

Abstract
Hodgkin lymphoma (HL) is a lymphoid malignancy that is typically derived from germinal-center B cells. EBV infection, mutations in NF-κB pathway genes, and genetic susceptibility are known risk factors for developing HL. CD30 and NF-κB have been identified as potential biomarkers in pediatric HL patients, and these molecules may represent therapeutic targets. Although current risk adapted and response based treatment approaches yield overall survival rates of >95%, treatment of relapse or refractory patients remains challenging. Targeted HL therapy with the antibody-drug conjugate Brentuximab vedotin (Bv) has proven to be superior to conventional salvage chemotherapy and clinical trials are being conducted to incorporate Bv into frontline therapy that substitutes Bv for alkylating agents to minimize secondary malignancies. The appearance of secondary malignancies has been a concern in pediatric HL, as these patients are at highest risk among all childhood cancer survivors. The risk of developing secondary leukemia following childhood HL treatment is 10.4 to 174.8 times greater than the risk in the general pediatric population and the prognosis is significantly poorer than the other hematological malignancies with a mortality rate of nearly 100%. Therefore, identifying clinically valuable biomarkers is of utmost importance to stratify and select patients who may or may not need intensive regimens to maintain optimal balance between maximal survival rates and averting late effects. Here we discuss epidemiology, risk factors, staging, molecular and genetic prognostic biomarkers, treatment for low and high-risk patients, and the late occurrence of secondary malignancies in pediatric HL.
Keywords: Hodgkin lymphoma, pediatric, adolescent, biomarker, tumor microenvironment
INTRODUCTION
Hodgkin lymphoma (HL) is a unique monoclonal lymphoid malignancy that is characterized by the presence of typical bi/multinucleated Reed-Sternberg cells and their variants, mononucleated Hodgkin cells, collectively known as Hodgkin Reed-Sternberg cells (HRS). Most HRS cells express CD15 and CD30 (85% and 100%, respectively in HL cases). HRS cells have a B-cell ancestry as revealed by single cell PCR detection of Ig gene rearrangements [1]; and somatic mutations in the variable region of heavy and light chain Ig genes are suggestive of a late germinal or post-germinal center B-cell origin [2–4]. Despite their B-cell origin, HRS cells typically lack core B cell features, including specific signaling molecules that are associated with the B-cell lineage, which are either absent or expressed by only a small subset of cells [5–7]. In addition to the variable B-cell marker expression shown by ∼95% of classical Hodgkin lymphoma (cHL) cases, HRS cells can express markers that are characteristic of other hematopoietic lineages, including dendritic cells, monocytes, and T-cells (5-15% cHL cases) [8–10]. Though rarely expressed, T-cell markers on HRS cells have been reported to be independently associated with poor prognosis [10, 11].
Biologically, HL is similar if not identical in children and adults except for the relative incidence of specific histological subtypes and distinct immune response against HRS cells in tumor microenvironment. HL can be divided into two broad classes based on histologic features and phenotypes: (1) Classical Hodgkin lymphoma (cHL) and (2) Nodular lymphocyte predominant Hodgkin lymphoma (NLPHL). cHL can be further divided into four subtypes: lymphocyte rich (LR), nodular sclerosis (NS), mixed cellularity (MC), and lymphocyte depleted (LD). NLPHL is considered to be a distinct disease entity that is more similar to B-cell non-HL than to cHL [12]. Therefore, this review will focus on cHL.
With the advent of recent advanced treatment and imaging technologies, a cure rate of >95% has been achieved for pediatric HL patients. However, the prognosis of relapse/refractory (RR) patients is dismal The secondary malignancy and cardiopulmonary toxicity that can be caused by HL treatment motivated greater focus on therapy refinements that would minimize toxicity while maintaining high cure rates. As such, identification of prognostic biomarkers in pediatric patients that correlate with clinical outcome will increase the understanding of HL pathology and likely influence therapeutic approaches. In this review, we discuss the main characteristics and prognostic biomarkers in pediatric cHL as well as current treatments and emerging targeted therapies. We also briefly present treatment complications and secondary malignant neoplasms (SMNs) that can occur in long-term survivors of HL.
EPIDEMIOLOGY AND RISK FACTORS
Childhood HL represents 6% of all cancers and has an incidence rate of 12 cases/million/year in the 0-14 year age group with a typical male predominance [13]. Among the demographics of different age groups, HL shows a characteristic bimodal distribution, with the first, larger peak seen for adolescents and young adults (15-24 year age group) and a second, smaller peak occurring for adults (around 59 years) (Table 1) [14, 15]. The MC histological subtype, mostly associated with Epstein-Barr virus (EBV), is observed primarily in young children and represents about 20% of HL; whereas, the NS subtype is seen predominantly in adolescents and young adults and represents about 75% of HL [16]. Although EBV positivity is not a direct finding of HL, tumor cells are infected with EBV in approximately 30% and 90% of all HL cases in developed and developing countries, respectively [16, 17]. Two key EBV genes involved in the etiology of HL are: latent membrane protein (LMP) 1 (lmp1), which induces constitutive nuclear factor-kappaB (NF-κB) activation by mimicking CD40 receptor [18], and lmp2A, which can take over the function of the B-cell receptor (BCR) [19]. Various findings revealed raised antibody titers and EBV DNA detection in HL patients that are suggestive of association of EBV with HL [20, 21]. Additionally, the increased deregulation of the NF-κB pathway in EBV+ patients relative to EBV- patients is consistent with a role for EBV in the development of HL [22]. In EBV- cases signaling events are complemented by mutations in tumor necrosis factor alpha-induced protein 3 (TNFAIP3), which encodes the NF-κB inhibitor A20 [23]. Despite these findings, the prognostic significance of EBV positivity is puzzling, and is less well investigated in pediatric HL patients. Only a few studies have reported a direct prognostic significance of EBV positivity in HL [24–26], whereas numerous studies reported either no association or better clinical outcome of EBV+ pediatric HL [27–29]. The different outcomes might be attributed to variations in the presence of EBV+ HL that is related to geography, age, ethnicity, and histological type.
Table 1. Comparison of demographic and clinical characteristics between different age groups of Hodgkin lymphoma (HL) patients.
| Characteristics [Ref] | Pediatric HL | AYA (Adolescent and young adult) HL | Adult HL |
|---|---|---|---|
| Age | 0-14 | 15-34 | >35 |
| Prevalence [13] | 6/million/yr | 32/million/yr | 28/million/yr |
| Predominant gender [13, 149] | Male | Female (15-19), Male (<20) | Male (common: 55-74 yr) |
| Histology [16] | Mixed-cellularity Hodgkin lymphoma (22%) | Nodular sclerosis Hodgkin lymphoma (76%) | Nodular sclerosis Hodgkin lymphoma (61%) |
| Predominant stage at Diagnosis [16] | II | II | II |
| Most common symptoms [16, 150] | Painless adenopathy involving supraclavicular or cervical area (80%) | Asymptomatic mediastinal disease | Asymptomatic lymphadenopathy (<80%), B symptoms (40%), Intermittent fever (35%) |
| EBV association [151] | Yes (<10yr: 80%) | No (<30%) | No (20-50 yr), Yes (>60yr; 70%) |
| Treatment outcome (5 yr OSa) [16, 149] | 90-95% | 90% | 85.90% |
OS, Overall Survival
Epidemiologic studies linked the risk of childhood HL to limited social contact, fewer siblings, low housing density, immunodeficiency, viral infection, high fetal growth rate, and familial HL (4.5% of cases) [30–32]. Genetic susceptibility to HL is strongly supported by a twin study of lymphomas, wherein, monozygotic twins of HL patients had nearly a 100-fold increased risk of HL, but dizygotic twins had no excess risk suggestive of co-inheritance of genetic variants [33]. The co-inheritance of genetic variants and shared childhood environmental effects could confer HL susceptibility was further proven by the 3-fold elevated risk among first-degree relatives of HL patients [32]. The highest familial risk (81-fold) was found in first-degree relatives with a concordant LR subtype [32]. Other genetic risk factors that are associated with familial predisposition to HL, including single nucleotide polymorphisms, are reviewed elsewhere [34].
CURRENT STAGING
The Ann Arbor staging system with Cotswolds' modification is the current standard for staging HL in children. This system includes four stages wherein Stages I and II indicate limited disease while stages III and IV as advanced disease. Stage I constitutes 19%, stage II 49%, stage III 19%, and stage IV 13% of all pediatric HL cases [16]. HL patients are further stratified into 3 risk groups based on disease stage and extent, disease bulk, and systemic B symptoms (e.g., unexplained persistent fevers, weight loss, or drenching night sweats). According to the Children's Oncology Group (COG) classification, the low-risk group includes patients with stage IA/IIA without bulk or extranodal extension (E) while patients at intermediate-risk are included in a broad group that encompasses stage IA bulk or E, IB, IIA bulk or E, IIB, IIIA and IVA. High-risk includes all stage IIIB and IVB patients.
Staging and monitoring of pediatric HL patients is often done using fluorodeoxyglucose (FDG) positron emission tomography (PET)-computed tomography, which integrates functional and anatomic tumor characteristics to help delineate radiotherapy (RT) margins and provide a baseline scan for subsequent response assessment. The primary goals of FDG-PET for interim assessment after initial cycles of chemotherapy are to further refine risk classification and to identify patients who are either cured and do not require RT or need escalated treatment. However, there is no consensus definition and optimum time point for response assessment. In advanced-stage patients, bilateral bone marrow aspirate and biopsies are also required for staging of pediatric HL. Surveillance scans are recommended after completion of therapy to identify early relapse in high-risk patients. The Childhood Hodgkin Lymphoma International Prognostic Score (CHIPS) system described four clinical factors that are predictive of worse event-free survival (EFS): stage IV disease, large mediastinal adenopathy, albumin level < 3.5 g/dL, and fever [35]. These findings provided the basis for prospective COG clinical trials. Most pediatric HL research groups also follow the presence of molecular and genetic factors for risk stratification, which informs treatment strategies and improves outcomes.
MOLECULAR AND GENETIC PROGNOSTIC BIOMARKERS
Although prognostic biomarkers for pediatric HL have been investigated (Table 2), none have been sufficiently informative to be of significant value in clinical practice. Thus, molecular markers that can identify high-risk patients and predict risk of treatment failure are needed. Moreover, the availability of predictive biomarkers could aid the development of possible therapeutic targets that would help balance treatment outcomes and late effects of therapy. Several putative prognostic factors linked to pediatric HL are discussed below (also see Table 2).
Table 2. Prognostic biomarkers in pediatric Hodgkin lymphoma.
| Biomarker | Function | Patients (n) | Median/ mean Age (years) | Specimen type | Method | Levels/Expression pattern | Prognosis | Associated with | Ref |
|---|---|---|---|---|---|---|---|---|---|
| p53 | cell cycle and apoptosis | 54 | 8 | FFPE | IHC | 81% patients overexpressed | No significant difference | - | [37] |
| bcl-2 | anti- and pro-apoptosis | 104 | 14 | TMA | IHC | 52.4% patients positive | (↓) 0.8 fold 10 yr EFS (P = 0.048) in high risk | unfavorable risk | [36] |
| Ki-67 | proliferation | 121 | 8 | pretreatment LN biopsy | IHC | 100% cases nuclear positive expression | (↓) 0.7 fold 5 yr FFS in low vs. high PI (cutoff 74%, P = 0.016) | - | [43] |
| Ki-67 | proliferation | 224 | 13.7 | biopsy | IHC | 100% patients positive | No significant difference | No significant correlation | [41] |
| IL-10/IL-12 | anti-inflammatory/pro-inflammatory | 30 vs. 30 controls | 15.4 | pretreatment serum | ELISA | (↑) > 2 fold in IL10 and IL-12 levels in tumors vs. healthy control (p < 0.00001) | - | general symptoms | [44] |
| CD30 + cells | proliferation | 96 | 14 | TMA | IHC | 45% cases positive with >5% cellularity | (↓) EFS in high vs. low CD30+ cells (cutoff 5%, P = 0.048) | - | [46] |
| sCD30 | proliferation | 303 | - | pretreatment serum | ELISA | (↑) 78.2% patients (> 20 U/mL) | (↓) 0.6 fold EFS high vs. low CD30 levels (cutoff 100 U/mL, P < 0.001) | stage, B symptoms, tumor burden | [47] |
| ICAM-1 (CD-54) | cell-cell adhesion, cell-mediated cytotoxicity | 69 vs. 32 | 14 | pretreatment serum | ELISA | (↑) 2 fold in tumors vs. normal controls (P = .0001); (↓) in patients from diagnosis to CR (P < 0.0001) | (↓) DFS in high vs. low ICAM-1 levels (cutoff 500 ng/ml, P = 0.016) | advanced disease, B symptom, higher ESR, relapse | [51] |
| ICAM-1 (CD-54) | cell-cell adhesion, cell-mediated cytotoxicity | 12 vs. 8 controls | 7.4 | pretreatment serum | ELISA | (↑) ∼7 fold in tumors vs. control (p < 0.000) | (↑) bad outcome (death) in high ICAM-1 levels (1,894.9 +/− 149.8 ng/ml, P = 0.009) | B symptoms, LDH levels | [52] |
| ICAM-1 (CD-54) | cell-cell adhesion, cell-mediated cytotoxicity | 10 vs. 12 controls | - | pretreatment serum | ELISA | (↑) 2 fold in tumors vs. control (p < 0.01) | (↓) 0.4 fold 3 yr survival in high vs low ICAM-1 levels (median 286.4 ng/ml, P < .05) | high ESR | [50] |
| CD-44 | metastasis | 16 vs. 12 controls | FFPE | IHC | 75% positivity in advanced stage | - | high serum levels | [54] | |
| pretreatment serum | ELISA | (↑) ∼2 fold in tumors vs. control (p < 0.001); (↓) in patients from diagnosis to CR (P < 0.05) | (↓) 0.2 fold 3 yr OS in high vs. low ICAM-1 levels (median 1627 ng/ml, P = 0.03) | high ESR, B-symptoms, advanced-stage disease | |||||
| CD-44 | metastasis | 18 vs. 20 controls | - | pretreatment serum | ELISA | (↑) ∼2 fold in tumors vs. control (p = 0.001) | - | LDH levels, advanced disease | [53] |
| α-1-antitrypsin | protease inhibitor | 22 | 14.7 | pretreatment serum | SELDI | (↑) 3.5 fold mean Intensity in stage IV sera vs. stage II | - | advanced stage | [56] |
| NK cells | immunosurveillance | 38 | 8.5 | tissue sections | IHC | Mean CD-57+ cell number 173.42; (↓) 2 fold in relapsed cases | (↓) EFS in low vs. high CD-57+ cells (cutoff 150, P = 0.0207). | - | [57] |
| NF-kB | lymphocyte proliferation and survival | 102 | 15 | pretreatment LN biopsy | IHC | (↑) cytoplasmic NF-κB2 in H/RS cells vs. controls | (↓) EFS in increased Rel-B (P = 0.009), NIK (P = 0.015), and A20 (P = 0.03) expression | slow response to therapy | [22] |
| Heparanase | metastasis and angiogenesis | 19 | - | pre and post treatment blood | ELISA | (↑) 6 fold Heparanase in patients vs. control; (↓) 1.7 fold at restaging (P = .035) | - | treatment response | [67] |
| VEGEF | angiogenesis | 22 vs. 20 controls | 13 | pretreatment blood | ELISA | (↑) > 4 fold VEGEF in patients vs. controls (P = 0.0001)* | (↑) 5 fold unsuccessful treatment in high vs. low VEGEF level (cutoff 33.4 pg/ml, P = 0.01)* | - | [70] |
| VEGEF | angiogenesis | 19 | 10.3 | pre and post treatment blood | ELISA | (↑) > 6 fold plasma levels at diagnosis vs. controls (p < .0001) | - | treatment response | [66] |
| CD-68+ cells | survival and metastasis | 74 | - | TMA | IHC | 100% cases >5% CD68+ macrophages; 86% cases >25% macrophages | - | EBV positivity | [74] |
| CD-163+ cells | survival and metastasis | 100 | 14 | TMA | IHC | CD68+ve cells number 290.81 cells/mm2; CD163+ve cells number 164.1 cells/mm2 | (↓) PFS in EBV- cases with high CD163+ cells; (↓) 5 year OS in high vs. low CD163/CD8 ratio (cutoff 2, P = 0.005) | histological type: MC | [72] |
Abbreviations : CR, Complete Remission; DFS, Disease-free Survival; EBV, Epstein-Barr Virus; EFS, Event Free Survival; ELISA, Enzyme-linked Immunosorbent Assay; ESR, Erythrocyte Sedimentation Rate; FFPE, Formalin-fixed, Paraffin-embedded; H/RS, Hodgkin and Reed Sternberg; HL, Hodgkin Lymphoma; IHC, Immunohistochemistry; ISH, In Situ Hybridization; LDH, Lactate Dehydrogenase; LN, Lymph Node; MC, Mixed Cellularity; NF-kB, Nuclear Factor Kappa-light-chain-enhancer of Activated B Cells; OS, Overall Survival; PFS, Progression Free Survival; RT-qPCR, Quantitative Reverse Transcription Polymerase Chain Reaction; SD, Standard Deviation; SELDI, Surface-enhanced Laser Desorption/Ionization; TMA, tumor associated macrophages
Combined results of HL and NHL
Bcl-2
B-cell leukemia/lymphoma 2 (Bcl-2) confers a protective advantage by mediating LMP-1-driven immortalization and enabling cells to evade programmed cell death. Approximately 50% of pediatric HL patients express Bcl-2 [36], and this Bcl-2 positivity is associated with decreased EFS in low-risk patients, and unfavorable risk patients in multivariate analysis [36]. However, some reports suggested that Bcl-2 expression did not affect outcomes [24, 37], and thus the prognostic value of Bcl-2 expression in HL is unclear.
