

The human pathogen Toxoplasma gondii builds a remarkable structure, the conoid, from the ubiquitous cytoskeletal protein tubulin but arranges it into a completely novel form. Deleting the T. gondii gene for a DCX-domain protein disrupts the structure of the conoid and cripples the ability of the parasite to invade host cells.
Abstract
The ∼6000 species in phylum Apicomplexa are single-celled obligate intracellular parasites. Their defining characteristic is the apical complex—membranous and cytoskeletal elements at the apical end of the cell that participate in host-cell invasion. The apical complex of Toxoplasma gondii and some other apicomplexans includes a cone-shaped assembly, the conoid, which in T. gondii comprises 14 spirally arranged fibers that are nontubular polymers of tubulin. The tubulin dimers of the conoid fibers make canonical microtubules elsewhere in the same cell, suggesting that nontubulin protein dictates their special arrangement in the conoid fibers. One candidate for this role is TgDCX, which has a doublecortin (DCX) domain and a TPPP/P25-α domain, both of which are known modulators of tubulin polymer structure. Loss of TgDCX radically disrupts the structure of the conoid, severely impairs host-cell invasion, and slows growth. Both the conoid structural defects and the impaired invasion of TgDCX-null parasites are corrected by reintroduction of a TgDCX coding sequence. The nontubular polymeric form of tubulin found in the conoid is not found in the host cell, suggesting that TgDCX may be an attractive target for new parasite-specific chemotherapeutic agents.
INTRODUCTION
Infection with Toxoplasma gondii can cause severe illness when the infection is contracted congenitally or is reactivated in immunosuppressed hosts. T. gondii is one of ∼6000 species of intracellular protozoan parasites in the phylum Apicomplexa, all members of which are parasitic, including various important human/animal pathogens, such as Plasmodium (causative agents of malaria), Cryptosporidium (cryptosporidiosis), Theileria and Babesia (important pathogens of cattle), and Eimeria (pathogens of poultry and cattle).
The apicomplexan parasites belong to the superphylum Alveolata, organisms that have alveolae—adjoining membrane sacs constituting two additional layers of membrane underlying the plasma membrane. Apicomplexa is one of the three major clades of Alveolate, along with dinoflagellates, which diverged from the apicomplexans several hundred million years ago, and ciliates, which diverged from other alveolates up to 1 billion years ago (Leander and Keeling, 2003). A number of organisms belonging to sister clades of apicomplexans and dinoflagellates have been identified. These include marine photosynthetic relatives of the apicomplexans (chromerids), free-living relatives of the apicomplexans that prey on other cells by myzocytosis (colpodellids), and marine parasitic relatives of dinoflagellates that can be cultured in a free-living form (perkinsids). Morphologically, the perkinsids and the chromerids are unified with the apicomplexans not only by the cortical membrane sacs shared by all the alveolates, but also by a striking membrane-cytoskeletal assembly called the apical complex, which consists of electron-dense, elongated vesicles associated with a cone-shaped or flattened array of tubulin polymers located at the apical end of the cell. In perkinsids and chromerids, the apical complex contains a half-closed cone structure (pseudoconoid) formed of a sheet of microtubules (30–35 in the case of Chromera velia; Portman et al., 2014). In T. gondii, the cytoskeletal apical complex includes three rings (the polar rings), a pair of short, straight microtubules (the intraconoid microtubules), and the cone-shaped conoid, a left-handed spiral of 14 fibers made from a nontubular tubulin polymer (Figure 1). Unlike typical microtubules, conoid fibers are not closed tubes and contain only approximately nine protofilaments (Hu et al., 2002b). How are the structural changes in the conoid that occurred along the evolutionary path connecting Toxoplasma with the chromerids and perkinsids related to the transition from a free-living to a parasitic lifestyle? The answer to that question seems likely to provide important insights into the functional significance of apical complex components in the pathogenesis of toxoplasmosis and thus provide useful guides to developing effective antiparasitic agents.
FIGURE 1:
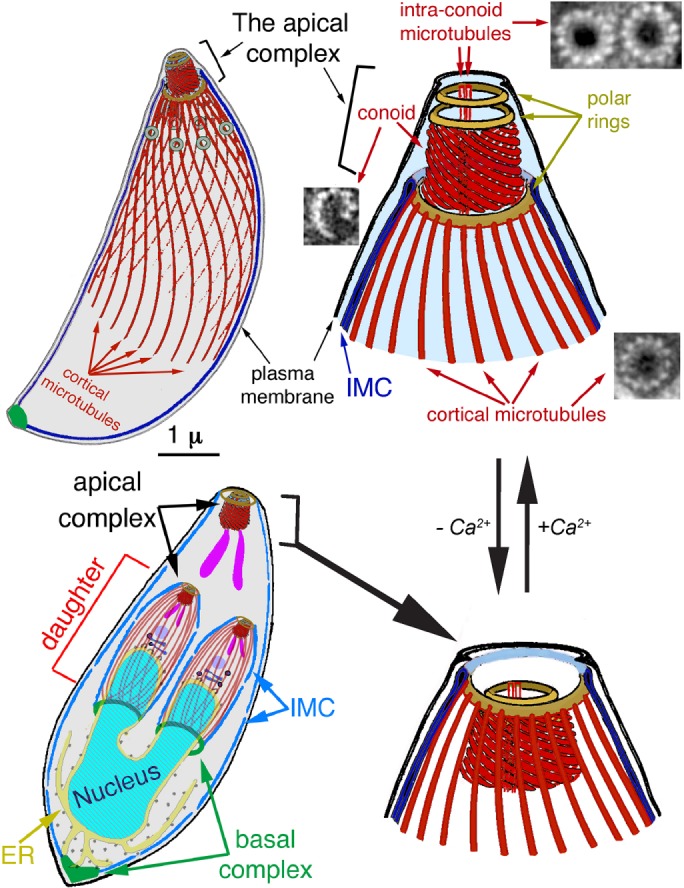
Diagram of the T. gondii cytoskeleton. From Liu et al. (2016). The 22 cortical microtubules, 2 intraconoid microtubules, and 14 conoid fibers, which are nontubular tubulin polymers, are shown in red. EM images of each of those polymers are also shown in cross section (Hu et al., 2002b). IMC, inner membrane complex. Bottom left, a replicating parasite, with daughter parasites being built inside the mother. The cortical microtubules of the adult are omitted for clarity. At this stage of daughter formation, most of the membrane-bound organelles have been produced or duplicated and partitioned into daughters, except for the mitochondrion (Nishi et al., 2008), although only the Golgi stack (dark blue), endoplasmic reticulum (yellow), apicoplast (a plastid-like organelle; light purple), and rhoptries (one of the specialized organelles for invasion; purple) are shown here. In intracellular parasites, the conoid is normally retracted, as shown in the bottom diagrams. An increase in cytoplasmic [Ca2+], which normally accompanies egress from the host cell and reinvasion, triggers extension of the conoid through the apical polar ring and a change in pitch of the conoid fibers, changing the conoid from a truncated cone into a straight-walled cylinder, as in the top diagrams.
The tubulin dimers that form the conoid fibers are used elsewhere in the same cell to make canonical microtubules, suggesting that the special arrangement of tubulin in the conoid fibers is dictated by nontubulin proteins. Previously, we partially purified the apical complex of T. gondii and identified its protein components (Hu et al., 2006). One of the proteins highly enriched in the apical complex fraction is TgDCX, which contains domains belonging to two protein families (P25-α and DCX) that are ubiquitous among metazoans. All sequenced apicomplexan genomes (Nagayasu et al., 2006; Orosz, 2009), as well as the genomes of chromerids and perkinsids (Orosz, 2016), have orthologues of TgDCX, recognizable by having DCX and P25-α domains together in the same molecule. Outside this group, this domain arrangement is found only in an early-diverging metazoan (Trichoplax adhaerens, phylum Placozoa; Orosz, 2009).
Proteins containing either the ∼40–amino acid (aa) P25-α domain or the ∼70-aa DCX domain are generally involved in interactions with microtubules. In vertebrates, proteins containing either of these domains are restricted to brain tissue (Gleeson et al., 1999; Liliom et al., 1999). Antibodies against doublecortin are widely used as a marker for identifying neuronal lineage cells. In humans, mutations in the X-linked doublecortin gene, which codes for a protein containing tandem DCX domains, disturb neuronal migration to the cortex in the developing CNS, resulting in double-cortex syndrome in females and the more severe X-linked lissencephaly in males (des Portes et al., 1998).
Doublecortin, by binding to the groove between protofilaments in microtubules, is believed to play a role in preferentially stabilizing particular microtubule structures (Moores et al., 2004; Fourniol et al., 2010). P25-α–domain proteins also modulate the structure of polymers formed from tubulin (Hlavanda et al., 2002). Although there has been significant progress in unraveling how these proteins might function, there are enormous gaps in our understanding. For instance, whereas the binding site on microtubules for one of the DCX domains in human doublecortin has been convincingly determined by cryo–electron microscopy (cryoEM), three-fourths of the doublecortin molecule, including the second DCX domain, is missing from the three-dimensional (3D) reconstruction (Fourniol et al., 2010). However, the entire doublecortin molecule is highly conserved, suggesting that the missing DCX domain does have an important function and implying that our understanding of how doublecortin interacts with microtubules is seriously incomplete. The cryoEM reconstruction shows a DCX domain snugly embedded in 12 of the 13 valleys between protofilaments, absent only at the seam where the microtubule lattice adopts the A rather than the B form. That binding pattern implies a preference for straight protofilaments, suggesting a rationale for doublecortin’s ability to nucleate 13 protofilament microtubules (which have straight protofilaments) in preference to 14-protofilament or other forms, in which the protofilaments have a twist. On the other hand, observations by light microscopy of binding to microtubules in vitro suggest that doublecortin binds more strongly to curved regions of microtubules, particularly the splayed protofilaments at the end of the polymer (Bechstedt and Brouhard, 2012; Bechstedt et al., 2014). With either mechanism, there is no satisfactory explanation for why either DCX or P25-α proteins would be necessary for microtubule formation/stabilization in neuronal cells but not in any other cell type. Also unclear is the connection between the putative microtubule-modifying actions of doublecortin and the defects in neuronal migration that occur when it is mutated. Further information about the function of DCX domains, particularly in a nonneuronal context, would be welcome.
Given the evidence that P25-α and DCX domain proteins can influence the structure of tubulin polymers, a protein such as TgDCX that contains one of each domain is thus a prime candidate for specifying the arrangement of tubulin dimers in the conoid fibers, an arrangement that differs radically from canonical microtubules and all other known tubulin polymers. Here we show that loss of TgDCX radically disrupts the structure of the conoid, with severe consequences for host-cell invasion and parasite growth.
RESULTS
TgDCX gene structure and cloning
TgDCX was initially identified in the apical complex as one of ∼170 proteins in a comparative proteomics analysis of conoid-enriched and conoid-depleted fractions of T. gondii (Hu et al., 2006). The TgDCX gene was cloned from a T. gondii RH strain cDNA library. The 5′ and 3′ cDNA ends were determined by rapid amplification of cDNA ends (RACE) experiments. Five (5′RACE) or four (3′RACE) clones were analyzed. Although there was clone-to-clone variation, the differences were small, <20 base pairs at each end. The 5′ end was mapped at −532 to −513 base pairs from the putative translational start site of TgDCX, and the 3′ end was mapped at 679–685 base pairs from the last base of the stop codon. The transcript from T. gondii is predicted to code for a 256-aa protein of 29.2 kDa. Comparison of gDNA and cDNA sequences indicated that there are six exons in this gene in T. gondii (Figure 2A), and gene models for most of the apicomplexans also predict six exons. The estimated molecular weights of TgDCX orthologues range from 19.1 kDa in Trichoplax to 33.3 kDa in Cryptosporidium. There is a well-conserved ∼80-aa DCX domain (a brain-specific, microtubule-interacting domain; Gleeson et al., 1999), part of the ubiquitin superfamily (Kim et al., 2003), near the C-terminus, which makes up slightly less than one-third of the entire protein length in TgDCX. Another well-conserved region is present in the middle of TgDCX, rich in charged residues, with similarity to a portion of TPPP/P25-α, a brain-specific, tubulin polymerization–promoting protein (Liliom et al., 1999; Hlavanda et al., 2002). The amino-terminal part is the least conserved.
