

Abstract
Key points
Methamphetamine (METH) abuse is escalating worldwide, with the most common cause of death resulting from cardiovascular failure and hyperthermia; however, the underlying physiological mechanisms are poorly understood.
Systemic administration of METH in anaesthetised rats reduced the effectiveness of some protective cardiorespiratory reflexes, increased central respiratory activity independently of metabolic function, and increased heart rate, metabolism and respiration in a pattern indicating that non‐shivering thermogenesis contributes to the well‐described hyperthermia.
In animals that showed METH‐induced behavioural sensitisation following chronic METH treatment, no changes were evident in baseline cardiovascular, respiratory and metabolic measures and the METH‐evoked effects in these parameters were similar to those seen in saline‐treated or drug naïve animals.
Physiological effects evoked by METH were retained but were neither facilitated nor depressed following chronic treatment with METH.
These data highlight and identify potential mechanisms for targeted intervention in patients vulnerable to METH overdose.
Abstract
Methamphetamine (METH) is known to promote cardiovascular failure or life‐threatening hyperthermia; however, there is still limited understanding of the mechanisms responsible for evoking the physiological changes. In this study, we systematically determined the effects on both autonomic and respiratory outflows, as well as reflex function, following acute and repeated administration of METH, which enhances behavioural responses. Arterial pressure, heart rate, phrenic nerve discharge amplitude and frequency, lumbar and splanchnic sympathetic nerve discharge, interscapular brown adipose tissue and core temperatures, and expired CO2 were measured in urethane‐anaesthetised male Sprague‐Dawley rats. Novel findings include potent increases in central inspiratory drive and frequency that are not dependent on METH‐evoked increases in expired CO2 levels. Increases in non‐shivering thermogenesis correlate with well‐described increases in body temperature and heart rate. Unexpectedly, METH evoked minor effects on both sympathetic outflows and mean arterial pressure. METH modified cardiorespiratory reflex function in response to hypoxia, hypercapnia and baroreceptor unloading. Chronically METH‐treated rats failed to exhibit changes in baseline sympathetic, cardiovascular, respiratory and metabolic parameters. The tonic and reflex cardiovascular, respiratory and metabolic responses to METH challenge were similar to those seen in saline‐treated and drug naive animals. Overall, these findings describe independent and compound associations between physiological systems evoked by METH and serve to highlight that a single dose of METH can significantly impact basic homeostatic systems and protective functions. These effects of METH persist even following chronic METH treatment.
Key points
Methamphetamine (METH) abuse is escalating worldwide, with the most common cause of death resulting from cardiovascular failure and hyperthermia; however, the underlying physiological mechanisms are poorly understood.
Systemic administration of METH in anaesthetised rats reduced the effectiveness of some protective cardiorespiratory reflexes, increased central respiratory activity independently of metabolic function, and increased heart rate, metabolism and respiration in a pattern indicating that non‐shivering thermogenesis contributes to the well‐described hyperthermia.
In animals that showed METH‐induced behavioural sensitisation following chronic METH treatment, no changes were evident in baseline cardiovascular, respiratory and metabolic measures and the METH‐evoked effects in these parameters were similar to those seen in saline‐treated or drug naïve animals.
Physiological effects evoked by METH were retained but were neither facilitated nor depressed following chronic treatment with METH.
These data highlight and identify potential mechanisms for targeted intervention in patients vulnerable to METH overdose.
Abbreviations
- AP
arterial pressure
- HR
heart rate
- iBAT
interscapular brown adipose tissue
- lSNA
lumbar sympathetic nerve activity
- MAP
mean arterial pressure
- METH
methamphetamine
- PNA
phrenic nerve activity
- PNamp
phrenic nerve amplitude
- PNf
phrenic nerve frequency
- SNA
sympathetic nerve activity
- sSNA
splanchnic sympathetic nerve activity
Introduction
Methamphetamine (METH) distributes rapidly throughout the brain and body to block and reverse monoamine transporters and as a result, enhances synaptic and extra‐synaptic levels of dopamine, noradrenaline and serotonin (Brown et al. 2002; Sulzer et al. 2005; Cruickshank & Dyer, 2009). While in humans, well‐described psychogenic effects are accompanied by a range of physiological changes that include tachycardia, hypertension, tachypnoea and hyperthermia (Schep et al. 2010), the type and degree of change reported are impacted by the drug dose, length of drug use, and varying patient populations whose histories of METH use (binge/occasional) are complicated by poly‐drug use (Carvalho et al. 2012). Initial attempts in conscious animals to describe the physiological effects evoked by METH indicate that METH increases arterial pressure (AP), heart rate (HR), locomotor activity and body temperature (Yoshida et al. 1993; Sprague et al. 2004; Rusyniak et al. 2012); however, these earlier studies are limited by a narrow range of physiological measures. As such, we do not yet understand the full range and degree of physiological effects of acute METH administration, or their underlying mechanisms. Importantly, identifying precisely how and what extent these systems are altered may expose damage incurred by chronic METH use and factors that underpin patient mortality.
As METH administration promotes neuronal plasticity (Guilarte et al. 2003; Jedynak et al. 2007) and/or neurotoxicity (Bowyer & Holson, 1995; Cadet & Krasnova, 2009; Krasnova & Cadet, 2009) depending upon the dose, and overdose is typically associated with significant cardiac effects and life threatening hyperthermia, a comprehensive study to delineate the physiological changes evoked by repeated METH use is also required. Chronic use of METH is associated with altered psychological profiles and can evoke acute psychosis that is indistinguishable from schizophrenia (Srisurapanont et al. 2003; Zweben et al. 2004; Kim et al. 2009; Glasner‐Edwards et al. 2010; Krasnova et al. 2010). Many of these psychological changes induced by METH are associated with abnormalities within the brain (Steketee, 2003) and these neural changes are detectable through behavioural examination. Repeated exposure to METH followed by a period of abstinence evokes a facilitated behavioural response in locomotor activity and/or stereotypy when subsequently challenged with METH, a process known as behavioural sensitization (Pierce & Kalivas, 1997), while the locomotor enhancement can further be attenuated by pretreatment with antipsychotic drugs (Abekawa et al. 2008). The neurobiological changes associated with repeated METH use are increasingly described, but the effects of METH sensitisation on autonomic and respiratory function are unknown.
Thus, the objectives of this study were twofold. The first aim was to define the changes in cardiovascular, sympathetic, respiratory and metabolic function, as well as the effects on cardiorespiratory reflex function, that are evoked by increasing concentrations of systemically administered METH. The second aim was to determine if basal physiological and cardiorespiratory reflex functions were changed in a model of behavioural sensitisation, and whether or not there is enhanced sympathetic, cardiovascular, metabolic or respiratory sensitivity to METH challenge. Experiments were conducted in urethane‐anaesthetised rats in order to permit a comprehensive assessment of central inspiratory drive, non‐shivering thermogenesis, multiple sympathetic nerve activities, blood pressure and heart rate, together with central and peripheral chemoreceptor and baroreceptor reflex function unencumbered by changes in locomotor activity, to investigate specific reflex pathways and systems altered by METH administration.
Methods
Ethical approval
All procedures conform to the regulations detailed in the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes 7th edition (2004) and were approved by the Macquarie University Animal Ethics Committee (Animal Research Authority Number 2010/045, 2010/013).
Housing
Male Sprague‐Dawley rats obtained from the Animal Resources Centre (Perth, Australia) were housed at Macquarie University vivarium in groups of four in standard plastic cages (64 cm (L) × 40 cm (W) × 20 cm (H)) lined with wood shavings and environmental enrichment material and were kept in a humidity‐ and temperature‐controlled room (21°C, 60% humidity) maintained on a 12 h light–12 h dark cycle (lights on at 06.00 h). All experimentation was carried out during the light period. Food and water were available ad libitum in the home cages.
Drugs
(±) Methamphetamine HCl (METH) was obtained from the Australian Government Analytical Laboratories (AGAL, Pymble, Australia). METH was dissolved in isotonic saline and injected into the intraperitoneal cavity (i.p.) at a volume of 1 ml kg−1. Control rats were treated with isotonic saline.
General surgical procedures
Anaesthesia and surgical preparation was conducted as previously described by Hassan et al. (2015). Briefly, rats were anaesthetised with urethane (10% in saline, 1.3 g kg−1 i.p.; Sigma‐Aldrich, St Louis, MO, USA). Right femoral artery and veins were cannulated for arterial pressure (AP) and drug administration, respectively. Tracheostomy permitted artificial ventilation and measurement of expired CO2. Colonic temperature was recorded and maintained at 36.5–37.5°C using a thermoregulated heating blanket set to turn off at 37.0°C. Whole nerve recordings were made from left phrenic, splanchnic and lumbar sympathetic nerves. Interscapular brown adipose tissue temperature was recorded. The left and right vagi were cut and neuromuscular blockade was maintained with pancuronium bromide (0.8 mg i.v. induction, 0.4mg h−1 i.v. maintenance; Astra Pharmaceuticals Pty Ltd, Sydney, Australia). Rats were positioned prone in a stereotaxic frame. Nerve recordings were amplified (CWE Inc., Ardmore, PA, USA), band pass filtered (0.1–3 kHz), sampled at 5 kHz (1401, CED Ltd, Cambridge, UK) and recorded on a computer using Spike2 software (CED Ltd). Core and iBAT temperatures and end‐tidal CO2 were sampled at 2 kHz and recorded similarly. Heart rate (HR) was derived from the AP signal. Phrenic nerve burst frequency (PNf) was derived from PNA. All parameters were continuously recorded for the duration of experiment with additional doses of urethane administered as required. At the end of each experiment, rats were killed with 3 m KCl i.v.
