

Abstract
Cancer arises from complex, multi-layer interactions between diverse genetic and environmental factors. Genetic studies have identified multiple loci associated with tumor susceptibility. However, little is known about how germline polymorphisms interact with one another and with somatic mutations within a tumor to mediate acquisition of cancer traits. Here, we survey recent studies showing gene-gene interactions, also known as epistases, affecting genetic susceptibility in colorectal, gastric and esophageal cancers. We also catalog epistasis types and cancer hallmarks with respect to the interacting genes. A total of 22 gene variation pairs displayed all levels of statistical epistasis, including synergistic, redundant, suppressive and co-suppressive interactions. Five genes primarily involved in base excision repair formed a linear topology in the interaction network, MUTYH-OGG1-XRCC1-PARP1-MMP2, and three genes in mTOR cell-proliferation pathway formed another linear network, PRKAG2-RPS6KB1-PIK3CA. Discrete pairwise epistasis was also found in nucleotide excision repair, detoxification, proliferation, TP53, TGF-β and other pathways. We propose that three modes of biological interaction underlie the molecular mechanisms for statistical epistasis. The direct binding, linear pathway and convergence modes can exhibit any level of statistical epistasis in susceptibility to gastrointestinal cancers, and this is likely true for other complex diseases as well. This review highlights the link between cancer hallmarks and susceptibility genes.
Keywords: colorectal cancer, epistasis, esophageal cancer, gastric cancer, gene-gene interaction
INTRODUCTION
Mutation of oncogenes and tumor suppressor genes is a primary force underlying oncogenesis and cancer progression. To identify genetic factors involved in tumor traits, most studies have focused on tumor-associated loci that are presumed to include gene(s) responsible for cancer development. This type of research reveals germline tumor susceptibility elements. However, because tumors acquire many malignant traits through somatic mutations, the success of such approaches depends on identifying genetic variations in known oncogenes and tumor suppressor genes, and such variations strongly influencing gene expression or function.
As is common for complex diseases, development of cancer traits occurs through the interaction of multiple genes and environmental factors. Additionally, tumors actively engage with nearby cells, such as stromal and immune cells recruited to the tumor tissue, allowing tumors to acquire traits that promote survival and malignancy. Given the importance of such interactions with respect to cancer susceptibility, development and progression, we need to better understand the molecular events and cellular contexts underlying epistasis. Knowledge of how environmental elements affect disease and how gene-gene and/or gene-environment interactions promote tumorigenesis, along with phenotypic categorization of tumor-associated hallmarks, has provided a framework for understanding how genetic factors contribute to oncogenesis [1, 2].
Epistasis in disease susceptibility
Bateson coined the term ‘epistasis’ to explain interactions between two genes [3]. In this situation, the observable phenotype of one gene is masked by the other gene's effect, and the masking gene is said to be epistatic to the masked gene. Epistasis leads to deviation from simple Mendelian segregation ratios and emergence of novel phenotypes in a combination of single gene alleles. Since its first use, this term has been employed with diverse, and sometimes obscure, meaning.
In contrast to Bateson's perspective of biological epistasis, Fisher used quantitative genetics to explain the interaction between genetic loci that determine quantitative traits, rather than discrete binary traits within a group of genetically heterogeneous backgrounds [4]. Fisher's term, ‘epistacy,’ indicates a deviation from the addition of quantitative effects or phenotypes of two alleles in a given population.
In ‘statistical epistasis,’ the observed phenotype resulting from the interaction of two genes can be influenced both by allele frequency within a population and penetrance of the effects, which should still be visible after averaging multi-locus effects. Because the phenotype of interest, e.g. disease incidence, is arbitrarily determined by the investigator, genes in epistatic relationships can be discovered only when the effect of the combined genes is great enough to reach a penetrance threshold, resulting in a visible phenotype.
In this review, we investigate the interaction between two genes with statistically defined epistasis. We set the measure of individual function or effect of each allele as the disease susceptibility in a population, thus making it an odds ratio (OR). When the effects of particular alleles in two genes are OR1 and OR2, the phenotype observed with a combination of both alleles is represented as ORcombined, while the epistatic deviation from the simple multiplication of two ORs for the individual gene alleles is represented as ORinteraction = ORcombined / (OR1 × OR2).
Synergism and antagonism in epistasis
Epistasis can be either ‘synergistic’ or ‘antagonistic,’ if not ‘null,’ (Figure 1) according to whether the outcome of the genetic interaction potentiates or diminishes the effect of the individual gene alleles [4–6]. Antagonistic interactions can be further divided into ‘redundant,’ ‘suppressive’ and ‘co-suppressive’ interactions depending on the extent to which ORcombined departs from the two individual ORs. Thus, levels of non-null epistasis can be described as synergistic, redundant, suppressive or co-suppressive (Figure 1).
Figure 1. Quantitative levels of statistical epistasis.
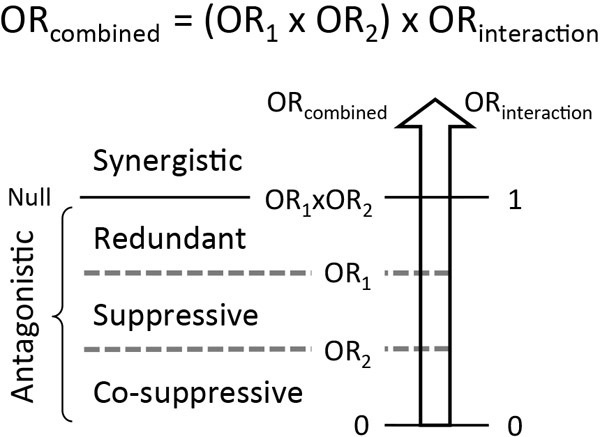
Epistasis can be synergistic or antagonistic, if not null, and antagonistic epistasis can be redundant, suppressive or co-suppressive. OR represents the effect of each gene allele on disease susceptibility. ORcombined is OR observed for a combination of two gene alleles. ORinteraction is deviation of ORcombined from (OR1 × OR2). If ORcombined = (OR1 × OR2), or ORinteraction = 1, the interaction is ‘null,’ as the two individual gene allele effects are each additive to the other, with no interaction between them. Therefore, they act independently of each other. If ORcombined > (OR1×OR2), or ORinteraction > 1, the interaction is considered ‘synergistic,’ as the two individual alleles, when combined, act synergistically. Conversely, if ORcombined < (OR1×OR2), or ORinteraction < 1, two gene alleles have an ‘antagonistic’ effect. Antagonistic interactions can be further subcategorized into redundant, suppressive and co-suppressive interactions depending on the extent to which ORcombined departs from the two individual ORs, OR1 and OR2. If ORcombined ≥ OR1 ≥ OR2, the interaction is considered ‘redundant,’ as one gene allele effect is redundant with the other gene allele effect. If OR1 > ORcombined ≥ OR2, the interaction is considered ‘suppressive’ because one gene allele suppresses the other's effect. Lastly, if ORcombined ≤ OR2 < OR1, the interaction is ‘co-suppressive,’ as both gene alleles suppress each other.