FIGURE 2:

TgDCX gene, protein, and localization in T. gondii. (A) Arrangement of TgDCX (TGGT1_256030; www.toxodb.org) on Toxoplasma strain TGGT1 chromosome VIIb (top), structure of the mRNA (middle), and protein domain organization (bottom). Noncoding regions of the mRNA are indicated by the white boxes. Coding sequence is in yellow. The arrow indicates an alternative start codon. The stop codon is marked by an asterisk. Regions of the protein with significant homology to known eukaryotic proteins are marked by the purple boxes. The arrow indicates the alternative translation initiation site 15 aa residues from the first methionine. Numbers indicate aa residues counting from the first methionine. (B) Western blot with rabbit antibody raised against bacterially expressed recombinant TgDCX. Lane 1 (T.g.), whole-cell lysate of RH parasites, 10 µg of protein, 7 × 106 cells. Lane 2 (HFF), 10 µg of protein from whole-cell lysate of uninfected host cells (HFFs). (C) Images of a transgenic RH line expressing EGFP-β3-tubulin (green) and stained with rabbit anti-TgDCX antibody (red). Top, intracellular parasites fixed and permeabilized with methanol and briefly treated with 10 mM sodium deoxycholate. With this protocol or using nonionic detergent permeabilization, the developing daughter conoids are labeled with anti-TgDCX (yellow arrows), but antibody does not penetrate the adult conoid (magenta arrows). Anti-GFP antibody behaves similarly; in parasites expressing TgDCX-EGFP, daughter conoids can be stained with a GFP antibody, but adult conoids are not labeled (unpublished data), even though the adult conoids are brightly fluorescent from the DCX-EGFP they contain, as in the top left. Bottom, using longer deoxycholate treatment to permeabilize/extract extracellular parasites allows the antibody to penetrate the adult conoid (magenta arrow), but the architecture of the parasite is mostly disrupted. (D) Three EM images of deoxycholate-extracted RH parasites stained with anti-TgDCX antibody and 1.4-nm-gold secondary antibody and silver enhanced. The silver/gold deposits are confined to the conoid. Cortical microtubules are not labeled by the antibody. Middle, the conoid is partially uncoiled, revealing individual conoid fibers with attached gold/silver deposits.
Localization of TgDCX in T. gondii by antibody staining
For immunochemical localization of TgDCX in T. gondii, an anti-TgDCX antibody was produced in rabbits by immunizing them with recombinant TgDCX protein purified from bacteria. Two bands of unequal intensity were detected by Western blotting analysis of T. gondii whole-cell lysate using this rabbit antibody (Figure 2B). The estimated molecular masses of the two bands were 30.3 and 28.3 kDa, close to the theoretical molecular masses calculated based on the two different translation initiation sites (29.2 and 27.5 kDa). Immunofluorescence staining of T. gondii showed localization at only the daughter and adult parasite apical complex (conoid) (Figure 2C). Immuno-EM labeling of detergent extracted extracellular parasites with rabbit anti-TgDCX and 1.4-nm gold particles with silver enhancement shows TgDCX only in the tubulin-containing conoid fibers (Figure 2D). No other structures are labeled. The labeling suggests that TgDCX is distributed all along the conoid fibers and not, for instance, confined to one end, as was suggested for human doublecortin binding to microtubules in vitro (Bechstedt and Brouhard, 2012; Bechstedt et al., 2014).
Localization of fluorescent protein–tagged TgDCX in T. gondii by live-cell imaging
Stable transformants of T. gondii were created by homologous recombination that expressed FP-tagged TgDCX (FP refers to any of the fluorescent proteins; e.g., enhanced green fluorescent protein [EGFP], mCherryFP, mNeonGreenFP) as a replacement for the endogenous gene under control of the native promoter (TgDCX knock-in parasites). Fluorescence from FP-tagged TgDCX in adult parasites and developing daughters of the homologous recombinant lines is restricted to the conoid (Figure 3). Homologous recombinants expressing any of mCherryFP-TgDCX, TgDCX-mCherryFP, or TgDCX-mNeonGreenFP all showed the same localization pattern (i.e., conoid only), and their growth rate was indistinguishable from that of the parental line (RHΔku80Δhx).
FIGURE 3:

Creation of a knock-in parasite line in which TgDCX was replaced with mCherryFP-TgDCX by homologous recombination. (A) Fluorescence and DIC images of mCherryFP-TgDCX knock-in parasites. The field of view includes one vacuole containing four parasites, each with two forming daughters, and another vacuole with one parasite with no daughter. The mCherryFP fluorescence is located in a single spot at the apical end of both adult (arrow) and developing daughter (arrowheads) parasites. For reference, a cartoon of a parasite with two daughters is included in the inset (see also Figure 1). One parasite of the four-parasite vacuole is enclosed by the dotted white line in the fluorescence and overlay panels. For clarity, the other four parasites are outlined in the overlay channel only (yellow, green, magenta, and cyan dotted lines). (B) Map of a TgDCX knock-in plasmid. Sandwiched between two LoxP sites are a selection marker (HXGPRT expression cassette), the coding sequence for mCherryFP-tagged TgDCX, and the 3′-UTR from the T. gondii GRA2 gene. Flanking the LoxP sites are 3′ and 5′ sequences, ∼900 base pairs each, copied from T. gondii genomic DNA that direct integration of the LoxP sandwich into the genome by homologous integration at the TgDCX locus, replacing the entire TgDCX coding region, exons, and introns. The plasmid is linearized by NotI cutting before electroporation into a Δhxgprt line of T. gondii (e.g., RHΔhx or RHΔku80Δhx). The EGFP expression cassette allows FACS or manual selection against nonhomologous recombinants. Integration at the TgDCX locus by homologous recombination with double crossover removes the region of the plasmid not framed by the 5′ and 3′ homology regions, including the EGFP expression cassette. After homologous recombination, expressing Cre recombinase in knock-in parasites deletes TgDCX and HXGPRT coding sequences from the genome (Figure 5).
TgDCX appears very early in daughter development
To determine the location and timing of TgDCX incorporation into developing daughters, we used structured illumination microscopy (SIM) to visualize FP-tagged TgDCX over the course of daughter development. Earlier work with parasites expressing FP-tagged tubulin showed that the initiation of the nascent daughter apical complex is marked by the appearance of a bright spot of tubulin distinct from the tubulin located in the spindle pole/spindle (Hu, 2002, 2008; Nishi et al., 2008). With the advantage of SIM, that spot of tubulin can now be resolved into a five-petaled flower-like arrangement (Figure 4), as previously observed (Liu et al., 2016). Coexpressing FP-tagged tubulin in the mCherryFP-TgDCX knock-in line reveals that in early daughter development, a spot of TgDCX appears in the nascent daughter apical cytoskeleton, at the center of the five-petaled flower (Figure 4), soon after tubulin (10–15 min after; the complete parasite cell cycle extends over ∼8 h).
FIGURE 4:

SIM images of mCherryFP-TgDCX (red) and NeonGreenFP-β-tubulin (green) in mCherryFP-TgDCX knock-in parasites. (A) A Z-projection of a 3D stack showing two parasites at the initiation of daughter formation. A 0.5-μm-thick subregion of the stack containing one nascent daughter (dashed cyan border) has been cut out and shown enlarged to the right. The nascent daughters appear as a central bright spot of tubulin with five surrounding lobes, a “five-petaled flower,” which will develop into the daughter apical cytoskeleton. A trace of TgDCX, marking the beginning of daughter conoid formation, is seen just beneath the spot of tubulin. (B) Two parasites with developing daughters at a slightly later stage (roughly 5–10 min) of daughter formation. Microtubules of the spindle are now visible (white arrows). Right, enlargements of the regions bordered by the dashed cyan and magenta borders. The cyan-bordered images are from a 1.6-μm-thick subregion that includes the five-petaled flowers superimposed on the underlying spindle and spindle poles. One daughter is viewed along the future apical basal axis (gray circle), and the other is viewed from the side (gray oval). A thinner subregion (0.125 μm, a single optical section) including only the five-petaled flower of one daughter is shown in the magenta-bordered images. The flower is now larger, and the amount of mCherryFP-TgDCX is increased compared with A. Note that the red channel of the images in the enlargements of A and B have been strongly contrast enhanced to make the small spot of mCherryFP-TgDCX visible. Its true intensity at this stage of daughter formation is ∼1% of the adult mCherryFP-TgDCX spot, that is, not visible in this display without contrast enhancement. (C) Two parasites with much larger daughters, roughly 1 h after B (estimated based on Hu et al., 2002a). The cortical microtubules in the daughters have grown to about one-half of their adult length, and the daughter conoids contain almost the full adult content of TgDCX. (D) Cartoon of a parasite showing the relationship of the developing daughters (green blobs) to the nucleus (purple) and mitotic spindle (cyan rods) during the closed-nuclear division of Toxoplasma. The nucleus appears in B as a dark region from which the diffuse cytoplasmic fluorescence is excluded.
Creation and phenotype of TgDCX-null mutant lines of T. gondii
A Cre-recombinase method described earlier (Heaslip et al., 2010, 2011; Liu et al., 2016) was used in conjunction with the three homologous recombinant (knock-in) lines described above to excise TgDCX from the genome. A population of slow-growing but viable parasites was obtained (TgDCX-knockout parasites), from which 10 clonal lines were established: two clones derived from the mCherryFP-TgDCX knock-in line, four from the TgDCX-mCherryFP knock-in, and four from the TgDCX-mNeonGreenFP knock-in. The loss of TgDCX protein was confirmed by the disappearance of the fluorescence due to FP-tagged TgDCX. The deletion of the TgDCX coding sequence from the genome was verified by Southern blotting (Figure 5). The growth of these TgDCX-null lines was much slower than that of the parental line. Under conditions in which the parental line showed a doubling time (Materials and Methods) of ∼7.5 h, the TgDCX-knockout line doubled in ∼17 h. However, with continued culture, some adaptation occurred, with the growth rate stabilizing after ∼4 wk of serial passage in host cells. In two independent clones of the TgDCX-knockout parasite line maintained in continuous culture for several months, the doubling time decreased to ∼13 h. In a plaque assay performed after ∼10 wk of continuous culture, the TgDCX-knockout parasite line formed approximately one-fourth as many plaques as the parental line, and the plaques were about one-third the size (Figure 6). Complementation of the null mutant with an ectopic copy of the TgDCX CDS tagged with EGFP and expressed under control of the Toxoplasma α-tubulin promoter restored the number of plaques to approximately the same as in the mCherryFP-TgDCX knock-in line (Figure 6 and Table 1).