Experimental protocols
Experiment 1: acute METH
Male Sprague‐Dawley rats (350–550 g, n = 6) were used. A stabilisation period of 10–20 min was allowed. Baroreceptor function (i.v. bolus of 10 μg sodium nitroprusside and 10 μg phenylephrine; Sigma Aldrich) and then peripheral and central chemoreflex function (hypoxia: 14 s 100% nitrogen; hypercapnia: 60 s of 10% carbon dioxide) were tested. METH was then administered (i.p.) cumulatively at concentrations of 0.03, 0.1, 0.3, 1 and 5 mg kg−1. The peak responses in MAP, HR, splanchnic and lumbar sympathetic nerve activities (sSNA and lSNA, respectively), phrenic nerve burst amplitude (PNamp) and PNf, iBAT and core temperatures, and expired CO2 were determined at each dose with a minimum period of 30 min between doses. Assessment of baroreceptor and then chemoreflex function was repeated at the time immediately after the peak response was reached following each dose, with each stimulus challenge presented twice and the response averaged. Saline vehicle (1ml kg−1 i.p.) was administered as control (n = 4).
In order to investigate whether the physiological effects evoked by METH were dependent or independent of the hypercapnia produced (in paralysed, ventilated animals), an additional group of animals (n = 5) received a single dose of METH (5 mg kg−1 i.p.) and the METH‐evoked hypercapnia was immediately compensated by increasing the ventilation rate to maintain the expired CO2 level within 0.1% of control levels.
Experiment 2: chronic METH sensitisation
The behavioural sensitisation protocol is as described in Wearne et al. (2015). Male Sprague‐Dawley rats (277 ± 7g at the start of testing (n = 26)) were used. Rats were acclimated to the vivarium for 1 week. On Day 1, all rats were given 15 min to explore a locomotor chamber. Rats then received an injection of saline (1 ml kg−1 i.p.), were placed back into the locomotor chamber and their activity measured for 1 h. Rats were then allocated to treatment groups (METH (n = 13) or saline (n = 13)) based on baseline locomotor activity such that there was no significant difference between groups prior to the commencement of the drug schedule (P = 0.21).
METH‐treated rats received once daily 1 mg kg−1 ml−1 METH (i.p.) on Days 2 and 8 for locomotor testing, and once daily injections of 5 mg kg−1 ml−1 METH (i.p.) on Days 3–7 in the home cages. Saline rats received once daily injections of physiological 1 ml kg−1 saline (i.p.) from Days 2–8. METH doses used were low to moderate and would be unlikely to cause neurotoxicity, consistent with previous methods (Iwazaki et al. 2008; Morshedi & Meredith, 2008). Locomotor activity was measured on Days 2 and 8 in all rats and recorded as the number of beam breaks per 60 min following drug injection (total time in chamber = 75 min). At the end of the 8 day drug schedule, rats were left for a withdrawal period of 14–16 days in their home cages prior to locomotor testing (METH n = 6, saline n = 6) or electrophysiological assessment (METH n = 7, saline n = 7).
Behavioural measures
Locomotor activity in all rats was recorded on Days 1, 2 and 8 of the drug regime. Twelve standard chambers (250 (L) × 310 (W) × 500 (H) mm) consisting of aluminum top and side panels and plexi‐glass front and back panels with metal rod floor (16 rods, 6 mm diameter, 15 mm apart) were used. The chambers were housed in individual wooden sound attenuation boxes with ventilation fans providing a masking noise. Each chamber was equipped with four infrared photobeam detectors (Quantum PIR motion sensor, part no. 890‐087‐2, NESS Security Products, Australia) positioned on the front and back panels approximately 50 mm apart and 30 mm above the floor. Locomotor activity was quantified as the number of photobeam interruptions and recorded via an attached Macintosh computer equipped with Med‐IV PC software (Med Associates). Rats were placed in the test chamber 15 min prior to drug injection to reduce environment‐induced increases in activity. The test room was maintained under low light conditions at an ambient temperature of 21°C.
Following the 14–16 day withdrawal period, rats undergoing locomotor testing only (METH n = 6, saline n = 6) were injected with 1.0 ml kg−1 saline (i.p.) to test for conditioned baseline responding. The next day (Day 24), all rats were assessed for METH‐induced sensitisation via a challenge dose of 1 mg kg−1 ml−1 METH (i.p.). This procedure provides a behavioural correlate to the physiological changes measured in the remaining cohort of animals described below.
Electrophysiological measures
Following the 14–16 day withdrawal period the remaining animals from each treatment group (METH n = 7, saline n = 7) were assessed for cardiovascular, respiratory and thermoregulatory function and cardiorespiratory reflexes as described in Experiment 1. Behavioural sensitisation is maintained for up to 1 year (Paulson et al. 1991) and therefore these rats were assessed on alternate days between Days 21 and 34 (1 rat day−1). Rats were collected and anaesthetised at the same time on each day.
Electrophysiological data analysis
Nerve recordings were rectified and averaged using a 2 s time constant. Baseline values were obtained by averaging 5 min of data in 60 s bins prior to the injection of METH. Peak changes are expressed as peak value or percentage (%) of baseline activity for each variable. The peak change data are expressed as means ± standard error of the mean (SEM). Non‐linear regression was used to curve fit the dose–response data.
To assess sympathetic baroreflex function the effects of sodium nitroprusside (10 μg) and phenylephrine (10 μg) were assessed on splanchnic SNA and MAP. X–Y plots of SNA vs. MAP were determined and the data normalised setting 100% SNA to baseline levels of SNA. Data were then fitted using non‐linear regression to obtain, top, bottom, V 50 and slope. The first derivative of the curve was calculated in order to determine the maximum baroreflex gain (%ΔSNA/mmHg). To assess cardiac sympathetic baroreflex function maximum and minimum HR values generated in response to sodium nitroprusside and phenylephrine were determined. One‐way ANOVA with Dunnett's multiple comparison test was used to compare doses to control responses, significance was indicated if P < 0.05.
For peripheral and central chemoreflex challenge, peak values of HR, PNf and sSNA were determined following METH and compared to controls.
Statistical analysis
Statistical calculations were performed using SPSS version 17 or GraphPad 9. For behavioural analysis statistical evaluations were made using two‐tailed unpaired t tests. One‐way ANOVA (Dunnett's multiple comparison test) was used to compare dose–response to control in physiological data. Curves were fitted using non‐linear regression where possible. Pearson's correlation analysis examined iBAT temperatures against other autonomic parameters, plots were then assessed for linear regression where possible. For chemoreflex and baroreflex analysis, two‐tailed unpaired t tests and/or one‐way ANOVA (Dunnett's multiple comparison test) was used. Two‐way ANOVA (Bonferroni multiple comparison test) was used to compare between treatment groups and doses. Significance was indicated if P < 0.05.
Results
Experiment 1: acute METH
Effects of METH on cardiovascular, respiratory and metabolic outflows
To determine the effects of METH on sSNA, lSNA, MAP, HR, iBAT and core temperatures, expired CO2 and PNamp and PNf, METH was cumulatively administered at 0.03, 0.1, 0.3, 1 and 5 mg kg−1 doses (Fig. 1). Very small if any response was evoked in sSNA, lSNA and MAP. In contrast, clear dose‐related responses were generated in HR, iBAT and core temperatures, expired CO2, PNamp and PNf, with thresholds consistently between 0.1 and 0.3 mg kg−1. The maximal response was typically generated between 1 and 5 mg kg−1 for respiratory and metabolic parameters.
Figure 1. Effects of methamphetamine (METH) on respiratory, cardiovascular and metabolic outputs .

Grouped data showing the cumulative effects of METH at 0.03, 0.1, 0.3, 1 and 5 mg kg−1 administered intraperitoneally on splanchnic and lumbar sympathetic nerve activity (sSNA and lSNA), mean arterial pressure (MAP), heart rate (HR), interscapular brown adipose tissue (iBAT) and core temperatures, expired CO2, phrenic nerve (PN) amplitude and frequency. Grouped data are presented as means ± SEM; non‐linear regression curves are shown where possible.
Figure 2 A shows a representative example of the physiological effects evoked by METH (5mg kg−1). Decreases in sSNA, lSNA and AP (only seen at this dose) were accompanied by increases in HR, PNamp, PNf, expired CO2, iBAT and core temperatures. Effects were of varying duration, with some increases in parameters ≥150 min except for PNf, which returned to baseline levels within <20 min.
Figure 2. Co‐ordinated respiratory, cardiovascular and metabolic effects evoked following METH administration .