Ki-67
Several studies documented that high expression levels of the cell proliferation marker Ki-67 are common in HL [38–40] and can negatively impact outcome [40]. However, an immunohistochemical study of 224 pediatric patients enrolled in the German Society for Pediatric Oncology and Hematology (GPOH) HD-90 and HD-95 trials found that high Ki-67 expression in HRS cells as well as lymphocytic and histiocytic cells was not related to either advanced clinical stage or poor clinical outcome. Meanwhile, expression of the cell cycle marker repp86, which generally becomes detectable at the G1-S boundary, was low, implying that HRS or lymphocytic and histiocytic cells are arrested in the G1 phase of the cell cycle [41]. Better outcomes in pediatric HL cases that have a high proliferative index are attributed to increased susceptibility of proliferative cells to chemotherapeutic drugs [17, 42, 43]. Therefore, the role of Ki-67 and other cell cycle antigens in HL prognosis remains elusive and requires further investigation.
IL-10 and IL-12
The Th2 cytokine IL-10 is expressed in about 30-50% of HL cases. IL-12 is involved in Th1 differentiation and is expressed in primary cHL tumors. IL-10 and IL-12 have antagonistic actions and shifts in Th1/Th2 cytokine ratios to Th2 predominance are common in cancer patients. (Figure 1). Elevated IL-10 and IL-12 levels in pediatric patients were found to have prognostic significance and were correlated with poor response to therapy, relapse, and shorter EFS and overall survival (OS) (Table 2) [44]. Additionally, IL-10/IL-12 ratios were significantly higher (p = 0.044) among symptomatic patients relative to asymptomatic patients, suggesting a role for these cytokines in HL.
Figure 1. Potential molecular biomarkers in pediatric Hodgkin lymphoma.
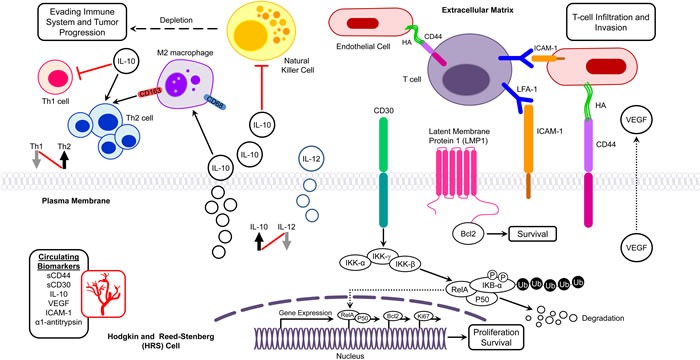
Interplay of biomarkers in different molecular networks in the tumor microenvironment of Hodgkin lymphoma presenting with infiltration, evasion, and metastasis. Bcl-2, B-cell lymphoma 2; HA, Hyaluronic acid; ICAM-1, Intercellular Adhesion Molecule 1; IκB, Inhibitor of κB; IKK, IκB kinase; LFA-1, Lymphocyte Function Antigen-1; LMP-1, latent membrane protein 1; Th, T helper; VEGEF, Vascular Growth Endothelial Factor.
CD30
Overexpression of CD30 results in ligand-independent constitutive signaling that activates the transcription factors NF-κB and activator protein-1 (AP-1), which is critical for HRS cell survival (Figure 1) [45]. Under normal physiologic conditions, CD30 is not typically expressed on most human tissues and thus its selective expression on HRS cells makes it an optimal target for directed therapy. The association of high numbers of CD30+ RS cells with poor survival is suggestive of the potential prognostic value of CD30 expression in pediatric HL [46]. The extracellular region of CD30 is proteolytically cleaved from CD30+ cells, possibly on activation by CD30L, to produce a soluble form of CD30, sCD30, which can be detected in the serum. A study by Nadali et al. showed that the presence of sCD30 in HL at presentation correlates with several clinical features, including advanced stage, B symptoms, and tumor bulkiness [47]. They also found that sCD30 serum levels > 100 U/mL at diagnosis entailed a significantly higher risk of treatment failure [47]. A comparison of low-risk patients (stage I and II) with sCD30 < 100 U/mL to those of clinically advanced stage (III and IV) patients with sCD30 levels ≥ 100 U/mL, showed a 42% decrease in EFS for those patients with higher sCD30 levels (P < 0.001) (Table 2). The elevated sCD30 levels in HL reflect the functional behavior of HRS cells, and suggest that sCD30 may play a pathophysiologic role by mediating interactions between cytokines and the tumor microenvironment.
Intercellular adhesion molecule-1 (ICAM-1, CD54)
ICAM-1 is involved in the development and progression of the malignant phenotype, and is overexpressed by HRS cells (Figure 1) [48, 49]. Serum ICAM-1 levels have been found to increase by 2-7 fold in pediatric HL patients, and levels decline or reach normal with complete remission (CR) (Table 2) [50, 51]. High serum ICAM-1 levels are also associated with advanced stages, B symptoms, higher ESR, relapses, and poor outcome [50–52]. Moreover, elevated ICAM-1 levels in patients with advanced stage disease may represent an increased host immune response to tumor cells or simply reflect a larger tumor burden.
CD44
CD44 is a hyaluronic acid receptor that is involved in tumor development (Figure 1). Elevated serum sCD44 levels in pediatric HL patients are associated with B-symptoms, advanced stages, and poor survival [53, 54]. Additionally, higher expression levels of CD44 in tumor tissues are correlated with higher serum sCD44 levels, suggesting that at least a part of sCD44 found in HL patients originated from tumor cells [54].
Alpha-1-antitrypsin (AAT)
AAT is a systemic inhibitor of neutrophil elastase (NE). An imbalance between AAT and NE may lead to tissue damage that results in tumorigenesis, invasion, and metastasis [55]. Alpha-1-antitrypsin was identified as a biomarker of tumor stage severity using surface enhanced laser desorption/ionization (SELDI-TOF) in 22 pediatric HL patients [56]. A fragment corresponding to 11.7 kDa identified as AAT was expressed to a greater extent in stage IV HL patients than in stage II patients (P = 0.03). An observed cluster of peaks was thought to represent allelic variants of AAT, which have been implicated in increased risk of multiple forms of cancer. The role of AAT in HL would benefit from additional analysis.
Natural Killer (NK) cells
Due to their cytotoxic activity against tumors in vivo, NK cells have been evaluated as prognostic markers in different cancers (Figure 1). In an immunohistochemical analysis of NK cells in 38 pediatric HL patients, a significant decrease in EFS was found among patients with low CD57+ cell counts (NK cells) compared to those with high CD 57+ cell counts [57]. In contrast, another pediatric HL study (n = 17) found no correlation between NK cell activity and prognosis [58]. The difference in these results could be attributed to the different detection methods that were used to identify NK cells.
Nuclear Factor-κB (NF-κB)
NF-κB proteins represent a family of inducible transcription factors that includes p65 (RelA), RelB, c-Rel, NF-κB1 (p105/p50), and NF-κB2 (p100/p52) [59]. These transcription factors mediate expression of genes involved in the immune response, cell proliferation, tumor metastasis, inflammation, and viral replication. NF-κB proteins form various homodimers and heterodimers, and are retained in an inactive form by cytoplasmic association with the IκB-α inhibitory protein [59]. Canonical and non-canonical pathways both lead to NF-κB activation, and both require activation of the IKK complex that consists of catalytic kinase subunits (IKKα/IKKβ) and the regulatory scaffold protein IKKγ [60].
The canonical NF-κB pathway is triggered by numerous signals, including those mediated by innate and adaptive immune receptors. Tumor necrosis factor (TNF-α) activates NF-κB via canonical phosphorylation of IKK that is mediated by the TNFRSF1A associated via death domain (TRADD), TNF receptor associated factor 2 (TRAF2), and NF-κB inducing kinase (NIK) [61]. When a cell receives a stimulatory signal, IκB-α is phosphorylated by IKK (IκB kinase) to induce IκB-α polyubiquitination and proteolytic degradation (Figure 1). Following IκB-degradation, NF-κB dimers translocate into the nucleus to activate gene transcription [62]. In the non-canonical pathway NF-κB2 (p100/RelB) complexes are inactive in the cytoplasm. The non-canonical pathway of NF-κB activation operates in response to the ligation of only certain TNFR superfamily members. In the non-canonical pathway, TRAF stimulates NIK, which subsequently activates IKK-α that in turn phosphorylates p100 leading to the processing and liberation of the p52/RelB active heterodimer [60].
Dysregulated NF-κB signaling leads to constitutive NF-κB activation, which is common in various lymphoid malignancies, and is a hallmark of HRS cells [62, 63]. In adult HL patients, studies have reported that NF-κB mutations promote constitutive activity of NF-κB signaling pathways [63, 64], but in pediatric HL, only a few studies have investigated the NF-κB pathway. The COG clinical trial AHOD0031 that examined intermediate-risk pediatric patient samples (n = 102) to clarify the role of NF-κB pathway proteins in HL found increased protein expression and nuclear localization of proteins in both the classical and alternative NF-κB pathways in pediatric patients compared to controls [22]. NF-κB pathway protein expression was dysregulated in EBV+ tumors and in patients with a slow early response to therapy. Five NF-κB proteins, including nuclear Rel-B, NIK, and A20, along with cytoplasmic Rel-A and IKK-β, were significantly associated with decreased EFS in multivariate analysis (Table 2). Notably, elevated levels of the non-canonical pathway protein NIK were also associated with a slow response to therapy (P = 0.005) [22]. In a phase II clinical trial of the proteasome inhibitor bortezomib (see below) in combination with ifosfamide/vinorelbine, nuclear phospho-RelA and RelB NF-κB subunits, as well as cytoplasmic RelB, were overexpressed in all relapsed HL patients compared to either non-malignant tissue or to HL patients at original diagnosis, but expression of these proteins was not associated with EBV status [65]. These findings suggest that the NF-κB pathway plays an essential role in pediatric HL high-risk patients.
Heparanase
Heparanase is an endoglycosidase that cleaves heparan sulfate proteoglycans (HSPG) to alter the structure of the extracellular matrix (ECM), and also plays an important role in tumor metastasis. The prognostic significance of heparanase in pediatric HL patients was demonstrated by a 6-fold increase in heparanase level at diagnosis and a subsequent decrease associated with complete remission (CR) or a good partial response (Table 2) [66, 67]. Heparanase levels remained stable in patients with poor treatment response and tumor progression, indicating its association with tumor burden.
Vascular growth endothelial factor (VEGF)
VEGF is produced by cancer cells, and its activation promotes proliferation and migration of endothelial cells with formation of new blood vessels. High levels of circulating VEGF are a well-established indicator of poor prognosis [68]. Serum VEGF levels have been reported to be elevated by 4-6 fold in pediatric HL patients compared to normal controls (P < 0.0001) (Table 2) [69, 70]. VEGF levels in 9 children correlated with the treatment response assessed after two cycles of chemotherapy, and post-therapy levels were decreased by 50% in children who were in good partial remission or CR [69]. A similar analysis revealed that the median VEGF concentration in the group with unsuccessful treatment (partial remission, progressive disease, and early relapse) was significantly higher than that in the subgroup of children who achieved CR (P = 0.02) [70]. Baseline serum VEGF concentrations among pediatric HL patients thus appear to be a promising predictive marker of response to treatment.
Tumor associated macrophages
In HL, scant HRS cells (1% of the cell population) are surrounded by a dense microenvironment consisting of a variety of reactive cells (99% of the cell population) that include T cells, B cells, plasma cells, macrophages, mast cells, dendritic cells, neutrophils, eosinophils, and fibroblasts. HRS cells attract infiltrating cells by secreting cytokines and chemokines that in turn play a role in HRS cell survival, proliferation, and inflammatory reaction [12]. HRS cells also secrete lymphotoxin-α (LTα), which stimulates endothelial cells to upregulate ICAM-1 and hyaluronan levels to enhance recruitment of T cells into the tumor milieu [71]. Recruitment of infiltrating cells is also stimulated by reactive cells- particularly macrophages and mast cells. In a Th2-shifted immunosuppressive tumor microenvironment, HRS cells are rescued from attack by Th1 cells, which consequently affect HL pathogenesis and prognosis (Figure 1).
The prognostic impact of tumor associated macrophages (TAMs) was assessed by Barros et al. and Gupta et al. in 100 and 96 pediatric HL samples, respectively [46, 72]. In contrast to the HL study of adult patients by Steidl et al. [73], neither of the two studies found any significant impact of CD68+ macrophages on disease outcome in pediatric populations. In another study, a high number of CD68+ cells in the tumor microenvironment of patients, which may be associated with the presence of EBV, was associated with a good prognosis [74]. However, Barros et al. showed that high numbers of CD163+ macrophages were associated with worse progression-free survival in EBV- cases but not in EBV+ cases (Table 2) [72]. These results indicate that the composition of TAMs in the tumor microenvironment is distinct in pediatric HL patients. In EBV+ cases, these macrophages are M1 polarized and therefore may mediate effective immune surveillance [75]. Further studies that use a combination of tumor milieu components in a comparable series of pediatric HL patients may be helpful to understand the role of TAMs in tumor progression and prognosis. The role of EBV and age as potential confounding variables should also be investigated.
A Comparison of the results of biomarker studies in pediatric HL patients indicates that NF-κB, sCD30, and ICAM-1, are most likely to be independent prognostic markers for clinical outcome predictions (Table 2), although ICAM-1 requires validation with a larger cohort. A study by Horie et al. [45] revealed that CD30 overexpression in HRS cells results in ligand independent signaling and constitutive NF-κB activation. The study also demonstrated that an adenovirus vector carrying either a decoy CD30 that lacks the cytoplasmic region or a dominant negative IkBα mutant could block NF-κB activation and induce apoptosis. Therefore, therapeutic approaches that focus on blocking CD30 and downregulating NF-κB expression could be effective therapeutic approaches for pediatric HL. Assessing levels of a combination of biomarkers such as NF-κB, sCD30, and ICAM-1 in pediatric HL patients would be helpful to gain insight into disease progression throughout the course of tumor management. A COG study (NCT01366157/AHOD11B1) is underway to assess the correlation between human GC-associated lymphoma (HGAL) protein levels and early response in low-risk pediatric HL. A comparison of HGAL protein markers in low-risk and intermediate-risk HL might provide insights into the early transforming events that lead to tumorigenesis.