FIGURE 5:
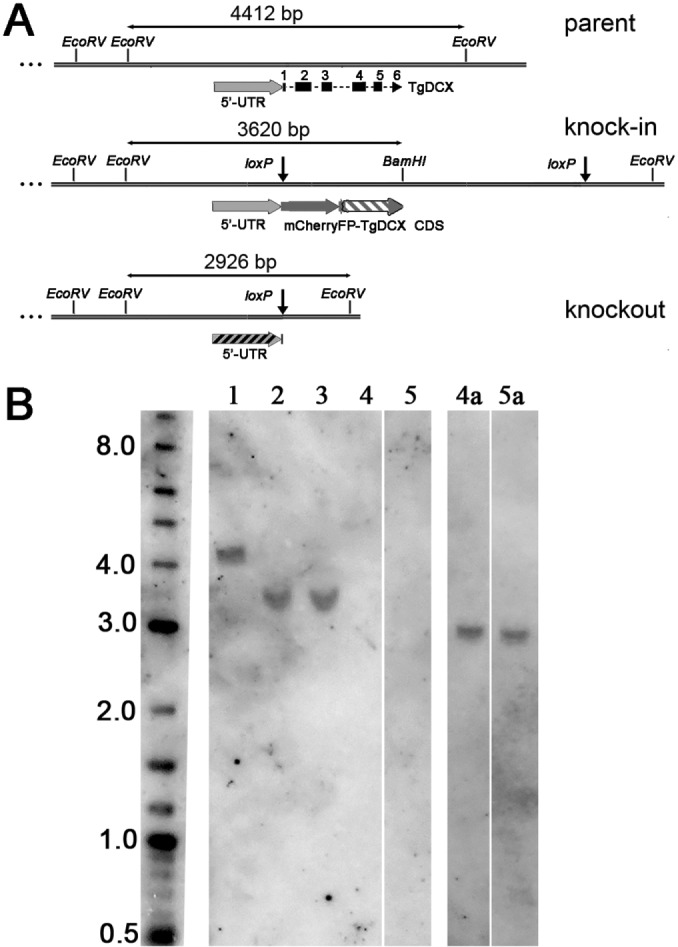
Southern blot with T. gondii genomic DNA digested with BamHI plus EcoRV. (A) Map of the TgDCX genomic region in parental (RHΔku80Δhx), knock-in (FP-TgDCX or TgDCX-FP), and knockout (TgDCX null) parasites. The sizes of the BamHI-EcoRV or EcoRV-EcoRV fragments containing the hybridization targets are indicated. Sequence regions included in the probes are indicated by the striped regions; exons 2–5 probe, gray-white stripes in the knock-in map; 5′-UTR probe, gray-black stripes in the knockout map. The 5′-UTR region is identical in the three parasite genomes. (B) Southern blot. Left to right, size marker (length in kilo base pairs); lanes 1–5: BamHI-EcoRV–digested genomic DNA hybridized with a probe against exons 2–5 of TgDCX CDS. Lane 1, RHΔku80Δhx parasites, expected size, 4412 base pairs; lanes 2 and 3, mCherryFP-TgDCX and TgDCX-NeonGreenFP knock-in parasites, expected size, 3620 base pairs; lanes 4 and 5, clones of knockout parasites derived from mCherryFP-TgDCX and TgDCX-NeonGreenFP knock-in parasites, respectively, no band expected. Lanes 4a and 5a are lanes 4 and 5, respectively, of the original blot after it was stripped and rehybridized with a probe against the 5′-UTR region of the TgDCX gene; expected size, 2926 base pairs.
FIGURE 6:

Loss of TgDCX dramatically slows growth and hinders host-cell invasion. Left, plaque assay (Materials and Methods). Top to bottom, wild type (WT), parental (RHΔku80Δhx); k-in, mCherryFP-TgDCX knock-in; ΔDCX, knockout parasites (TgDCX null); comp, knockout parasites complemented with TgDCX-EGFP expressed under control of the T. gondii α-tubulin promoter. The TgDCX-knockout parasites form many fewer and much smaller plaques than the WT, knock-in, or complemented knockout lines. Right, host-cell invasion assay (Materials and Methods). The red channel shows parasites outside the host cell, that is, those that adhered to the host cells but failed to invade. Next the red channel has been overlaid on a green channel that marks all parasites, inside as well as outside the host cell. In this overlay, parasites that invaded the host cell are green, and parasites that failed to invade are yellow. Only a small fraction of the TgDCX-knockout parasites are able to invade. Red and green fluorescence channels are both overlaid on a DIC image of the same field in the last column.
TABLE 1:
Host cell lysis and invasion by T. gondii lines.
| Plaque assay | Invasion assay | ||||
|---|---|---|---|---|---|
| Number of plaques | Plaque size (mm2) | Cytolytic efficiency (%) | Number invaded | Percentage of parental | |
| RHΔku80Δhx | 61 ± 7 | 21 ± 1 | 100 | 2435 ± 84 | 100 |
| mCherryFP-DCX | 58 ± 3 | 20 ± 0.3 | 92 | 1199 ± 85 | 49 |
| ΔDCX | 18 ± 1 | 8 ± 1 | 12 | 628 ± 43 | 26 |
| ΔDCX plus EGFP-DCX | 52 ± 6 | 14 ± 0.4 | 55 | 1244 ± 79 | 51 |
Average number and size of plaques (±SE) formed in three replicate experiments. Cytolytic efficiency is defined as the ratio of the total area lysed by a line to the total area lysed by the parental line, expressed as a percentage.
To further characterize the growth defect seen in TgDCX-knockout parasites, we assayed host-cell invasion (Carey et al., 2004) by the parental, knock-in, knockout, and “complemented” knockout parasite lines (Figure 6 and Table 1). In this assay, monolayer cultures of host cells are exposed to a suspension of parasites for a short time and then rinsed with fresh medium, fixed, and labeled with an antibody against a Toxoplasma surface antigen and secondary antibody coupled to a red fluorophore. Parasites that had adhered to host cells but had not yet invaded are accessible to the antibody and therefore labeled with red fluorescence. Parasites that had invaded into host cells are not accessible to the antibody and so are not stained. The host cells are then permeabilized with detergent, and the antibody labeling is repeated, this time using a secondary antibody conjugated with a green fluorophore. Thus, when the assay is complete, intracellular parasites are green, but those parasites on the outside of the host cell are both red and green (Figure 6), enabling easy discrimination between completed invasion events and mere adhesion to the host cell surface.
Replacing the endogenous TgDCX with mCherryFP-TgDCX (TgDCX knock-in parasites) reduced host-cell invasion in our assay by approximately twofold. Complete loss of TgDCX reduced invasion by fourfold compared with wild type. Adding back a copy of the TgDCX CDS tagged with EGFP randomly inserted into the genome restored host-cell invasion to the same level as in the mCherryFP-TgDCX knock-in parasites. Note that whereas the native TgDCX promoter drives expression in the mCherryFP-TgDCX knock-in parasites, in the TgDCX-EGFP complemented knockout parasite, expression is driven by a mismatched promoter (T. gondii α-tubulin gene).
Loss of TgDCX is associated with a loss of tubulin in the apical complex
Examination of TgDCX-knockout parasites in which all tubulin-containing structures were made visible by transforming with a plasmid driving expression of NeonGreenFP-β1-tubulin revealed that the apical complex region of the TgDCX null parasites was abnormal, with a much reduced or sometimes complete absence of the normally observed bright apical spot of fluorescence due to tubulin in the conoid. Figure 7 shows SIM images of NeonGreenFP-β1-tubulin fluorescence in wild-type and knockout parasites. Fluorescence arising from the cortical microtubules in the adult and daughter knockout parasites appeared normal, as did the mitotic spindle, spindle pole, and centrioles. The only noticeable difference is in the conoid, which is clearly resolved in slices through the apical region of parasites whose long axis happened to be oriented perpendicular to the optical axis. In single slices through the middle of the conoid region of appropriately oriented parasites, the walls of the conoid appear as two short bars of enhanced fluorescence extending down ∼0.2 μm from the arch of cortical microtubules, as seen in the enlargements on the right side of Figure 7. These bars are much less prominent or absent in images from TgDCX-knockout parasites.
FIGURE 7:

Tubulin is lost from the conoid in TgDCX-knockout parasites. SIM images of parental wild-type (top) and TgDCX-null (bottom) parasites expressing mNeonGreenFP-β1-tubulin. Left, Z-projections of the entire 3D stacks of images. Middle, subregions from single slices that happened to pass through the middle of a conoid region were cut out and superimposed on the Z-projection of the entire stack. Right, 4× magnified views of the conoid regions of three wild-type (top) and two knockout (bottom) parasites from those single slices. In these single optical slices through the middle of a conoid, the walls appear as two short bars of brighter fluorescence extending down from the arch of the cortical microtubules. In the knockout parasites, the bars are much less prominent or absent. The red arrowheads point at the walls of the conoid in one of the wild-type parasites (1) and toward the same location in one of the knockout parasites (a). Scale bars, 5 μm (left and middle), 1 μm (right).
Of interest, at least two other proteins normally found in the apical complex, TgAKMT (Heaslip et al., 2011) and TgCAP2 (Leveque et al., 2016), are not obviously affected by the loss of TgDCX, as judged by immunofluorescence localization in knockout parasites by wide-field epifluorescence (Figure 8). Extension of the conoid/apical complex induced by calcium-ionophore treatment of the TgDCX-knockout parasites, as judged by phase-contrast light microscopy, was also indistinguishable from that observed in the parental wild-type parasites (Figure 8; parental, 88.4 ± 1.6% of treated parasites had extended conoids/apical complex; TgDCX knockout, 87.4 ± 1.4% had extended conoids/apical complex).
FIGURE 8:
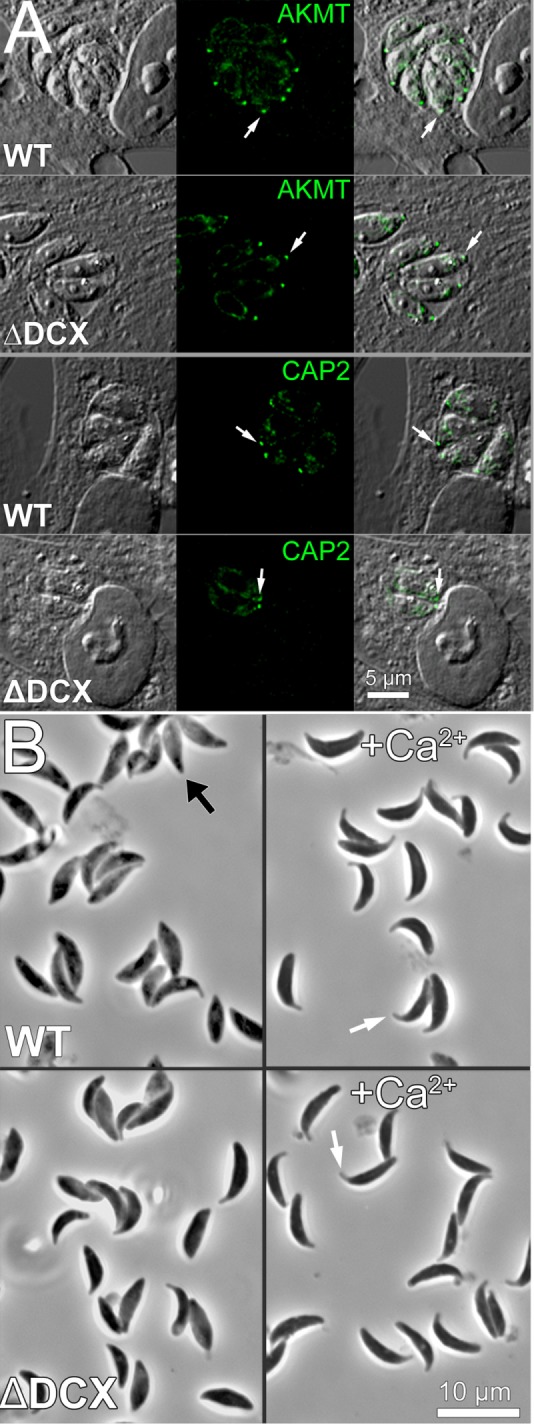
The localizations of some conoid proteins and conoid extension are unaffected by removal of TgDCX. (A) DIC (grayscale) and fluorescence (green) images showing immunolocalization of two conoid-associated proteins, TgAKMT and TgCAP2, in WT (parental line RHΔku80Δhx) and TgDCX-knockout parasites (ΔDCX). The fluorescent spot corresponding to one conoid is marked by the white arrow. Localization of both proteins to the conoid region is unaffected by loss of TgDCX. (B) Conoid/apical complex extension induced by treatment with Ca ionophore A23187. Top (WT), parental line RHΔku80Δhx; bottom (ΔDCX), TgDCX-knockout parasites. Left, no treatment; most parasites have not extended their conoid/apical complex, as indicated in one case by the black arrow. Right (+Ca2+), treated with 5 µM A23187. In essentially all parasites of both strains, the conoid/apical complex appears to be extended. Two examples are indicated by the white arrows.