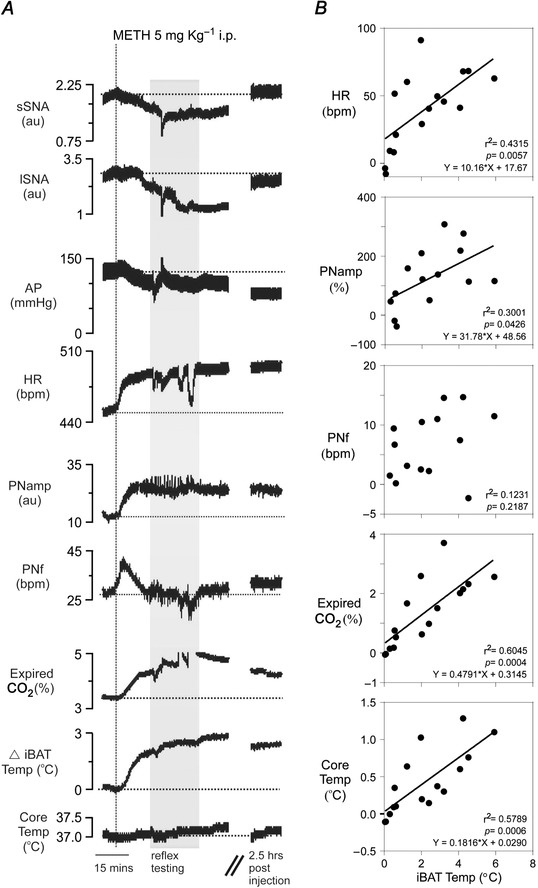
A, representative example of the effects of systemic METH (5 mg kg−1) on lSNA, sSNA, AP, HR, PNamp, PNf, expired CO2, iBAT and core temperature (respectively). Grey box indicates period in which cardiorespiratory reflexes were tested. B, correlation scatterplots showing the relationship between METH‐evoked responses in HR, PNamp, PNf, expired CO2 and core temperature, with respect to METH‐evoked changes in iBAT temperature (n = 15). Strong correlations are seen between HR, expired CO2, core temperature and iBAT temperature.
A co‐ordinated response in iBAT temperature, HR, expired CO2 and core temperature was consistently observed following METH administration. Figure 2 B shows the correlation plots of changes in iBAT temperature evoked by METH with changes in HR, PNamp, PNf, expired CO2, and core temperature; correlation coefficients are indicated. A strong linear correlation was observed between iBAT temperature and HR (r 2 = 0.4315, P = 0.0057), expired CO2 (r 2 = 0.6045, P = 0.0004), and core temperature (r 2 = 0.5789, P = 0.0006). Increases in iBAT temperature were weakly associated with increases in PNamp (r 2 = 0.3001, P = 0.0426) but not PNf (r 2 = 0.1231).
Figure 2 A shows that the increase in PNamp appeared to precede the increase in expired CO2 so we aimed to determine whether increases in respiratory parameters resulted from elevated levels of expired CO2 induced by METH administration. To test this, another group of animals was administered METH (5 mg kg−1) and expired CO2 levels in each animal were tightly preserved at control levels by increasing the ventilation rate; such that subsequent changes in expired CO2 levels in the cohort were small (0.038 ± 0.047% expired CO2, n = 5, compensated). Figure 3 Aa shows representative examples during the control period and METH‐evoked increases in central respiratory drive (PNAmp and PNf) when the expired CO2 was compensated for by increasing the ventilation rate (right‐hand side Y axis, Fig. 3 Ab). Figure 3 B shows that METH‐evoked increases in PNf and PNAmp (independent of CO2 stimulus) resulted from reductions of the expiratory period, with little change to the inspiratory period. The peak responses in all parameters evoked by vehicle, METH (5 mg kg−1) and ‘METH (5 mg kg−1) + compensated’ are compared (Fig. 3 C). PNamp and PNf were still increased by >3‐fold following METH, despite similar levels of expired CO2 during compensation (Fig. 3 C ). Compensating the increasing levels of CO2 after METH (5 mg kg−1) did not significantly alter the HR, PNamp, PNf and iBAT and core temperature responses evoked by METH. Significant changes were evoked by both METH (5 mg kg−1) (M) alone as well as ‘METH (5 mg kg−1) + compensated’ (M*) compared to vehicle in HR (one‐way ANOVA, F (2,12) = 21.99, P < 0.0001; Dunnett's multiple comparison test; M, P < 0.001; M*, P < 0.0001), PNamp (F (2, 11) = 6.664, P = 0.0127; M, P < 0.05; M*, P < 0.05), PNf (F (2,10) = 5.006, P = 0.0312; M*, P < 0.05), expired CO2 (F (2,12) = 125.2, P < 0.0001; M, P < 0.0001), iBAT temperature (F (2,12) = 19.90, P = 0.0002; M, P < 0.0001; M*, P < 0.01) and core temperature (F (2,12) = 8.934, P = 0.0042; M, P < 0.05; M*, P < 0.01). SSNA and lSNA were unchanged following administration of 5 mg kg−1 METH alone or 5 mg kg−1 METH when expired CO2 was compensated.
Figure 3. Physiological effects of systemic methamphetamine (METH) before and after compensating METH‐evoked increases in expired CO2 .
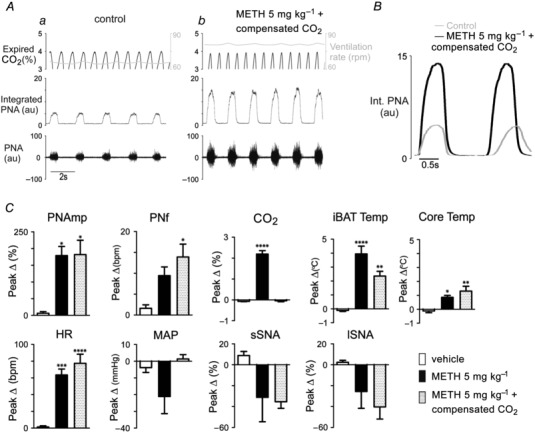
A, representative example of expired CO2, ventilation rate (right axis, grey), integrated and raw phrenic nerve activity during (a) control period and (b) after METH (5 mg kg−1)‐evoked changes in expired CO2 were compensated by increasing ventilation rate. B, comparison of integrated phrenic nerve activity respiratory period in control (grey line) and following METH (5 mg kg−1) with compensation of expired CO2 (continuous line). C, group data comparing peak responses following vehicle (open bar), METH (filled bar) and METH with CO2 compensation (shaded bar) and. Grouped data are presented as means ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Effects of METH on cardiorespiratory reflex function: peripheral and central chemoreceptor reflexes
HR, PNf and sSNA responses to hypoxia and hypercapnia are shown in Fig. 4, with responses in PNamp, lSNA and MAP collected but not shown.
Figure 4. Physiological effects evoked by cardiorespiratory reflex activation following acute systemic methamphetamine (METH) .
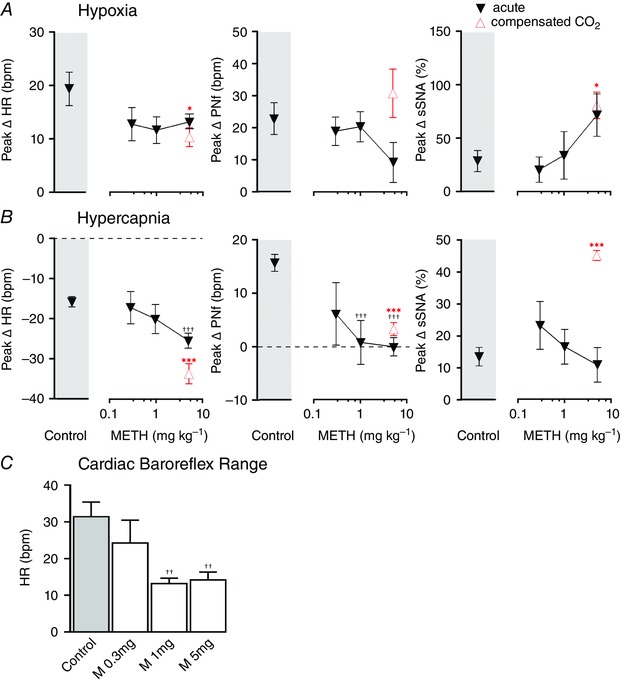
Response to hypoxia (A) and hypercapnia (B) before (grey boxes) and after acute METH (0.3, 1 and 5 mg kg−1) with expired CO2 compensation (open symbol). C, comparison of heart rate range in response to sodium nitroprusside and phenylephrine before (shaded bar) and after METH (0.3, 1 and 5 mg kg−1) (open bar). Grouped data are presented as means ± SEM; *P < 0.05, ***P < 0.001 vs. control (two‐tailed unpaired t tests), † P < 0.05, †† P < 0.01, ††† P < 0.001 vs. control (one‐way ANOVA, Dunnett's multiple comparison test).