Other potential biomarkers that have been investigated in adult HL are Thymus and activation-regulated chemokine (TARC), Syndecan-1 (SDC1), and Fibroblast growth factor-2 (FGF2). In adult HL, TARC correlates with disease stage, tumor burden, PET positivity, and poorer outcome, which makes it an ideal HL biomarker [76–78]. SDC-1 expression on HRS cells ranges from completely negative to 45%-100% [2, 79, 80]. Increased serum levels or expression of SDC-1 have been associated with poor outcome and adverse prognosis [81, 82]. Fibroblast growth factor-2 (FGF2) has been shown to be overexpressed in putative circulating CD15+/CD30+ cells from poor outcome HL patients [81]. The levels of FGF2 and SDC1 were 245- and 91-fold higher, respectively, in the poor outcome group compared to the good outcome group [81]. However, in pediatric patients the prognostic significance of TARC, SDC-1, and FGF2 expression has not been evaluated. Therefore, it may be useful to extrapolate the findings from adult HL patients to pediatric HL patients to help improve clinical management of high-risk patients in particular.
Genetic polymorphism
Single nucleotide polymorphisms (SNPs) within the promoter regions of cytokine genes have been associated with differential transcription levels, and may have opposing pleiotropic effects in different cancer types. Clinical and pathological features of cHL often result from cytokine imbalances that produce aberrant immune responses. Due to their effects on cytokine expression, cytokine promoter region SNPs are likely mediators of inherited risk. In an analysis of SNPs in various cytokines among 37 pediatric HL patients, the CD14-159 (C>T) SNP was suggested to be associated with pediatric HL [83]. Soluble CD14 (sCD14) is a regulatory molecule that modulates cellular and humoral responses by interacting with T and B cells [84]. Notably, CD14 is located in the same cluster on chromosome 5q31 as other Th2 cytokines, suggesting that CD14 gene polymorphisms may be involved in regulating Th2-type responses [85]. This possibility highlights the importance of a Th2 biased tumor microenvironment. In another example, a polymorphism in the untranslated region (UTR) of IL-12 (1188 3′ UTR A>C) is associated with a significant (2.8-fold) increase in risk in adolescent HL patients [86]. Moreover, IL-12 levels were lower in probands and their unaffected twins compared to controls, suggesting that decreased IL-12 levels are a potential risk factor for HL. Also, germline mutations in IL-2 inducible T cell kinase (ITK) reportedly cause EBV-associated lympho-proliferation. The novel homozygous nonsense mutation C1764G in ITK exon 16 found in 3 HL patients in a single family was shown to be a potential candidate for a heritable risk factor for EBV-associated HL [87]. Another study identified two heterozygous mutations, C49T and 922delG, in ITK genes in a pediatric patient who presented with B-cell hyperplasia followed by EBV-associated HL [88]. ITK is involved in NK cell maturation and survival, and plays a pivotal role in EBV infection responses. Mutations in the ITK gene lead to production of a truncated protein that produces aberrant immune system responses with severe clinical presentation. Although these findings suggest that ITK mutations may serve as a marker for EBV-associated HL in pediatric patients, how these mutations influence clinical and histopathological features of pediatric EBV-associated HL is currently under investigation (NCT01490801).
HL survivors are at significant risk for developing secondary malignancies. Two independent genome wide association studies (GWAS) reported that variants at chromosome 6q21 in pediatric HL patients were associated with radiation induced second malignant neoplasms (SMNs) [89, 90]. Best and colleagues revealed significant associations between SNPs on chromosome 6q21, rs4946728 (P = 0.002), and rs1040411 (P = 0.03) and the odds of developing SMNs, which increased by >3-fold and >2-fold per copy of the major allele rs4946728 and rs1040411, respectively [89]. These SNPs were also associated with decreased basal expression of the transcriptional repressor PRDM1 (PR domain containing 1, with ZNF domain, also known as BLIMP1) and impaired induction of PRDM1 expression after radiation exposure, suggesting a novel role for PRDM1 as a radiation-responsive tumor suppressor.
The glutathione S-transferase M1 (GSTM1) gene is polymorphic in humans. The frequency of the GSTM1 null genotype is approximately 50% in the U.S. and Europe. A childhood cancer survivor study (CCSS) of 650 childhood HL survivors in the U.S. and Canada demonstrated that individuals lacking GSTM1 have a significantly higher risk of developing SMN [91]. However, the magnitude of this effect was small, indicating that multiple genes may mediate genetic susceptibility to DNA damage. Therefore, additional studies will be needed to strengthen the associations of these putative risk factors with risk of SMN.
TREATMENT
The standard chemotherapy regimen of MOPP (nitrogen mustard (mechlorethamine), vincristine, procarbazine and prednisolone) provided high therapeutic success rates in the 1970s. However, two decades later adverse complications became apparent, including risks of secondary malignancy, gonadal toxicity, and sterility in females because of the inclusion of an alkylating agent (mechlorethamine) in this regimen. The combination of ABVD (adriamycin (doxorubicin), bleomycin, vinblastine, and dacarbazine) proved to be more effective than MOPP and had a lower incidence of subsequent leukemia and infertility, and eventually replaced MOPP as the standard of care in the United States. However, ABVD therapy has its own set of complications, including cardiopulmonary toxicity caused by doxorubicin and bleomycin that are subject to the cumulative dose of chemotherapeutic agents and addition of RT. To reduce the risk of long-term toxicities in younger patients, pediatric HL treatment regimens have modified from those used to treat adult HL. Contemporary therapeutic approaches for pediatric HL are based on the refinement of risk group stratification that titrate the length and intensity of chemotherapy, as well as radiation dose using response assessment made through interim or post-chemotherapy PET/CT analysis.
Treatment for favorable-risk pediatric patients
Various European and North American study groups used ABVD derivatives such as ABVE (doxorubicin, bleomycin, vincristine, etoposide) [92], OEPA (vincristine, etoposide, prednisone, and doxorubicin) for boys, OPPA (vincristine, procarbazine, prednisone, and doxorubicin) for girls [93], and VAMP (vinblastine, doxorubicin, methotrexate, and prednisone) in an effort to limit the toxicities caused by doxorubicin, bleomycin and procarbazine [94]. Based on the response to these contemporary multi-agent chemotherapy regimens, patients could also be optionally treated with low dose (15-30 Gy) involved field radiation (IFRT), which provided an EFS >90% and overall survival (OS) >95% without significant radiation or alkylator toxicity (Table 3). The occurrence of complications from high-dose RT such as cardiovascular disease, second malignancy, hypothyroidism, infertility, and impaired musculoskeletal development in pediatric survivors [95–97] motivated groups to evaluate treatment methods that omit radiation altogether for low-risk patients. Indeed, several clinical trials, evaluating response based chemotherapy by computed tomography, magnetic resonance imaging, gallium scans, and PET scans, demonstrated that radiation therapy may not be needed for low-risk patients who achieve CR after 2 cycles of chemotherapy (Table 3) [94, 98, 99]. The CCG 5942 randomized trial was the first study in children and young adults with HL that evaluated the effect of omitting RT for patients who achieved a complete response to initial COPP/ABV chemotherapy [99]. The long-term outcome of this study showed a 10-year EFS rate of 91.2% for the IFRT group and 82.9% for the no further therapy group (P = 0.004). However, the overall survival probability was similar in both randomization arms (P = 0.50) (Table 3). In a study of favorable risk patients, VAMP chemotherapy, which eliminates the alkylating agents, epipodophyllotoxins and bleomycin, resulted in a 5-year EFS of 89%, with no significant difference in outcome between those who were treated with RT (93%) and those who were not (89%) (P = 0.61), and no patient developed SMNs. However, this finding awaits long-term validation in larger cohorts [94] (Table 3). In the GPOH-HD95 trial [98], the 10-year PFS rates in low-risk patients were not statistically different for the group without RT (97.0%) compared to the RT group (92.2%) (P = 0.21), and the OS rates in all treatment groups were also promising and similar. However, the 10 year PFS rates were unsatisfactory for intermediate and advanced stage disease patients who did not receive RT. The COG trial used Rapid Early Response as a measure of chemosensitivity, which has been a promising predictor of EFS [100]. PET-guided early interim imaging is also a promising approach to reduce treatment-related toxicities among patients undergoing chemotherapy [101, 102]. The results of these studies indicate that reduced RT and modified chemotherapy regimens (cumulative doses of anthracyclines and alkylating agents) produced fewer complications, and are workable strategies to develop balanced treatment protocols through which favorable-risk patients can avoid late sequelae. Of note, the latency and frequency of secondary tumors is comparable in children treated with low dose (15-25 Gy) and high-dose IFRT [103]. Taken together, these findings appear to justify the omission of RT in early stage pediatric HL patients.
Table 3. Summary of clinical trials in low, intermediate, and high risk pediatric Hodgkin lymphoma.
| Trial Identifier | Pts. (n) | Years of Study | Chemotherapy | RT (Gy) | Risk groups | EFS, PFS % | OS % | Toxicity, RR patients (%) | Ref |
|---|---|---|---|---|---|---|---|---|---|
| NCT00025259/AHOD0031 (III) | 1712 | 2002-2012 | 4 ABVE-PC ± 2 DECA (SER patients) | RER: CR no RT; SER: 21, IF | Intermediate risk/high risk IB, IAE, IIB, IIAE, IIIA, IVA bulk/no bulk; IA, IIA with bulk | 4 yrs 85.0 4yrs 86.9 RER | 4 yrs 97.8 4 yrs 98.5 RER | hematologic (83.8); febrile neutropenia (24.9); RR patients (14.1); SMN (0.8) | [102] |
| 4 yrs 77.4 SER (P < 0.001) | 4 yrs 95.3 SER (P < 0.001) | ||||||||
| GPOHHD-95 | 1018 | 1995-2001 | 2 OPPA (girls, Group I) 2 OEPA (Boys Group II) Additional 2-4 COPP for Group 2 and 3 | CR: No RT 20 >75% tumor regression 30 <75% tumor regression | Low Risk (Group I: IA/B, IIA) | 10 yrs PFS 93.2 | 10 yrs 98.8 | RR patients (5.99); SMN (1.86) | [98] |
| Intermediate/high risk (Group 2: IIB, IIIA, IAE, IBE, IIAE) | 10 yrs PFS 85.5 | 10 yrs 95 | |||||||
| high risk (Group 3:IIIB, IV, IIBE, IIIAE, IIIBE) | |||||||||
| NCT00145600/HOD99 (II) | 88 | 2000-2008 | 4 VAMP | CR: no RT; PR: 25.5, IF | low I and II | 2 yrs 90.8 | 2 yrs 100 | Neutropenia (60) | [94] |
| NCT00002827/P9426 | 255 | 1996-2000 | 2-4 ABVE ±DRZ | 25.5, IF | low I, IIA,IIIA | 8 yrs 86.0 85.7 no DRZ 86.8 DRZ (P=0.70) | 8 yrs 97 | no DRZ (54); DRZ (68.8) Neutropenia; RR patients (11.4); SMN (1.96) | [152] |
| CCG-5942 | 826 | 1995-1998 | Group 1 : 4COPP/ABV | CR: no RT, PR: 21, IF | Low risk (Group 1:I and II) | 10 yrs 89.1 no RT 100 RT | 10 yrs 95.9 no RT 97.1 RT | RR patients (13.1); SMN (0.36) | [99] |
| Group 2: 6COPP/ABV | Intermediate Risk (Group 2: I, II, III) | 10 yrs 78.95 no RT 86.25 RT | |||||||
| Group 3: 6COPP/ABV | High Risk (Group 3: IV) | ||||||||
| NCT00004010/COG-59704 | 98 | 1999-2003 | 4 BEACOPP2 (all patients), RER: 4 COPP/ABV (Females), 2 ABVD (Males) SER: 4 BEACOPP2 | RER: Females no RT, male 21, IF SER: both females and males 21, IF | High risk IV, II/III with B symptoms and bulk | 5 yrs 94 | 5 yrs 97 | Thrombocytopenia (62); Neutropenia (83); RR patients (3.0); SMN (2.0) | [105] |
| GPOH-HD-2002 | 573 | 2002-2005 | 2 OEPA (Boys) 2 OPPA (Girls) | CR: no RT | Low Risk: (Group 1: IA, IB, IIA) | 5 yrs 92.0 | 5 yrs 99.5 | OEPA (70.5)/ OPPA (52.4) Leukopenia; OEPA (81.5)/ OPPA (57.1) Neutropenia; RR patients (1.74); SMN (1.9) | [93] |
| 2 OEPA-COPDAC (Boys) 2 OPPA-COPP (Girls) | 19.8-35, IF | Intermediate Risk: (Group 2: IE, IIB, IIAE, IIIA) | 5 yrs 87.7 | 5 yrs 96.2 | |||||
| 4 OEPA-COPDAC (Boys) 4 OPPA-COPP (Girls) | high risk: (Group 3: IIBE, IIIAE, IIIB, IVA, IVB, IVE) | ||||||||
| NCT00005578/P9425 | 216 | 1997-2004 | 5 ABVE-PC DRZ | 21 | Intermediate Risk: IB, IIA/IIIA1,IIIA2 High risk: IIB, IIIB, IV | 5 yrs 84.3 | 5 yrs 95.4 | no DRZ (29.6/ DRZ (72.6) Thrombocytopenia; no DRZ (77.8)/ DRZ (93.4) Neutropenia; no DRZ (0.9), DRZ (2.8) RR patients (11.5) | [100] |
ABVD, Doxorubicin, Bleomycin, Vinblastine, and Dacarbazine; ABVE, Doxorubicin, Bleomycin, Vincristine, and Etoposide; BEACOPP, Bleomycin, Etoposide, Doxorubicin, Cyclophosphamide, Vincristine, Procarbazine, Prednisone; COPDAC, Cyclophosphamide, Vincristine, Prednisone, and Dacarbazine; COPP, Cyclophosphamide, Vincristine, Procarbazine, and Prednisone; CR, Complete Remission; DECA, Dexamethasone, Etoposide, Cisplatin, and Cytarabine; DRZ, Dexrazoxane; IF, Involve Field; OEPA, Vincristine, Etoposide, Prednisone, and Doxorubicin; OPPA, Vincristine, Procarbazine, Prednisone, and Doxorubicin; OS, Overall survival; PFS, Progression Free Survival; PR, Partial Remission; RER, Rapid Early Response; RR, Relapsed/Refractory; RT, Radiotherapy; SER, Slow Early Response; SMN, Second Malignant Neoplasm; VAMP, Vinblastine, Doxorubicin, Methotrexate, and Prednisone
Treatment for intermediate/high-risk pediatric patients
Many pediatric oncology groups consider that the combined chemotherapy and RT modality for high-risk patients is superior to chemotherapy-only protocols as a means to avoid reaching the toxicity threshold for high cumulative doses of alkylating agents, bleomycin, and anthracyclines, as well as to reduce relapses and subsequent need for toxic salvage therapy. Adjuvant RT has been demonstrated to be effective in many clinical trials for pediatric high-risk HL patients. The Stanford/St. Jude/Dana Farber consortium investigated VAMP/COP and response-based IFRT for high-risk HL patients [104]. The poor outcomes of the study indicated that limited cumulative doses of alkylating agents and anthracycline chemotherapy in combination with low-dose IFRT are not sufficient for disease control in high-risk patients. The incorporation of dose-intensification strategies such as BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, procarbazine, and prednisone) [105] and ABVE-PC [102] (doxorubicin, bleomycin, vincristine, etoposide, prednisone, and cyclophosphamide) in intermediate/high-risk patients have shown promising outcomes, although BEACOPP has not been widely used because of its various toxicities (e.g., hematologic toxicity, cardiopulmonary toxicity, secondary malignancy, and infertility) (Table 3). However, an escalated BEACOPP regimen in adult HL patients was recently evaluated using PET-guided interim response assessment to identify poor outcome patients and direct subsequent treatment modifications [106]. In this study, all patients received two cycles of escalated BEACOPP, then underwent PET imaging. In the PET arm, patients who were PET-positive received four additional cycles of escalated BEACOPP and PET-negative patients received four cycles of ABVD. In the standard arm, patients received 6 cycles of escalated BEACOPP regardless of PET status. The estimated 2-year PFS was similar in both groups: 91.6% in the standard arm and 88.3% in the PET-driven arm (p = 0.79). Future studies will be needed to examine the feasibility of this strategy for pediatric patients with PET-positive advanced stage HL. The ABVE-PC regimen excludes procarbazine and limits the cumulative dose of epipodophyllotoxins and anthracyclines, thereby lowering the number of SMNs (0.8%) [102]. This regimen was used in a COG trial where it showed excellent 4 year EFS (85%) and OS (98%), although the relapse rate was 14% (Table 3). Pediatric Oncology Group used this dose-dense regimen in its P9425 trial with or without dexrazoxane (topoisomerase inhibitor), as a cardioprotectant to prevent anthracycline-associated cardiac toxicity during treatment. The study in advanced HL patients proved to be effective in disease control with a 5-year EFS of 84% and OS of 95%, however dexrazoxane could possibly be associated with increased risk of second malignancies and acute myeloid leukemia (AML)/myelodysplastic syndrome when used with ABVE-PC regimen [107]. The study evaluating long-term effects of dexrazoxane on secondary cancer and cardiac health among childhood HL survivors is needed to validate these results. In the GPOH-HD-2002 trial, gender-stratified chemotherapy with OEPA-COPDAC for boys and OPPA-COPP for girls was used to treat intermediate and advanced stage HL patients. In this trial, procarbazine in COPP was replaced by dacarbazine, which has reduced gonadotoxicity and has seen wide use in ABVD regimens. Response based IFRT was an integral part of the treatment for high-risk groups, even in patients who achieved CR after chemotherapy. The trial showed 5-year EFS of 89% and OS of 97.4% with SMNs occurring in ∼ 2% of cases. Both regimens were found to be interchangeable among male and female pediatric patients with intermediate and advanced stage cHL (Table 3) [93]. The Euronet trial (NCT00433459) is currently underway to compare the efficacy OEPA-COPDAC and OEPA-COPP treatments using PET-guided response assessment after two cycles of chemotherapy. In light of these clinical trial results, the challenge is to prevent relapse in early stage patients who did not receive RT and to limit the dose of alkylating agents and RT to minimize the incidence of SMNs.