Deletion of TgDCX causes severe structural defects in the conoid
Electron microscopy of negatively stained TgDCX-knockout parasites revealed gross structural abnormalities in the conoid. Figures 9 and 10 compare normal conoids from wild-type parasites with conoids from the TgDCX–knock-in line, two of the TgDCX-knockout lines, and the same two TgDCX-knockout lines after rescue by transfecting with a plasmid expressing TgDCX. The parasites were treated with calcium ionophore to induce conoid extension and briefly with mild detergent to permit visualization of the conoid by negative staining. The conoid of the mCherryFP-TgDCX–knock-in parasites, from which the knockout parasites were derived, is indistinguishable from wild type at this resolution, but the TgDCX knockout is abnormal.
FIGURE 9:

EM images of the conoid region of negatively stained T. gondii. All parasites in all rows, except for the one labeled WT retracted conoid (left, middle), have extended conoids. Top, WT, three examples of extended conoids from the parental line RHΔku80Δhx showing their consistent appearance. Middle, WT retracted conoid, from a parental parasite that had not been treated to induce conoid extension. The location of the conoid is indicated by the white brackets; ΔDCX-A1 and ΔDCX-B7, parasites from two different lines of TgDCX-knockout parasites, A1 and B7. Bottom, cherry-DCX kin, an mCherryFP-TgDCX knock-in parasite; A1 complement and B7 complement, the same two lines of knockout parasites after rescue by transformation with a plasmid driving expression of TgDCX-EGFP.
FIGURE 10:

Montage of EM images of the conoid region of 16 randomly chosen negatively stained TgDCX-knockout parasites. All parasites in all rows have extended conoids. Unlike wild-type parasites (Figure 9), the appearance of the conoid is variable. Compared to wild type, the TgDCX-knockout conoid is generally shorter and less rectangular, often more of a truncated cone than a rectangle after extension. The basket-weave stripe pattern arising from superposition of the conoid fibers is much weaker or sometimes absent and looks disordered.
When the conoid of wild-type parasites extends, it loses most of its conical taper and becomes an almost straight-walled cylinder, presenting a nearly rectangular profile, ∼450 nm wide and 320 nm tall in negatively stained samples. The wild-type conoid is also characterized by a prominent basket-weave stripe pattern, arising from superposition of the front and back sides of the spiral of 14 conoid fibers (Hu et al., 2002b). In TgDCX-knockout parasites, the overall shape of the conoid is variable (Figure 10) but generally shorter and less rectangular in profile than the wild type, often more of a truncated cone than a rectangle after extension, similar to the shape of retracted conoids of wild-type parasites. The basket-weave stripe pattern is weaker or sometimes absent, less regular, and less extensive in conoids from the knockout lines. Whereas the wild-type conoid looks much the same in every cell (Figure 9), the knockout conoid is quite variable in appearance. In some cases, the overall shape of the conoid in the knockout parasites resembles a partially extended conoid in normal parasites, but in detail they are not alike. Figure 9 shows that in wild-type parasites, even with a retracted conoid, the conoid fibers are still visible and appear well ordered, unlike the disheveled appearance of the knockout.
Figure 10 shows a montage of conoid images from randomly selected TgDCX-knockout parasites to illustrate the range of variation. Some are less disrupted than others, but in an extended survey of several hundred TgDCX-knockout parasites, we did not observe one that appeared normal. Nevertheless, some structure remains, and in some cases, a stripe pattern is observed that suggests the presence of some remnant conoid fibers. We interpret this variability to mean that the DCX-less conoid is metastable, which hints that there may be other, as-yet-unidentified proteins that cooperate with TgDCX to fully stabilize it.
The structural defects in the conoid are reversed by expression of TgDCX protein
To confirm that the observed structural defect in the conoid is indeed due to the loss of DCX protein, we transformed TgDCX-knockout parasites with a plasmid containing the TgDCX CDS fused to EGFP. By fluorescence microscopy, the TgDCX-EGFP is localized to a bright spot at the apical end of these “complemented knockout” parasites (unpublished data). Figure 9 shows that the structural defect in the conoid of the TgDCX-knockout parasites is completely reversed by expression of the TgDCX CDS. It is important to note that this rescue must be due solely to TgDCX protein. The rescue plasmid contains the protein coding portions only of the TgDCX gene, not the untranslated regions (UTRs). The 5′-UTR (promoter) for the rescue plasmid was taken from the T. gondii α-tubulin gene and the 3′-UTR from the T. gondii dihydrofolate reductase gene. Thus the complementation of the conoid structural defect of knockout parasites, as well as reversal of their defects in growth and invasion (Table 1 and Figure 6), is achieved solely by making TgDCX protein available.
The several tubulin-based macromolecular assemblies in T. gondii can be independently targeted for disassembly
The conoid fibers contain tubulin dimers that are essentially identical in primary amino acid sequence to the tubulin dimers in the cortical microtubules, intraconoid microtubules, mitotic spindle, and other tubulin-containing structures in the cell, and all of these structures are assembled contemporaneously during daughter formation. Tubulin expressed from extra copies of the α1-tubulin gene or any of the three β-tubulin isoforms (Hu et al., 2004b), even human tubulin expressed in T. gondii (Nagayasu et al., 2006), is incorporated promiscuously into all tubulin-containing structures, and yet TgDCX is normally found only on the conoid fibers, and removal of TgDCX affects only the conoid fibers, not any other tubulin-containing structure. This surprising independence extends in both directions. Figure 11 shows images from a recently described (Liu et al., 2016) parasite line lacking three microtubule-associated proteins (TLAP2, SPM1, TLAP3). In this triple-knockout line, the cortical and intraconoid microtubules are absent in the Triton X-100–extracted parasites, but the conoid fibers are completely normal.
FIGURE 11:

EM images of the conoid region from a wild-type T. gondii (WT) and two Δtlap2Δspm1Δtlap3 parasites (tko), which lack three microtubule-associated proteins (Liu et al., 2016). The 22 cortical (black arrows) and 2 intraconoid (white arrows) microtubules are missing in the triple-knockout parasites, but the conoid fibers are intact.
The exquisite precision revealed by the differential localization and independent effects of these tubulin-binding proteins is remarkable. As seen in Figure 4, the assembly of the cortical microtubules, conoid fibers, intraconoid microtubules, and spindle microtubules overlap in time during daughter parasite formation and occur within ∼2 μm of each other, a distance over which the concentrations of proteins of this size would be equilibrated by diffusion in ≪1 s. This observation immediately raises the question of how TgDCX is normally targeted exclusively to conoid fibers, even though numerous other potential binding sites are available. Reciprocally, it is known that several other tubulin/microtubule binding proteins are simultaneously being targeted to their own specific subset of the available tubulin polymers but never to the conoid fibers (Liu et al., 2016).
The exclusion of TgDCX from all nonconoidal tubulin sites is not due to inaccessibility. In parasite lines overexpressing FP-tagged TgDCX (i.e., from randomly integrated extra copies of the gene), fluorescence is observed in other locations (cortical microtubules, spindle/spindle poles) in addition to the conoid during early daughter development, but this extraconoidal fluorescence disappears as the daughters mature (unpublished data). The localization to sites other than the conoid is an artifact of overexpression, but nevertheless the artifact is very informative because it proves that during daughter development, TgDCX does have access to all of the tubulin polymers that are being assembled contemporaneously, not just to conoid fibers.
DISCUSSION
The common morphological feature of all members of the phylum Apicomplexa is the presence of an apical complex, an assemblage of cytoskeletal elements and secretory organelles at one end of the cell (Figure 1). Although the exact structures of the apical complex vary among phylum members, all are believed to serve important functions in both invasion of host cells and replication of the parasites. Many members of the phylum have in their apical complex a distinctive organelle, the conoid, an ∼380-nm-diameter motile organelle, consisting in T. gondii of 14 spirally wound conoid fibers, which are nonmicrotubule polymers built from tubulin subunits. It has been hypothesized that the apical complex is structurally related to the flagellum, as a number of flagellar components, such as dynein light chain and SAS6-like protein, are found in the apical complex in T. gondii (Hu et al., 2006; de Leon et al., 2013). Further, organisms in the sister clades that possess a pseudoconoid invariably have flagella rooted close to the apical complex (Portman and Slapeta, 2014). T. gondii also forms a flagellum during its sexual stage when the parasite differentiates into microgametes in the epithelial cells of the small intestine of a cat. However, the structural relationship between the apical complex and the flagellum is unknown in T. gondii because it is difficult to obtain microgametes. It is also not known whether the apical complex and the flagella in chromerids and perkinsids share common structural origins or are simply unrelated structures that happen to be located in the vicinity of each other.
The conoids of T. gondii and its close relatives in the Apicomplexa differ from those of more distant relatives not only in being closed cones rather than half-closed cones or flat ribbons, but also in the size of the organelle and consequent curvature of the fibers composing it. In T. gondii, the conoid fibers form the wall of the ∼380-nm-diameter conoid. Therefore the fibers are bent into an arc with a radius of curvature of ∼280 nm. The rigidity of microtubules in cells is increased to some extent by binding of associated proteins (Kurachi et al., 1995), but even when completely bare, ordinary microtubules break when bent into arcs with radii <∼500 nm (Amos and Amos, 1991). Thus a transition from the large conoids of other alveolates to the compact conoid of apicomplexans may require special modifications of the conoid fibers to decrease their flexural rigidity. Among those modifications is presumably the change from a tube (ordinary microtubules) to a convex ribbon (T. gondii conoid fibers). However, such a structural modification is not possible using tubulin alone, because the tubulin dimers in a convex ribbon cannot all be in equivalent local environments. Whereas it is possible in principle to make a cylindrical (helical) polymer of protofilaments of tubulin dimers in which every dimer is equivalent, this is not possible with a convex ribbon. The energy to support this sort of nonequivalence among identical building blocks must be supplied in some form, most commonly in the form of the binding energy of nontubulin-associated proteins. If that source of extra energy were to be removed, for instance, by removal of the associated protein, the polymer with nonequivalent subunits would become less stable. This seems a reasonable explanation for the destabilization of the conoid fibers and loss of tubulin from the conoid in the TgDCX-knockout parasites. This explanation predicts that tubulin polymerized in the presence of TgDCX would assemble into some structure other than a canonical microtubule, perhaps a curved convex ribbon. Unfortunately it has not yet been possible to test this prediction because bacterially expressed TgDCX remains in solution only under denaturing conditions, but planned future experiments using an Sf9 expression system may provide the opportunity.
Does T. gondii make the 9- to10-protofilament conoid fibers in the absence of the TgDCX protein? Albeit unstable, are they perhaps present in reduced number or transiently during parasite replication when the apical complex of the daughters is formed? At first thought, one might hope to answer this question by looking at sections of fixed, embedded, DCX-knockout parasites, and in fact we did that experiment and did not see convincing 9- to 10-protofilament fibers, but a consideration of what is required to see that definitive structure strongly suggests that such images will always be inconclusive. The conoid fibers can be seen only over a very narrow range of orientations and plane of sections. For that reason, conoid fibers with protofilaments are not seen in the overwhelming majority of images even for wild-type conoids. Thus failing to see conoid fiber protofilaments in sections of a somewhat disordered conoid is inconclusive with respect to whether they actually exist.