Hypoxia (14 s of 100% N2) evoked well‐described responses (Sun & Reis, 1994) that included tachycardia, tachypnoea and sympathoexcitation (Fig. 4 A). After each dose of METH, hypoxia evoked similar effects in HR and in PNf (ns, one‐way ANOVA) (Fig. 4 A). The PNamp response to hypoxia was significantly reduced at 5 mg kg‐1 METH (one‐way ANOVA, F (3,13) = 4.146, P = 0.0288; control, 122.8 ± 21.9%; METH: 0.3 mg kg−1, 67.0 ± 45.8%; 1 mg kg−1, 49.3 ± 24.7%; 5 mg kg−1, 28.3 ± 4.9%, P < 0.01). Sympathoexcitation in lSNA was significantly enhanced following METH 5 mg kg−1 (F (3,18) = 3.959, P = 0.0249; lSNA; control, 22.0 ± 7.1%; METH: 0.3 mg kg−1, 18.2 ± 13.1%; 1 mg kg−1, 32.9 ± 22.6%; 5 mg kg−1, 73.9 ± 17.1%, P < 0.05) but unchanged in sSNA. MAP was not significantly affected by hypoxia (control, −2.9 ± 4.4 mmHg; METH: 0.3 mg kg−1, 3.9 ± 1.0 mmHg; 1 mg kg−1, 6.9 ± 6.9 mmHg; 5 mg kg−1, 2.6 ± 6.5 mmHg, ns).
Hypercapnia (60 s of 10% carbon dioxide) also evoked well‐described responses (Makeham et al. 2004), which included bradycardia, tachypnoea and sympathoexcitation (Fig. 4 B). METH significantly potentiated the bradycardic response (F (3,19) = 6.188, P = 0.0041; METH 5 mg kg−1, P < 0.01), whereas, the increase in respiratory drive (PNf) was significantly reduced (F (3,17) = 10.84, P = 0.003; METH 1 mg kg−1, P < 0.01) or abolished (METH 5 mg kg−1, P < 0.001) (Fig. 4 B). No differences were observed in the PNamp response (control, 66.3 ± 25.6%; METH: 0.3 mg kg−1, 141.0 ± 118.1%; 1 mg kg−1, 36.3 ± 19.9%; 5 mg kg−1, 22.5 ± 4.9%, ns). Similarly, sympathoexcitation was unaffected, but the small pressor response was blunted at 5 mg kg−1 METH (F (3,18) = 3.453, P = 0.0385; control, 9.0 ± 2.4 mmHg; METH: 0.3 mg kg−1, 3.0 ± 4.2; 1 mg kg−1; 3.7 ± 1.8; 5 mg kg−1, −1.2 ± 1.6 mmHg, P < 0.05).
Peripheral and central chemoreflex function was also assessed following METH (5 mg kg−1, n = 5) and CO2 compensation (Fig. 4 A and B, open triangles). Hypoxia elicited a slightly reduced tachycardia (two‐tailed unpaired t test, P = 0.0375, Fig. 4 A) compared to control and a sympathoexcitation that was similar to that evoked by METH alone, which was significantly larger than that evoked at baseline (P = 0.0165, Fig. 4 A). Hypercapnia following METH (5 mg kg−1) during compensated CO2 conditions evoked a greater bradycardia (P < 0.001, Fig. 4 B) and maintained reductions in tachypnoea (P = 0.0005, Fig. 4 B). Compensating the expired CO2 levels did not alter the effects of METH on PNamp for either hypoxia or hypercapnia, which were similar to control (59.7 ± 17.5% and 46.2 ± 10.2%, respectively, ns). MAP was similarly unaffected (6.2 ± 4.7 mmHg and 2.7 ± 2.1 mmHg, respectively, ns). Surprisingly sympathoexcitation was markedly increased by hypercapnia following METH with CO2 compensation (P = 0.0001).
Sympathetic baroreceptor reflex
Cardiac sympathetic baroreflex function was assessed by determining the total range in HR change which could be evoked by infusion of SNP and PE (Fig. 4 C). Increasing doses of METH significantly attenuated the HR range in response to SNP and PE (one‐way ANOVA, F (3,16) = 6.568, P = 0.0042) with Dunnett's test showing significant effects of both 1 mg kg−1 and 5 mg kg−1 METH (P < 0.01) (Fig. 4 C).
Sympathetic baroreflex function was also assessed from the splanchnic nerve. No significant differences in sympathetic baroreflex range or gain were seen following METH administration (Table 1, acute).
Table 1.
Baroreflex
| METH | |||||
|---|---|---|---|---|---|
| Control | 0.3 mg | 1 mg | 5 mg | ||
| A: upper plateau (%sSNA) | Acute | 128.8 ± 14.6 | 174.7 ± 31.7 | 185.5 ± 34.3 | 146.0 ± 20.3 |
| Saline treated | 133.0 ± 8.0 | 150.7 ± 10.6 | 171.9 ± 12.3 | 168.2 ± 21.9 | |
| METH treated | 126.8 ± 4.2 | 172.4 ± 19.3 | 186.6 ± 20.4 | 162.3 ± 25.9 | |
| B: lower plateau (%sSNA) | Acute | 0.2 ± 3.3 | 17.9 ± 5.5 | 12.9 ± 15.4 | 17.4 ± 4.6 |
| Saline treated | 10.6 ± 4.2 | 12.1 ± 6.2 | 13.7 ± 4.3 | 16.9 ± 4.6 | |
| METH treated | 10.6 ± 4.2 | 11.22 ± 3.8 | 9.3 ± 2.6 | 5.1 ± 3.6 | |
| C: gain (%sSNA/mmHg) | Acute | −2.4 ± 0.7 | −2.9 ± 0.6 | −2.4 ± 0.2 | −2.4 ± 0.2 |
| Saline treated | −2.3 ± 0.3 | −1.9 ± 0.1 | −2.6 ± 0.3 | −2.8 ± 0.4 | |
| METH Treated | −1.9 ± 0.2 | −3.0 ± 0.3 | −2.9 ± 0.5 | −2.8 ± 0.6 | |
Experiment 2: chronic METH sensitisation
Behavioural sensitisation
Injections of METH were given daily for 7 days and locomotor activity recorded on Days 2–8. Locomotor activity significant increased (two‐tailed unpaired t test, P = 0.0001) in the METH‐treated group on Day 8 (n = 9) compared with Day 2 (n = 9) (Fig. 5 A). Moreover, the METH‐treated animals were significantly more active than saline‐treated animals on Day 8 (P < 0.0001), with no significant differences seen within the saline‐treated group.
Figure 5. Behavioural sensitisation following chronic methamphetamine (METH) .

A, locomotor activity on Days 2 and 8 of the drug schedule (see text) in saline‐treated (open bar) and METH‐treated (filled bar) rats. B, locomotor activity following METH (1 mg kg−1) challenge after 14 days of abstinence in aligned cohort of rats which did not undergo physiological assessment (grey boxed area). Grouped data presented as means ± SEM; *P < 0.05, ***P < 0.001, ****P < 0.0001; METH treated vs. saline treated.
On Day 24 (saline and METH treatment completed Day 8) locomotor activity to METH challenge was measured in half of each cohort (Challenge, Fig. 5 B). Both treated groups showed increased locomotor activity on Day 24 in response to METH challenge compared to locomotor activity measured on Day 8 (two‐tailed unpaired t test; saline treated, P = 0.0371; METH P = 0.0314). Sensitisation of the METH‐treated group was indicated by a greater increase in locomotor activity in METH‐treated animals (n = 6) compared to saline‐treated animals (n = 6) (P = 0.0199). As a result the remaining animals in each cohort were assumed to demonstrate behavioural sensitization, so we then determined whether autonomic and respiratory sensitisation to METH challenge occurred.
Respiratory, cardiovascular and metabolic effects following chronic METH sensitisation protocol
Following chronic METH sensitisation but prior to METH challenge, resting cardiorespiratory and metabolic measurements and cardiorespiratory reflex function were determined and compared (Tables 1 and 2, Fig. 7 ). Chronic METH treatment had no significant effect on baseline variables: MAP, HR, PNf, iBAT and core temperatures and sSNA.
Table 2.
Baseline physiological values
| MAP (mmHg) | HR (bpm) | PNf (bpm) | Core (°C) | iBAT (°C) | sSNA (μV) | |
|---|---|---|---|---|---|---|
| Saline treated | 88.3 ± 4.4 | 449.6 ± 4.5 | 31.7 ± 1.8 | 36.72 ± 0.19 | 34.15 ± 0.47 | 3.24 ± 0.50 |
| METH treated | 90.1 ± 3.6 | 438.6 ± 7.2 | 35.4 ± 2.8 | 36.80 ± 0.18 | 34.17 ± 0.59 | 4.24 ± 1.39 |
Figure 7. Physiological effects evoked by cardiorespiratory reflex activation following chronic administration of methamphetamine (METH) or saline .

Response to hypoxia (A) and hypercapnia (B) before (grey boxes) and after different doses of METH (0.3, 1 and 5 mg kg−1) in saline (open symbols) and METH (filled symbol)‐treated animals. C, comparison of heart rate range in response to sodium nitroprusside and phenylephrine following METH challenge (0.3, 1 and 5 mg kg−1) in saline (open bars) and METH (dark shaded bar)‐treated animals, compared to control. Grouped data are presented as means ± SEM; *P < 0.05 (two‐tailed unpaired t tests). ♦, METH treated; †, saline treated; ♦/† P < 0.05, ♦♦/†† P < 0.01, ♦♦♦/††† P < 0.001, †††† P < 0.0001 vs. control (one‐way ANOVA, Dunnett's multiple comparison test). # P < 0.05, ## P < 0.01 (two‐way ANOVA).