Salvage therapy for relapsed/refractory (RR) pediatric patients
Although there has been remarkable progress in the treatment of pediatric HL that resulted in high cure rates, 10-25% of cases are relapse/refractory (RR) patients, which present greater challenges for clinical management. There is currently no gold standard for second line treatment, but the retrieval regimen for RR patients includes re-induction or salvage chemotherapy and, thereafter, high dose chemotherapy (HDCT) followed by autologous stem cell transplantation (ASCT). This regimen is associated with remission rates of approximately 50-65% [108, 109]. However, low-risk patients with late relapse and limited stage may be retrieved with standard dose chemotherapy (SDCT) and RT. The most common prognostic factors are chemosensitivity to initial salvage therapy [110], primary progressive disease [111, 112], time to relapse [113], and extranodal disease [114]. As with adult patients, pediatric patients also show poor prognosis, associated with time from diagnosis to first relapse of <1 year [115]. Likewise, primary progressive HL that remains refractory has a poor outcome with HDCT/ASCT in children. Therefore, alternative treatment approaches, including allogeneic stem cell transplant (alloSCT) or novel biological and targeted therapies, should be considered for these patients. In recent years, patients were more often retrieved with mini-BEAM [BCNU (carmustine), etoposide, cytarabine, melphalan] (BEAM chemotherapy in lower doses) [116]; MINE (mitoguazone, ifosfamide, vinorelbine, and etoposide) [113], or alternating IEP-ABVD (ifosfamide, etoposide, prednisolone) [109]. Reinduction regimen, ICE (Ifosfamide, carboplatin, and etoposide) in RR pediatric and adult patients has shown a combined response rate of 88% [117]. However, ICE poses a risk for myelosuppression and secondary malignancy because of alkylating agents and epipodophyllotoxins. Contemporary non-etoposide retrieval regimens in RR patients, such as the combination of ifosfamide with vinorelbine (IV) [118] or gemcitabine in combination with vinorelbine (GV) [119] resulted in overall response rates of 72-76% (Table 4). However, pediatric patients with early relapse or inadequate response rate are unlikely to achieve long-term remission with standard salvage therapy, and their survival remains very poor, ranging from 18-41% [109, 110, 115]. This low survival rate presents a therapeutic challenge for treating RR patients, and reinforces the need to test novel immune and non-immune strategies to combat HL.
Table 4. Summary of completed and ongoing clinical trials involving relapsed/refractory pediatric Hodgkin lymphoma.
| Trial Identifier | Phase, status | Agent (trade name) | Manufacturer | Patients (N) | Response (%) | Ref |
|---|---|---|---|---|---|---|
| NCT00070304/ AHOD0321 | II, completed | Gemcitabine (Gemzar), Vinorelbine (Navelbine) | Eli Lilly | 30 | 76% (1 yr EFS 59.5%; OS 86.0%) | [119] |
| NCT00006760/ AHOD00P1 | II, completed | Ifosfamide (Ifex), Vinorelbine (Navelbine) | Bristol-Myers Squibb; Pierre Fabre Pharmaceuticals, Inc. | 66 | 72% (5 yr EFS 57.2%; OS 73.9%) | [118] |
| NCT00381940 | II, completed | Bortezomib (Velcade) | Millennium | 26 | 83 | [65] |
| NCT01492088 | I/II, ongoing/recruiting | Brentuximab Vedotin (Adcetris; SGN-35) | Seattle Genetics | 16 | 64 | [153] |
| NCT01920932 | II, ongoing/ recruiting | Brentuximab Vedotin (Adcetris; SGN-35) | Seattle Genetics | - | - | |
| NCT02166463 | III, ongoing/recruiting | Brentuximab Vedotin (Adcetris; SGN-35) | Seattle Genetics | - | - | |
| NCT00994500/ ADVL0916 | I, completed | Vorinostat (Zolinza); Bortezomib (Velcade) | Merck | - | - | |
| NCT01321346 | I, completed | Panobinostat/LBH-589 (Farydak) | Novartis | - | - | |
| NCT01748721 | I, completed | MORAb-004 (Ontuxizumab) | Morphotek | - | - | |
| NCT02304458 | I/II, ongoing/recruiting | Nivolumab | Bristol-Myers Squibb | - | - |
EFS, Event Free Survival; HDAC, Histone Deacetylase; NF-kB, Nuclear Factor Kappa-light-chain-enhancer of Activated B Cells; OS, Overall Survival; PD-L1, Programmed Death-ligand 1; TEM1, Tumor Endothelial Marker 1
Targeted therapies
Due to the limited success rates of HDCT with ASCT, and the association of these regimens with significant risk of secondary malignancies, there is a need to develop alternative therapeutic approaches that are efficacious and safe for RR pediatric patients. Novel therapeutic approaches include the use of monoclonal antibodies, signal transduction inhibitors, immunotherapy, epigenetic agents such as histone deacetylase (HDAC) inhibitors and demethylating agents, and agents that target the tumor microenvironment [120]. Unfortunately, randomized clinical trials of these approaches have not yet been conducted in pediatric patients.
Brentuximab vedotin (Bv) (SGN-35) is an anti-CD30 antibody-drug conjugate that delivers the anti-tubulin agent monomethyl auristatin E (MMAE) to HRS cells where it binds tubulin to disrupt the microtubule network and induce apoptosis [121] (Figure 2). Compared to standard salvage regimens, Bv alone (as a primary salvage chemotherapy) or combined with standard therapy (as a first line therapy in treatment-naive patients) produced better outcomes (ORR ∼ 65%) and reduced toxicity in RR patients [122, 123]. In a phase I/II trial, 16 pediatric patients with RR HL were treated with Bv to achieve ORR (CR+ PR) of 64% and 21% CR. These preliminary treatment results provide hope that Bv treatment may also benefit RR pediatric HL patients. Using the same guidelines as for adult patients, several contemporary pediatric clinical trials are underway to test the effectiveness of Bv as a front-line treatment: i) Bv substituting for vincristine in the OEPA/COPDAC regimen (NCT01920932); ii) Bv in combination with AVEPC or ABVE-PC alone (NCT02166463/AHOD1331 (Table 4). Also, the combination of Bv with gemcitabine is being assessed in a phase 1/2 trial for RR pediatric and young adult HL (NCT01780662/AHOD1221). The objective of these approaches is to reduce toxicity by using a combination of agents that have distinct mechanisms of action against tumor cells.
Figure 2. Molecular targets and agents affecting specific targets in pediatric Hodgkin lymphoma.
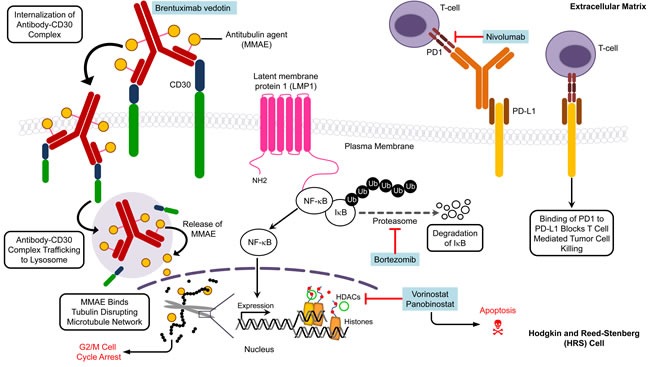
Multiple pathways are implicated in Hodgkin lymphoma and thus present potential targets for therapy. The various targets and agents illustrate the need for future clinical trials to focus on synergistic action of inhibitors to kill tumor cells. The drugs are listed in blue boxes adjacent to the corresponding target. HDAC, histone deacetylase; MMAE, monomethyl auristatin E; PD1, programmed cell death protein 1; PD-L1, programmed death-ligand 1.
Proteasome inhibitors have been used to treat HL based on their ability to block degradation of IκBα that in turn inhibits NF-κB activation. Bortezomib (Velcade, PS341) selectively inhibits the 26S proteasome to stabilize proteins that are degraded by the ubiquitin-proteasome system, including the NF-κB inhibitor IκB [124] (Figure 2). The results from a phase 2 clinical trial (AHOD0521) investigating the tolerability and efficacy of bortezomib in combination with ifosfamide and vinorelbine (IV) in 23 evaluable RR pediatric HL patients demonstrated an ORR approaching 83%, which is a marked improvement over the poor response seen with this combination in adult HL patients (Table 4) [65, 125].
To date, Bv and bortezomib have been shown to be the most effective agents to treat RR pediatric patients (Table 4) and highlights the essential role of the CD30 and NF-κB biomarkers in HL. The combination of Bv and Bortezomib may exert a synergistic cytotoxic effect that promotes downregulation of NF-κB expression (see Biomarkers section), and may represent a novel therapeutic approach to improve CD30-based targeted therapy. Moreover, an in vitro and xenograft model study revealed the synergistic cytotoxic effect of 5F11 (anti-CD30 mAb) and bortezomib in HRS cells, which again emphasizes the importance of CD30 in particular as a HL biomarker [126].
The HDAC inhibitors vorinostat and panobinostat have favorable antiproliferative activity that is manifested through cell cycle arrest and apoptosis (Figure 2). Interestingly, the anti-tumor activity of vorinostat is associated with decreased production of Th2 cytokines and chemokines, including TARC, that confers immunosuppression and protects tumor cells from cytotoxic T cells [127]. Therefore, vorinostat may sufficiently alter the tumor microenvironment to restore the Th1 anti-tumor response, and consequently suppress tumor progression and metastasis in HL patients. Panobinostat also inhibits T cell PD-1 expression in HL cell lines and RR HL patients, suggesting that this agent may promote tumor cell recognition and elimination by effector lymphocytes [128]. However, the clinical use of vorinostat and panobinostat as single agents has been of limited value in adult HL patients, and produced response rates of only 4% [129] and 27% [130], respectively. Thus, contemporary clinical trials are exploring their use in combination with other standard salvage agents in RR pediatric HL patients (NCT00994500; NCT01321346). HDAC inhibitors have been shown to interact synergistically with proteasome inhibitors to induce apoptosis [131], and studies are also underway to evaluate these findings in patients (Table 4) since combination therapies involving HDAC inhibitors and proteasome inhibitors may provide meaningful clinical outcomes.
Ontuxizumab (MORAb-004) is a recently developed humanized recombinant mouse antibody (Ab) directed against endosialin (TEM-1, CD248) [132]is predominantly expressed on the surface of cells with a mesenchymal origin, including tumor vasculature, tumor pericytes, and active tumor stromal fibroblasts [133, 134]. Inhibiting endosialin by monoclonal Ab can decrease tumor growth and tumor metastasis [135]. As such, clinical trials aimed to determine safety profiles and the optimal dose for endosialin-targeted drugs are underway in RR pediatric HL patients (NCT01748721).
Nivolumab (BMS-936558) is a monoclonal antibody that may block the PD-1 receptor expressed on peritumoral T cells. Programmed death-ligand 1 (PD-L1) is aberrantly expressed on HRS cells and interacts with PD-1 to contribute to evasion of immune detection by inhibiting T cell receptor signaling (Figure 2). Promising results from an early study in adult patients [136] prompted further investigation of nivolumab in RR pediatric HL patients (NCT02304458).
SECOND MALIGNANT NEOPLASMS (SMNS) - FOCUS ON SECONDARY LEUKEMIA
SMNs have long been known to be a major complication in long-term survivors of HL. SMNs have a serious impact, particularly in children who, at the time of treatment, are still in the growth phase and thus are more sensitive to mutagenic compounds and radiations than adults [137, 138]. Among all childhood cancer survivors, HL survivors exhibit the highest risk for SMNs, with an absolute SMN excess of 5 to 8 per 1,000 person years of follow-up [139–141]. HL survivors are at increased risk for developing secondary sarcomas and carcinomas including breast, thyroid, gastrointestinal, lung, and non-Hodgkin lymphoma (NHL) caused by RT that can occur 10-20 years after HL therapy [139, 140]. HL survivors are also more susceptible to secondary leukemia that is attributed to alkylating agents (mechlorethamine and procarbazine) or epipodophyllotoxin (topoisomerase II inhibitor- etoposide) [142].
Table 5 lists the cumulative incidences, SIR values, and mortality rates for secondary leukemia among childhood HL survivors. Notably, up to 37% of secondary malignancies that occur after HL treatment are leukemias, and of these, 70-90% are AML (Table 5). In CCSS, secondary AML affected 1.8% of HL patients, but only those cases who survived at least 5 years after primary cancer diagnosis, while leukemia can emerge as early as 2 years after HL treatment [141]. The overall cumulative incidence of secondary leukemia up to 30 years after treatment for childhood HL ranges from 0.8% to 2.1%, depending on the therapy (Table 5). The risk of developing leukemia peaks at 4 to 6 years of follow-up, declines over the next 10 years, and then plateaus at around 15-20 years [139, 140]. However, the latency period between initiating treatment with topoisomerase II inhibitors and leukemia onset is even shorter, with a median of 2 to 3 years [143]. The risk of developing secondary leukemia, including AML, following childhood treatment for HL is 10.4- to 174.8-fold greater than the risk for the general pediatric population (Table 5). However, variability in cohort size, patient classification, treatment approach, and duration of follow-up complicates the comparison of these studies. Dorffel et al. reported relatively lower standardized incidence ratio (SIR) value for secondary leukemia apparently because none of the patients in this cohort were given the alkylating agent mechlorethamine in chemotherapeutic DAL/GPOH protocols for HL (Table 5) [144, 145]. Dose intense chemotherapy [105] or ASCT [146] elevates the risk of secondary leukemias, thus the incidence of secondary leukemia is higher in relapsed patients who require prolonged maintenance treatment with multiple courses of combination chemotherapy and transplantation.