Human doublecortin, which has two DCX domains instead of one DCX plus one P25-α as found in TgDCX, binds to microtubules in vitro with 8-nm periodicity, that is, one doublecortin for each tubulin dimer. However, in the cryoEM reconstruction of microtubules with bound doublecortin, three-fourths of the doublecortin molecule is invisible. What is seen is only one DCX domain (although that is seen beautifully) binding to four tubulin dimers in the valley between protofilaments, the other DCX domain presumably lost because it has no consistent localization under those conditions (Fourniol et al., 2010). This, however, is at odds with the observation that doublecortin readily bundles microtubules (as does TgDCX when expressed in mammalian cells; unpublished data): that is, both DCX domains of doublecortin are capable of forming stable bonds with tubulin. As mentioned in the cryoEM structure report (Fourniol et al., 2010), it remains possible that under some conditions, the two DCX domains would bind alternately along a microtubule, yielding a stoichiometry of one doublecortin per two tubulin dimers. In this connection, it is interesting to note that the linker between the two DCX domains in human doublecortin is 38 aa, long enough to allow both domains to bind simultaneously, which is quite similar in length to the distance between the single DCX domain and the P25-α domain in TgDCX (45 aa).
Orthologues of TgDCX are present in all sequenced apicomplexan genomes, even in those lacking a recognizable conoid. Its ubiquitous presence suggests that the acquisition of the DCX gene occurred early in apicomplexan evolution. The continued existence of the TgDCX orthologues in apicomplexans that lack distinctive conoid structures, such as Cryptosporidium, poses interesting questions. Part of the answer may lie in differences between life-cycle stages. For instance, although it seems that haematozoea species (haemosporidians) have lost the conoid from their apical complexes in their merozoite stage, the ookinete and sporozooite stages in some species retain conoids (Desser, 1970; Patra and Vinetz, 2012). This suggests that it would be worth looking carefully in all of those species for some remnant or radically modified form of conoid. Localization studies of DCX domain proteins in those seemingly conoid-less species, particularly in their ookinete and sporozooite stages, could provide valuable insight into this interesting question.
Deletion of TgDCX significantly decreases the rate of host-cell invasion. In our assay, in which parasites are presented with host cell targets for a limited time (1 h), four times as many wild-type parasites complete invasion as do TgDCX-knockout parasites. In the less exacting setting of a plaque assay or in continuous culture, where the time allowed for invasion is essentially unlimited, the difference between wild-type and knockout parasites is reduced, resulting in an overall difference in apparent growth rate of only twofold. However, both of these situations are artificial and not representative of the situation faced by the parasite attempting to survive and spread in an intact animal with a functioning immune system. In that situation, the parasite is protected when enclosed in its intracellular vacuole but highly vulnerable when extracellular. Thus there must be enormous selection pressure acting to make reinvasion after egress as fast as possible.
Our experiments do not provide any insight into exactly how TgDCX facilitates host-cell invasion. It might be that its stabilization of the conoid fibers provides some beneficial mechanical advantage for host-cell penetration or, alternatively, render the intraconoid passageway a better conduit for secretion from the parasite of the effector molecules known to be important during invasion. Further work is required to distinguish between these possibilities, but the knock-in, knockout, and complemented knockout parasite lines we have established provide the tools needed to make the distinction experimentally.
MATERIALS AND METHODS
Culture, harvest, and transformation of T. gondii
T. gondii tachyzoites were used in all experiments and grown in monolayers of human foreskin fibroblast (HFF) cells (Roos et al., 1994). Parasites were harvested from culture supernatant by centrifugation at 3600 × g for 1 min when the monolayer was ∼80% lysed. For purification, the parasite suspension was passed through a 3-μm Nuclepore filter (110612; Whatman), centrifuged, and resuspended in Dulbecco’s phosphate-buffered saline (DPBS) at ∼4 × 108/ml. Transfection of T. gondii tachyzoites was carried out as previously described (Heaslip et al., 2009) using 30–40 µg of plasmid DNA in “cytomix” buffer (120 mM KCl, 0.15 mM CaCl2, 10 mM KH2PO4/K2HPO4, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 2 mM ethylene glycol tetraacetic acid, 5 mM MgCl2, 2 mM K2ATP, 5 mM glutathione, pH 7.6).
Cloning of TgDCX
All PCR primers are listed in Table 2.
TABLE 2:
Oligonucleotides used in this study.
| Name | Sequence |
|---|---|
| AS1 | GCTAGTCGACTTACACAACGAAGAACTGGCTC |
| AS2 | GCTACTTAAGTTAGACAACGGGTTTTCGAGGTC |
| AS3 | GGGAAGGCATGGCCTCGTGA |
| AS4 | GGCGGATAGGGCGGAGGAAA |
| AS5 | GCTAAGATCTCACAACGAAGAACTGGCTCG |
| AS6 | ACTGCTTAAGCTACGCGCCTTCCTCTGCACCCATCTCG |
| AS7 | ACTGGAAGACTCTTAAGTCACTCGGCGCCTTCACCCTCTGCAGCTC |
| AS8 | GGGCAGCTTCTGTTTACTTAAGCTGTATGCTAGCGGATCTAAAAGGGAATTC |
| AS9 | TGACCCACCGGAACCAGTTCCACCAGACCCGGTACCTCCCTTGTACAGCTCGTCCA |
| AS10 | GGGCAGCTTCTGTTTACTTAAGCTACGCGCCTTCCTCTGC |
| AS11 | CACTTTCGTCGTAGTCGGATCCGAATTCTTACTTGTACAGCTCGTCCATGCC |
| AS12 | TCGCCCTTGCTCACCATTTTGGCATAACTTCGTATAATGT |
| AS13 | CCTCCACTAGAACCTCCCTTGTACAGCTCGTCC |
| AS14 | GATGCCGCACGCAGTCGACCCGCC |
| AS15 | TCGTCGTAGTCGGATCCTTACACAACGAAGAACTGGC |
| AS19 | GCTAGAATTCTTACACAACGAAGAACTGGCTC |
| AS20 | GTTCTCCAGAGGTACACAAGTGTTTCGCGCCATCAACAATG |
| AS21 | AGTACGCTTAAGTTACTTGTACAGCTCGTCCATGCCGAG |
| S1 | ACTTTGCCTCTAACGGAGCATTT |
| S2 | ATCGAGATCTACCCCCGGTACCTTGGGTATC |
| S3 | CACAACGAAGAACTGGCTCGGAAC |
| S5 | ATCGGCTAGCATGGCGACACGACAGGCAG |
| S6 | ACTGAGATCTAGAGAAATCGTCCACGTTCAGGGTGGC |
| S7 | ACTGGGATCCAGAGAAATTGTACACATTCAGGGAGGCC |
| S8 | GAATTCCCTTTTAGATCCGCTAGCATACAGCTTAAGTAAACAGAAGCTGCCC |
| S9 | GAATTCCCTTTTAGATCCGCTAGCAAAATGGTGAGCAAGGGC |
| S10 | GGTTCCGGTGGGTCAAGAGAAATCGTCCACG |
| S11 | GGACTAGTTCTAGAGGCGCCGGCGGTGTGAGCAAGGGCGAGGAGG |
| S12 | CGTTGGCTACACTCGCAAGCTGGACTT |
| S13 | CATTATACGAAGTTATGCCAAAATGGTGAGCAAGGGCGAG |
| S14 | TGGACGAGCTGTACAAGGGTGGGAGCTCTGGTG |
| S15 | GAAGTGGCGGATCTACTGCGTGCGGCATCC |
| S19 | ATCGGGATCCGATGGCGACACGACAGGCA |
| S21 | AACCTGAAGATCTGGAGTGAGCAAGGGCGAGGAG |
| SAS1 | GACCACGCGTATCGATGTCGACTTTTTTTTTTTTTTTA |
| SAS2 | GACCACGCGTATCGATGTCGACTTTTTTTTTTTTTTTC |
| SAS3 | GACCACGCGTATCGATGTCGACTTTTTTTTTTTTTTTG |
| SAS4 | GACCACGCGTATCGATGTCGAC |
5′RACE.
At the time TgDCX was cloned (Nagayasu et al., 2006), the various T. gondii genome annotations contained conflicting gene models for a hypothetical protein, varying in length from 131 to 1109 aa. A template cDNA was synthesized from T. gondii RH strain total RNA by SuperScriptII RT (Invitrogen, Carlsbad, CA) using reverse primer AS1 (all oligonucleotides are listed in Table 2), complementary to nucleotides 705–726 counted from the then putative translational start of the hypothetical protein, 750–771 from the start of the ultimately cloned TgDCX (all subsequent numbers refer to the current TgDCX gene model), followed by dA-tailing using dATP and terminal transferase. The product was subjected to three rounds of heminested PCR. For the first round, oligo(dT)-anchor primers (SAS1, SAS2, and SAS3), an anchor primer without oligo(dT) (SAS4), and a gene-specific primer AS2 (position 382–402) were used. For the second and third rounds, the same anchor primer (SAS4) was used in combination with either AS3 (for the second round, position 167–186) or AS4 (for the third round, position 82–101). The final PCR products were purified on a 1.0% agarose gel and then cloned into the vector pCR2.1 TOPO (Invitrogen). Five clones were sequenced and compared with the genomic DNA sequence obtained from a public database (www.toxodb.org).
3′RACE.
3′RACE-ready cDNA was synthesized using T. gondii total RNA and the oligo(dT) anchor primers (SAS1/SAS2/SAS3) by SuperScriptII RT. Three rounds of heminested PCR were performed using the synthesized cDNA, forward primers S1 (first round, position −194 to −172), S2 (second round, position 403–423), and then S3 (third round, position 745–768) in combination with the anchor primer (SAS4). The final PCR products were gel-purified, cloned, and then sequenced as for the 5′RACE products.
Plasmid construction
After construction, plasmids were used to transform chemically competent TOP10 cells by heat shock or electrocompetent DH5α cells (NEB C2989) by electroporation. Plasmid DNA was isolated by standard procedures, and the constructions were verified by DNA sequencing.
ptub-TgDCX-EGFP is a derivative of the plasmid ptub-H2b–yellow fluorescent protein (YFP; Hu et al., 2004a). Converting from ptub-H2b-YFP to ptub-TgDCX-EGFP entailed removing the H2b sequence between the NheI and BglII sites and replacing it with the coding region of TgDCX extending from the second methionine to the amino acid before the stop codon, obtained by reverse transcription PCR (RT-PCR) using T. gondii total RNA and primer pair S5/AS5. YFP was swapped with EGFP via the BglII and AflII restriction sites and a coding sequence of EGFP PCR amplified using primers S21 and AS21. In the final expressed product, TgDCX is coupled to the N-terminus of EGFP (lacking its initial methionine) via the three-residue aa sequence RSG. Another version of ptub-TgDCX-EGFP was also constructed, with amino acid substitution Y219H in the TgDCX CDS (numbered according to position in the full-length protein), corresponding to a doublecortin mutation giving rise to lissencephaly in humans (human Y125H). By light microscopy, the localization of these two forms of TgDCX-EGFP did not differ significantly from each other or from wild type. Both were effective in complementing the structural defects of the conoid in TgDCX-knockout parasites by EM analysis and in complementing its growth defect by plaque assay.
ptub-EGFP-TgtubB1 and ptub-EGFP-TgtubB3 are both based on the ptub vector backbone. In both cases, the EGFP coding sequence is inserted between the NheI and BglII sites, and the tubulin coding sequence lacking the initial methionine is inserted between the BglII and AflII sites. The β1-tubulin coding sequence was obtained by RT-PCR using T. gondii total RNA and primer pair S6/AS6, cut with BglII and AflII, and ligated into the vector backbone. The β3-tubulin coding sequence was amplified by RT-PCR from T. gondii total RNA with primer pair S7/AS7, cloned into vector pCR4-TOPO (Invitrogen), cut with BamHI and BbsI to give BglII-AflII–compatible ends, and ligated into the BglII-AflII cut vector backbone. In the expressed protein product, the EGFP is in both cases linked to the tubulin by the five-aa sequence SGLRS.
ptubg-mNeonGreenFP-TgtubB1 was derived from the plasmid ptubg. Plasmid ptubg was created from ptub-EGFP-TgtubB1 by a two-component assembly using NEBuilder HiFi DNA Assembly reagents (E2621S; NEB), following the manufacturer’s recommended protocol. Component 1 was prepared from ptub-EGFP-TgtubB1 by cutting out the 2088 base pair NheI-AflII fragment. Component 2 was prepared by mixing equal volumes of oligonucleotides S8 and AS8, 10 μM each, heating to 97°C for 2 min, and cooling slowly to room temperature. Plasmid ptubg-mNeonGreenFP-TgtubB1 was constructed by a three-component NEBuilder HiFi assembly. Component 1 was prepared by cutting out the 12 base pair stuffer from ptubg using NheI and AflII. Component 2 was prepared by PCR amplifying the mNeonGreenFP coding sequence from pmNeonGreen-N1 (Shaner et al., 2013) using primers S9 and AS9. Component 3 was prepared by PCR amplification of the T. gondii β1-tubulin coding sequence from plasmid ptub-EGFP-TgtubB1 with primers S10 and AS10. In the expressed protein product, mNeonGreenFP is coupled to the N-terminus of β1-tubulin (lacking its initial methionine residue) by the 13-aa flexible linker G-(GTGSG)2-GS.