In saline (open circles) and METH (filled squares)‐treated groups the effects of METH challenge (0.1, 0.3, 1 and 5 mg kg−1) on sSNA, lSNA, MAP, HR, iBAT, core temperature, expired CO2, PN amplitude and frequency were assessed (Fig. 6). The lowest doses of METH evoked little or no response in any parameter in either group. Treatment effects were only observed in expired CO2 (two‐way ANOVA, F (1,44) = 18.81, P < 0.0001) with significant dose effects (F (3,44) = 4.255, P = 0.0101) observed at 1 mg kg−1 (P < 0.05) and 5 mg kg−1 METH (P < 0.001). Dose related increases to METH were maintained in both treatment groups in HR (F (3,43) = 37.77, P < 0.0001), iBAT (F (3,44) = 98.99, P < 0.0001), core temperature (F (3,44) = 23.52, P < 0.0001), expired CO2 (F (3,44) = 122.1, P < 0.0001), PNamp (F (3,41) = 16.87, P < 0.0001) and PNf (F (3,44) = 4.259, P = 0.01). METH challenge evoked responses in sSNA, lSNA, MAP, HR, iBAT, core temperature, PN amplitude and frequency in METH‐treated animals (Fig. 6) were also similar to those seen in treatment naïve animals (Fig. 1).
Figure 6. Effects of methamphetamine (METH) challenge on respiratory, cardiovascular and metabolic outputs following chronic treatment with METH or saline .

Dose–response curves to METH (0.1, 0.3, 1 and 5 mg kg−1) in saline (open symbols) and METH (filled symbols)‐treated animals showing effects on splanchnic and lumbar sympathetic nerve activity (sSNA and lSNA), mean arterial pressure (MAP), heart rate (HR), interscapular brown adipose tissue (iBAT) and core temperatures, expired CO2, phrenic (PN) amplitude and frequency. Grouped data are presented as means ± SEM; non‐linear regression curves are shown where possible.
Peripheral and central chemoreceptor reflex function following METH sensitisation protocol
In both the saline and METH‐treated groups responses to hypoxia and hypercapnia were obtained at baseline prior to METH administration and at the peak of METH‐evoked effects (Fig. 7). Hypoxia at baseline evoked characteristic tachycardia, tachypnoea and sympathoexcitation, with no significant differences between the groups (two‐tailed unpaired t tests) (Fig. 7 A). Tachycardia and tachypnoea to hypoxia were unaffected throughout the dose range of METH challenge within or between treatment groups. METH‐evoked sympathoexcitation was significantly increased in both saline (one‐way ANOVA, F (3,24) = 20.61, P < 0.0001) and METH (F (3,24) = 5.007, P = 0.0078)‐treated animals, with significant increases at 5 mg kg−1 METH in both saline‐treated (P < 0.0001) and METH‐treated (P < 0.01) animals compared to their respective baseline responses. Surprisingly, saline‐treated animals showed exaggerated sympathoexcitation (two‐way ANOVA, F (3,104) = 4.77, P = 0.0037) in response to hypoxia with increasing METH doses (F (3,104) = 53.57, P < 0.0001).
Hypercapnia at baseline evoked bradycardia, tachypnoea and sympathoexcitation, with no significant differences between the two treatment groups observed except in HR (two‐tailed unpaired t tests, P = 0.0235) (Fig. 7 B). Dose‐related increases in the bradycardic response with increasing doses of METH were retained in saline‐treated animals (one‐way ANOVA, F (3,23) = 10.13, P = 0.0002) and METH (F (3,15) = 14.61, P < 0.0001), with significant effects at 1 mg kg−1 (P < 0.01) and at 5 mg kg−1 (P < 0.001) METH in saline‐treated animals, and at 5 mg kg−1 METH (P < 0.0001) in METH‐treated animals. Increasing doses of METH also blunted the tachypnoea response in both saline (F (3,24) = 8.564, P = 0.0005) and METH (F (3,19) = 4.971, P = 0.0103)‐treated animals, with significant effects at 0.3 mg kg−1 METH (P < 0.05), 1 mg kg−1 METH (P < 0.01) and 5 mg kg−1 METH (P < 0.001) in saline‐treated and 1 mg kg−1 METH (P < 0.05) and 5 mg kg−1 METH (P < 0.05) in METH‐treated animals. Tachypnoea responses to hypercapnia were significantly different between treatment groups (two‐way ANOVA, F (1,43) = 6.88, P = 0.0120), with greater dose‐dependent reductions in the METH‐treated group (F (3,43) = 12.74, P < 0.0001). In contrast, sympathoexcitation was similar in both treatment groups.
Sympathetic baroreceptor reflexes following METH sensitisation protocol
Sympathetic cardiac baroreflex range was reduced in the METH‐treated group at baseline control (two‐tailed unpaired t tests, P = 0.0387, Fig. 7 C). HR range following METH challenge was significantly different to baseline in both saline (one‐way ANOVA, F (3,24) = 12.66, P < 0.0001) and METH groups (F (3,21) = 9.828, P = 0.0003), with significant reductions in HR following METH 0.3 mg kg−1 (saline treated; P < 0.01), 1 mg kg−1 (saline treated, P < 0.01; METH treated, P < 0.05) and METH 5 mg kg−1 (saline treated, P < 0.0001; METH treated, P < 0.001). However, there was no significant difference between treatment groups (two‐way ANOVA, Fig. 7 C).
Upper and lower plateau values and sympathetic baroreflex gains following 0.3, 1 and 5 mg kg−1 METH compared to control are shown in (Table 1 A–C). No differences in sympathetic baroreflex function were observed within or between treatment groups following METH challenge (one‐way ANOVA/two‐way ANOVA).
Discussion
The principal findings of this study are that systemic METH administration (a) increased HR, iBAT temperature, expired CO2 and inspiratory drive and frequency in a dose‐dependent manner, with little effect on MAP or the sympathetic outflows to the viscera or hindlimb, (b) increased centrally mediated ventilatory drive independently of changes in CO2, (c) modified protective cardiorespiratory reflexes, and (d) was not associated with facilitated sympathetic, respiratory, metabolic, cardiovascular responses or cardiorespiratory reflex function following a behaviourally validated METH sensitisation protocol.
The patterned physiological response of increased HR and expired CO2 which was correlated with iBAT temperature and accompanied by increased respiratory drive, is typical of non‐shivering thermogenesis (Morrison, 2011). This suggests that METH triggers non‐shivering thermogenesis which contributes to the well‐described METH‐evoked hyperthermia. The simplest explanation is that METH acts to release noradrenaline from BAT sympathetic postganglionic terminals, but other effectors may be possible as centrally mediated release at other sympathetic outflows was not evident. In addition, METH drives centrally evoked increases in ventilatory drive independent of metabolically induced changes in CO2 which underlies the tachypnoea described in the clinic. The data further indicate that METH‐evoked behavioural sensitisation is mediated independently of mechanisms which evoke sympathetic, cardiovascular, respiratory and metabolic changes. Finally, acutely administered METH alters tonic and reflex control of cardiovascular, respiratory and metabolic systems in such a way that does not provide an optimal environment to withstand cardiorespiratory challenge.
Methodological considerations
Several studies have reported the effects of METH on a limited range of physiological variables able to be measured in conscious rodents (MAP, HR and body temperature; Yoshida et al. 1993; Sprague et al. 2004; Myles et al. 2008; Rusyniak et al. 2012; Varner et al. 2013). An anaesthetised model was chosen for the current study, permitting the concurrent measurement of multiple variables, thus providing a greater understanding of the physiological systems altered by METH administration. However, the effects of urethane anaesthesia on neurotransmitter function in the brain (Hara & Harris, 2002) and specifically the inhibitory effects when urethane is directly applied to sympathetic premotor centres in the medulla (Sun & Reis, 1995) may mask METH‐induced physiological effects seen in conscious but not in anaesthetised rats. The lack of effect on the sympathetic outflows measured was also surprising considering METH concentrates in brain regions containing aminergic presympathetic populations, potentially driving these outflows at least at very high doses of the drug (Li et al. 2012 b). The threshold dose of METH for variables measured in this study was between 0.1 and 0.3 mg kg−1, in keeping with studies in conscious animals (Yoshida et al. 1993; Varner et al. 2013).
Physiological effects of acute METH
Thermoregulation
METH increased core temperature as described previously (Bowyer et al. 1992; Callaway & Clark, 1994; Makisumi et al. 1998; Arora et al. 2001; Rusyniak et al. 2012), with our data indicating that non‐shivering thermogenesis contributes to METH‐evoked hyperthermia. METH evoked an increase in iBAT temperature of up to 4°C, which was strongly correlated with increases in core temperature, despite the use of a thermoregulated heating blanket. METH also evoked tachycardia and an increase in expired CO2, which were positively correlated both temporally and with the magnitude of the iBAT response. Non‐shivering thermogenesis is consistently accompanied by increased HR, probably increasing BAT blood flow in order to distribute heat, and increased expired CO2 resulting from increased BAT metabolism (Morrison, 2004; Morrison et al. 2012). Our findings are supported by a recent study showing that iBAT ablation reduced METH‐induced hyperthermia in conscious mice (Sanchez‐Alavez et al. 2013) and is in keeping with a previous report in which systemic amphetamine in female rats evoked an increase in iBAT temperature (Wellman, 1983). However, no other parameters were measured in this study so the effects could not be reliably attributed to increased thermogenesis and changes in blood flow could strongly influence iBAT temperature. Non‐shivering thermogenesis is likely to be evoked by METH increasing the availability of biogenic amines directly at brown adipocytes by noradrenaline release from BAT sympathetic postganglionic terminals evoked either at the level of the synapse or centrally as iBAT denervation reduces METH‐induced mitochondrial dysfunction in iBAT (Sanchez‐Alavez et al. 2013), or possibly via indirect mechanisms.