To achieve synergism or antagonism, cooperation or a feedback loop is triggered when the combined effect of two interacting gene alleles surpasses a threshold necessary for activation of the feedback loop. In this feedback threshold model, a positive feedback loop potentiates the system and results in synergism, whereas a negative feedback loop diminishes the combined effect of the two alleles and leads to antagonism.
Synergism and antagonism may also be determined by qualitative regulation. When the interaction of two gene alleles, but not the individual activities of these alleles, results in a ‘gain of function’ with a novel ‘Gestalt’ product or activity that either enhances or suppresses the system, synergism or antagonism may ensue, respectively. The interaction scheme whereby two gene products operate in a linear pathway and the upstream gene allele effect is dominant over the downstream gene allele effect (i.e., if the observed ORcombined is the same as the OR of the dominant gene allele) is explained by a ‘dominance’ model in which the upstream gene allele effect masks the downstream gene allele effect.
Review scope
We searched the PubMed, Google Scholar and Web of Science databases for studies associating statistical epistasis with susceptibility to gastrointestinal cancers, such as colorectal (colon, rectal or both), gastric (stomach) and esophageal (gullet) cancers. Sixteen studies met our selection criteria among those published before July 8, 2015. Search criteria and results are summarized in Table 1, where all 22 reported epistasis pairs are grouped into three categories: (i) interactions between cancer-associated genes, (ii) interactions between cancer-associated and non-associated genes and (iii) interactions between non-associated genes.
Table 1. Gene-gene interactions in gastrointestinal cancer susceptibility.
| Gene 1 | Gene 2 | Cancer | n (case/control) | ||||
|---|---|---|---|---|---|---|---|
| Gene name (variation)a | ORb | Gene name (variation)a | ORb | ORcombinedb | ORinteractionc | typed | ethnicity [reference]e |
| Interaction between cancer-associated gene variations | |||||||
| PARP1 (p.Val762Ala) | 2.17 | XRCC1 (p.Arg399Gln) | 1.61 | 6.43 | 1.84 | GC | 500/1000 Chinese [17] |
| ADH1B (p.Arg47His) | (3.34) | ALDH2 (p.Glu504Lys) | (2.51) | 13.46 (8.08) | 1.60' (0.96) | EC | 4220/8946 Asian [30] |
| ADH1B (p.Arg47His) | 1.85 | ALDH2 (p.Glu504Lys) | 1.66 | 6.79 | 2.21 | EC | 1070/2836 Japanese [29] |
| MDM2 (rs2279744 T>G) | 1.94 | TP53 (p.Arg72Pro) | 1.72 | 5.05 | 1.51 | GC | 500/1000 Chinese [59] |
| MUTYH (p.Gln324His) | 3.35 | OGG1 (p.Ser326Cys) | 1.83 | 8.31 | 1.36 | CRC | 182/200 Polish [9] |
| XRCC1 (p.Arg399Gln) | 2.03 | OGG1 (p.Ser326Cys) | 1.83 | 4.97 | 1.34 | CRC | 182/200 Polish [9] |
| TP53 (p.Arg72Pro) | (1.78) | MDM2 (rs2279744 T>G) | (1.46) | 3.10 | 1.19' | EC | 758/1420 Chinese [58] |
| CHEK2 (quadruple)f | (1.38) | CDKN1B (p.Val109Gly) | (1.34) | (2.14) | (1.16) | CC | 872/3812 Polish [72] |
| TPH2 (rs10879357 G>A) | (1.22) | CASC20 (rs1571218 G>T) | (1.20) | 1.63 (1.55) | 1.11' (1.06) | CRC | 10907/13216 mixed [81] |
| SMAD7 (rs11874392 T>A) | 1.47 | TGFBR1 (rs6478972 A>G) | 1.38 | 1.44 (1.50) | 0.71 (0.74) | CRC | 794/842 Chinese [69] |
| Interaction between cancer-associated and non-associated gene variations | |||||||
| CYP1A1 (p.Ile462Val) | (2.87) | CYP2E1 (rs2031920 C>T)g | (1.62) | (11.25) | (1.85) | EC | 526/526 Kashmir [48] |
| PRKAG2 (rs1104897 G>A) | (1.30) | RPS6KB1 (rs180515 A>G) | (1.05) | (1.63) | (1.19) | RC | 791/999 mixedh[73] |
| NQO1 (p.Pro187Ser) | 6.65 | NQO2 (rs2070999 G>A) | 1.48 | 11.41 | 1.16 | EC | 135/195 Kashmiri [37] |
| MUTYH (p.Gln324His) | (4.05) | XRCC1 (p.Arg194Trp) | (1.24) | (5.65) | (1.13) | CRC | 182/200 Polish [9] |
| GSTT1 (Δ) | (1.81) | APEX1 (p.Asp148Glu) | (1.21) | 1.84 | 0.84' | GC | 314/548 Italian [23] |
| PIK3CA (rs7640662 C>G) | (1.31) | RPS6KB1 (rs180519 G>A) | (1.13) | (1.19) | (0.80) | RC | 791/999 mixedh [73] |
| GSTM1 (Δ) and GSTT1 (Δ) | (2.50) | APEX1 (p.Asp148Glu) | (1.21) | 2.32 | 0.77' | GC | 314/548 Italian [23] |
| GSTM1 (Δ) | (2.24) | NAT2 (p.Arg197Gln) | (1.81) | (2.82) | (0.70) | CRC | 150/162 Romanian [45] |
| PARP1 (p.Val762Ala) | (2.19) | MMP2 (rs243865 C>T) | (1.05) | (1.38) | (0.60) | GC | 59/320 Korean [79] |
| OGG1 (p.Ser326Cys) | (1.71) | XRCC1 (p.Arg194Trp) | (1.22) | (0.92) | (0.44) | CRC | 182/200 Polish [9] |
| Interaction between non-associated gene variations | |||||||
| XPC (PAT S>L) | (1.20) | XPA (rs1800975 G>A) | (1.02) | 2.15 (1.55) | 1.76' (1.27) | GC | 314/548 Italian [23] |
| MDM2 (rs2279744 T>G) | (1.11) | TP53 (p.Arg72Pro) | (1.06) | 1.78 (1.76) | 1.52' (1.50) | CRC | 444/569 Han Chinese [57] |
The interacting gene variant pairs are listed in descending order of ORinteraction value, shown in the sixth column. Nonsynonymous SNPs are denoted by the encoded amino acid changes, while the other variations are denoted by the reference SNP (rs) numbers. Risk-associated alleles that are more frequent in cancer cases than in healthy controls are shown in bold.