The plasmid pTKO4-TgDCX-mCherryFP was constructed in a backbone (pTKO4) designed for replacement of genes in T. gondii with mCherryFP-tagged versions of the genes by homologous recombination. This backbone was derived from pTKO2_II (Heaslip et al., 2010) by several modifications. 1) The EGFP coding sequence was replaced by a synthesized version of EGFP in which restriction sites for AflII, BpmI, MfeI, NcoI, and NsiI were removed by silent mutagenesis. 2) The three multiple-cloning-site (MCS) sequences were replaced (see later description). 3) mCherryFP was added, positioned so that it can be fused to the C-terminus of the target CDS.
MCS1 in pTKO2_II, which is used for insertion of a piece of the genomic DNA upstream of the target gene just in front of a LoxP site, encoded sites for NotI, PstI, KpnI, XhoI, and EcoRV. This was replaced with a synthesized sequence encoding sites for SgrDI, BsiWI, NotI, AflII, HindIII, EcoRI, and BglII.
MCS2 in pTKO2_II, which is used for insertion of the coding region of the target gene on the other side of the LoxP site, encoded sites for EcoRI, BglII, PmeI, AsiSI, RsrII, and StuI. This was replaced with a synthesized sequence encoding sites for PacI, NsiI, SphI, AsiSI, NheI, MfeI, PmeI, SpeI, XbaI, and MreI. Also inserted downstream of this new MCS2 was a synthesized sequence for mCherryFP minus the initial methionine codon. The last G of the MreI site of MCS2 is also the first base of the valine codon (GTG) in the N-terminal amino acid sequence (MVSKGEE) of mCherryFP. The sequence TAAGGATCC was also added on the 3′ end of the mCherryFP coding sequence (stop codon plus BamHI site).
MCS3 in pTKO2_II, which is used for insertion of a piece of the genomic DNA downstream of the target gene, is separated from MCS2 by the mCherryFP coding sequence, a portion of the 3′-UTR of TgGRA2 (serves as 3′-UTR for the target gene after homologous recombination), the expression cassette for TgHXGPRT (for selection of transfected cells with 80 µM mycophenolic acid plus 330 µM xanthine), and a second LoxP site to allow knockout of the target gene by Cre recombinase (Heaslip et al., 2010). MCS3 encoded sites for HindIII, NheI, HpaI, ApaI, and PspOMI. This was replaced with a synthesized sequence encoding sites for BclI, SacI, AscI, XhoI, AbsI, KpnI, SmaI, XmaI, FseI, SfiI, ApaI, and PspOMI.
To construct pTKO4-TgDCX-mCherryFP, the 887 base pair sequence of T. gondii RH genomic DNA immediately preceding the methionine initiation codon of TgDCX was inserted into MCS1 between the AflII and EcoRI sites. The sequence between the last base of the EcoRI site of MCSI and the last base of the PmeI site of MCS2 was replaced with a synthesized piece that included a LoxP site (34 base pairs), a T. gondii consensus Kozak sequence (6 base pairs), and the complete TgDCX CDS (768 base pairs). With this insertion, the TgDCX CDS was then fused to the N-terminus of mCherryFP (lacking initial ATG) by a 9-aa linker, PGTSSRGAG. Finally, a synthesized sequence extending from the last 8 base pairs of the TgDCX exon 6 in T. gondii RH genomic DNA, through the stop codon and the first 871 base pairs of the downstream genomic region, flanked by 5′-AscI and 3′-AbsI recognition sites, was inserted using the AscI and AbsI sites of MCS3.
To construct pTKO4-TgDCX-mNeonGreenFP, mCherryFP was removed from pTKO4-TgDCX-mCherryFP by cutting with BamHI and MreI and replaced by a PCR-amplified copy of the NeonGreenFP (Shaner et al., 2013) coding sequence (primers S11 and AS11) using NEBuilder HiFi DNA Assembly. In the final construct, the TgDCX CDS was fused to the N-terminus of mNeonGreenFP (lacking initial ATG) by a 10-aa linker, PGTSSRGAGG.
pTKO4-mCherryFP-TgDCX was constructed from pTKO4-TgDCX-mCherryFP by a five-component assembly using the NEBuilder HiFi assembly reagents. Component 1 was obtained from pTKO4-TgDCX-mCherryFP by removing the 1757 base pair BamHI-NheI fragment. Component 2 was obtained by PCR copying the piece of TgDCX 5′-UTR and adjacent LoxP site from pTKO4-TgDCX-mCherryFP using primers AS12 and S12. Component 3 was obtained by PCR amplifying an mCherryFP coding sequence (from which XcmI, HinCII, PvuII, BbsI, BtgI, PstI, and MscI sites had previously been removed by silent mutagenesis) using primers S13 and AS13. Component 4 was obtained by PCR amplifying a 69 base pair synthetic linker sequence using primers S14 and AS14. Component 5 was obtained by PCR copying the complete TgDCX CDS plus stop codon, minus the initial ATG codon, from pTKO4-TgDCX-mCherryFP using primers AS15 and S15. In the final construct, mCherryFP was coupled to the N-terminus of TgDCX (minus its initial methionine) via a 23 base pair flexible linker, GGSS-(GGS)6-T.
Creating mCherryFP-TgDCX, TgDCX-mCherryFP, and TgDCX mNeonGreenFP knock-in parasites
Plasmids pTKO4-TgDCX-mCherryFP, pTKO4-TgDCX-mNeonGreenFP, and pTKO4-mCherryFP-TgDCX (40 µg each) were linearized by cutting with NotI and separately introduced into extracellular RHΔku80Δhx parasites by electroporation. After 24 h, 80 µM mycophenolic acid and 330 µM xanthine were added to the culture medium, thereby initiating selection for parasites containing a functional HXGPRT derived from the introduced plasmid. After 10 d of drug selection, a stable population was obtained, from which 27 individual non–GFP–expressing (i.e., potentially homologous recombinants) clones were isolated by limiting dilution. Two of the candidate knock-in clones were eliminated by PCR screening for the intron-containing genomic version of TgDCX. Southern blotting was used to confirm that the remaining 25 were true homologous replacements.
Creating a TgDCX-knockout parasite
The pmin-Cre-EGFP_Gra-mCherry plasmid (Liu et al., 2016) was introduced into extracellular mCherryFP-TgDCX, TgDCX-mCherryFP, and TgDCX-mNeonGreenFP knock-in parasites by electroporation. This plasmid drives expression of both Cre recombinase and mCherryFP. The mCherryFP is diffusely cytoplasmic and much brighter than the small dot of mCherryFP-TgDCX at the conoid, making it easy to recognize transfected parasites. Three cytoplasmic mCherryFP-positive parasites were sorted by fluorescence-activated cell sorting (FACS) into each well of 96-well plates containing culture medium plus 0.5 mM 6-thioxanthine to select against parasites containing a functional HXGPRT. After plaques were visible, the wells containing single plagues were consolidated into a new 96-well plate and grown with no drug selection. Clones were screened for the absence of fluorescence. Ten “dark” clones were expanded and used to isolate genomic DNA. The absence of the TgDCX coding region was confirmed by Southern blotting.
ptub-TgDCX-EGFP complemented parasites
To generate the complemented lines, the ptub-TgDCX-EGFP plasmid was electroporated into TgDCX-knockout parasites. The populations were then subjected to chloramphenicol selection for ∼1 mo and taken off drug selection for ∼2 wk before the growth of the populations was compared with the knockout, knock-in, and RHΔku80Δhx parasites using plaque and invasion assays as shown in Figure 6 and Table 1.
Recombinant TgDCX protein
The coding region of TgDCX extending from the second methionine to the stop codon, corresponding in size to the major band seen in Western blots of T. gondii whole-cell extract (Figure 2), was obtained by RT-PCR with RH parasite total RNA and primer pairs S19 and AS19, then cloned into the BamHI-EcoRI site of pRSET-B vector (Invitrogen), resulting in a fusion of 33 aa (hexahistidine tag, T7 gene10 leader sequence, and Xpress epitope tag) to the N-terminus of the protein. The recombinant protein was produced in BL21 codon plus (DE3) RIL Escherichia coli (Stratagene, La Jolla, CA), and purified using Talon metal affinity resin (Clontech) under denaturing condition. Under nondenaturing conditions, the protein was found only in insoluble inclusion bodies. Extensive trials were undertaken in an attempt to prepare refolded protein that is soluble in physiological buffers, without success.
Antibody production
Polyclonal antiserum against the recombinant fusion protein was produced in rabbits by Cocalico Biologicals (Reamstown, PA). Although in some respects the results obtained with this antiserum were quite variable, in one indispensable characteristic it proved reliable: in immunofluorescence, the only structure that it consistently labeled was the conoid, the same pattern seen with FP-tagged TgDCX, and in Western blots, it always stained the recombinant or endogenous TgDCX bands. Under most conditions, in Western blots, the antiserum also reacted with some higher–molecular weight bands, even in extracts of host cells that were not infected with T. gondii. The reactivity against those extra bands could be attenuated or removed by several methods, including affinity purification, preabsorption against fixed (Toxo-free) host cell monolayers, and preabsorption against bovine serum albumin (BSA) or other unrelated proteins bound to nitrocellulose membrane. The recipes given later are those that were most successful in removing the nonspecific reactivity.
Anti-TgDCX was affinity purified from the antiserum using the recombinant protein bound to nitrocellulose as follows. Recombinant TgDCX, 160 µg, was run on a polyacrylamide gel and transferred onto a nitrocellulose membrane. The area of membrane that had the recombinant protein band was visualized by reversibly staining with Ponceau S, cut into small pieces, and incubated with 1 ml of the crude antiserum against TgDCX for 2 h at room temperature. The membrane pieces were washed twice with 0.1% Tween 20/phosphate-buffered saline (PBS; 3 × 5 min). Bound antibodies were eluted in 900 µl of 100 mM glycine (pH 2.5). The solution was neutralized with 100 µl of 1 M Tris (pH 8.0).
The affinity-purified antibody solution was then subjected to the following preadsorption procedure to remove residual nonspecific reactivity. HFF cells grown in a 75-cm2 flask were rinsed with PBS and then fixed with cold methanol for 5 min at −20°C. The cells were rehydrated with PBS 3 × 5 min, blocked with 3% BSA and 0.01% Tween 20/PBS for 1 h at room temperature, and then washed once with PBS. Affinity purified antibody solution, 30 µl, was diluted 1:100 in PBS and then incubated with the cells for 2 h at room temperature. The preadsorbed antibody solution was collected from the flask, and sodium azide was added to a final concentration of 0.02% for preservation at 4°C. This residual nonspecific reactivity is removed equally well by preabsorption with BSA or other proteins bound to nitrocellulose membrane.