Table 5. Secondary leukemia frequency, incidence, mortality rate in pediatric Hodgkin lymphoma.
| Patients (N) | Patients with SMN/ SMN no. (%) | Patients with leukemia (n) | % leukemia of total SMNs | Mean/ Median latency (leukemia) (yrs.) | Primary HL diagnosis period | CI of leukemia at 30 yrs. unless specified | SIR (Leukemia) | Mortality (leukemia) n (%) | Ref |
|---|---|---|---|---|---|---|---|---|---|
| 2548 | 147 (5.8) | 7 (5 AML, 1 ALL, 1 CML) | 4.76 | 5.3 | 1978-2002 | 1.5* | 10.4 | 6 (85%) | [145] |
| 110 | 18 (16.3) | 4 (3 AML, 1 ALL) | 22.2 | 6.9 | 1970-90 | 4 (15 yrs plateau) | 90.9 | 4 (100%) | [103] |
| 930 | 102 (11) | 9 (7 AML, 2 CML) | 8.82 | 6.4 | 1960-90 | - | 21.49 | 9 (100%) | [154] |
| 1380 | 73 (5.3) | 27 | 36.9 | 4.3 | 1955-86 | 2.1 | 174.8 | 25 (93%) | [139] |
| 2739 | 195 (7.1) | 26 | 13.3 | 6.6 | 1935-94 | - | 33.3 | - | [140] |
| 182 | 28 (15.3) | 2 (1 AML, 1 erythroleukemia) | 7.1 | 10.68 | 1960-89 | 1.98 | 21.96 | - | [155] |
| 694 | 59 (8.5) | 8 (7 AML) | 13.55 | 4.3 | 1960-95 | - | - | 8 (100%) | [156] |
| 1380 | 79 (5.7) | 26 (24 AML, 1 ALL, 1 CML) | 32.9 | 4.4 | 1955-86 | 2.8 (15 yrs plateau) | 99.6 | 25 (96%) | [157] |
| 1641 | 62 (3.8) | 7 | 11.29 | - | 1943-87 | 0.8 | 17 | - | [158] |
ALL, Acute Lymphocytic Leukemia; AML, Acute Myeloid Leukemia; CI, Cumulative Incidence; CML, Chronic Myeloid Leukemia; SIR, Standardized Incidence Ratio; SMN, Second Malignant Neoplasm
Cumulative incidence for leukemia and NHL together
The prognosis of secondary leukemia is significantly poorer compared to other hematological malignancies with a median survival of 2.5 to 4.5 months after diagnosis, and mortality rates that often reach 100% (Table 5). The OS and EFS at 3 years, specifically for therapy-related myelodysplastic syndrome/acute myeloid leukemia (tMDS/AML) secondary to primary cancers, are about 50% lower than that for de novo AML/MDS [147]. An increased risk of tMDS/AML is associated with mutations in genes that encode detoxifying enzymes (e.g., GSTM1, GSST1, and GSTP1), drug metabolizing enzymes (cP450), and proteins involved in DNA repair and apoptosis (XRCC1, p53) [91] (see section on genetic polymorphisms). In addition to genetic factors and chemotherapeutic agents, other unknown factors are likely involved in the development of secondary AML, as suggested by the occurrence of leukemias in seven HL patients who had not received mechlorethamine or etoposide [145]. In this regard, FGFR1 overexpression has been observed in lymph node biopsies of HL patients who developed AML [148]. Thus, assessment of FGFR as a potential molecular marker in pediatric HL patients who develop leukemia would be of value. Moreover, FGF2 can be overexpressed in adult HL patients [81], which highlights the importance of this signaling pathway, particularly in high-risk patients who are more likely to develop secondary AML.
Although recent successes in treating HL have been nothing short of revolutionary, we should not overlook the incidence of SMNs. Importantly, clinicians should comprehensively evaluate risks and benefits of available therapeutic approaches, and patients must be educated about the trade-offs between different treatment regimens and the importance of follow-up. Additionally, less aggressive regimens with low oncogenic potential that involve targeted therapies and lower doses of radiation should be given serious consideration when selecting HL treatment regimens. Genetic screening for potential SNPs that are implicated in HL may also offer progress toward personalized treatments for HL.
CONCLUDING REMARKS
Most pediatric HL patients are cured with current first line risk-adapted and response-based chemotherapy. This high cure rate represents a success in the treatment of childhood cancer. Despite the high overall cure rates for HL patients, patients with early relapse or inadequate response are unlikely to achieve long-term remission with standard salvage therapy, and their prognosis remains grim. Therefore, identification of novel therapeutic targets to improve outcomes in this group remains an important challenge. The primary long-term complication of HL treatment is the development of secondary malignancy, which remains a cause for concern, especially for pediatric HL patients. As a means to avoid future side effects, recent clinical trials examined the effect of omitting RT in early stage patients. Although therapeutic sequelae and genetic factors are important in the development of secondary malignancies, other factors are involved, and, therefore, further studies are needed to identify biomarkers that indicate high-risk patients who will likely experience poor outcomes and are predisposed to develop secondary malignancies. We also need to understand the underlying mechanisms that are involved in the pathophysiology of high-risk HL and incorporate novel agents that can be personalized to achieve an initial cure and minimize late effects of HL therapy.
Acknowledgments
The authors thank V. Desai for helping with reference organization, and J. Nagpal for helpful discussions. This work was supported by grants from the Lisa B. Fishman Foundation and The John Theurer Cancer Center of Hackensack University Medical Center.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- 1.Kuppers R, Rajewsky K, Zhao M, Simons G, Laumann R, Fischer R, Hansmann ML. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A. 1994;91:10962–10966. doi: 10.1073/pnas.91.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bräuninger A, Wacker HH, Rajewsky K, Küppers R, Hansmann ML. Typing the histogenetic origin of the tumor cells of lymphocyte-rich classical Hodgkin's lymphoma in relation to tumor cells of classical and lymphocyte-predominance Hodgkin's lymphoma. Cancer Res. 2003;63:1644–1651. [PubMed] [Google Scholar]
- 3.Kanzler H, Kuppers R, Hansmann ML, Rajewsky K. Hodgkin and Reed-Sternberg cells in Hodgkin's disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. The Journal of experimental medicine. 1996;184:1495–1505. doi: 10.1084/jem.184.4.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuppers R, Schwering I, Brauninger A, Rajewsky K, Hansmann ML. Biology of Hodgkin's lymphoma. Ann Oncol. 2002;13(Suppl 1):11–18. doi: 10.1093/annonc/13.s1.11. [DOI] [PubMed] [Google Scholar]
- 5.Schwering I, Brauninger A, Klein U, Jungnickel B, Tinguely M, Diehl V, Hansmann ML, Dalla-Favera R, Rajewsky K, Kuppers R. Loss of the B-lineage-specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2003;101:1505–1512. doi: 10.1182/blood-2002-03-0839. [DOI] [PubMed] [Google Scholar]
- 6.Stein H, Marafioti T, Foss HD, Laumen H, Hummel M, Anagnostopoulos I, Wirth T, Demel G, Falini B. Down-regulation of BOB.1/OBF.1 and Oct2 in classical Hodgkin disease but not in lymphocyte predominant Hodgkin disease correlates with immunoglobulin transcription. Blood. 2001;97:496–501. doi: 10.1182/blood.v97.2.496. [DOI] [PubMed] [Google Scholar]
- 7.Tzankov A, Zimpfer A, Pehrs AC, Lugli A, Went P, Maurer R, Pileri S, Dirnhofer S. Expression of B-cell markers in classical hodgkin lymphoma: a tissue microarray analysis of 330 cases. Mod Pathol. 2003;16:1141–1147. doi: 10.1097/01.MP.0000093627.51090.3F. [DOI] [PubMed] [Google Scholar]
- 8.Muschen M, Rajewsky K, Brauninger A, Baur AS, Oudejans JJ, Roers A, Hansmann ML, Kuppers R. Rare occurrence of classical Hodgkin's disease as a T cell lymphoma. The Journal of experimental medicine. 2000;191:387–394. doi: 10.1084/jem.191.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinkus GS, Pinkus JL, Langhoff E, Matsumura F, Yamashiro S, Mosialos G, Said JW. Fascin, a sensitive new marker for Reed-Sternberg cells of hodgkin's disease. Evidence for a dendritic or B cell derivation? The American journal of pathology. 1997;150:543–562. [PMC free article] [PubMed] [Google Scholar]
- 10.Tzankov A, Bourgau C, Kaiser A, Zimpfer A, Maurer R, Pileri SA, Went P, Dirnhofer S. Rare expression of T-cell markers in classical Hodgkin's lymphoma. Mod Pathol. 2005;18:1542–1549. doi: 10.1038/modpathol.3800473. [DOI] [PubMed] [Google Scholar]
- 11.Venkataraman G, Song JY, Tzankov A, Dirnhofer S, Heinze G, Kohl M, Traverse-Glehen A, Eberle FC, Hanson JC, Raffeld MA, Pittaluga S, Jaffe ES. Aberrant T-cell antigen expression in classical Hodgkin lymphoma is associated with decreased event-free survival and overall survival. Blood. 2013;121:1795–1804. doi: 10.1182/blood-2012-06-439455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Küppers R. Molecular biology of Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program. 2009:491–496. doi: 10.1182/asheducation-2009.1.491. [DOI] [PubMed] [Google Scholar]
- 13.Howlader N NA, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, et al. SEER Cancer Statistics Review. National Cancer Institute; Bethesda, MD: 1975-2012. [Google Scholar]
- 14.Foltz LM, Song KW, Connors JM. Hodgkin's lymphoma in adolescents. J Clin Oncol. 2006;24:2520–2526. doi: 10.1200/JCO.2005.04.5823. [DOI] [PubMed] [Google Scholar]
- 15.Barista I, Varan A, Ozyar E. Bimodal age distribution in Hodgkin's disease and nasopharyngeal carcinoma. Med Hypotheses. 2007;68:1421. doi: 10.1016/j.mehy.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 16.Bazzeh F, Rihani R, Howard S, Sultan I. Comparing adult and pediatric Hodgkin lymphoma in the Surveillance, Epidemiology and End Results Program, 1988-2005: an analysis of 21 734 cases. Leukemia & lymphoma. 2010;51:2198–2207. doi: 10.3109/10428194.2010.525724. [DOI] [PubMed] [Google Scholar]
- 17.Naresh KN, Johnson J, Srinivas V, Soman CS, Saikia T, Advani SH, Badwe RA, Dinshaw KA, Muckaden M, Magrath I, Bhatia K. Epstein-Barr virus association in classical Hodgkin's disease provides survival advantage to patients and correlates with higher expression of proliferation markers in Reed-Sternberg cells. Ann Oncol. 2000;11:91–96. doi: 10.1023/a:1008337100424. [DOI] [PubMed] [Google Scholar]
- 18.Kilger E, Kieser A, Baumann M, Hammerschmidt W. Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J. 1998;17:1700–1709. doi: 10.1093/emboj/17.6.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caldwell RG, Wilson JB, Anderson SJ, Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. 1998;9:405–411. doi: 10.1016/s1074-7613(00)80623-8. [DOI] [PubMed] [Google Scholar]
- 20.Axdorph U, Porwit-MacDonald A, Sjoberg J, Grimfors G, Ekman M, Wang W, Biberfeld P, Bjorkholm M. Epstein-Barr virus expression in Hodgkin's disease in relation to patient characteristics, serum factors and blood lymphocyte function. Br J Cancer. 1999;81:1182–1187. doi: 10.1038/sj.bjc.6690827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weiss LM, Movahed LA, Warnke RA, Sklar J. Detection of Epstein-Barr viral genomes in Reed-Sternberg cells of Hodgkin's disease. N Engl J Med. 1989;320:502–506. doi: 10.1056/NEJM198902233200806. [DOI] [PubMed] [Google Scholar]
- 22.Horton TM, Sheehan AM, Lopez-Terrada D, Hutchison RE, Narendra S, Wu MF, Liu H. Analysis of NF- B pathway proteins in pediatric Hodgkin lymphoma: Correlations with EBV status and clinical outcome - A Children's Oncology Group study. Lymphoma. 2012;2012 doi: 10.1155/2012/341629. (no pagination)(341629) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmitz R, Hansmann ML, Bohle V, Martin-Subero JI, Hartmann S, Mechtersheimer G, Klapper W, Vater I, Giefing M, Gesk S, Stanelle J, Siebert R, Küppers R. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. The Journal of experimental medicine. 2009;206:981–989. doi: 10.1084/jem.20090528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akta S, Kargi A, Olgun N, Diniz G, Erbay A, Vergin C. Prognostic significance of cell proliferation and apoptosis-regulating proteins in Epstein-Barr virus positive and negative pediatric Hodgkin lymphoma. Lymphat Res Biol. 2007;5:175–182. doi: 10.1089/lrb.2007.5305. [DOI] [PubMed] [Google Scholar]
- 25.Cainap S, Rachisan A, Fetica B, Cosnarovici R, Mihut E, Popa G, Gheban D, Cainap C. EBV in pediatric neoplasia—intensity of infection as independent prognostic factor. Journal of medicine and life. 2012;5:283–287. [PMC free article] [PubMed] [Google Scholar]
- 26.Claviez A, Tiemann M, Lüders H, Krams M, Parwaresch R, Schellong G, Dörffel W. Impact of latent Epstein-Barr virus infection on outcome in children and adolescents with Hodgkin's lymphoma. J Clin Oncol. 2005;23:4048–4056. doi: 10.1200/JCO.2005.01.701. [DOI] [PubMed] [Google Scholar]
- 27.Chabay PA, Barros MH, Hassan R, De Matteo E, Rey G, Carrico MK, Renault IZ, Preciado MV. Pediatric Hodgkin lymphoma in 2 South American series: a distinctive epidemiologic pattern and lack of association of Epstein-Barr virus with clinical outcome. J Pediatr Hematol Oncol. 2008;30:285–291. doi: 10.1097/MPH.0b013e3181647bc3. [DOI] [PubMed] [Google Scholar]
- 28.Engel M, Essop MF, Close P, Hartley P, Pallesen G, Sinclair-Smith C. Improved prognosis of Epstein-Barr virus associated childhood Hodgkin's lymphoma: study of 47 South African cases. J Clin Pathol. 2000;53:182–186. doi: 10.1136/jcp.53.3.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JH, Kim Y, Choi JW, Kim YS. Prevalence and prognostic significance of Epstein-Barr virus infection in classical Hodgkin's lymphoma: a meta-analysis. Arch Med Res. 2014;45:417–431. doi: 10.1016/j.arcmed.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 30.Alexander FE, Ricketts TJ, McKinney PA, Cartwright RA. Community lifestyle characteristics and incidence of Hodgkin's disease in young people. Int J Cancer. 1991;48:10–14. doi: 10.1002/ijc.2910480103. [DOI] [PubMed] [Google Scholar]