Odds ratios (ORs) of the risk-associated alleles for cancer susceptibility are shown. ORs of non-significant associations are italicized. If the publication presented OR of non-risk allele rather than risk allele, crude OR of risk allele was calculated using the χ2 test and is shown here in parenthesis.
ORinteraction = ORcombined ÷ (OR1 × OR2). When an adjusted ORinteraction value was not reported in the publication, OR'interaction (marked with a prime) was calculated using the reported adjusted ORcombined and the crude OR1 and OR2 that were calculated using the reported genotype data. Alternatively, crude ORinteraction values were calculated using crude ORcombined, OR1 and OR2 values and are parenthesized here.
CC = colon cancer, CRC = colorectal cancer, EC = esophageal cancer, GC = gastric cancer, RC = rectal cancer.
Criteria for publication search and study selection. This review included only studies where ORinteraction shown in the sixth column is >1.10 or <0.91. A search for articles in PubMed, Google Scholar and Web of Science, published through July 8, 2015, was conducted using the following keywords: ‘epistasis’ or ‘gene-gene interaction’ or ‘epistatic interaction’; and ‘esophageal cancer’ or ‘gastric cancer’ or ‘colorectal cancer’ or 'colon cancer' or 'rectal cancer' or ‘intestine cancer’.
The quadruple variation of CHEK2 includes del5395, 1100delC, c.444+1G>A and I157T [72].
Only rs2031920 (C>T) of CYP2E1, perfectly correlated (r2 = 1) with rs3813867 (G>C), is shown here [48].
The mixed population includes non-Hispanic Caucasians, Hispanics, American Indians, African Americans and Asians [73].
In principle, both synergistic and antagonistic interactions can be observed in each category, although underlying regulatory mechanisms may be different within distinct categories. In the cases where two variants interact, but are not individually associated with cancer risk, or where only one variant is associated with risk, effects are likely to be qualitative. In contrast, the interaction between two risk-associated variants may exert effects through both quantitative and qualitative mechanisms.
Here, we summarize previous studies demonstrating the role of epistasis in susceptibility to gastrointestinal cancers. Only cancers arising in the digestive tract, but not in accessary organs such as liver, are surveyed in this review. We also list and define the types of epistatic relationships between cancer susceptibility genes, and categorize the genes based on their functions and corresponding cancer hallmarks (Table 2).
Table 2. Predicted hallmarks and modes of biological interaction.
| Partner 1 | Partner 2 | Direct binding | Linear pathway | Convergence |
|---|---|---|---|---|
| Genome instability hallmark | ||||
| OGG1 | XRCC1 | Synergistic and Co-suppressivea | ||
| PARP1 | XRCC1 | Synergistic | ||
| XPA | XPC | Synergistic | ||
| TP53 | MDM2 | Synergistic | ||
| TGFBR1 | SMAD7 | Suppressiveb | ||
| MUTYH | OGG1 | Synergistic | ||
| MUTYH | XRCC1 | Synergistic | ||
| ADH1B | ALDH2 | Synergistic | ||
| NQO1 | NQO2 | Synergistic | ||
| CYP1A1 | CYP2E1 | Synergistic | ||
| GSTT1 | APEX1 | Redundant | ||
| GSTT1/GSTM1 | APEX1 | Suppressive | ||
| GSTM1 | NAT2 | Redundant | ||
| Proliferation hallmark | ||||
| RPS6KB1 | PRKAG2 | Synergistic | ||
| RPS6KB1 | PIK3CA | Suppressive | ||
| CDKN1B | CHEK2 | Synergistic | ||
| Multiple hallmarksc | ||||
| MMP2 | PARP1 | Suppressive |
An OGG1 SNP showed a synergistic interaction with p.Arg399Gln SNP (rs25487) but a co-suppressive interaction with p.Arg194Trp SNP (rs1799782) of XRCC1 [9].
The interaction was suppressive with the adjusted ORcombined, but redundant with the crude ORcombined [69].
Multiple hallmarks include inflammation, proliferation, insensitivity, resistance, immortality and angiogenesis for MMP2, and mutation for PARP1.
EPISTASIS OF GENOME INSTABILITY AND MUTATION
The ten current hallmarks of cancer include two cancer-enabling characteristics and eight cancer-associated features [2]. One enabling characteristic is genome instability and mutation. DNA can be damaged by genotoxic stresses from environmental agents, such as UV, ionizing radiation and DNA-altering chemicals. Endogenous agents that originate from normal cellular metabolism, such as reactive oxygen species, can also cause DNA damage. Defects in genome maintenance and DNA repair are observed in numerous tumors and may lead to genetic errors and tumorigenesis.
Base excision repair
The base excision repair system recognizes small DNA lesions in the form of single-strand nicks or aberrant bases modified by oxidation, reduction or methylation. Damaged bases are first removed by DNA glycosylases. 8-Oxoguanine (8-oxoG), an oxidized derivative of guanine (G) made by reactive oxygen species, can pair with adenine (A) rather than cytosine (C) during replication, leading to G:C to T:A conversion if not repaired. This A is excised by adenine glycosylase MUTYH and replaced with C in subsequent base excision repair reactions. The resulting 8-oxoG:C pair is then excised by glycosylase OGG1 [7].
The rs1052134 single-nucleotide polymorphism (SNP) in the OGG1 gene encodes p.Ser326Cys, a substitution of Ser for Cys at amino acid 326 of the OGG1 protein. This OGG1 SNP interacts synergistically with MUTYH SNP rs3219489, encoding p.Gln324His and affecting glycosylase activity, to increase colorectal cancer risk (ORinteraction = 1.36) [8, 9]. The OGG1 Cys variant had multiple defects in DNA binding, repair and/or nuclear localization [10–12]. This OGG1-MUTYH interaction demonstrates the possibility that two genes within the same pathway can be mutually influential in altering cancer susceptibility, although their gene products do not necessarily physically interact with each other.
OGG1 directly interacts with XRCC1 [13, 14], and the OGG1 rs1052134 showed epistasis with two XRCC1 SNPs in colorectal cancer risk [9]. These included a synergistic interaction (ORinteraction = 1.34) with XRCC1 rs25487 encoding p.Arg399Gln, and a co-suppressive interaction (crude ORinteraction = 0.44) with XRCC1 rs1799782 encoding p.Arg194Trp. (Whereas ORinteraction values were calculated using adjusted OR1, OR2 and ORcombined values for the risk-associated alleles, ‘crude ORinteraction’ values were calculated using crude OR1, OR2 and ORcombined values from the reported data when their adjusted values for the risk-associated alleles were not available. Crude OR values are parenthesized in Table 1.) Incidentally, the XRCC1 rs1799782 synergistically interacted (crude ORinteraction = 1.13) with the MUTYH rs3219489, albeit weakly, in colorectal cancer risk [9].