Western blotting analysis
Whole-cell extract from 7 × 106 parasites or 10 µg of protein from uninfected HFF cells was mixed with NuPage sample buffer (Invitrogen) containing a final concentration of 10 mM dithiothreitol (DTT) and run on 4–12% NuPage Bis-Tris gel (Invitrogen). The separated proteins were transferred to a nitrocellulose or polyvinylidene fluoride membrane. The membrane was blocked in 5% nonfat dry milk in PBS, incubated with rabbit anti-TgDCX antibody diluted 1:5000 or preabsorbed antibody diluted to 1:100 in blocking buffer plus 0.1% Tween 20 for 1 h at room temperature, and washed in PBS-T (PBS plus 0.1% Tween 20). The membrane was then incubated with horseradish peroxidase–conjugated anti-rabbit immunoglobulin G (IgG; Amersham) diluted 1:10,000 in blocking buffer plus 0.1% Tween 20 for 1 h at room temperature and washed with PBS-T and then PBS. Chemiluminescence detection was performed using ECL-plus (Amersham).
Immunofluorescence assays
HFF cells grown in 35-mm glass-bottom dishes (P35G-1.5-14-C or P35G-1.5-20-C; MatTek) were infected with wild-type or transgenic parasites expressing EGFP-Tgβ3-tubulin. About 16 h postinoculation, cells were fixed with 0.5% formaldehyde in PBS for 5 min at room temperature, rinsed with PBS twice, and refixed/permeabilized with cold methanol for 5 min at −20°C. After being rinsed twice with PBS, cells were further extracted with 10 mM deoxycholate in water for 5 min at room temperature with gentle rocking. Samples were rinsed with PBS twice and then blocked with 3% BSA and 0.01% Tween 20/PBS, followed by incubation with rabbit anti-TgDCX for 1 h at room temperature. Cells were washed with 0.01% Tween 20/PBS (3 × 5 min) and then incubated with goat anti-rabbit IgG conjugated with Alexa Fluor 555 (Molecular Probes; diluted 1:500 in blocking buffer) for 1 h at room temperature. Cells were washed with 0.01% Tween 20/PBS (3 × 5 min) and then mounted using ProLong anti-fade reagent (P7481; Molecular Probes).
Antibodies penetrate into adult conoids very poorly, making detection of antigens in adult conoids unreliable with typical immunostaining procedures, including the foregoing protocol, which incorporates brief treatment with 10 mM deoxycholate. To circumvent this problem, a different procedure was developed using extracellular parasites and more vigorous deoxycholate treatment. Freshly lysed-out parasites were harvested and passed through a 3-µm-pore filter (111112; Whatman). Cells were washed once with calcium saline (20 mM HEPES, 2.7 mM KCl, 138 mM NaCl, and 5 mM CaCl2), pelleted again, resuspended in 200 µl of warm conoid extension buffer (2 µM A23187 in calcium saline), and incubated for 10 min at room temperature. The cell suspension was transferred to a glass-bottom dish, and the cells were allowed to settle. The supernatant was carefully removed and replaced with 500 µl of extraction buffer (10 mM deoxycholate in water supplemented with DNaseI [18068-015; Invitrogen]) and then incubated for 15 min at room temperature. Cells were further extracted with 2 ml of the extraction buffer without DNaseI for an additional hour with gentle rocking. Blocking and primary and secondary antibody reactions were performed similarly to the first immunostaining procedure.
Immuno–electron microscopy
Immunogold labeling used essentially the second immunostaining procedure described earlier. Extracellular parasites of the wild-type RH strain were passed through a 3-µm-pore filter, washed once with calcium saline, pelleted, resuspended in 30 µl of the warm conoid extension buffer, and then incubated for 10 min at room temperature. Parasites were adsorbed onto a carbon-film-coated nickel grid for 10 min, incubated with 30 µl of extraction buffer (10 mM deoxycholate) with DNaseI for 15 min at room temperature, and then incubated with 2 ml of the extraction buffer without DNaseI for an additional hour with gentle shaking. The extracted parasites were blocked with 5% BSA and 0.1% fish gelatin/PBS for 10 min at room temperature, rinsed once with incubation buffer (0.8% BSA, 0.1% fish gelatin in PBS), and then incubated overnight with rabbit anti-TgDCX. Grids were then washed with PBS (3 × 5 min; then 1 × 15 min), incubated with secondary antibody (anti-rabbit IgG conjugated with 1.4-nm gold; Nanoprobes, Yaphank, NY), diluted 1:160 in incubation buffer, incubated for 3 h at room temperature, and then washed with PBS (3 × 5 min, then 1 × 15 min). The sample was fixed for 5 min with 1% glutaraldehyde in PBS and washed in distilled water (3 × 5 min). Silver enhancement was carried out using the HQ silver enhancement kit (Nanoprobes) by floating grids on a mixture of the initiator, activator, and modulator for 2 min. The grid was washed with distilled water and then stained using 2% phosphotungstic acid (pH 7.0).
Southern blot
Nick translation was used to synthesize single-stranded, biotin-labeled probe DNA. The labeling reaction included 5 U of Klenow fragment (3′-5′ exo minus; M0212; NEB), 3 μM biotinylated random octamers, 25 mM Tris-HCl, 100 mM HEPES, 1 mM DTT, 2.5 mM MgCl2, 0.1 mM dCTP, 0.1 mM dGTP, 0.1 mM dTTP, 0.084 mM dATP, and 0.016 mM biotin-14-dATP, pH 7.0, at 25°C. Template DNA (100–500 ng) was denatured in boiling water for 5 min, put on ice for a few minutes, and then incubated with the labeling reaction mix at 37°C for 6 h. Labeled probe was isolated and purified by ethanol precipitation. Template DNA for the exon 2-5 probe was made by PCR amplification of a 641 base pair fragment of the TgDCX CDS, spanning the region from the codon 4 of exon 2 (M16) to the codon 2 of exon 6 (E225) using primers S5 and AS20. Template DNA for the 5′-UTR probe was a synthesized sequence corresponding to the 893 base pairs immediately preceding the first methionine codon of TgDCX in T. gondii genomic DNA.
Approximately 5 µg of T. gondii genomic DNA was digested with BamHI plus EcoRV, run on an agarose gel with a biotinylated DNA ladder, transferred, and ultraviolet cross-linked to a positively charged nylon membrane. The blot was prehybridized for 1–2 h and then hybridized for 12–18 h with 10 ng/ml biotinylated probe. Detection of bound probe used 1 µg/ml streptavidin (N7021; NEB) and 0.1 μg/ml biotinylated alkaline phosphatase (B-2005-1; Vector Labs). The chemiluminescence signal generated with substrate CDP-Star reagent (12-041-677-001; Roche) was recorded on Kodak blue-sensitive x-ray film.
Plaque assay and overall doubling time
Plaque assays were performed as previously described, with some modifications (Liu et al., 2016). A total of 50, 100, or 200 parasites was added to each well of a six-well plate containing a confluent HFF monolayer and incubated at 37°C for 10 d undisturbed. Infected monolayers were then fixed with cold methanol for 10 min with gentle shaking, washed with DPBS, stained with 0.5% crystal violet in 20% methanol for 30 min, gently rinsed with double-distilled H2O, air dried, and scanned.
An approximate value for the overall doubling time of parasite lines in culture was estimated from the ratio of inoculum volume to total volume of medium in the flask and the time required for complete lysis of the host cell monolayer. For example, inoculating 70 μl of culture supernatant from a freshly lysed flask of wild-type parasites into a new flask containing 4.0 ml of culture medium results in complete lysis of the new flask 44 ± 4 h later. This is an amplification of 58-fold (4.07/0.07), that is, 25.86-fold, in 44 h, corresponding to a doubling time of ∼7.5 (44/5.86) h. Note that the doubling time measured in this way is a composite of all the events of the lytic cycle as they occur in cultures, including time to invade, cell cycle time, time required for host cell lysis, and time spent searching for a new host cell.
Host-cell invasion assay
To maximize the proportion of vigorously active parasites, parasites were harvested when the host cell monolayer was <50% lysed. Initial inoculation of the cultures was timed so that all five parasite lines to be examined would be ready at the same time. The monolayers were scraped off the flask, suspended in culture medium, and passed through a 23-gauge needle. The concentration of parasites was determined by counting in a hemocytometer, and 107 parasites of each line were inoculated into a nearly confluent layer of African green monkey kidney epithelial cells (BS-C-1; American Type Culture Collection [ATCC] CCL-26) growing in 35-mm plastic culture dishes with a glass coverslip bottom (MatTek). The dishes were incubated at 37°C for 1 h, washed twice with DPBS, and then fixed with 3.7% formaldehyde and 0.06% glutaraldehyde for 15 min. The dishes were then washed five times with DPBS and blocked with 1% BSA in DPBS for 30–45 min. Parasites that had attached to the host cell surface but had not yet invaded were labeled by incubation with a mouse antibody against SAG1 for 30–45 min, washed in DPBS five times, and then incubated with Alexa 568 goat anti-mouse IgG. After permeabilization with 0.5% (vol/vol) Triton X-100 in DPBS for 15 min, washing five times with DPBS, and blocking for 30 min with 1% BSA in DPBS, all parasites were labeled by incubating with a rabbit antibody against SAG1 and a Cy2 goat anti-rabbit IgG.
The 3D stacks of images of the red and green fluorescent parasites were acquired with a 20×/numerical aperture (NA) 0.75 objective lens along with a reference differential interference contrast (DIC) image from the middle of the stack. Images were collected at 15 different locations in each dish for each line, and the entire experiment was repeated three times. The fluorescent images were projected along the optic axis and deconvolved with a two-dimensional optical transfer function for the objective lens. The image processing program Fiji (Schindelin et al., 2012; Schneider et al., 2012) was used to automate counting of the red and green fluorescent parasites in each of the 450 images.
Conoid/apical complex extension assay
The assay was performed as described by Mondragon and Frixione (1996), with the following modifications. Parasites from a T12.5 flask were harvested when the host cell layer was ∼80% lysed. The monolayers were scraped off the flask, suspended in culture medium, passed through a 23-gauge needle, filtered through a 3-μm-pore filter, and collected by centrifugation at 1500 × g for 5 min. The parasite pellet was resuspended in CO2 Independent Medium (18045-088; Life Technologies) supplemented with 0.5 mM CaCl2, rotated at room temperature for 30 min, and centrifuged at 3600 × g for 1 min. The pellet was resuspended in 0.5 ml of the same solution plus 5 μM Ca ionophore A23187 and rotated at room temperature for 5 min. The parasites were fixed by adding 0.5 ml of freshly prepared CO2 Independent Medium plus 0.25 M glutaraldehyde. After 5 min, residual aldehyde groups were blocked (to prevent clumping of the parasites) by adding 0.5 ml of 1.0 M glycine, pH 8.5, and rotating for 5 min. The parasites were collected by centrifugation, washed once with water, suspended in 80% glycerol, mounted as a thin layer under a coverslip, and imaged by phase contrast microscopy with a 60× objective lens. Parasites oriented with their apical end clearly visible were counted as either “conoid extended” or “conoid retracted” as indicated in Figure 8. Approximately 300 parasites were scored for each parasite line, and the entire experiment was carried out three times.
Light microscopy
Parasites were grown on a subconfluent layer of HFF, rat aorta cells (A7r5, putative smooth muscle; ATCC CRL-1444), or BS-C-1 cells in a 35-mm plastic dish with #1.5 glass coverslip bottom (P35G-1.5-10-C; MatTek). Just before transfer to the microscope, the medium was replaced with CO2-independent medium. The dish was maintained at 37°C in a humidified environmental chamber surrounding the microscope stage. The 3D image stacks were collected at z-increments of 0.3 µm on an Applied Precision DeltaVision workstation using a 60×/NA 1.3 silicone immersion lens. Brightly fluorescent 0.2-μm beads placed in the dish were used to adjust the correction collar of the lens to minimize spherical aberration. Deconvolved images were computed using the point-spread functions and software supplied by the manufacturer. SIM images were collected at z-increments of 0.125 μm on an Applied Precision OMX 3D-SIM system using a 100×/NA 1.4 objective lens and processed using the manufacturer’s software. All quantitative measurements were carried out using the raw image data, but in the final displayed images, the relative brightness of different fluorophores is altered as needed to best use the gamut of a computer monitor LCD display.
