exercise in human skeletal muscle. J Physiol 577, 1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H & Johansen T (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 171, 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandauer J, Vienberg SG, Andersen MA, Ringholm S, Risis S, Larsen PS, Kristensen JM, Frosig C, Leick L, Fentz J, Jorgensen S, Kiens B, Wojtaszewski JF, Richter EA, Zierath JR, Goodyear LJ, Pilegaard H & Treebak JT (2013). AMP‐activated protein kinase regulates nicotinamide phosphoribosyl transferase expression in skeletal muscle. J Physiol 591, 5207–5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujak AL, Crane JD, Lally JS, Ford RJ, Kang SJ, Rebalka IA, Green AE, Kemp BE, Hawke TJ, Schertzer JD & Steinberg GR (2015). AMPK activation of muscle autophagy prevents fasting‐induced hypoglycemia and myopathy during aging. Cell Metab 21, 883–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartee GD, Young DA, Sleeper MD, Zierath J, Wallberg‐Henriksson H & Holloszy JO (1989). Prolonged increase in insulin‐stimulated glucose transport in muscle after exercise. Am J Physiol 256, E494–E499. [DOI] [PubMed] [Google Scholar]
- Castets P, Lin S, Rion N, Di FS, Romanino K, Guridi M, Frank S, Tintignac LA, Sinnreich M & Ruegg MA (2013). Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation‐induced autophagy and causes a severe, late‐onset myopathy. Cell Metab 17, 731–744. [DOI] [PubMed] [Google Scholar]
- Chomczynski P & Sacchi N (1987). Single‐step method of RNAisolation by acid guanidiniumthiocyanate‐phenol‐chloroform extraction. Anal Biochem 162, 156–159. [DOI] [PubMed] [Google Scholar]
- Dan HC, Ebbs A, Pasparakis M, Van DT, Basseres DS & Baldwin AS (2014). Akt‐dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IκB kinase α (IKK α). J Biol Chem 289, 25227–25240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dela F, Larsen JJ, Mikines KJ, Ploug T, Petersen LN & Galbo H (1995). Insulin‐stimulated muscle glucose clearance in patients with NIDDM. Effects of one‐legged physical training. Diabetes 44, 1010–1020. [DOI] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M & Shaw RJ (2011). Phosphorylation of ULK1 (hATG1) by AMP‐activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frosig C, Jorgensen SB, Hardie DG, Richter EA & Wojtaszewski JF (2004). 5′‐AMP‐activated protein kinase activity and protein expression are regulated by endurance training in human skeletal muscle. Am J Physiol Endocrinol Metab 286, E411–E417. [DOI] [PubMed] [Google Scholar]
- Frosig C, Roepstorff C, Brandt N, Maarbjerg SJ, Birk JB, Wojtaszewski JF, Richter EA & Kiens B (2009). Reduced malonyl‐CoA content in recovery from exercise correlates with improved insulin‐stimulated glucose uptake in human skeletal muscle. Am J Physiol Endocrinol Metab 296, E787–E795. [DOI] [PubMed] [Google Scholar]
- Frosig C, Rose AJ, Treebak JT, Kiens B, Richter EA & Wojtaszewski JF (2007. a). Effects of endurance exercise training on insulin signaling in human skeletal muscle: interactions at the level of phosphatidylinositol 3‐kinase, Akt, and AS160. Diabetes 56, 2093–2102. [DOI] [PubMed] [Google Scholar]
- Frosig C, Sajan MP, Maarbjerg SJ, Brandt N, Roepstorff C, Wojtaszewski JF, Kiens B, Farese RV & Richter EA (2007. b). Exercise improves phosphatidylinositol‐3,4,5‐trisphosphate responsiveness of atypical protein kinase C and interacts with insulin signalling to peptide elongation in human skeletal muscle. J Physiol 582, 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollnick PD, Piehl K & Saltin B (1974). Selective glycogen depletion pattern in human muscle fibres after exercise of varying intensity and at varying pedalling rates. J Physiol 241, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, Maraldi NM, Bernardi P, Sandri M & Bonaldo P (2010). Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 16, 1313–1320. [DOI] [PubMed] [Google Scholar]