PARP1 is activated following sensing of nicked DNA and is auto-ribosylated. It recruits XRCC1-ligase 3 to regions of single-strand nicks, where the ribosyltransferase activity of PARP1 is required. PARP1 directly binds XRCC1 at a central region containing the BRCT domain [15, 16]. Susceptibility to gastric cancer is associated with PARP1 rs1136410 encoding p.Val762Ala [17], along with the Ala variant, which displayed diminished enzymatic activity [18]. The PARP1 rs1136410 synergistically interacted (ORinteraction = 1.84) with XRCC1 rs25487 encoding p.Arg399Gln in gastric cancer risk [17]. Both risk-associated PARP1 and XRCC1 substitutions are expected to decrease base excision repair through reduced PARP1 ribosylation and XRCC1 recruitment. Because PARP1 can also induce cell death by enhancing nuclear translocation of apoptosis-inducing factor, PARP1 modifications merit further investigation.
The above findings demonstrate multiple epistatic relationships between MUTYH, OGG1, XRCC1 and PARP1 of the base excision repair system. These interactions, excluding the weak interaction between MUTYH and XRCC1 (rs1799782), may shape a linear network topology in the form of MUTYH-OGG1-XRCC1-PARP1 (Figure 2A), although the three interaction links were observed in two different studies for different cancers (two links in Polish colorectal cancer [9] and one in Chinese gastric cancer [17]). Every direct link in this linear network was synergistic. Therefore, super-synergism can be expected for carriage of all risk-associated alleles in these four genes.
Figure 2. Topology of epistasis networks.
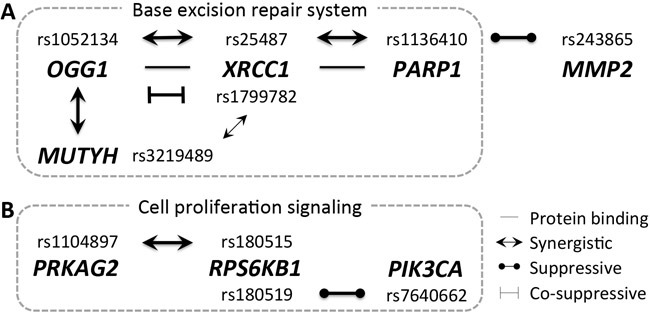
A. Five genes, primarily part of the base excision repair system, form a linear or lariat epistasis network. B. Three genes involved in mTOR signaling of cell proliferation form a linear epistasis network.
Nucleotide excision repair
The nucleotide excision repair system recognizes bulky DNA lesions, including UV-induced pyrimidine dimers, photoadducts and chemical adducts. Damaged DNA is recognized by a complex of XPA, XPC and RPA [19]. The SNP, rs1800975, in the 5′ untranslated region of XPA, just four nucleotides upstream of the AUG start codon, is part of the Kozak sequence, which affects translation initiation efficiency. The major allele G was associated with a higher DNA repair capacity than the minor allele A in Caucasians [20–22].
This XPA rs1800975 synergistically interacted (OR’interaction = 1.76) with an XPC polymorphism, PAT S>L, in gastric cancer risk [23]. (Here, an OR’interaction value, marked with a prime symbol, was calculated using a reported adjusted ORcombined value and the crude OR1 and OR2 values that we calculated using the reported genotype data.) The XPC PAT polymorphism alone was not associated with cancer risk, but enhanced the effect of XPA rs1800975. The XPC PAT polymorphism is in linkage disequilibrium with XPC rs2228001 encoding p.Lys939Gln. This nonsynonymous SNP did not affect the XPC function in vitro [24], and the effect of the PAT polymorphism here remains to be determined.
Detoxification in alcohol metabolism
Alcohol consumption is an environmental risk factor for certain cancers. Alcohol is metabolized into acetate through two steps. First, ethanol is oxidized to acetaldehyde by alcohol dehydrogenase, primarily ADH1B. Then, acetaldehyde is further oxidized to acetate by acetaldehyde dehydrogenase ALDH2. With respect to ADH1B rs1229984 encoding p.Arg47His, the Arg allele enzyme has lower catalytic activity for ethanol metabolism than the His allele enzyme [25, 26]. Regarding ALDH2 rs671 encoding p.Glu504Lys (also known as Glu487Lys), the Lys allele enzyme has lower activity than the Glu allele enzyme [27].
SNPs in both ADH1B and ALDH2 are associated with susceptibility to esophageal and gastric cancers in alcohol drinkers [28]. Synergistic interaction between ADH1B and ALDH2 SNPs was observed in a study of Japanese esophageal cancer (ORinteraction = 2.21) [29], and also in a subsequent meta-analysis (OR’interaction = 1.60) [30]. Further study is required to determine how the interaction between the two lower activity ADH1B (47Arg) and ALDH2 (504Lys) variants leads to increased cancer risk. One interesting possibility is that un-metabolized ethanol reaches the intestinal mucosa, where it is converted to harmful acetaldehyde by gut microflora and thereby increases colon cancer risk [31].
Detoxification in quinone metabolism
Another example of environmental factors in cancer risk is NQO1 and NQO2 of quinone metabolism. Carcinogenic quinones and polycyclic aromatic hydrocarbons are detoxified by these flavoprotein enzymes, which thereby protect cells from redox cycling and oxidative stress. Polycyclic aromatic hydrocarbons are contained in exogenous chemicals such as tobacco smoke, automobile exhaust and burnt foods.
Several studies have associated NQO1 polymorphisms with susceptibility to esophageal and cutaneous (skin) cancers [32, 33]. The Ser variant of NQO1 rs1800566 encoding p.Pro187Ser has reduced enzymatic activity [34, 35] and is associated with higher benzene toxicity compared with the Pro variant [36]. This NQO1 SNP was associated with esophageal adenocarcinoma risk and synergistically interacted (ORinteraction = 1.16) with a promoter SNP of NQO2, rs2070999, which was not associated with esophageal adenocarcinoma risk [37]. The functional role of this NQO2 SNP in gastrointestinal cancer remains to be elucidated.
Detoxification by glutathione conjugation
Glutathione S-transferases (GSTs) form a large family of detoxifying enzymes that neutralize electrophiles and radicals by conjugating environmental carcinogens to reduced glutathione. Within the GST family genes, GSTT1 and GSTM1 are frequently deleted (referred to as the null genotype) in the general population. Carriers of the null genotype were more susceptible to cancer than non-carriers [38].
A case-control study separately associated GSTT1- and GSTM1-null genotypes with increased gastric cancer risk [39]. Carriers of the double-null genotype, lacking both GSTT1 and GSTM1, display increased gastric cancer risk relative to non-carriers in an Italian population and by a meta-analysis [40, 41]. Deletion of GSTs might be associated with high levels of Helicobacter pylori-induced reactive oxygen species and thus increase susceptibility to inflammation-related cancer through enhanced DNA damage.