The METH‐evoked temperature changes were independent of physical exertion, which may contribute to hyperthermia measured under conscious conditions (Yoshida et al. 1993; Sprague et al. 2004; Rusyniak et al. 2012). The poor temporal relationship between temperature and locomotor‐induced activity has previously suggested that factors other than locomotion contribute to the hyperthermia (Phelps et al. 2010; Rusyniak et al. 2012). Data described here provide the first measurement of iBAT temperature indicating that it contributes to METH‐evoked hyperthermia, adding to previous reports that also implicate glucocorticoids (Makisumi et al. 1998), the thyroid gland and hypermetabolism in skeletal muscle (Makisumi et al. 1998; Sprague et al. 2004).
Respiration
We demonstrate for the first time that METH evokes centrally mediated increases in inspiratory drive and frequency, which were independent of METH‐driven metabolic increases in plasma CO2 levels. The increased inspiratory drive and reduced expiratory period underlie the tachypnoea often reported in clinical case and animal studies following METH use (Lin et al. 1980; Mendelson et al. 2006; Cruickshank & Dyer, 2009). These changes may be mediated via activation of central adrenergic receptors, as central β‐adrenoreceptor blockade abolishes the tachypnoea seen following amphetamine administration (Mediavilla et al. 1979; Mora et al. 1983). Whether central respiratory centres are directly affected is yet to be determined, but it is of interest that METH when administered at lethal concentrations has been reported to concentrate in brainstem regions (Li et al. 2012 b) where major respiratory centres are located (Richter & Smith, 2014).
Cardiovascular
In the present study, METH consistently evoked tachycardia but had no effect on blood pressure except at 5 mg kg−1 where depressor responses were evoked. METH‐evoked tachycardia is consistently reported in conscious and anaesthetised animal studies (Yoshida et al. 1993; Arora et al. 2001) and in clinical reports (Chan et al. 1994; West et al. 2010). This is probably mediated via β1 adrenoreceptors (Schindler et al. 1992) by direct actions of METH at the heart and/or via activation of the cardiac sympathetic outflow (as the animals reported here were vagotomised), or as a consequence of the increased thermogenesis as indicated above.
The depressor response evoked at high doses of METH in anaesthetised rats could be indicative of approaching thresholds of METH toxicity, which at 12 mg kg−1 i.v. can lead to cardiovascular collapse and death in conscious animals (Chan et al. 1994; Kaye et al. 2007, 2008; Li et al. 2012 a,b). The lack of effect of lower doses of METH on MAP contrasts strongly to the pressor responses evoked in conscious animals (Yoshida et al. 1993) and thus may result from the anaesthesia (see Methodological considerations above). It is possible that in non‐vagotomised conscious animals the METH‐evoked tachycardia, which is significantly greater than in vagotomised anaesthetised rats used here, contributed to the relatively modest pressor effect (∼17 mmHg at 1 mg kg−1 METH) seen in conscious rats or, alternatively, that MAP changes seen in conscious animals simply reflect those required to accommodate the increased locomotor activity and altered behaviour elicited by METH (Shoblock et al. 2003; Hall et al. 2008).
Cardiorespiratory reflexes
We addressed the effects of METH on chemo‐ and baroreceptor reflexes which act to maintain blood pressure, appropriate tissue perfusion and gas exchange. METH had little effect on the response to hypoxia except for a reduction in respiratory drive, likely to be due to a ceiling effect resulting from METH‐evoked CO2 load. In contrast, METH altered the response to hypercapnia, facilitating the bradycardia and the sympathoexcitation (under CO2 compensation), but abolishing the tachypnoea.
The sympathetic baroreflex was unaffected by METH, but METH reduced the range of the sympathetic cardiac baroreflex. This effect could be mediated centrally or directly at the heart, although the effect may simply result from a ceiling effect, as METH evoked a large increase in HR which may not be further increased under the anaesthetised and paralysed conditions here. Nevertheless, it is interesting to note that reduced HR variability indicative of baroreceptor dysfunction is common in those suffering METH addiction (Henry et al. 2012) and in a model of METH overdose using very high levels of METH, baroreceptor function is severely attenuated in those animals that succumbed to fatal doses of METH (Li et al. 2012 b).
These data indicate that respiratory and blood pressure challenges assessed here following METH administration fail to elicit appropriate protective reflex responses.
Physiological effects of acute METH following METH‐induced behavioural sensitisation
In line with others, rats that received repeated METH showed increased locomotor activity compared to those that received saline (Stewart & Badiani, 1993; McDaid et al. 2006). Following 14 days of abstinence, locomotor activity was further exaggerated as previously described (Paulson et al. 1991; McDaid et al. 2006). Evidence suggests this is mediated by long‐term neuroadaptative changes in the brain (Cornish & Kalivas, 2001), particularly associated with the mesolimbic dopamine system (Robinson & Becker, 1986).
Despite this significant behavioural sensitisation we found no evidence either of changes in baseline sympathetic, cardiovascular, respiratory or metabolic variables or cardiorespiratory reflex function. Furthermore, exaggerated respiratory or autonomic responses were not seen at any dose of METH administered in animals chronically treated with METH. Yoshida and colleagues (1993) showed that repeated administration of METH in telemetered animals evoked an enhanced response in MAP, body temperature, as well as activity (but not HR) following i.p. METH challenge. However, they showed that only activity was sensitised following intracerebroventricular METH challenge, so it is possible that the enhanced locomotor activity could have contributed to these larger changes in MAP and body temperature. Also, rats receiving an escalating dose of METH or saline for 12 days and then challenged with METH (10 mg kg−1) on Day 13 showed no change in temperature within 120 min in the METH‐treated group, with a large rise in core temperature exhibited in the saline‐treated group (Myles & Sabol, 2008). However, we cannot rule out that significant differences due to METH administration regimes (both time course and route of administration) or the contribution of anaesthesia could account for the differences between all these studies. Nevertheless, using a protocol which demonstrated behavioural sensitisation following a period of abstinence in our conscious animal cohort, there was no facilitation of METH‐evoked autonomic responses in the anaesthetised animals.
The sensitised locomotor response is likely to be mediated by the mesolimbic dopamine system (Pierce & Kalivas, 1997; Kelly et al. 2008). Although little evidence is available to describe the neurotransmitters involved in mediating central respiratory and autonomic effects of METH, we have shown, at least in the prefrontal cortex, that noradrenaline rather than dopamine best mimics the physiological effects of systemic METH administration (Hassan et al. 2015). It is possible that sensitisation to repeated METH administration does not occur in all systems. However, we have also demonstrated that METH sensitisation alters proteins associated with the GABAergic inhibitory network (Wearne et al. 2015), one of many neurotransmitter systems targeted by the general anaesthetic used here (Hara & Harris, 2002). It is conceivable that such an interaction could limit measures of physiological facilitation induced by chronic METH exposure.
In response to peripheral and central chemoreceptor stimulation, METH‐pretreated animals responded very similarly to both saline‐pretreated and naïve animals, with only small differences in a few parameters between treatment groups. Curiously, the sympathoexcitation evoked by hypoxia following METH challenge was seen in both treatment groups and in naive animals. Chronic METH treatment does not alter the responses to hypoxia or hypercapnia at baseline but was similarly deleterious in both METH‐pretreated and drug naïve animals during METH challenge. Likewise, the cardiac sympathetic baroreflex range was reduced in the METH‐pretreated group, which mimicked the effect in drug naïve animals. The effect of all doses of METH on the physiological measures here were equivalent across all treated groups.
This lack of sensitisation of cardiovascular, respiratory and metabolic effects following chronic METH is in contrast to that seen following other chronic stimuli. Sustained hypoxia and chronic intermittent hypoxia (CIH) evoke increased baseline ventilation (Reeves et al. 2003) and CIH causes increased sympathetic vasomotor tone, MAP, and haematocrit, with enhanced peripheral and central chemoreflex sympathetic and respiratory responses as well as augmentation of other sympathoexcitatory reflexes (Greenberg et al. 1999; Zoccal et al. 2009; Silva & Schreihofer, 2011). Furthermore, these changes were seen in anaesthetised preparations. Physiological adjustments to chronic exposure to low environmental temperatures include maintenance of lower levels of body temperature and a shift from shivering to non‐shivering thermogenesis (Cadena & Tattersall, 2014). Following cessation of all these chronic insults the physiological changes do recover over time, so that it is possible that immediately following a chronic exposure to METH physiological effects may be detected, but this remains to be determined. Alternatively, the mechanisms affected by METH to evoke physiological change may not be amenable to such plasticity.