- Grumati P, Coletto L, Schiavinato A, Castagnaro S, Bertaggia E, Sandri M & Bonaldo P (2011). Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI‐deficient muscles. Autophagy 7, 1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel‐Duby R, Scherer PE & Levine B (2012). Exercise‐induced BCL2‐regulated autophagy is required for muscle glucose homeostasis. Nature 481, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero‐Martin G, Hoyer‐Hansen M, Garcia‐Garcia C, Fumarola C, Farkas T, Lopez‐Rivas A & Jaattela M (2009). TAK1 activates AMPK‐dependent cytoprotective autophagy in TRAIL‐treated epithelial cells. EMBO J 28, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamart C, Francaux M, Millet GY, Deldicque L, Frere D & Feasson L (2012). Modulation of autophagy and ubiquitin‐proteasome pathways during ultra‐endurance running. J Appl Physiol (1985) 112, 1529–1537. [DOI] [PubMed] [Google Scholar]
- Jamart C, Naslain D, Gilson H & Francaux M (2013). Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am J Physiol Endocrinol Metab 305, E964–E974. [DOI] [PubMed] [Google Scholar]
- James DE, Kraegen EW & Chisholm DJ (1985). Effects of exercise training on in vivo insulin action in individual tissues of the rat. J Clin Invest 76, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TE, Rose AJ, Jorgensen SB, Brandt N, Schjerling P, Wojtaszewski JF & Richter EA (2007). Possible CaMKK‐dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am J Physiol Endocrinol Metab 292, E1308–E1317. [DOI] [PubMed] [Google Scholar]
- Jensen TE, Ross FA, Kleinert M, Sylow L, Knudsen JR, Gowans GJ, Hardie DG & Richter EA (2015). PT‐1 selectively activates AMPK‐γ1 complexes in mouse skeletal muscle, but activates all three γ subunit complexes in cultured human cells by inhibiting the respiratory chain. Biochem J 467, 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Chen K, Lu X, Gao HJ, Qin ZH & Lin F (2014). Exercise ameliorates the detrimental effect of chloroquine on skeletal muscles in mice via restoring autophagy flux. Acta Pharmacol Sin 35, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Varadhachary AS, Miller SE & Weihl CC (2010). Quantitation of “autophagic flux” in mature skeletal muscle. Autophagy 6, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Ro SH, Cao J, Otto NM & Kim DH (2010). mTOR regulation of autophagy. FEBS Lett 584, 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B & Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YA, Kim YS, Oh SL, Kim HJ & Song W (2013). Autophagic response to exercise training in skeletal muscle with age. J Physiol Biochem 69, 697–705. [DOI] [PubMed] [Google Scholar]
- Kim YA, Kim YS & Song W (2012). Autophagic response to a single bout of moderate exercise in murine skeletal muscle. J Physiol Biochem 68, 229–235. [DOI] [PubMed] [Google Scholar]
- Kleinert M, Sylow L, Fazakerley DJ, Krycer JR, Thomas KC, Oxboll AJ, Jordy AB, Jensen TE, Yang G, Schjerling P, Kiens B, James DE, Ruegg MA & Richter EA (2014). Acute mTOR inhibition induces insulin resistance and alters substrate utilization in vivo. Mol Metab 3, 630–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo‐Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre‐Ghiso JA, Ahn HJ, et al, (2012). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse R, Vind BF, Petersson SJ, Kristensen JM & Hojlund K (2015). Markers of autophagy are adapted to hyperglycaemia in skeletal muscle in type 2 diabetes. Diabetologia 58, 2087–2095. [DOI] [PubMed] [Google Scholar]
- Lee JW, Park S, Takahashi Y & Wang HG (2010). The association of AMPK with ULK1 regulates autophagy. PLoS One 5, e15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kim JH, Hong Y, Lee SR, Chang KT & Hong Y (2012). Prophylactic effects of swimming exercise on autophagy‐induced muscle atrophy in diabetic rats. Lab Anim Res 28, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B & Kroemer G (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM & Mills GB (2007). The energy sensing LKB1‐AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 9, 218–224. [DOI] [PubMed] [Google Scholar]
- Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, Hoehn KL & Yan Z (2013). Autophagy is required for exercise training‐induced skeletal muscle adaptation and improvement of physical performance. FASEB J 27, 4184–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Niu Y, Yuan H, Huang J & Fu L (2015). AMPK binds to Sestrins and mediates the effect of exercise to increase insulin‐sensitivity through autophagy. Metabolism 64, 658–665. [DOI] [PubMed] [Google Scholar]
- Luo L, Lu AM, Wang Y, Hong A, Chen Y, Hu J, Li X & Qin ZH (2013). Chronic resistance training activates autophagy and reduces apoptosis of muscle cells by modulating IGF‐1 and its receptors, Akt/mTOR and Akt/FOXO3a signaling in aged rats. Exp Gerontol 48, 427–436. [DOI] [PubMed] [Google Scholar]
- Manning BD & Cantley LC (2007). AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S & Sandri M (2009). Autophagy is required to maintain muscle mass. Cell Metab 10, 507–515. [DOI] [PubMed] [Google Scholar]
- Masschelein E, Van TR, D'Hulst G, Hespel P, Thomis M & Deldicque L (2014). Acute environmental hypoxia induces LC3 lipidation in a genotype‐dependent manner. FASEB J 28, 1022–1034. [DOI] [PubMed] [Google Scholar]
- Meley D, Bauvy C, Houben‐Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P & Meijer AJ (2006). AMP‐activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem 281, 34870–34879. [DOI] [PubMed] [Google Scholar]
- Mizushima N & Komatsu M (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. [DOI] [PubMed] [Google Scholar]
- Mizushima N & Yoshimori T (2007). How to interpret LC3 immunoblotting. Autophagy 3, 542–545. [DOI] [PubMed] [Google Scholar]
- Moller AB, Vendelbo MH, Christensen B, Clasen BF, Bak AM, Jorgensen JO, Moller N & Jessen N (2015). Physical exercise increases autophagic signaling through ULK1 in human skeletal muscle. J Appl Physiol (1985) 118, 971–979. [DOI] [PubMed] [Google Scholar]
- Mu J, Brozinick JT, Jr , Valladares O, Bucan M & Birnbaum MJ (2001). A role for AMP‐activated protein kinase in contraction‐ and hypoxia‐regulated glucose transport in skeletal muscle. Mol Cell 7, 1085–1094. [DOI] [PubMed] [Google Scholar]
- Ost M, Werner F, Dokas J, Klaus S & Voigt A (2014). Activation of AMPKalpha2 is not crucial for mitochondrial uncoupling‐induced metabolic effects but required to maintain skeletal muscle integrity. PLoS One 9, e94689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano AF, Py G, Bernardi H, Candau RB & Sanchez AM (2014). Autophagy and protein turnover signaling in slow‐twitch muscle during exercise. Med Sci Sports Exerc 46, 1314–1325. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G & Johansen T (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282, 24131–24145. [DOI] [PubMed] [Google Scholar]
- Pilegaard H, Ordway GA, Saltin B & Neufer PD (2000). Transcriptional regulation of gene expression in humanskeletal muscle during recovery from exercise. Am J Physiol Endocrinol Metab 279, E806–E814. [DOI] [PubMed] [Google Scholar]
- Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N, Ralston E & Plotz P (2008). Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet 17, 3897–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter EA, Garetto LP, Goodman MN & Ruderman NB (1982). Muscle glucose metabolism following exercise in the rat: increased sensitivity to insulin. J Clin Invest 69, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter EA, Jensen P, Kiens B & Kristiansen S (1998). Sarcolemmal glucose transport and GLUT‐4 translocation during exercise are diminished by endurance training. Am J Physiol 274, E89–E95. [DOI] [PubMed] [Google Scholar]
- Richter EA, Mikines KJ, Galbo H & Kiens B (1989). Effect of exercise on insulin action in human skeletal muscle. J Appl Physiol (1985) 66, 876–885. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Frosig C, Kiens B, Wojtaszewski JF & Richter EA (2007). Effect of endurance exercise training on Ca2+ calmodulin‐dependent protein kinase II expression and signalling in skeletal muscle of humans. J Physiol 583, 785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S & Klionsky DJ (2009). In search of an “autophagomometer”. Autophagy 5, 585–589. [DOI] [PubMed] [Google Scholar]