APEX1 rs1130409 encoding p.Asp148Glu showed redundant interaction (OR’interaction = 0.85) with the GSTT1-null genotype, but suppressive interaction (OR’interaction = 0.77) with the GSTT1-GSTM1 double-null genotype in gastric cancer susceptibility [23]. APEX1 is a key enzyme involved in base excision repair, which regulates chemosensitivity. APEX1 expression is increased with H. pylori infection, which plays a critical role in gastric cancer development [42]. However, this nonsynonymous APEX1 SNP did not alter protein function or structure [43].
Alone, APEX1 rs1130409 was not associated with gastric cancer risk, suggesting that this SNP has an impact only in conjunction with the GSTT1- or double-null genotype. Given that GSTs protect against DNA damage and that APEX1 participates in DNA repair, cooperationbetween DNA damage prevention and repair appears critical for maintenance of genome integrity and underlies inter-individual variability in gastric cancer risk.
The GSTM1 null genotype was associated with higher risk for colorectal and gastric cancers [7, 44]. Additionally, this null genotype redundantly interacted (crude ORinteraction = 0.70) in sporadic colorectal cancer risk with NAT2 rs1799930 encoding p.Arg197Gln [45]. NAT2 mediates acetylation of aromatic and heterocyclic carcinogenic amines [46], which can detoxify or activate the chemicals.
Pro-carcinogens present in tobacco, such as nitrosamines and polycyclic aromatic hydrocarbons, are metabolized into active carcinogens by cytochrome P450 proteins (CYPs). For example, CYP1A1, also known as aryl hydrocarbon hydroxylase, catalyzes metabolic activation of benzo(a)pyrene into a carcinogenic epoxide [47]. Synergistic interaction between CYP1A1 rs1048943 encoding p.Ile462Val and CYP2E1 rs2031920 was observed in Kashmir esophageal cancer (crude ORinteraction = 1.85) [48].
TP53 tumor suppressor pathway
Transcription factor TP53 is involved in DNA repair, cell-cycle arrest, senescence and apoptosis in response to various cellular stresses. Somatic, attenuating TP53 mutations are found in many human cancers [49]. However, tumors can also arise from mutations in genes regulating the TP53 pathway without TP53 mutation itself [50]. For example, expression of MDM2, encoding an E3 ubiquitin ligase, is elevated in various human tumors, and MDM2 negatively regulates TP53 transcriptional activity either directly or indirectly [51].
TP53 and MDM2 polymorphisms have been studied in regards to both susceptibility to cancer and responsiveness to cancer therapy. The Pro variant of TP53 rs1042522 encoding p.Arg72Pro is less potent in inducing apoptosis than the Arg variant [52, 53]. However, the Pro allele confers higher apoptotic capacity during chemotherapy in the presence of some somatic tumor-associated TP53 mutations, suggesting that the success of chemotherapy in inducing TP53-mediated apoptosis is dependent on a given patient's combined somatic and germline TP53 modifications [54].
For MDM2 rs2279744, the minor allele G has higher affinity for transcription factor SP1 than the major allele T, increases MDM2 expression and attenuates TP53-mediated apoptosis [55]. Higher MDM2 expression and consequent TP53 pathway attenuation are associated with susceptibility to lung and esophageal cancers [56, 57]. While MDM2 and TP53 function within the same pathway, synergistic interactions were found between the minor alleles of MDM2 rs2279744 and TP53 rs1042522 in increasing the risk of colorectal (OR’interaction = 1.52), gastric (ORinteraction = 1.51) and esophageal (OR’interaction = 1.19) cancers [57–59].
Transforming growth factor pathway
Transforming growth factor TGF-β is multifunctional. It inhibits the cell cycle by suppressing c-Myc expression and enhancing expression of cyclin-dependent kinase inhibitors such as CDKN2B and CDKN1A [60]. Similar to TP53, TGF-β has both intrinsic effects on genomic stability, cell differentiation, senescence and apoptosis, and extrinsic effects on suppression of inflammation and mitogens.
Contrary to the tumor-suppressive role of TGF-β at early tumor development stages, malignant cells at late stages downregulate expression of TGF-β receptors and become resistant to TGF-β-mediated growth inhibition. Moreover, TGF-β signaling activation at late stages paradoxically promotes cancer cell metastasis by activating the epithelial-to-mesenchymal transition, modulating microenvironments and suppressing anti-tumor immune responses [61]. Therefore, the consequence of TGF-β signaling activation varies depending on cell type and context.
TGF-β signaling is initiated by its binding to a heterodimeric complex of type-I and type-II TGF-β receptors, TGFBRI and TGFBRII. This signaling is negatively regulated by SMAD7, which competes with R-SMADs for receptor binding and mediates degradation of the receptors via ubiquitination [62, 63]. Some TGF-β signaling genes were associated with colorectal cancer risk [64–68], and specifically TGFBR1 rs6478972 and SMAD7 rs11874392 exhibited suppressive epistasis (ORinteraction = 0.71) [69].
TGFBR1 rs6478972 is in linkage disequilibrium with rs334348, which is located in the 3′ untranslated region and might affect microRNA binding and consequently TGFBR1 protein levels [70, 71]. Accordingly, the two interacting SNPs could affect expression of the two cancer-associated proteins, TGFBR1 and SMAD7, participating in the same TGF-β signaling pathway, but affecting multiple cancer hallmarks.
EPISTASIS IN SUSTAINING PROLIFERATIVE SIGNALING
The next cancer hallmarks exhibiting epistasis are sustaining growth signals and insensitivity to anti-growth signals. These characteristics are related to the autonomous capability of cancer to increase cell numbers. The CDKN1B and CHEK2 pair showed epistasis in colorectal cancer susceptibility. Among the four CHEK2 polymorphisms associated with colon cancer risk, three variants (del5395, 1100delC and c.444+1G>A) produce truncated proteins, and one (I157T) is a missense mutant. These four CHEK2 variations synergistically interacted (crude ORinteraction = 1.16) with CDKN1B rs2066827 encoding p.Val109Gly in colon cancer risk [72].
Cyclin-dependent kinases (CDKs) drive cell cycle progression, which is counter-regulated by CDK inhibitors. Pauses at the G1-S cell cycle checkpoint are mediated by CDKN1B, a cell cycle inhibitor, and CHEK2, a checkpoint protein activated by DNA damage and replication inhibition. CDKN1B inhibits the CDK2-cyclin E complex, preventing cells from progressing into S phase. CHEK2 functions, such as phosphorylating CDC25C phosphatase and stabilizing TP53, converge in inhibition of the CDK2-cyclin E and CDK4-cyclin D complexes. Therefore, CDKN1B and CHEK2 have different targets, but the same effect, namely, regulation of the G1-S checkpoint.