- 31.Crump C, Sundquist K, Sieh W, Winkleby MA, Sundquist J. Perinatal and family risk factors for Hodgkin lymphoma in childhood through young adulthood. Am J Epidemiol. 2012;176:1147–1158. doi: 10.1093/aje/kws212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kharazmi E, Fallah M, Pukkala E, Olsen JH, Tryggvadottir L, Sundquist K, Tretli S, Hemminki K. Risk of familial classical Hodgkin lymphoma by relationship, histology, age, and sex: a joint study from five Nordic countries. Blood. 2015;126:1990–1995. doi: 10.1182/blood-2015-04-639781. [DOI] [PubMed] [Google Scholar]
- 33.Mack TM, Cozen W, Shibata DK, Weiss LM, Nathwani BN, Hernandez AM, Taylor CR, Hamilton AS, Deapen DM, Rappaport EB. Concordance for Hodgkin's disease in identical twins suggesting genetic susceptibility to the young-adult form of the disease. N Engl J Med. 1995;332:413–418. doi: 10.1056/NEJM199502163320701. [DOI] [PubMed] [Google Scholar]
- 34.Cerhan JR, Slager SL. Familial predisposition and genetic risk factors for lymphoma. Blood. 2015;126:2265–2273. doi: 10.1182/blood-2015-04-537498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwartz CL, Friedman DL, McCarten K, Wolden SL, Voss S, Constine LS, Chen L. Predictors of early response and event-free survival in Hodgkin lymphoma (HL): PET versus CT imaging. Journal of Clinical Oncology Conference: ASCO Annual Meeting. 2011:29. [Google Scholar]
- 36.Barros MH, Scheliga A, De Matteo E, Minnicelli C, Soares FA, Zalcberg IR, Hassan R. Cell cycle characteristics and Epstein-Barr virus are differentially associated with aggressive and non-aggressive subsets of Hodgkin lymphoma in pediatric patients. Leukemia & lymphoma. 2010;51:1513–1522. doi: 10.3109/10428194.2010.489243. [DOI] [PubMed] [Google Scholar]
- 37.Chabay P, Pesce P, De Matteo E, Lombardi MG, Rey G, Preciado MV. No Influence of bcl-2, p53, and p21waf1 protein expression on the outcome of pediatric Hodgkin lymphomas. J Pediatr Hematol Oncol. 2006;28:552–558. doi: 10.1097/01.mph.0000212955.43350.bb. [DOI] [PubMed] [Google Scholar]
- 38.Claviez A, Tiemann M, Peters J, Kreipe H, Schneppenheim R, Parwaresch R. The impact of EBV, proliferation rate, and Bcl-2 expression in Hodgkin's disease in childhood. Ann Hematol. 1994;68:61–66. doi: 10.1007/BF01715132. [DOI] [PubMed] [Google Scholar]
- 39.Joensuu H, Klemi PJ, Korkeila E. Prognostic value of DNA ploidy and proliferative activity in Hodgkin's disease. Am J Clin Pathol. 1988;90:670–673. doi: 10.1093/ajcp/90.6.670. [DOI] [PubMed] [Google Scholar]
- 40.Morente MM, Piris MA, Abraira V, Acevedo A, Aguilera B, Bellas C, Fraga M, Garcia-Del-Moral R, Gomez-Marcos F, Menarguez J, Oliva H, Sanchez-Beato M, Montalban C. Adverse clinical outcome in Hodgkin's disease is associated with loss of retinoblastoma protein expression, high Ki67 proliferation index, and absence of Epstein-Barr virus-latent membrane protein 1 expression. Blood. 1997;90:2429–2436. [PubMed] [Google Scholar]
- 41.Tiemann M, Claviez A, Lüders H, Zimmermann M, Schellong G, Dörffel W, Parwaresch R. Proliferation characteristics in pediatric Hodgkin's lymphoma point to a cell cycle arrest in the G(1) phase. Mod Pathol. 2005;18:1440–1447. doi: 10.1038/modpathol.3800466. [DOI] [PubMed] [Google Scholar]
- 42.Barros MH, Zalcberg IR, Hassan R. Prognostic impact of CD15 expression and proliferative index in the outcome of children with classical Hodgkin lymphoma. Pediatr Blood Cancer. 2008;50:428–429. doi: 10.1002/pbc.21380. author reply 430. [DOI] [PubMed] [Google Scholar]
- 43.Dinand V, Malik A, Unni R, Arya LS, Pandey RM, Dawar R. Proliferative index and CD15 expression in pediatric classical Hodgkin lymphoma. Pediatr Blood Cancer. 2008;50:280–283. doi: 10.1002/pbc.21204. [DOI] [PubMed] [Google Scholar]
- 44.Bien E, Balcerska A, Adamkiewicz-Drozynska E, Rapala M, Krawczyk M, Stepinski J. Pre-treatment serum levels of interleukin-10, interleukin-12 and their ratio predict response to therapy and probability of event-free and overall survival in childhood soft tissue sarcomas, Hodgkin's lymphomas and acute lymphoblastic leukemias. Clin Biochem. 2009;42:1144–1157. doi: 10.1016/j.clinbiochem.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 45.Horie R, Watanabe T, Morishita Y, Ito K, Ishida T, Kanegae Y, Saito I, Higashihara M, Mori S, Kadin ME. Ligand-independent signaling by overexpressed CD30 drives NF-kappaB activation in Hodgkin-Reed-Sternberg cells. Oncogene. 2002;21:2493–2503. doi: 10.1038/sj.onc.1205337. [DOI] [PubMed] [Google Scholar]
- 46.Gupta S, Yeh S, Chami R, Punnett A, Chung C. The prognostic impact of tumour-associated macrophages and Reed-Sternberg cells in paediatric Hodgkin lymphoma. Eur J Cancer. 2013;49:3255–3261. doi: 10.1016/j.ejca.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 47.Nadali G, Tavecchia L, Zanolin E, Bonfante V, Viviani S, Camerini E, Musto P, Di Renzo N, Carotenuto M, Chilosi M, Krampera M, Pizzolo G. Serum level of the soluble form of the CD30 molecule identifies patients with Hodgkin's disease at high risk of unfavorable outcome. Blood. 1998;91:3011–3016. [PubMed] [Google Scholar]
- 48.Albelda SM. Role of integrins and other cell adhesion molecules in tumor progression and metastasis. Lab Invest. 1993;68:4–17. [PubMed] [Google Scholar]
- 49.Ruco LP, Pomponi D, Pigott R, Gearing AJ, Baiocchini A, Baroni CD. Expression and cell distribution of the intercellular adhesion molecule, vascular cell adhesion molecule, endothelial leukocyte adhesion molecule, and endothelial cell adhesion molecule (CD31) in reactive human lymph nodes and in Hodgkin's disease. The American journal of pathology. 1992;140:1337–1344. [PMC free article] [PubMed] [Google Scholar]
- 50.Tacyildiz N, Yavuz G, Gozdasoglu S, Unal E, Ertem U, Duru F, Ikinciogullari A, Babacan E, Ensari A, Okcuoglu-Cavdar A. Serum levels and differential expression of intercellular adhesion molecule-1 in childhood leukemia and malignant lymphoma: prognostic importance and relationship with survival. Pediatr Hematol Oncol. 1999;16:149–158. doi: 10.1080/088800199277470. [DOI] [PubMed] [Google Scholar]
- 51.Pui CH, Hudson M, Luo X, Wilimas J, Evans W, Crist WM. Serum interleukin-2 receptor levels in Hodgkin disease and other solid tumors of childhood. Leukemia. 1993;7:1242–1244. [PubMed] [Google Scholar]
- 52.Abdelrazik N, Fouda M, Zaghloul MH, Abbas D. Serum level of intercellular adhesion molecule-1 in children with malignant lymphoma. Med Princ Pract. 2008;17:233–238. doi: 10.1159/000117798. [DOI] [PubMed] [Google Scholar]
- 53.Elli M, Dagdemir A, Bozkurt C, Pinarli FG, Duzgun A, Ozmen ZC, Ertem U, Acar S. Serum osteopontin and CD44 levels in lymphoreticular malignancies in children. Bratisl Lek Listy. 2012;113:534–538. doi: 10.4149/bll_2012_120. [DOI] [PubMed] [Google Scholar]
- 54.Taçyildiz N, Cavdar AO, Yavuz G, Gözdaoglu S, Unal E, Ertem U, Duru F, Ikinciogullari A, Babacan E, Kuzu I, Cin S. Serum levels and differential expression of CD44 in childhood leukemia and malignant lymphoma: correlation with prognostic criteria and survival. Pediatr Int. 2001;43:354–360. doi: 10.1046/j.1442-200x.2001.01415.x. [DOI] [PubMed] [Google Scholar]
- 55.Sun Z, Yang P. Role of imbalance between neutrophil elastase and alpha 1-antitrypsin in cancer development and progression. Lancet Oncol. 2004;5:182–190. doi: 10.1016/S1470-2045(04)01414-7. [DOI] [PubMed] [Google Scholar]
- 56.Qi L, Cazares L, Johnson C, de Alarcon P, Kupfer GM, Semmes OJ. Serum protein expression profiling in pediatric Hodgkin lymphoma: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2008;51:216–221. doi: 10.1002/pbc.21581. [DOI] [PubMed] [Google Scholar]
- 57.Ortaç R, Akta S, Diniz G, Erbay A, Vergin C. Prognostic role of natural killer cells in pediatric mixed cellularity and nodular sclerosing Hodgkin's disease. Anal Quant Cytol Histol. 2002;24:249–253. [PubMed] [Google Scholar]
- 58.Ikincioullari A, Dou F, Babacan E, Oflaz G, Ertem U, Yavuz G, Unal E, Gözdaolu S, Taçyildiz N, Dademir A, Cavdar AO. Natural killer cell numbers and cytotoxic activity in pediatric Hodgkin disease. Pediatr Hematol Oncol. 2000;17:133–139. doi: 10.1080/088800100276488. [DOI] [PubMed] [Google Scholar]
- 59.Sun SC, Ley SC. New insights into NF-kappaB regulation and function. Trends Immunol. 2008;29:469–478. doi: 10.1016/j.it.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev. 2012;246:125–140. doi: 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ding WX, Yin XM. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J Cell Mol Med. 2004;8:445–454. doi: 10.1111/j.1582-4934.2004.tb00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jost PJ, Ruland J. Aberrant NF-kappaB signaling in lymphoma: mechanisms, consequences, and therapeutic implications. Blood. 2007;109:2700–2707. doi: 10.1182/blood-2006-07-025809. [DOI] [PubMed] [Google Scholar]
- 63.Bargou RC, Leng C, Krappmann D, Emmerich F, Mapara MY, Bommert K, Royer HD, Scheidereit C, Dörken B. High-level nuclear NF-kappa B and Oct-2 is a common feature of cultured Hodgkin/Reed-Sternberg cells. Blood. 1996;87:4340–4347. [PubMed] [Google Scholar]
- 64.Emmerich F, Meiser M, Hummel M, Demel G, Foss HD, Jundt F, Mathas S, Krappmann D, Scheidereit C, Stein H, Dörken B. Overexpression of I kappa B alpha without inhibition of NF-kappaB activity and mutations in the I kappa B alpha gene in Reed-Sternberg cells. Blood. 1999;94:3129–3134. [PubMed] [Google Scholar]
- 65.Horton TM, Drachtman RA, Chen L, Cole PD, McCarten K, Voss S, Guillerman RP, Buxton A, Howard SC, Hogan SM, Sheehan AM, López-Terrada D, Mrazek MD, Agrawal N, Wu MF, Liu H, et al. A phase 2 study of bortezomib in combination with ifosfamide/vinorelbine in paediatric patients and young adults with refractory/recurrent Hodgkin lymphoma: a Children's Oncology Group study. Br J Haematol. 2015;170:118–122. doi: 10.1111/bjh.13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ben Arush MW, Shafat I, Ben Barak A, Shalom RB, Vlodavsky I, Vlodavsky E, Ilan N. Plasma heparanase as a significant marker of treatment response in children with Hodgkin lymphoma: pilot study. Pediatr Hematol Oncol. 2009;26:157–164. doi: 10.1080/08880010902754917. [DOI] [PubMed] [Google Scholar]
- 67.Shafat I, Barak AB, Postovsky S, Elhasid R, Ilan N, Vlodavsky I, Arush MW. Heparanase levels are elevated in the plasma of pediatric cancer patients and correlate with response to anticancer treatment. Neoplasia. 2007;9:909–916. doi: 10.1593/neo.07673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jelkmann W. Pitfalls in the measurement of circulating vascular endothelial growth factor. Clin Chem. 2001;47:617–623. [PubMed] [Google Scholar]
- 69.Ben Arush MW, Ben Barak A, Maurice S, Livne E. Serum VEGF as a significant marker of treatment response in hodgkin lymphoma. Pediatr Hematol Oncol. 2007;24:111–115. doi: 10.1080/08880010601052381. [DOI] [PubMed] [Google Scholar]
- 70.Mizia-Malarz A, Sobol G, Janowska J, Wos H, Zahorska-Markiewicz B. Prognostic value of proangiogenic cytokines in children with lymphomas. Pediatr Blood Cancer. 2009;53:1195–1199. doi: 10.1002/pbc.22179. [DOI] [PubMed] [Google Scholar]
- 71.Fhu CW, Graham AM, Yap CT, Al-Salam S, Castella A, Chong SM, Lim YC. Reed-Sternberg cell-derived lymphotoxin- activates endothelial cells to enhance T-cell recruitment in classical Hodgkin lymphoma. Blood. 2014;124:2973–2982. doi: 10.1182/blood-2014-05-576140. [DOI] [PubMed] [Google Scholar]
- 72.Barros MH, Hassan R, Niedobitek G. Tumor-associated macrophages in pediatric classical Hodgkin lymphoma: association with Epstein-Barr virus, lymphocyte subsets, and prognostic impact. Clinical cancer research. 2012;18:3762–3771. doi: 10.1158/1078-0432.CCR-12-0129. [DOI] [PubMed] [Google Scholar]
- 73.Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, Delaney A, Jones SJ, Iqbal J, Weisenburger DD, Bast MA, Rosenwald A, Muller-Hermelink HK, Rimsza LM, Campo E, Delabie J, et al. Tumor-associated macrophages and survival in classic Hodgkin's lymphoma. N Engl J Med. 2010;362:875–885. doi: 10.1056/NEJMoa0905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zameer MA, Premalata CS, Arunakumari B, Appaji L, Rao CR. Pediatric Hodgkin lymphoma in a South Indian regional cancer center: its immunomorphology, tumor-associated macrophages, and association with Epstein-Barr virus. Pediatr Hematol Oncol. 2015;32:229–238. doi: 10.3109/08880018.2014.954071. [DOI] [PubMed] [Google Scholar]
- 75.Barros MH, Segges P, Vera-Lozada G, Hassan R, Niedobitek G. Macrophage polarization reflects T cell composition of tumor microenvironment in pediatric classical Hodgkin lymphoma and has impact on survival. PLoS One. 2015;10:e0124531. doi: 10.1371/journal.pone.0124531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Farina L, Rezzonico F, Spina F, Dodero A, Mazzocchi A, Crippa F, Alessi A, Dalto S, Viviani S, Corradini P. Serum thymus and activation-regulated chemokine level monitoring may predict disease relapse detected by PET scan after reduced-intensity allogeneic stem cell transplantation in patients with Hodgkin lymphoma. Biol Blood Marrow Transplant. 2014;20:1982–1988. doi: 10.1016/j.bbmt.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 77.Plattel WJ, van den Berg A, Visser L, van der Graaf AM, Pruim J, Vos H, Hepkema B, Diepstra A, van Imhoff GW. Plasma thymus and activation-regulated chemokine as an early response marker in classical Hodgkin's lymphoma. Haematologica. 2012;97:410–415. doi: 10.3324/haematol.2011.053199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jones K, Vari F, Keane C, Crooks P, Nourse JP, Seymour LA, Gottlieb D, Ritchie D, Gill D, Gandhi MK. Serum CD163 and TARC as disease response biomarkers in classical Hodgkin lymphoma. Clinical cancer research. 2013;19:731–742. doi: 10.1158/1078-0432.CCR-12-2693. [DOI] [PubMed] [Google Scholar]