- Sanchez AM, Csibi A, Raibon A, Cornille K, Gay S, Bernardi H & Candau R (2012). AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J Cell Biochem 113, 695–710. [DOI] [PubMed] [Google Scholar]
- Schwalm C, Jamart C, Benoit N, Naslain D, Premont C, Prevet J, Van TR, Deldicque L & Francaux M (2015). Activation of autophagy in human skeletal muscle is dependent on exercise intensity and AMPK activation. FASEB J 29, 3515–3526. [DOI] [PubMed] [Google Scholar]
- Shi L, Zhang T, Zhou Y, Zeng X, Ran L, Zhang Q, Zhu J & Mi M (2015). Dihydromyricetin improves skeletal muscle insulin sensitivity by inducing autophagy via the AMPK‐PGC‐1α‐Sirt3 signaling pathway. Endocrine 50, 378–389. [DOI] [PubMed] [Google Scholar]
- Tam BT, Pei XM, Yu AP, Sin TK, Leung KK, Au KK, Chong JT, Yung BY, Yip SP, Chan LW, Wong CS & Siu PM (2015). Autophagic adaptation is associated with exercise‐induced fibre‐type shifting in skeletal muscle. Acta Physiol (Oxf) 214, 221–236. [DOI] [PubMed] [Google Scholar]
- Tanida I, Minematsu‐Ikeguchi N, Ueno T & Kominami E (2005). Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 1, 84–91. [DOI] [PubMed] [Google Scholar]
- Treebak JT, Frosig C, Pehmoller C, Chen S, Maarbjerg SJ, Brandt N, MacKintosh C, Zierath JR, Hardie DG, Kiens B, Richter EA, Pilegaard H & Wojtaszewski JF (2009). Potential role of TBC1D4 in enhanced post‐exercise insulin action in human skeletal muscle. Diabetologia 52, 891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treebak JT, Pehmoller C, Kristensen JM, Kjobsted R, Birk JB, Schjerling P, Richter EA, Goodyear LJ & Wojtaszewski JF (2014). Acute exercise and physiological insulin induce distinct phosphorylation signatures on TBC1D1 and TBC1D4 proteins in human skeletal muscle. J Physiol 592, 351–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbricht A, Gehlert S, Leciejewski B, Schiffer T, Bloch W & Hohfeld J (2015). Induction and adaptation of chaperone‐assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy 11, 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainshtein A, Tryon LD, Pauly M & Hood DA (2015). Role of PGC‐1α during acute exercise‐induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol 308, C710–C719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendelbo MH, Moller AB, Christensen B, Nellemann B, Clasen BF, Nair KS, Jorgensen JO, Jessen N & Moller N (2014). Fasting increases human skeletal muscle net phenylalanine release and this is associated with decreased mTOR signaling. PLoS One 9, e102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeux V, Chandakkar P, Zhao H, d'Abramo C, Davies P & Marambaud P (2011). Novel synthetic small‐molecule activators of AMPK as enhancers of autophagy and amyloid‐β peptide degradation. FASEB J 25, 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder WW & Hardie DG (1996). Inactivation of acetyl‐CoA carboxylase and activation of AMP‐activated protein kinase in muscle during exercise. Am J Physiol 270, E299–E304. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Hansen BF, Gade, Kiens B , Markuns JF, Goodyear LJ & Richter EA (2000. a). Insulin signaling and insulin sensitivity after exercise in human skeletal muscle. Diabetes 49, 325–331. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Hansen BF, Kiens B & Richter EA (1997). Insulin signaling in human skeletal muscle: time course and effect of exercise. Diabetes 46, 1775–1781. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA & Kiens B (2000. b). Isoform‐specific and exercise intensity‐dependent activation of 5′‐AMP‐activated protein kinase in human skeletal muscle. J Physiol 528, 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, Calay ES & Hotamisligil GS (2010). Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 11, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A & Sabatini DM (2011). mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]