Additionally, RPS6KB1 rs180515 exhibited synergistic epistasis (crude ORinteraction = 1.19) with PRKAG2 rs1104897, but RPS6KB1 rs180519 showed suppressive epistasis (crude ORinteraction = 0.80) with PIK3CA rs7640662 in rectal cancer susceptibility [73]. Although two different RPS6KB1 SNPs are involved, PRKAG2 may interact with PIK3CA indirectly through RPS6KB1, forming the linear interaction network, PRKAG2-RPS6KB1-PIK3CA (Figure 2B).
RPS6KB has two isoforms, RPS6KB1 and RPS6KB2, and is involved in protein synthesis necessary for cell growth in response to various growth factors, insulin and nutrients. RPS6KB is phosphorylated by the mTOR complex, which serves as a regulatory axis for cell growth and proliferation [74]. The mTOR pathway and RPS6KB are activated by PIK3CA and inhibited by PRKAG2 [75]. This sharing of a common regulatory target explains the linear interaction network, PRKAG2-RPS6KB1-PIK3CA.
EPISTASIS OF OTHER COMPLEX HALLMARKS
Epistasis between different cancer hallmark pathways
For tumor cell invasion and metastasis, the extracellular matrix and basement membrane must be degraded, and this in turn facilitates angiogenesis. The altered tissue environment around tumor cells is called the tumor microenvironment, and matrix metalloproteinases (MMPs) are crucial for shaping this microenvironment and promoting tumor progression [76].
Specifically, MMP2 expression is associated with gastric cancer progression and lymph node metastasis [77]. An MMP2 promoter SNP, rs243865, affects gene expression by altering SP1 transcription factor binding [78]. This functional SNP was not associated with gastric cancer susceptibility, but its minor allele suppressed the gastric cancer risk-enhancing effect of the minor allele in the above-mentioned PARP1 rs1136410 (crude ORinteraction = 0.60) [79].
The interaction between PARP1 and MMP2 would extend the MUTYH-OGG1-XRCC1-PARP1 linear interaction network to MUTYH-OGG1-XRCC1-PARP1-MMP2 (Figure 2A). However, the PARP1-MMP2 link in this network is suppressive, unlike the other three synergistic interactions, and has been observed in different studies (Korean gastric cancer [79] versus Chinese gastric cancer [17] and Polish colorectal cancer [9]).
PARP1 participates in the base excision repair system, but not in any pathway known to involve MMP2. This is the only case reported to date where two statistically interacting genes are involved in apparently distinct pathways, but crosstalk has not been detected. If the statistical epistasis between PARP1 and MMP2 is related to cancer risk, there must be crosstalk or merging of the two pathways, incorporating disparate cancer traits. Although multiple MMP-conferred effects add complexity to the understanding of epistasis in cancer, dissecting this link at the molecular level could be relevant for improved therapeutics.
Epistasis of noncoding RNA
Lastly, there was a weakly synergistic interaction (OR’interaction = 1.11) between intronic rs10879357 of TPH2 and intergenic rs1571218 at human chromosome 20p12.3 in colorectal cancer susceptibility [81]. Because both rs1571218 and nearby rs961253 are associated with colorectal cancer risk [81, 82], this locus cancer risk association appears to be replicated. These two intergenic SNPs are both closer to the long noncoding RNA gene CASC20 than to nearby BMP2, but no function is yet known for CASC20.
Meanwhile, TPH2 participates in serotonin synthesis and lack of serotonin has been associated with tumor growth reductions in a mouse model of colon cancer allografts [83]. It will be worthwhile to determine whether CASC20 regulates TPH2, because many long noncoding RNAs regulate diverse gene expression steps [84].
MODE OF BIOLOGICAL INTERACTION FOR STATISTICAL EPISTASIS
All four levels of epistasis, i.e. synergistic, redundant, suppressive and co-suppressive (Figure 1), have been observed for susceptibility to gastrointestinal cancers (Table 2). We propose here that all statistical epistases can be explained by three different modes of biological interaction: ‘direct binding,’ ‘linear pathway’ and ‘convergence’ modes (Figure 3).
Figure 3. Three plausible modes of biological interaction between two statistically epistatic gene products.
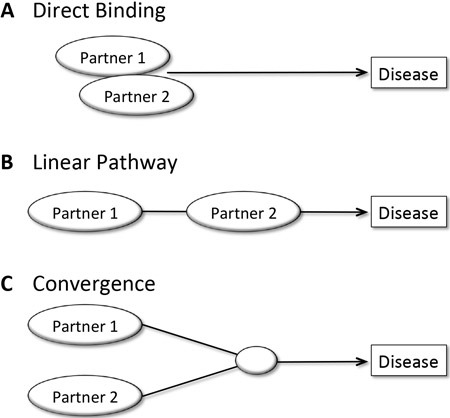
A. In the direct binding mode, two partners physically bind to each other and function together to affect disease susceptibility. B. In the linear pathway mode, two partners work sequentially. C. In the convergence mode, two partner-involving pathways converge to promote disease susceptibility.
In the direct binding mode, two gene products (proteins or noncoding RNAs) physically bind to each other and the final combined effect is determined by combinatorial codes. Therefore, their interaction would be expected to be synergistic or antagonistic in altering disease susceptibility. This principle is exemplified by the five direct-binding pairs, where one partner is already known to directly control the other; OGG1-XRCC1, XRCC1-PARP1, XPA-XPC, TP53-MDM2 and TGFBR1-SMAD7 (Table 2)
Without direct binding between their products, two genes could still be epistatic by participating in the same pathway (linear pathway mode) or two distinct, but converging pathways (convergence mode). When one partner precedes the other in a linear pathway leading to cancer development, their cooperation increases cancer susceptibility. For example, OGG1-MUTYH-XRCC1 in base excision repair, ADH1B-ALDH2 in alcohol metabolism, NQO1-NQO2 in quinone metabolism and PRKAG2-RPS6KB1-PIK3CA in the mTOR pathway all exhibit the linear pathway mode of interaction (Table 2).
Convergence mode, where two pathways merge and proceed together in a single pathway, is also observed (Table 2). CDKN1B and CHEK2 each have distinct target proteins, but both eventually participate in the G1-S checkpoint, regulating the cell cycle. Both the GSTT1 and APEX1 pathways act in DNA repair, and GSTM1 and NAT2 promote detoxification. Notably, PARP1 and MMP2 are not known to participate in a single linear or converged pathway, but their interaction suggests as-yet unidentified crosstalk between their respective pathways.
EMERGING PERSPECTIVES ON CANCER GENETICS
Several new perspectives have surfaced from the last decade of cancer genetics research. First, some genes originally classified as part of a core cancer hallmark in fact exert pleiotropic effects on multiple hallmarks. For example, TP53 functions as a growth suppressor, a mediator for senescence and apoptosis, and a guardian of the genome [10]. TGF-β has either tumor-suppressing or tumor-promoting activities, depending on the context [14].