- 79.Thompson LD, Fisher SI, Chu WS, Nelson A, Abbondanzo SL. HIV-associated Hodgkin lymphoma: a clinicopathologic and immunophenotypic study of 45 cases. Am J Clin Pathol. 2004;121:727–738. doi: 10.1309/PNVQ-0PQG-XHVY-6L7G. [DOI] [PubMed] [Google Scholar]
- 80.Watanabe K, Yamashita Y, Nakayama A, Hasegawa Y, Kojima H, Nagasawa T, Mori N. Varied B-cell immunophenotypes of Hodgkin/Reed-Sternberg cells in classic Hodgkin's disease. Histopathology. 2000;36:353–361. doi: 10.1046/j.1365-2559.2000.00830.x. [DOI] [PubMed] [Google Scholar]
- 81.Gharbaran R, Goy A, Tanaka T, Park J, Kim C, Hasan N, Vemulapalli S, Sarojini S, Tuluc M, Nalley K, Bhattacharyya P, Pecora A, Suh KS. Fibroblast growth factor-2 (FGF2) and syndecan-1 (SDC1) are potential biomarkers for putative circulating CD15+/CD30+ cells in poor outcome Hodgkin lymphoma patients. J Hematol Oncol. 2013;6:62. doi: 10.1186/1756-8722-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vassilakopoulos TP, Kyrtsonis MC, Papadogiannis A, Nadali G, Angelopoulou MK, Tzenou T, Dimopoulou MN, Siakantaris MP, Kontopidou FN, Kalpadakis C, Kokoris SI, Dimitriadou EM, Tsaftaridis P, Pizzolo G, Pangalis GA. Serum levels of soluble syndecan-1 in Hodgkin's lymphoma. Anticancer Res. 2005;25:4743–4746. [PubMed] [Google Scholar]
- 83.Andrie E, Michos A, Kalampoki V, Pourtsidis A, Moschovi M, Polychronopoulou S, Athanasiadou-Piperopoulou F, Kalmanti M, Hatzakis A, Paraskevis D, Nieters A, Petridou ET. Genetic variants in immunoregulatory genes and risk for childhood lymphomas. Eur J Haematol. 2009;83:334–342. doi: 10.1111/j.1600-0609.2009.01288.x. [DOI] [PubMed] [Google Scholar]
- 84.Arias MA, Rey Nores JE, Vita N, Stelter F, Borysiewicz LK, Ferrara P, Labéta MO. Cutting edge: human B cell function is regulated by interaction with soluble CD14: opposite effects on IgG1 and IgE production. J Immunol. 2000;164:3480–3486. doi: 10.4049/jimmunol.164.7.3480. [DOI] [PubMed] [Google Scholar]
- 85.Baldini M, Lohman IC, Halonen M, Erickson RP, Holt PG, Martinez FD. A Polymorphism* in the 5′ flanking region of the CD14 gene is associated with circulating soluble CD14 levels and with total serum immunoglobulin E. Am J Respir Cell Mol Biol. 1999;20:976–983. doi: 10.1165/ajrcmb.20.5.3494. [DOI] [PubMed] [Google Scholar]
- 86.Cozen W, Gill PS, Salam MT, Nieters A, Masood R, Cockburn MG, Gauderman WJ, Martínez-Maza O, Nathwani BN, Pike MC, Van Den Berg DJ, Hamilton AS, Deapen DM, Mack TM. Interleukin-2, interleukin-12, and interferon-gamma levels and risk of young adult Hodgkin lymphoma. Blood. 2008;111:3377–3382. doi: 10.1182/blood-2007-08-106872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stepensky P, Weintraub M, Yanir A, Revel-Vilk S, Krux F, Huck K, Linka RM, Shaag A, Elpeleg O, Borkhardt A, Resnick IB. IL-2-inducible T-cell kinase deficiency: clinical presentation and therapeutic approach. Haematologica. 2011;96:472–476. doi: 10.3324/haematol.2010.033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Alme C, Satwani P, Alobeid B, Bhagat G, Kelly KM. Atypical Clinical Course in Pediatric Hodgkin Lymphoma: Association With Germline Mutations in Interleukin-2-inducible T-Cell Kinase. J Pediatr Hematol Oncol. 2015;37:507–508. doi: 10.1097/MPH.0000000000000366. [DOI] [PubMed] [Google Scholar]
- 89.Best T, Li D, Skol AD, Kirchhoff T, Jackson SA, Yasui Y, Bhatia S, Strong LC, Domchek SM, Nathanson KL, Olopade OI, Huang RS, Mack TM, Conti DV, Offit K, Cozen W, et al. Variants at 6q21 implicate PRDM1 in the etiology of therapy-induced second malignancies after Hodgkin's lymphoma. Nat Med. 2011;17:941–943. doi: 10.1038/nm.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Varszegi D, Duga B, Melegh BI, Sumegi K, Kisfali P, Maasz A, Melegh B. Hodgkin disease therapy induced second malignancy susceptibility 6q21 functional variants in roma and hungarian population samples. Pathol Oncol Res. 2014;20:529–533. doi: 10.1007/s12253-013-9724-z. [DOI] [PubMed] [Google Scholar]
- 91.Mertens AC, Mitby PA, Radloff G, Jones IM, Perentesis J, Kiffmeyer WR, Neglia JP, Meadows A, Potter JD, Friedman D, Yasui Y, Robison LL, Davies SM. XRCC1 and glutathione-S-transferase gene polymorphisms and susceptibility to radiotherapy-related malignancies in survivors of Hodgkin disease. Cancer. 2004;101:1463–1472. doi: 10.1002/cncr.20520. [DOI] [PubMed] [Google Scholar]
- 92.Tebbi CK, Mendenhall N, London WB, Williams JL, de Alarcon PA, Chauvenet AR, Group CsO Treatment of stage I, IIA, IIIA1 pediatric Hodgkin disease with doxorubicin, bleomycin, vincristine and etoposide (DBVE) and radiation: a Pediatric Oncology Group (POG) study. Pediatr Blood Cancer. 2006;46:198–202. doi: 10.1002/pbc.20546. [DOI] [PubMed] [Google Scholar]
- 93.Mauz-Körholz C, Hasenclever D, Dörffel W, Ruschke K, Pelz T, Voigt A, Stiefel M, Winkler M, Vilser C, Dieckmann K, Karlén J, Bergsträsser E, Fosså A, Mann G, Hummel M, Klapper W, et al. Procarbazine-free OEPA-COPDAC chemotherapy in boys and standard OPPA-COPP in girls have comparable effectiveness in pediatric Hodgkin's lymphoma: the GPOH-HD-2002 study. J Clin Oncol. 2010;28:3680–3686. doi: 10.1200/JCO.2009.26.9381. [DOI] [PubMed] [Google Scholar]
- 94.Metzger ML, Weinstein HJ, Hudson MM, Billett AL, Larsen EC, Friedmann A, Howard SC, Donaldson SS, Krasin MJ, Kun LE, Marcus KJ, Yock TI, Tarbell N, Billups CA, Wu J, Link MP. Association between radiotherapy vs no radiotherapy based on early response to VAMP chemotherapy and survival among children with favorable-risk Hodgkin lymphoma. JAMA. 2012;307:2609–2616. doi: 10.1001/jama.2012.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Castellino SM, Geiger AM, Mertens AC, Leisenring WM, Tooze JA, Goodman P, Stovall M, Robison LL, Hudson MM. Morbidity and mortality in long-term survivors of Hodgkin lymphoma: a report from the Childhood Cancer Survivor Study. Blood. 2011;117:1806–1816. doi: 10.1182/blood-2010-04-278796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Donaldson SS, Kaplan HS. Complications of treatment of Hodgkin's disease in children. Cancer Treat Rep. 1982;66:977–989. [PubMed] [Google Scholar]
- 97.Mauch PM, Weinstein H, Botnick L, Belli J, Cassady JR. An evaluation of long-term survival and treatment complications in children with Hodgkin's disease. Cancer. 1983;51:925–932. doi: 10.1002/1097-0142(19830301)51:5<925::aid-cncr2820510527>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 98.Dörffel W, Rühl U, Lüders H, Claviez A, Albrecht M, Bökkerink J, Holte H, Karlen J, Mann G, Marciniak H, Niggli F, Schmiegelow K, Schwarze EW, Pötter R, Wickmann L, Schellong G. Treatment of children and adolescents with Hodgkin lymphoma without radiotherapy for patients in complete remission after chemotherapy: final results of the multinational trial GPOH-HD95. J Clin Oncol. 2013;31:1562–1568. doi: 10.1200/JCO.2012.45.3266. [DOI] [PubMed] [Google Scholar]
- 99.Wolden SL, Chen L, Kelly KM, Herzog P, Gilchrist GS, Thomson J, Sposto R, Kadin ME, Hutchinson RJ, Nachman J. Long-term results of CCG 5942: a randomized comparison of chemotherapy with and without radiotherapy for children with Hodgkin's lymphoma—a report from the Children's Oncology Group. J Clin Oncol. 2012;30:3174–3180. doi: 10.1200/JCO.2011.41.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schwartz CL, Constine LS, Villaluna D, London WB, Hutchison RE, Sposto R, Lipshultz SE, Turner CS, deAlarcon PA, Chauvenet A. A risk-adapted, response-based approach using ABVE-PC for children and adolescents with intermediate- and high-risk Hodgkin lymphoma: the results of P9425. Blood. 2009;114:2051–2059. doi: 10.1182/blood-2008-10-184143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Engert A, Plütschow A, Eich HT, Lohri A, Dörken B, Borchmann P, Berger B, Greil R, Willborn KC, Wilhelm M, Debus J, Eble MJ, Sökler M, Ho A, Rank A, Ganser A, et al. Reduced treatment intensity in patients with early-stage Hodgkin's lymphoma. N Engl J Med. 2010;363:640–652. doi: 10.1056/NEJMoa1000067. [DOI] [PubMed] [Google Scholar]
- 102.Friedman DL, Chen L, Wolden S, Buxton A, McCarten K, FitzGerald TJ, Kessel S, De Alarcon PA, Chen AR, Kobrinsky N, Ehrlich P, Hutchison RE, Constine LS, Schwartz CL. Dose-intensive response-based chemotherapy and radiation therapy for children and adolescents with newly diagnosed intermediate-risk hodgkin lymphoma: a report from the Children's Oncology Group Study AHOD0031. J Clin Oncol. 2014;32:3651–3658. doi: 10.1200/JCO.2013.52.5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.O'Brien MM, Donaldson SS, Balise RR, Whittemore AS, Link MP. Second malignant neoplasms in survivors of pediatric Hodgkin's lymphoma treated with low-dose radiation and chemotherapy. J Clin Oncol. 2010;28:1232–1239. doi: 10.1200/JCO.2009.24.8062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hudson MM, Krasin M, Link MP, Donaldson SS, Billups C, Merchant TE, Kun L, Billet AL, Kaste S, Tarbell NJ, Howard S, Friedmann AM, Hurwitz CA, Young JA, Marcus KC, Rai S, et al. Risk-adapted, combined-modality therapy with VAMP/COP and response-based, involved-field radiation for unfavorable pediatric Hodgkin's disease. J Clin Oncol. 2004;22:4541–4550. doi: 10.1200/JCO.2004.02.139. [DOI] [PubMed] [Google Scholar]
- 105.Kelly KM, Sposto R, Hutchinson R, Massey V, McCarten K, Perkins S, Lones M, Villaluna D, Weiner M. BEACOPP chemotherapy is a highly effective regimen in children and adolescents with high-risk Hodgkin lymphoma: a report from the Children's Oncology Group. Blood. 2011;117:2596–2603. doi: 10.1182/blood-2010-05-285379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Casasnovas O, Brice P, Bouabdallah R, Salles AG, Stamatoullas A, Dupuis J, Reman O. Randomized Phase III Study Comparing an Early PET Driven Treatment De-Escalation to a Not PET-Monitored Strategy in Patients with Advanced Stages Hodgkin Lymphoma: Interim Analysis of the AHL2011 Lysa Study. 57th ASH Annual Meeting; Orlando, FL. 2015. [Google Scholar]
- 107.Tebbi CK, London WB, Friedman D, Villaluna D, De Alarcon PA, Constine LS, Mendenhall NP, Sposto R, Chauvenet A, Schwartz CL. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin's disease. J Clin Oncol. 2007;25:493–500. doi: 10.1200/JCO.2005.02.3879. [DOI] [PubMed] [Google Scholar]
- 108.Harris RE, Termuhlen AM, Smith LM, Lynch J, Henry MM, Perkins SL, Gross TG, Warkentin P, Vlachos A, Harrison L, Cairo MS. Autologous peripheral blood stem cell transplantation in children with refractory or relapsed lymphoma: results of Children's Oncology Group study A5962. Biol Blood Marrow Transplant. 2011;17:249–258. doi: 10.1016/j.bbmt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schellong G, Dörffel W, Claviez A, Körholz D, Mann G, Scheel-Walter HG, Bökkerink JP, Riepenhausen M, Lüders H, Pötter R, Rühl U, DAL/GPOH Salvage therapy of progressive and recurrent Hodgkin's disease: results from a multicenter study of the pediatric DAL/GPOH-HD study group. J Clin Oncol. 2005;23:6181–6189. doi: 10.1200/JCO.2005.07.930. [DOI] [PubMed] [Google Scholar]
- 110.Metzger ML, Hudson MM, Krasin MJ, Wu J, Kaste SC, Kun LE, Sandlund JT, Howard SC. Initial response to salvage therapy determines prognosis in relapsed pediatric Hodgkin lymphoma patients. Cancer. 2010;116:4376–4384. doi: 10.1002/cncr.25225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Claviez A, Klingebiel T, Beyer J, Nürnberger W, Ehninger G, Suttorp M, Dreger P, Dörffel W, Schmitz N. Allogeneic peripheral blood stem cell transplantation following fludarabine-based conditioning in six children with advanced Hodgkin's disease. Ann Hematol. 2004;83:237–241. doi: 10.1007/s00277-003-0814-y. [DOI] [PubMed] [Google Scholar]
- 112.Belgaumi A, Al-Kofide AA, Khafaga Y, Joseph N, Jamil-Malik R, Siddiqui KS, Sabbah RS. Clinical characteristics and outcome of pediatric patients with stage IV Hodgkin lymphoma. Hematology/oncology and stem cell therapy. 2009;2:278–284. doi: 10.1016/s1658-3876(09)50038-6. [DOI] [PubMed] [Google Scholar]
- 113.Gorde-Grosjean S, Oberlin O, Leblanc T, Pacquement H, Donadieu J, Lambilliotte A, Schell M, Dommange F, Munzer M, Paillard C, Schmitt C, Lutz P, Edan C, Ansoborlo S, Stephan JL, Michel G, et al. Outcome of children and adolescents with recurrent/refractory classical Hodgkin lymphoma, a study from the Société Française de Lutte contre le Cancer des Enfants et des Adolescents (SFCE) Br J Haematol. 2012;158:649–656. doi: 10.1111/j.1365-2141.2012.09199.x. [DOI] [PubMed] [Google Scholar]
- 114.Lieskovsky YE, Donaldson SS, Torres MA, Wong RM, Amylon MD, Link MP, Agarwal R. High-dose therapy and autologous hematopoietic stem-cell transplantation for recurrent or refractory pediatric Hodgkin's disease: results and prognostic indices. J Clin Oncol. 2004;22:4532–4540. doi: 10.1200/JCO.2004.02.121. [DOI] [PubMed] [Google Scholar]
- 115.Satwani P, Jin Z, Martin PL, Bhatia M, Garvin JH, George D, Chaudhury S, Talano J, Morris E, Harrison L, Sosna J, Peterson M, Militano O, Foley S, Kurtzberg J, Cairo MS. Sequential myeloablative autologous stem cell transplantation and reduced intensity allogeneic hematopoietic cell transplantation is safe and feasible in children, adolescents and young adults with poor-risk refractory or recurrent Hodgkin and non-Hodgkin lymphoma. Leukemia. 2015;29:448–455. doi: 10.1038/leu.2014.194. [DOI] [PubMed] [Google Scholar]