Electron microscopy of whole-mount Toxoplasma
Parasites from 1 ml of a freshly lysed culture were collected by centrifugation for 1 min at 3600 × g, washed once with HEPES-buffered, serum-free culture medium, and resuspended in 40 µl of the same medium containing 20 µM calcium ionophore A23187. After 2 min, 4 µl of parasite suspension was pipetted onto a carbon-coated electron microscope grid and placed in a humid chamber for 8 min. The grid was then placed parasite-side down onto a 50-µl drop of 0.5% Triton X-100 for 3 min and transferred to a drop of 0.002% Triton X-100 and 2% phosphotungstic acid, pH 7.3, for 2 min, and all of the liquid was wicked off by touching the grid to the edge of a piece of filter paper. Images were collected with a JEOL 1010 electron microscope operated at 80 keV.
Acknowledgments
We thank Lloyd Kasper (Dartmouth College, Hanover, NH) for the rabbit anti-SAG1 antibody, Richard Day (Indiana University School of Medicine, Indianapolis, IN) for a plasmid containing the mNeonGreenFP coding sequence, Jacqueline Leung (Indiana University, Bloomington, IN) for insightful discussions, and Amanda Rollins for technical support. This study was supported by funding from the March of Dimes (6-FY15-198) and the National Institutes of Health/National Institute of Allergy and Infectious Diseases (R01-AI098686), the Laura and Arthur Colwin Endowed Summer Research Fellowship Fund of the Marine Biological Laboratory (Woods Hole, MA) awarded to K.H., and facility funding from the Indiana Clinical and Translational Sciences Institute to K.H., funded in part by Grants UL1 TR001108 and TL1TR001107 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award.
Abbreviations used:
- aa
amino acid(s)
- CDS
coding sequence
- DPBS
Dulbecco’s phosphate-buffered saline
- EGFP
enhanced green fluorescent protein
- FP
fluorescent protein
- MCS
multiple cloning site
- RACE
rapid amplification of cDNA ends
- RT
reverse transcription, reverse transcriptase
- SIM
structured illumination microscopy
- YFP
yellow fluorescent protein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-08-0587) on December 8, 2016.
REFERENCES
- Amos LA, Amos WB. The bending of sliding microtubules imaged by confocal light microscopy and negative stain electron microscopy. J Cell Sci Suppl. 1991;14:95–101. doi: 10.1242/jcs.1991.supplement_14.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechstedt S, Brouhard GJ. Doublecortin recognizes the 13-protofilament microtubule cooperatively and tracks microtubule ends. Dev Cell. 2012;23:181–192. doi: 10.1016/j.devcel.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechstedt S, Lu K, Brouhard GJ. Doublecortin recognizes the longitudinal curvature of the microtubule end and lattice. Curr Biol. 2014;24:2366–2375. doi: 10.1016/j.cub.2014.08.039. [DOI] [PubMed] [Google Scholar]
- Carey KL, Westwood NJ, Mitchison TJ, Ward GE. A small-molecule approach to studying invasive mechanisms of Toxoplasma gondii. Proc Natl Acad Sci USA. 2004;101:7433–7438. doi: 10.1073/pnas.0307769101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leon JC, Scheumann N, Beatty W, Beck JR, Tran JQ, Yau C, Bradley PJ, Gull K, Wickstead B, Morrissette NS. A SAS-6-like protein suggests that the Toxoplasma conoid complex evolved from flagellar components. Eukaryot Cell. 2013;12:1009–1019. doi: 10.1128/EC.00096-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- des Portes V, Francis F, Pinard JM, Desguerre I, Moutard L M, Snoeck I, Meiners LC, Capron F, Cusmai R, Ricci S, et al. doublecortin is the major gene causing X-linked subcortical laminar heterotopia (SCLH) Hum Mol Genet. 1998;7:1063–1070. doi: 10.1093/hmg/7.7.1063. [DOI] [PubMed] [Google Scholar]
- Desser SS. The fine structure of Leucocytozoon simondi. III. The ookinete and mature sporozoite. Can J Zool. 1970;48:641–645. doi: 10.1139/z70-072. [DOI] [PubMed] [Google Scholar]
- Fourniol FJ, Sindelar CV, Amigues B, Clare DK, Thomas G, Perderiset M, Francis F, Houdusse A, Moores CA. Template-free 13-protofilament microtubule-MAP assembly visualized at 8 A resolution. J Cell Biol. 2010;191:463–470. doi: 10.1083/jcb.201007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Heaslip AT, Dzierszinski F, Stein B, Hu K. TgMORN1 is a key organizer for the basal complex of Toxoplasma gondii. PLoS Pathog. 2010;6:e1000754. doi: 10.1371/journal.ppat.1000754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaslip AT, Ems-McClung SC, Hu K. TgICMAP1 is a novel microtubule binding protein in Toxoplasma gondii. PLoS One. 2009;4:e7406. doi: 10.1371/journal.pone.0007406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaslip AT, Nishi M, Stein B, Hu K. The motility of a human parasite, Toxoplasma gondii, is regulated by a novel lysine methyltransferase. PLoS Pathog. 2011;7:e1002201. doi: 10.1371/journal.ppat.1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlavanda E, Kovacs J, Olah J, Orosz F, Medzihradszky KF, Ovadi J. Brain-specific p25 protein binds to tubulin and microtubules and induces aberrant microtubule assemblies at substoichiometric concentrations. Biochemistry. 2002;41:8657–8664. doi: 10.1021/bi020140g. [DOI] [PubMed] [Google Scholar]
- Hu K. Building a Parasite: The Study of the Cell Division and Cytoskeleton of Toxoplasma gondii. Dissertation. Philadelphia: University of Pennsylvania; 2002. [Google Scholar]
- Hu K. Organizational changes of the daughter basal complex during the parasite replication of Toxoplasma gondii. PLoS Pathog. 2008;4:e10. doi: 10.1371/journal.ppat.0040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K, Johnson J, Florens L, Fraunholz M, Suravajjala S, DiLullo C, Yates J, Roos DS, Murray JM. Cytoskeletal components of an invasion machine–the apical complex of Toxoplasma gondii. PLoS Pathog. 2006;2:121–138. doi: 10.1371/journal.ppat.0020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K, Mann T, Striepen B, Beckers CJ, Roos DS, Murray JM. Daughter cell assembly in the protozoan parasite Toxoplasma gondii. Mol Biol Cell. 2002a;13:593–606. doi: 10.1091/mbc.01-06-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K, Roos DS, Angel SO, Murray JM. Variability and heritability of cell division pathways in Toxoplasma gondii. J Cell Sci. 2004a;117:5697–5705. doi: 10.1242/jcs.01494. [DOI] [PubMed] [Google Scholar]
- Hu K, Roos DS, Murray JM. A novel polymer of tubulin forms the conoid of Toxoplasma gondii. J Cell Biol. 2002b;156:1039–1050. doi: 10.1083/jcb.200112086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K, Suravajjala S, DiLullo C, Roos DS, Murray JM. Functional specialization of tubulin isoforms in Toxoplasma gondii. 2004b. p. p424a. In: American Society for Cell Biology 44th Annual Meeting, Washington, DC. American Society for Cell Biology.
- Kim MH, Cierpicki T, Derewenda U, Krowarsch D, Feng Y, Devedjiev Y, Dauter Z, Walsh CA, Otlewski J, Bushweller JH, et al. The DCX-domain tandems of doublecortin and doublecortin-like kinase. Nat Struct Biol. 2003;10:324–333. doi: 10.1038/nsb918. [DOI] [PubMed] [Google Scholar]
- Kurachi M, Hoshi M, Tashiro H. Buckling of a single microtubule by optical trapping forces: direct measurement of microtubule rigidity. Cell Motil Cytoskeleton. 1995;30:221–228. doi: 10.1002/cm.970300306. [DOI] [PubMed] [Google Scholar]
- Leander BS, Keeling PJ. Morphostasis in alveolate evolution. Trends Ecol Evol. 2003;18:395–402. [Google Scholar]
- Leveque MF, Berry L, Besteiro S. An evolutionarily conserved SSNA1/DIP13 homologue is a component of both basal and apical complexes of Toxoplasma gondii. Sci Rep. 2016;6:27809. doi: 10.1038/srep27809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liliom K, Wagner G, Kovacs J, Comin B, Cascante M, Orosz F, Ovadi J. Combined enhancement of microtubule assembly and glucose metabolism in neuronal systems in vitro: decreased sensitivity to copper toxicity. Biochem Biophys Res Commun. 1999;264:605–610. doi: 10.1006/bbrc.1999.1547. [DOI] [PubMed] [Google Scholar]
- Liu J, He Y, Benmerzouga I, Sullivan WJ, Morrissette NS, Murray JM, Hu K. An ensemble of specifically targeted proteins stabilizes cortical microtubules in the human parasite Toxoplasma gondii. Mol Biol Cell. 2016;27:549–571. doi: 10.1091/mbc.E15-11-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragon R, Frixione E. Ca(2+)-dependence of conoid extrusion in Toxoplasma gondii tachyzoites. J. Eukaryot Microbiol. 1996;43:120–127. doi: 10.1111/j.1550-7408.1996.tb04491.x. [DOI] [PubMed] [Google Scholar]
- Moores CA, Perderiset M, Francis F, Chelly J, Houdusse A, Milligan RA. Mechanism of microtubule stabilization by doublecortin. Mol Cell. 2004;14:833–839. doi: 10.1016/j.molcel.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Nagayasu E, Zhang F, Hu K, Ananvoranich S, Murray JM. Cytoskeletal components of an invasion machine: The apical complex and conoid of Toxoplasma gondii. 2006. In: American Society for Cell Biology 46th Annual Meeting, Vol. 22, San Diego, CA, p. 293. [DOI] [PMC free article] [PubMed]
- Nishi M, Hu K, Murray JM, Roos DS. Organellar dynamics during the cell cycle of Toxoplasma gondii. J Cell Sci. 2008;121:1559–1568. doi: 10.1242/jcs.021089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orosz F. Apicortin, a unique protein, with a putative cytoskeletal role, shared only by apicomplexan parasites and the placozoan Trichoplax adhaerens. Infect Genet Evol. 2009;9:1275–1286. doi: 10.1016/j.meegid.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Orosz F. Wider than thought phylogenetic occurrence of apicortin, a characteristic protein of Apicomplexan parasites. J Mol Evol. 2016;82:303–314. doi: 10.1007/s00239-016-9749-5. [DOI] [PubMed] [Google Scholar]
- Patra KP, Vinetz JM. New ultrastructural analysis of the invasive apparatus of the Plasmodium ookinete. Am J Trop Med Hyg. 2012;87:412–417. doi: 10.4269/ajtmh.2012.11-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portman N, Foster C, Walker G, Slapeta J. Evidence of intraflagellar transport and apical complex formation in a free-living relative of the apicomplexa. Eukaryot Cell. 2014;13:10–20. doi: 10.1128/EC.00155-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portman N, Slapeta J. The flagellar contribution to the apical complex: a new tool for the eukaryotic Swiss Army knife. Trends Parasitol. 2014;30:58–64. doi: 10.1016/j.pt.2013.12.006. [DOI] [PubMed] [Google Scholar]
- Roos DS, Donald RG, Morrissette NS, Moulton AL. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol. 1994;45:27–63. doi: 10.1016/s0091-679x(08)61845-2. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods. 2013;10:407–409. doi: 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]