Second, crosstalk between different cancer hallmarks is increasingly explored. Epistasis in cancer susceptibilityis detected more frequently and with increasing statistical power. Some epistases support known biological crosstalk, but others suggest previously unidentified crosstalk, as exemplified by the PARP1-MMP2 interaction [79]. Dissection of crosstalk mechanisms among interacting gene pathways merits further investigation, and will provide a deeper understanding of cancer susceptibility.
Third, complex interaction network topological structures are expected, as point-to-point pairwise interactions are increasingly observed in disease susceptibility studies. For example, in this review, a linear topology is inferred for the interaction network, MUTYH-OGG1-XRCC1-PARP1-MMP2 (Figure 2A), although not all links were observed in the same cancer for a single ethnic population. Therefore, this network must be verified using the same study population for the same disease, incorporating a large enough number of samples to achieve sufficient statistical power. The topology does not need to be linear as in this example. Indeed, even this example could become a ring, mesh or other structure upon discovery of additional interaction links.
Fourth, epigenetic dysregulation is an emerging cancer characteristic. Comprehensive mapping of epigenetic landscapes and noncoding RNAs in cancer cells has revealed that they play essential roles in cell proliferation, apoptosis and metastasis [85]. Noncoding RNAs and epigenetic signatures representing DNA methylation, histone modification and nucleosome remodeling in gastric and colorectal cancers have been compiled [86–89] and applied to improve diagnoses, prognoses and therapeutic interventions [90, 91]. Epigenetic polymorphisms in cancers, as well as interactions between epigenetic and genetic polymorphisms in cancer susceptibility and hallmark genes, must be further investigated to identify meaningful epistatic interactions.
Fifth, multiple roles for a particular gene in various cancer traits can be clearly elucidated if the context in which the gene exerts its effect is understood. In tumor microenvironments, neoplastic cells interact with not only stromal cells, but also immune cells infiltrating tumor tissues, inducing an inflammatory response. Inflammation in the microenvironment can promote tumor cell proliferation, survival, vascularization, invasion, metastasis and epithelial-to-mesenchymal transition [11].
Finally, heterogeneity of somatic cells in tumor tissues is evident in whole-genome sequencing of individual tumor cells. Thus, single-cell analyses can reveal whether cancer susceptibility genes are mutated in an individual tumor cell, whether any germline polymorphisms are associated with somatic mutations of cancer hallmark genes, and whether there is epistasis between germline polymorphisms and somatic mutations in promotion of tumor development and progression.
CONCLUDING SUMMARY
In this review, we surveyed statistical gene-gene interactions reported for genetic susceptibility to colorectal, gastric and esophageal cancers (Table 1). We found multiple gene pairs participating in synergistic, redundant, suppressive or co-suppressive interactions (Table 2), as statistical epitasis is defined according to the degree and direction of interaction between genetic variants in disease susceptibility (Figure 1). We inferred that more than two genes could indirectly interact with one another in cancer susceptibility to form a topological network structure (Figure 2).
We also proposed three different modes of biological interaction as the underlying molecular mechanisms for statistical epistasis. The direct binding, linear pathway and convergence modes can exhibit any level of statistical epistasis in disease susceptibility (Figure 3). Finally, several perspectives are provided here regarding interactions among germline polymorphisms, epigenetic variations and somatic mutations in cancer susceptibility.
Acknowledgments
We thank Professors Dae-Sik Lim, Mi Young Kim and Ji-Joon Song for critical assessment of the manuscript. This work was supported by a grant (NRF-2011-0020334) from the National Research Foundation of Korea.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Bateson W. Mendel's Principles of Heredity. Cambridge University Press; Cambridge: 1909. pp. 79, 98 and 101. [Google Scholar]
- 4.Fisher RA. The correlation between relatives on the supposition of Mendelian inheritance. Trans R Soc Edin. 1918;52:399–433. doi: 10.1017/s0080456800012163. [DOI] [Google Scholar]
- 5.Crow JF, Kimura M. An Introduction to Population Genetics Theory. Harper & Row; New York: 1970. [Google Scholar]
- 6.Mackay TF. Epistasis and quantitative traits: using model organisms to study gene-gene interactions. Nat Rev Genet. 2014;15:22–33. doi: 10.1038/nrg3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wallace SS, Murphy DL, Sweasy JB. Base excision repair and cancer. Cancer Lett. 2012;327:73–89. doi: 10.1016/j.canlet.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ali M, Kim H, Cleary S, Cupples C, Gallinger S, Bristow R. Characterization of mutant MUTYH proteins associated with familial colorectal cancer. Gastroenterology. 2008;135:499–507. doi: 10.1053/j.gastro.2008.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Przybylowska K, Kabzinski J, Sygut A, Dziki L, Dziki A, Majsterek I. An association selected polymorphisms of XRCC1, OGG1 and MUTYH gene and the level of efficiency oxidative DNA damage repair with a risk of colorectal cancer. Mutat Res. 2013;745-746:6–15. doi: 10.1016/j.mrfmmm.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Hill JW, Evans MK. Dimerization and opposite base-dependent catalytic impairment of polymorphic S326C OGG1 glycosylase. Nucleic Acids Res. 2006;34:1620–32. doi: 10.1093/nar/gkl060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luna L, Rolseth V, Hildrestrand GA, Otterlei M, Dantzer F, Bjoras M, Seeberg E. Dynamic relocalization of hOGG1 during the cell cycle is disrupted in cells harbouring the hOGG1-Cys326 polymorphic variant. Nucleic Acids Res. 2005;33:1813–24. doi: 10.1093/nar/gki325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamane A, Kohno T, Ito K, Sunaga N, Aoki K, Yoshimura K, Murakami H, Nojima Y, Yokota J. Differential ability of polymorphic OGG1 proteins to suppress mutagenesis induced by 8-hydroxyguanine in human cell in vivo. Carcinogenesis. 2004;25:1689–94. doi: 10.1093/carcin/bgh166. [DOI] [PubMed] [Google Scholar]