- 116.Linch DC, Winfield D, Goldstone AH, Moir D, Hancock B, McMillan A, Chopra R, Milligan D, Hudson GV. Dose intensification with autologous bone-marrow transplantation in relapsed and resistant Hodgkin's disease: results of a BNLI randomised trial. Lancet. 1993;341:1051–1054. doi: 10.1016/0140-6736(93)92411-l. [DOI] [PubMed] [Google Scholar]
- 117.Moskowitz CH, Nimer SD, Zelenetz AD, Trippett T, Hedrick EE, Filippa DA, Louie D, Gonzales M, Walits J, Coady-Lyons N, Qin J, Frank R, Bertino JR, Goy A, Noy A, O'Brien JP, et al. A 2-step comprehensive high-dose chemoradiotherapy second-line program for relapsed and refractory Hodgkin disease: analysis by intent to treat and development of a prognostic model. Blood. 2001;97:616–623. doi: 10.1182/blood.v97.3.616. [DOI] [PubMed] [Google Scholar]
- 118.Trippett TM, Schwartz CL, Guillerman RP, Gamis AS, Gardner S, Hogan S, London WB, Chen L, de Alarcon P. Ifosfamide and vinorelbine is an effective reinduction regimen in children with refractory/relapsed Hodgkin lymphoma, AHOD00P1: a children's oncology group report. Pediatr Blood Cancer. 2015;62:60–64. doi: 10.1002/pbc.25205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cole PD, Schwartz CL, Drachtman RA, de Alarcon PA, Chen L, Trippett TM. Phase II study of weekly gemcitabine and vinorelbine for children with recurrent or refractory Hodgkin's disease: a children's oncology group report. J Clin Oncol. 2009;27:1456–1461. doi: 10.1200/JCO.2008.20.3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Diefenbach C, Advani R. Customized targeted therapy in Hodgkin lymphoma: hype or hope? Hematol Oncol Clin North Am. 2014;28:105–122. doi: 10.1016/j.hoc.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, Rejniak SX, Gordon KA, DeBlanc R, Toki BE, Law CL, Doronina SO, Siegall CB, Senter PD, Wahl AF. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102:1458–1465. doi: 10.1182/blood-2003-01-0039. [DOI] [PubMed] [Google Scholar]
- 122.Younes A, Connors JM, Park SI, Fanale M, O'Meara MM, Hunder NN, Huebner D, Ansell SM. Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin's lymphoma: a phase 1, open-label, dose-escalation study. Lancet Oncol. 2013;14:1348–1356. doi: 10.1016/S1470-2045(13)70501-1. [DOI] [PubMed] [Google Scholar]
- 123.Moskowitz CH, Nademanee A, Masszi T, Agura E, Holowiecki J, Abidi MH, Chen AI, Stiff P, Gianni AM, Carella A, Osmanov D, Bachanova V, Sweetenham J, Sureda A, Huebner D, Sievers EL, et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin's lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2015;385:1853–1862. doi: 10.1016/S0140-6736(15)60165-9. [DOI] [PubMed] [Google Scholar]
- 124.Cvek B, Dvorak Z. The ubiquitin-proteasome system (UPS) and the mechanism of action of bortezomib. Curr Pharm Des. 2011;17:1483–1499. doi: 10.2174/138161211796197124. [DOI] [PubMed] [Google Scholar]
- 125.Mendler JH, Kelly J, Voci S, Marquis D, Rich L, Rossi RM, Bernstein SH, Jordan CT, Liesveld J, Fisher RI, Friedberg JW. Bortezomib and gemcitabine in relapsed or refractory Hodgkin's lymphoma. Ann Oncol. 2008;19:1759–1764. doi: 10.1093/annonc/mdn365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Boll B, Hansen H, Heuck F, Reiners K, Borchmann P, Rothe A, Engert A, Pogge von Strandmann E. The fully human anti-CD30 antibody 5F11 activates NF-{kappa}B and sensitizes lymphoma cells to bortezomib-induced apoptosis. Blood. 2005;106:1839–1842. doi: 10.1182/blood-2005-01-0427. [DOI] [PubMed] [Google Scholar]
- 127.Buglio D, Georgakis GV, Hanabuchi S, Arima K, Khaskhely NM, Liu YJ, Younes A. Vorinostat inhibits STAT6-mediated TH2 cytokine and TARC production and induces cell death in Hodgkin lymphoma cell lines. Blood. 2008;112:1424–1433. doi: 10.1182/blood-2008-01-133769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Oki Y, Buglio D, Zhang J, Ying Y, Zhou S, Sureda A, Ben-Yehuda D, Zinzani PL, Prince HM, Harrison SJ, Kirschbaum M, Johnston PB, Shen A, von Tresckow B, Younes A. Immune regulatory effects of panobinostat in patients with Hodgkin lymphoma through modulation of serum cytokine levels and T-cell PD1 expression. Blood Cancer J. 2014;4:e236. doi: 10.1038/bcj.2014.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kirschbaum MH, Goldman BH, Zain JM, Cook JR, Rimsza LM, Forman SJ, Fisher RI. A phase 2 study of vorinostat for treatment of relapsed or refractory Hodgkin lymphoma: Southwest Oncology Group Study S0517. Leukemia & lymphoma. 2012;53:259–262. doi: 10.3109/10428194.2011.608448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Younes A, Sureda A, Ben-Yehuda D, Zinzani PL, Ong TC, Prince HM, Harrison SJ, Kirschbaum M, Johnston P, Gallagher J, Le Corre C, Shen A, Engert A. Panobinostat in patients with relapsed/refractory Hodgkin's lymphoma after autologous stem-cell transplantation: results of a phase II study. J Clin Oncol. 2012;30:2197–2203. doi: 10.1200/JCO.2011.38.1350. [DOI] [PubMed] [Google Scholar]
- 131.Zhang QL, Wang L, Zhang YW, Jiang XX, Yang F, Wu WL, Janin A, Chen Z, Shen ZX, Chen SJ, Zhao WL. The proteasome inhibitor bortezomib interacts synergistically with the histone deacetylase inhibitor suberoylanilide hydroxamic acid to induce T-leukemia/lymphoma cells apoptosis. Leukemia. 2009;23:1507–1514. doi: 10.1038/leu.2009.41. [DOI] [PubMed] [Google Scholar]
- 132.Diaz LA, Coughlin CM, Weil SC, Fishel J, Gounder MM, Lawrence S, Azad N, O'shannessy DJ, Grasso L, Wustner J, Ebel W, Carvajal RD. A first-in-human phase I study of MORAb-004, a monoclonal antibody to endosialin in patients with advanced solid tumors. Clinical cancer research. 2015;21:1281–1288. doi: 10.1158/1078-0432.CCR-14-1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.MacFadyen JR, Haworth O, Roberston D, Hardie D, Webster MT, Morris HR, Panico M, Sutton-Smith M, Dell A, van der Geer P, Wienke D, Buckley CD, Isacke CM. Endosialin (TEM1, CD248) is a marker of stromal fibroblasts and is not selectively expressed on tumour endothelium. FEBS Lett. 2005;579:2569–2575. doi: 10.1016/j.febslet.2005.03.071. [DOI] [PubMed] [Google Scholar]
- 134.Rouleau C, Curiel M, Weber W, Smale R, Kurtzberg L, Mascarello J, Berger C, Wallar G, Bagley R, Honma N, Hasegawa K, Ishida I, Kataoka S, Thurberg BL, Mehraein K, Horten B, et al. Endosialin protein expression and therapeutic target potential in human solid tumors: sarcoma versus carcinoma. Clinical cancer research. 2008;14:7223–7236. doi: 10.1158/1078-0432.CCR-08-0499. [DOI] [PubMed] [Google Scholar]
- 135.Rybinski K, Imtiyaz HZ, Mittica B, Drozdowski B, Fulmer J, Furuuchi K, Fernando S, Henry M, Chao Q, Kline B, Albone E, Wustner J, Lin J, Nicolaides NC, Grasso L, Zhou Y. Targeting endosialin/CD248 through antibody-mediated internalization results in impaired pericyte maturation and dysfunctional tumor microvasculature. Oncotarget. 2015;6:25429–25440. doi: 10.18632/oncotarget.4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, Rodig SJ, Chapuy B, Ligon AH, Zhu L, Grosso JF, Kim SY, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372:311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ng AK, Kenney LB, Gilbert ES, Travis LB. Secondary malignancies across the age spectrum. Semin Radiat Oncol. 2010;20:67–78. doi: 10.1016/j.semradonc.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Omer B, Kadan-Lottick NS, Roberts KB, Wang R, Demsky C, Kupfer GM, Cooper D, Seropian S, Ma X. Patterns of subsequent malignancies after Hodgkin lymphoma in children and adults. Br J Haematol. 2012;158:615–625. doi: 10.1111/j.1365-2141.2012.09211.x. [DOI] [PubMed] [Google Scholar]
- 139.Bhatia S, Yasui Y, Robison LL, Birch JM, Bogue MK, Diller L, DeLaat C, Fossati-Bellani F, Morgan E, Oberlin O, Reaman G, Ruymann FB, Tersak J, Meadows AT, Group LES. High risk of subsequent neoplasms continues with extended follow-up of childhood Hodgkin's disease: report from the Late Effects Study Group. J Clin Oncol. 2003;21:4386–4394. doi: 10.1200/JCO.2003.11.059. [DOI] [PubMed] [Google Scholar]
- 140.Metayer C, Lynch CF, Clarke EA, Glimelius B, Storm H, Pukkala E, Joensuu T, van Leeuwen FE, van't Veer MB, Curtis RE, Holowaty EJ, Andersson M, Wiklund T, Gospodarowicz M, Travis LB. Second cancers among long-term survivors of Hodgkin's disease diagnosed in childhood and adolescence. J Clin Oncol. 2000;18:2435–2443. doi: 10.1200/JCO.2000.18.12.2435. [DOI] [PubMed] [Google Scholar]
- 141.Neglia JP, Friedman DL, Yasui Y, Mertens AC, Hammond S, Stovall M, Donaldson SS, Meadows AT, Robison LL. Second malignant neoplasms in five-year survivors of childhood cancer: childhood cancer survivor study. J Natl Cancer Inst. 2001;93:618–629. doi: 10.1093/jnci/93.8.618. [DOI] [PubMed] [Google Scholar]
- 142.Bhatia S. Therapy-related myelodysplasia and acute myeloid leukemia. Semin Oncol. 2013;40:666–675. doi: 10.1053/j.seminoncol.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pedersen-Bjergaard J, Philip P. Balanced translocations involving chromosome bands 11q23 and 21q22 are highly characteristic of myelodysplasia and leukemia following therapy with cytostatic agents targeting at DNA-topoisomerase II. Blood. 1991;78:1147–1148. [PubMed] [Google Scholar]
- 144.Schellong G, Riepenhausen M. Late effects after therapy of Hodgkin's disease: update 2003/04 on overwhelming post-splenectomy infections and secondary malignancies. Klin Padiatr. 2004;216:364–369. doi: 10.1055/s-2004-832340. [DOI] [PubMed] [Google Scholar]
- 145.Dörffel W, Riepenhausenl M, Lüders H, Brämswig J, Schellong G. Secondary Malignancies Following Treatment for Hodgkin's Lymphoma in Childhood and Adolescence. Dtsch Arztebl Int. 2015;112:320–327. doi: 10.3238/arztebl.2015.0320. i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Baker KS, DeFor TE, Burns LJ, Ramsay NK, Neglia JP, Robison LL. New malignancies after blood or marrow stem-cell transplantation in children and adults: incidence and risk factors. J Clin Oncol. 2003;21:1352–1358. doi: 10.1200/JCO.2003.05.108. [DOI] [PubMed] [Google Scholar]
- 147.Barnard DR, Lange B, Alonzo TA, Buckley J, Kobrinsky JN, Gold S, Neudorf S, Sanders J, Burden L, Woods WG. Acute myeloid leukemia and myelodysplastic syndrome in children treated for cancer: comparison with primary presentation. Blood. 2002;100:427–434. doi: 10.1182/blood.v100.2.427. [DOI] [PubMed] [Google Scholar]
- 148.Brusa G, Zuffa E, Hattinger CM, Serra M, Remondini D, Castellani G, Righi S, Campidelli C, Pileri S, Zinzani PL, Gabriele A, Mancini M, Corrado P, Barbieri E, Santucci MA. Genomic imbalances associated with secondary acute leukemias in Hodgkin lymphoma. Oncol Rep. 2007;18:1427–1434. [PubMed] [Google Scholar]
- 149.Ries LAG SM, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR, editors. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program. NIH Pub. No. 99-4649; Bethesda, MD: 1999. [Google Scholar]
- 150.Board PDQPTE. PDQ Cancer Information Summaries. Bethesda (MD): National Cancer Institute (US); 2002. Childhood Hodgkin Lymphoma Treatment (PDQ(R)): Health Professional Version. [Google Scholar]
- 151.Armstrong AA, Alexander FE, Cartwright R, Angus B, Krajewski AS, Wright DH, Brown I, Lee F, Kane E, Jarrett RF. Epstein-Barr virus and Hodgkin's disease: further evidence for the three disease hypothesis. Leukemia. 1998;12:1272–1276. doi: 10.1038/sj.leu.2401097. [DOI] [PubMed] [Google Scholar]
- 152.Tebbi CK, Mendenhall NP, London WB, Williams JL, Hutchison RE, Fitzgerald TJ, de Alarcon PA, Schwartz C, Chauvenet A. Response-dependent and reduced treatment in lower risk Hodgkin lymphoma in children and adolescents, results of P9426: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2012;59:1259–1265. doi: 10.1002/pbc.24279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Locatelli F NK, Rosolen A, Landman-Parker J, Aladjidi N, Beishuizen A, Daw S, Gore L, Franklin A, Fasanmade A, Wang J, Sachs J, Mauz-Koerholz C. Phase 1/2 Study Of Brentuximab Vedotin In Pediatric Patients With Relapsed Or Refractory (R/R) Hodgkin Lymphoma (HL) Or Systemic Anaplastic Large-Cell Lymphoma (sALCL): Preliminary Phase 2 Data For Brentuximab Vedotin 1.8 Mg/Kg In The HL Study. Arm Blood. 2013:4378–4378. [Google Scholar]
- 154.Constine LS, Tarbell N, Hudson MM, Schwartz C, Fisher SG, Muhs AG, Basu SK, Kun LE, Ng A, Mauch P, Sandhu A, Culakova E, Lyman G, Mendenhall N. Subsequent malignancies in children treated for Hodgkin's disease: associations with gender and radiation dose. Int J Radiat Oncol Biol Phys. 2008;72:24–33. doi: 10.1016/j.ijrobp.2008.04.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Green DM, Hyland A, Barcos MP, Reynolds JA, Lee RJ, Hall BC, Zevon MA. Second malignant neoplasms after treatment for Hodgkin's disease in childhood or adolescence. J Clin Oncol. 2000;18:1492–1499. doi: 10.1200/JCO.2000.18.7.1492. [DOI] [PubMed] [Google Scholar]
- 156.Wolden SL, Lamborn KR, Cleary SF, Tate DJ, Donaldson SS. Second cancers following pediatric Hodgkin's disease. J Clin Oncol. 1998;16:536–544. doi: 10.1200/JCO.1998.16.2.536. [DOI] [PubMed] [Google Scholar]
- 157.Bhatia S, Robison LL, Oberlin O, Greenberg M, Bunin G, Fossati-Bellani F, Meadows AT. Breast cancer and other second neoplasms after childhood Hodgkin's disease. N Engl J Med. 1996;334:745–751. doi: 10.1056/NEJM199603213341201. [DOI] [PubMed] [Google Scholar]
- 158.Sankila R, Garwicz S, Olsen JH, Döllner H, Hertz H, Kreuger A, Langmark F, Lanning M, Möller T, Tulinius H. Risk of subsequent malignant neoplasms among 1,641 Hodgkin's disease patients diagnosed in childhood and adolescence: a population-based cohort study in the five Nordic countries. Association of the Nordic Cancer Registries and the Nordic Society of Pediatric Hematology and Oncology. J Clin Oncol. 1996;14:1442–1446. doi: 10.1200/JCO.1996.14.5.1442. [DOI] [PubMed] [Google Scholar]