Conclusions
In summary, acute METH administered in the anaesthetised animal increases cardiac, metabolic and respiratory function in a pattern that mimics activation of non‐shivering thermogenesis, indicating that thermogenesis probably contributes to METH‐evoked hyperthermia. METH increases ventilatory drive independently of changes in CO2. Acute METH administration also reduces the effectiveness of some components of protective cardiorespiratory reflexes, probably due to heightened METH‐evoked respiratory and cardiac functions. In contrast, chronic treatment with METH that facilitated METH‐evoked locomotor activity failed to modify basal physiological parameters or sympathetic, cardiovascular, respiratory and metabolic responses evoked by acute METH challenge. These findings highlight the potent and dangerous physiological effects of METH use and indicate some underlying mechanisms responsible for the increased cardiorespiratory vulnerability and lethal hyperthermia seen in those that abuse METH.
Additional information
Competing interests
None declared.
Author contributions
S.F.H., J.L.C. and A.K.G. designed the experiment. The experiments and analysis were conducted by S.F.H. and T.W. S.F.H. and A.K.G. interpreted the data. S.F.H. and A.K.G. wrote the manuscript. All authors critically reviewed content and approved the final version for publication. All agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The following grants supported the research reported in this manuscript: The Hillcrest Foundation (Perpetual Trustees FR2013/1308, FR2014/0781), National Health and Medical Research Council (APP1028183, APP1030301) and the Australian Research Council (DP120100920). S.F.H. was in receipt of an Australian Postgraduate Award.
Translational perspective
In the emergency room, the behavioural and physiological consequences of methamphetamine (METH) intoxication are commonly seen. The use of METH is increasing worldwide and the ease with which it can be manufactured is fuelling this. Death from an overdose of METH is due to the consequences of extreme temperatures generated in the body or from heart‐related events. METH evokes widespread physiological changes but the underlying mechanisms responsible are largely unknown. The data presented in this study provide the first comprehensive analysis of cardiovascular, respiratory and metabolic function altered by METH in the anaesthetised rat. The effects are described over a range of doses and following chronic administration. We demonstrate that non‐shivering thermogenesis due to the increased activity of brown adipose tissue, contributes to the METH‐induced increase in body temperature, potentially providing a site of action that could be targeted to lower the body temperature of patients who abuse METH. Furthermore, the data show that METH evokes centrally mediated increases in respiration which are independent of the metabolic consequences of the drug, and, further demonstrate a reduced capability of the heart and respiratory system to generate appropriate cardiorespiratory reflex functions which are normally protective. Such studies could ultimately pave the way for therapeutic intervention in patients suffering from METH overdose.
References
- Abekawa T, Ito K, Nakagawa S, Nakato Y & Koyama T (2008). Olanzapine and risperidone block a high dose of methamphetamine‐induced schizophrenia‐like behavioral abnormalities and accompanied apoptosis in the medial prefrontal cortex. Schizophr Res 101, 84–94. [DOI] [PubMed] [Google Scholar]
- Arora H, Owens SM & Gentry WB (2001). Intravenous (+)‐methamphetamine causes complex dose‐dependent physiologic changes in awake rats. Eur J Pharmacol 426, 81–87. [DOI] [PubMed] [Google Scholar]
- Bowyer J & Holson R (1995). Methamphetamine and amphetamine neurotoxicity. Neurological Disease And Therapy 36, 845–845. [Google Scholar]
- Bowyer JF, Tank AW, Newport GD, Slikker W, Jr. , Ali SF & Holson RR (1992). The influence of environmental temperature on the transient effects of methamphetamine on dopamine levels and dopamine release in rat striatum. J Pharmacol ExP Ther 260, 817–824. [PubMed] [Google Scholar]
- Brown JM, Riddle EL, Sandoval V, Weston RK, Hanson JE, Crosby MJ, Ugarte YV, Gibb JW, Hanson GR & Fleckenstein AE (2002). A single methamphetamine administration rapidly decreases vesicular dopamine uptake. J Pharmacol ExP Ther 302, 497–501. [DOI] [PubMed] [Google Scholar]
- Cadena V & Tattersall GJ (2014). Body temperature regulation during acclimation to cold and hypoxia in rats. J Therm Biol 46, 56–64. [DOI] [PubMed] [Google Scholar]
- Cadet JL & Krasnova IN (2009). Molecular bases of methamphetamine‐induced neurodegeneration. Int Rev Neurobiol 88, 101–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway CW & Clark RF (1994). Hyperthermia in psychostimulant overdose. Ann Emerg Med 24, 68–76. [DOI] [PubMed] [Google Scholar]
- Carvalho M, Carmo H, Costa VM, Capela JP, Pontes H, Remiao F, Carvalho F & Bastos Mde L (2012). Toxicity of amphetamines: an update. Arch Toxicol 86, 1167–1231. [DOI] [PubMed] [Google Scholar]
- Chan P, Chen JH, Lee MH & Deng JF (1994). Fatal and nonfatal methamphetamine intoxication in the intensive care unit. J Toxicol Clin Toxicol 32, 147–155. [DOI] [PubMed] [Google Scholar]
- Cornish JL & Kalivas PW (2001). Cocaine sensitization and craving: differing roles for dopamine and glutamate in the nucleus accumbens. J Addict Dis 20, 43–54. [DOI] [PubMed] [Google Scholar]
- Cruickshank CC & Dyer KR (2009). A review of the clinical pharmacology of methamphetamine. Addiction 104, 1085–1099. [DOI] [PubMed] [Google Scholar]
- Glasner‐Edwards S, Mooney LJ, Marinelli‐Casey P, Hillhouse M, Ang A, Rawson RA & Methamphetamine Treatment Project Corporate Authors (2010). Psychopathology in methamphetamine‐dependent adults 3 years after treatment. Drug Alcohol Rev 29, 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg HE, Sica A, Batson D & Scharf SM (1999). Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J Appl Physiol 85, 298–305. [DOI] [PubMed] [Google Scholar]
- Guilarte TR, Nihei MK, McGlothan JL & Howard AS (2003). Methamphetamine‐induced deficits of brain monoaminergic neuronal markers: distal axotomy or neuronal plasticity. Neuroscience 122, 499–513. [DOI] [PubMed] [Google Scholar]
- Hall DA, Stanis JJ, Marquez Avila H & Gulley JM (2008). A comparison of amphetamine‐ and methamphetamine‐induced locomotor activity in rats: evidence for qualitative differences in behavior. Psychopharmacology 195, 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K & Harris RA (2002). The anesthetic mechanism of urethane: the effects on neurotransmitter‐gated ion channels. Anesth Analg 94, 313–318. [DOI] [PubMed] [Google Scholar]
- Hassan SF, Zumut S, Burke PG, McMullan S, Cornish JL & Goodchild AK (2015). Comparison of noradrenaline, dopamine and serotonin in mediating the tachycardic and thermogenic effects of methamphetamine in the ventral medial prefrontal cortex. Neuroscience 295, 209–220. [DOI] [PubMed] [Google Scholar]
- Henry BL, Minassian A & Perry W (2012). Effect of methamphetamine dependence on heart rate variability. Addict Biol 17, 648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwazaki T, McGregor IS & Matsumoto I (2008). Protein expression profile in the amygdala of rats with methamphetamine‐induced behavioral sensitization. Neurosci Lett 435, 113–119. [DOI] [PubMed] [Google Scholar]
- Jedynak JP, Uslaner JM, Esteban JA & Robinson TE (2007). Methamphetamine‐induced structural plasticity in the dorsal striatum. Eur J Neurosci 25, 847–853. [DOI] [PubMed] [Google Scholar]
- Kaye S, Darke S, Duflou J & McKetin R (2008). Methamphetamine‐related fatalities in Australia: demographics, circumstances, toxicology and major organ pathology. Addiction 103, 1353–1360. [DOI] [PubMed] [Google Scholar]
- Kaye S, McKetin R, Duflou J & Darke S (2007). Methamphetamine and cardiovascular pathology: a review of the evidence. Addiction 102, 1204–1211. [DOI] [PubMed] [Google Scholar]
- Kelly MA, Low MJ, Rubinstein M & Phillips TJ (2008). Role of dopamine D1‐like receptors in methamphetamine locomotor responses of D2 receptor knockout mice. Gene Brain Behav 7, 568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YT, Lee SW, Kwon DH, Seo JH, Ahn BC & Lee J (2009). Dose‐dependent frontal hypometabolism on FDG‐PET in methamphetamine abusers. J Psychiatr Res 43, 1166–1170. [DOI] [PubMed] [Google Scholar]
- Krasnova IN & Cadet JL (2009). Methamphetamine toxicity and messengers of death. Brain Res Rev 60, 379–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnova IN, Justinova Z, Ladenheim B, Jayanthi S, McCoy MT, Barnes C, Warner JE, Goldberg SR & Cadet JL (2010). Methamphetamine self‐administration is associated with persistent biochemical alterations in striatal and cortical dopaminergic terminals in the rat. PloS One 5, e8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FC, Yen JC, Chan SH & Chang AY (2012. a). Bioenergetics failure and oxidative stress in brain stem mediates cardiovascular collapse associated with fatal methamphetamine intoxication. PloS One 7, e30589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FC, Yen JC, Chan SH & Chang AY (2012. b). Defunct brain stem cardiovascular regulation underlies cardiovascular collapse associated with methamphetamine intoxication. J Biomed Sci 19, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Chandra A, Chern YF & Tsay BL (1980). Effects of intracerebroventricular injection of d‐amphetamine on metabolic, respiratory, and vasomotor activities and body temperatures in the rat. Can J Physiol Pharmacol 58, 903–908. [DOI] [PubMed] [Google Scholar]
- McDaid J, Graham MP & Napier TC (2006). Methamphetamine‐induced sensitization differentially alters pCREB and DeltaFosB throughout the limbic circuit of the mammalian brain. Mol Pharmacol 70, 2064–2074. [DOI] [PubMed] [Google Scholar]
- Makeham JM, Goodchild AK, Costin NS & Pilowsky PM (2004). Hypercapnia selectively attenuates the somato‐sympathetic reflex. ResP Physiol Neurobiol 140, 133–143. [DOI] [PubMed] [Google Scholar]
- Makisumi T, Yoshida K, Watanabe T, Tan N, Murakami N & Morimoto A (1998). Sympatho‐adrenal involvement in methamphetamine‐induced hyperthermia through skeletal muscle hypermetabolism. Eur J Pharmacol 363, 107–112. [DOI] [PubMed] [Google Scholar]
- Mediavilla A, Feria M, Fernandez JF, Cagigas P, Pazos A & Florez J (1979). The stimulatory action of d‐amphetamine on the respiratory centre, and its mediation by a central alpha‐adrenergic mechanism. Neuropharmacology 18, 133–142. [DOI] [PubMed] [Google Scholar]
- Mendelson J, Uemura N, Harris D, Nath RP, Fernandez E, Jacob P 3rd, Everhart ET & Jones RT (2006). Human pharmacology of the methamphetamine stereoisomers. Clin Pharmacol Ther 80, 403–420. [DOI] [PubMed] [Google Scholar]
- Mora F, Lee TF & Myers RD (1983). Involvement of alpha‐ and beta‐adrenoreceptors in the central action of norepinephrine on temperature, metabolism, heart and respiratory rates of the conscious primate. Brain Res Bull 11, 613–616. [DOI] [PubMed] [Google Scholar]
- Morrison SF (2004). Central pathways controlling brown adipose tissue thermogenesis. News Physiol Sci 19, 67–74. [DOI] [PubMed] [Google Scholar]
- Morrison SF (2011). 2010 Carl Ludwig Distinguished Lectureship of the APS Neural Control and Autonomic Regulation Section: Central neural pathways for thermoregulatory cold defense. J Appl Physiol 110, 1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SF, Madden CJ & Tupone D (2012). Central control of brown adipose tissue thermogenesis. Front Endocrinol 3 pii: 00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morshedi MM & Meredith GE (2008). Repeated amphetamine administration induces Fos in prefrontal cortical neurons that project to the lateral hypothalamus but not the nucleus accumbens or basolateral amygdala. Psychopharmacology 197, 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myles BJ, Jarrett LA, Broom SL, Speaker HA & Sabol KE (2008). The effects of methamphetamine on core body temperature in the rat – part 1: chronic treatment and ambient temperature. Psychopharmacology 198, 301–311. [DOI] [PubMed] [Google Scholar]
- Paulson PE, Camp DM & Robinson TE (1991). Time course of transient behavioral depression and persistent behavioral sensitization in relation to regional brain monoamine concentrations during amphetamine withdrawal in rats. Psychopharmacology 103, 480–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps G, Speaker HA & Sabol KE (2010). Relationship between methamphetamine‐induced behavioral activation and hyperthermia. Brain Res 1357, 41–52. [DOI] [PubMed] [Google Scholar]
- Pierce RC & Kalivas PW (1997). A circuitry model of the expression of behavioral sensitization to amphetamine‐like psychostimulants. Brain Res Brain Res Rev 25, 192–216. [DOI] [PubMed] [Google Scholar]
- Reeves SR, Gozal E, Guo SZ, Sachleben LR Jr, Brittian KR, Lipton AJ & Gozal D (2003). Effect of long‐term intermittent and sustained hypoxia on hypoxic ventilatory and metabolic responses in the adult rat. J Appl Physiol 95, 1767–1774. [DOI] [PubMed] [Google Scholar]
- Richter DW & Smith JC (2014). Respiratory rhythm generation in vivo . Physiology 29, 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE & Becker JB (1986). Enduring changes in brain and behavior produced by chronic amphetamine administration: a review and evaluation of animal models of amphetamine psychosis. Brain Res 396, 157–198. [DOI] [PubMed] [Google Scholar]
- Rusyniak DE, Zaretsky DV, Zaretskaia MV, Durant PJ & Dimicco JA (2012). The orexin‐1 receptor antagonist SB‐334867 decreases sympathetic responses to a moderate dose of methamphetamine and stress. Physiol Behav 107, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Alavez M, Conti B, Wood MR, Bortell N, Bustamante E, Saez E, Fox HS & Marcondes MC (2013). ROS and sympathetically mediated mitochondria activation in brown adipose tissue contribute to methamphetamine‐induced hyperthermia. Fron Endocrinol 4, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep LJ, Slaughter RJ & Beasley DM (2010). The clinical toxicology of metamfetamine. Clin Toxicol 48, 675–694. [DOI] [PubMed] [Google Scholar]
- Schindler CW, Zheng JW, Tella SR & Goldberg SR (1992). Pharmacological mechanisms in the cardiovascular effects of methamphetamine in conscious squirrel monkeys. Pharmacol Biochem Behav 42, 791–796. [DOI] [PubMed] [Google Scholar]
- Shoblock JR, Sullivan EB, Maisonneuve IM & Glick SD (2003). Neurochemical and behavioral differences between d‐methamphetamine and d‐amphetamine in rats. Psychopharmacology 165, 359–369. [DOI] [PubMed] [Google Scholar]
- Silva AQ & Schreihofer AM (2011). Altered sympathetic reflexes and vascular reactivity in rats after exposure to chronic intermittent hypoxia. J Physiol 589, 1463–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague JE, Mallett NM, Rusyniak DE & Mills E (2004). UCP3 and thyroid hormone involvement in methamphetamine‐induced hyperthermia. Biochem Pharmacol 68, 1339–1343. [DOI] [PubMed] [Google Scholar]
- Srisurapanont M, Ali R, Marsden J, Sunga A, Wada K & Monteiro M (2003). Psychotic symptoms in methamphetamine psychotic in‐patients. Int J Neuropsychopharmacol 6, 347–352. [DOI] [PubMed] [Google Scholar]
- Steketee JD (2003). Neurotransmitter systems of the medial prefrontal cortex: potential role in sensitization to psychostimulants. Brain Res Brain Res Rev 41, 203–228. [DOI] [PubMed] [Google Scholar]
- Stewart J & Badiani A (1993). Tolerance and sensitization to the behavioral effects of drugs. Beha Pharmacol 4, 289–312. [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW & Galli A (2005). Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol 75, 406–433. [DOI] [PubMed] [Google Scholar]
- Sun MK & Reis DJ (1994). Hypoxia selectively excites vasomotor neurons of rostral ventrolateral medulla in rats. Am J Physiol 266, R245–256. [DOI] [PubMed] [Google Scholar]
- Varner KJ, Daigle K, Weed PF, Lewis PB, Mahne SE, Sankaranarayanan A & Winsauer PJ (2013). Comparison of the behavioral and cardiovascular effects of mephedrone with other drugs of abuse in rats. Psychopharmacology 225, 675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wearne TA, Mirzaei M, Franklin JL, Goodchild AK, Haynes PA & Cornish JL (2015). Methamphetamine‐induced sensitization is associated with alterations to the proteome of the prefrontal cortex: implications for the maintenance of psychotic disorders. J Proteome Res 14, 397–410. [DOI] [PubMed] [Google Scholar]
- Wellman PJ (1983). Influence of amphetamine on brown adipose thermogenesis. Res Commun Chem Pathol Pharmacol 41, 173–176. [PubMed] [Google Scholar]
- West PL, McKeown NJ & Hendrickson RG (2010). Methamphetamine body stuffers: an observational case series. Ann Emerg Med 55, 190–197. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Morimoto A, Makisumi T & Murakami N (1993). Cardiovascular, thermal and behavioral sensitization to methamphetamine in freely moving rats. J Pharmacol ExP Ther 267, 1538–1543. [PubMed] [Google Scholar]
- Zoccal DB, Bonagamba LG, Paton JF & Machado BH (2009). Sympathetic‐mediated hypertension of awake juvenile rats submitted to chronic intermittent hypoxia is not linked to baroreflex dysfunction. ExP Physiol 94, 972–983. [DOI] [PubMed] [Google Scholar]
- Zweben JE, Cohen JB, Christian D, Galloway GP, Salinardi M, Parent D, Iguchi M & Methamphetamine Treatment P (2004). Psychiatric symptoms in methamphetamine users. Am J Addict 13, 181–190. [DOI] [PubMed] [Google Scholar]