- 13.Campalans A, Marsin S, Nakabeppu Y, O'Connor TR, Boiteux S, Radicella JP. XRCC1 interactions with multiple DNA glycosylases: a model for its recruitment to base excision repair. DNA Repair. 2005;4:826–35. doi: 10.1016/j.dnarep.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 14.Marsin S, Vidal AE, Sossou M, Menissier-de Murcia J, Le Page F, Boiteux S, de Murcia G, Radicella JP. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J Biol Chem. 2003;278:44068–74. doi: 10.1074/jbc.M306160200. [DOI] [PubMed] [Google Scholar]
- 15.Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair. 2003;2:955–69. doi: 10.1016/s1568-7864(03)00118-6. [DOI] [PubMed] [Google Scholar]
- 16.Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, de Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol. 1998;18:3563–71. doi: 10.1128/MCB.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miao X, Zhang X, Zhang L, Guo Y, Hao B, Tan W, He F, Lin D. Adenosine diphosphate ribosyl transferase and x-ray repair cross-complementing 1 polymorphisms in gastric cardia cancer. Gastroenterology. 2006;131:420–7. doi: 10.1053/j.gastro.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 18.Wang XG, Wang ZQ, Tong WM, Shen Y. PARP1 Val762Ala polymorphism reduces enzymatic activity. Biochem Biophys Res Commun. 2007;354:122–6. doi: 10.1016/j.bbrc.2006.12.162. [DOI] [PubMed] [Google Scholar]
- 19.Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev. 2002;11:1513–30. http://cebp.aacrjournals.org/content/11/12/1513 [PubMed] [Google Scholar]
- 20.Lou Y, Li R, Zhang Y, Zhong R, Pei J, Xiong L, Zhang X, Han B. XPA gene rs1800975 single nucleotide polymorphism and lung cancer risk: a meta-analysis. Tumour Biol. 2014;35:6607–17. doi: 10.1007/s13277-014-1824-1. [DOI] [PubMed] [Google Scholar]
- 21.Park JY, Park SH, Choi JE, Lee SY, Jeon HS, Cha SI, Kim CH, Park JH, Kam S, Park RW, Kim IS, Jung TH. Polymorphisms of the DNA repair gene xeroderma pigmentosum group A and risk of primary lung cancer. Cancer Epidemiol Biomarkers Prev. 2002;11:993–7. http://cebp.aacrjournals.org/content/11/10/993 [PubMed] [Google Scholar]
- 22.Wu X, Zhao H, Wei Q, Amos CI, Zhang K, Guo Z, Qiao Y, Hong WK, Spitz MR. XPA polymorphism associated with reduced lung cancer risk and a modulating effect on nucleotide excision repair capacity. Carcinogenesis. 2003;24:505–9. doi: 10.1093/carcin/24.3.505. [DOI] [PubMed] [Google Scholar]
- 23.Palli D, Polidoro S, D'Errico M, Saieva C, Guarrera S, Calcagnile AS, Sera F, Allione A, Gemma S, Zanna I, Filomena A, Testai E, Caini S, et al. Polymorphic DNA repair and metabolic genes: a multigenic study on gastric cancer. Mutagenesis. 2010;25:569–75. doi: 10.1093/mutage/geq042. [DOI] [PubMed] [Google Scholar]
- 24.Khan SG, Metter EJ, Tarone RE, Bohr VA, Grossman L, Hedayati M, Bale SJ, Emmert S, Kraemer KH. A new xeroderma pigmentosum group C poly(AT) insertion/deletion polymorphism. Carcinogenesis. 2000;21:1821–5. doi: 10.1093/carcin/21.10.1821. [DOI] [PubMed] [Google Scholar]
- 25.Bosron WF, Li TK. Genetic polymorphism of human liver alcohol and aldehyde dehydrogenases, and their relationship to alcohol metabolism and alcoholism. Hepatology. 1986;6:502–10. doi: 10.1002/hep.1840060330. [DOI] [PubMed] [Google Scholar]
- 26.Smith M. Genetics of human alcohol and aldehyde dehydrogenases. Adv Hum Genet. 1986;15:249–90. doi: 10.1007/978-1-4615-8356-1_5. [DOI] [PubMed] [Google Scholar]
- 27.Crabb DW, Edenberg HJ, Bosron WF, Li TK. Genotypes for aldehyde dehydrogenase deficiency and alcohol sensitivity. The inactive ALDH2(2) allele is dominant. J Clin Invest. 1989;83:314–6. doi: 10.1172/jci113875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokoyama A, Muramatsu T, Omori T, Yokoyama T, Matsushita S, Higuchi S, Maruyama K, Ishii H. Alcohol and aldehyde dehydrogenase gene polymorphisms and oropharyngolaryngeal, esophageal and stomach cancers in Japanese alcoholics. Carcinogenesis. 2001;22:433–9. doi: 10.1093/carcin/22.3.433. [DOI] [PubMed] [Google Scholar]
- 29.Cui R, Kamatani Y, Takahashi A, Usami M, Hosono N, Kawaguchi T, Tsunoda T, Kamatani N, Kubo M, Nakamura Y, Matsuda K. Functional variants in ADH1B and ALDH2 coupled with alcohol and smoking synergistically enhance esophageal cancer risk. Gastroenterology. 2009;137:1768–75. doi: 10.1053/j.gastro.2009.07.070. [DOI] [PubMed] [Google Scholar]
- 30.Zhang G, Mai R, Huang B. ADH1B Arg47His polymorphism is associated with esophageal cancer risk in high-incidence Asian population: evidence from a meta-analysis. PLoS One. 2010;5:e13679. doi: 10.1371/journal.pone.0013679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Homann N, Tillonen J, Salaspuro M. Microbially produced acetaldehyde from ethanol may increase the risk of colon cancer via folate deficiency. Int J Cancer. 2000;86:169–73. doi: 10.1002/(SICI)1097-0215(20000415)86:2<169::AID-IJC4>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 32.Zhang JH, Li Y, Wang R, Geddert H, Guo W, Wen DG, Chen ZF, Wei LZ, Kuang G, He M, Zhang LW, Wu ML, Wang SJ. NQO1 C609T polymorphism associated with esophageal cancer and gastric cardiac carcinoma in North China. World J Gastroenterol. 2003;9:1390–3. doi: 10.3748/WJG.v9.i7.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang WC, Yin LH, Pu YP, Liang GY, Hu X, Liu YZ, Cui YS. [Relationship between quinone oxidoreductase1 gene ns-cSNP and genetic susceptibility of esophageal cancer] Zhonghua Yu Fang Yi Xue Za Zhi. 2006;40:324–7. doi: 10.3760/j:issn:0253-9624.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 34.Misra V, Grondin A, Klamut HJ, Rauth AM. Assessment of the relationship between genotypic status of a DT-diaphorase point mutation and enzymatic activity. Br J Cancer. 2000;83:998–1002. doi: 10.1054/bjoc.2000.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siegel D, McGuinness SM, Winski SL, Ross D. Genotype-phenotype relationships in studies of a polymorphism in NAD(P)H:quinone oxidoreductase 1. Pharmacogenetics. 1999;9:113–21. doi: 10.1097/00008571-199902000-00015. [DOI] [PubMed] [Google Scholar]
- 36.Nebert DW, Roe AL, Vandale SE, Bingham E, Oakley GG. NAD(P)H:quinone oxidoreductase (NQO1) polymorphism, exposure to benzene, and predisposition to disease: a HuGE review. Genet Med. 2002;4:62–70. doi: 10.1097/00125817-200203000-00003. [DOI] [PubMed] [Google Scholar]
- 37.Malik MA, Zargar SA, Mittal B. Role of NQO1 609C>T and NQO2 -3423G>A gene polymorphisms in esophageal cancer risk in Kashmir valley and meta analysis. Mol Biol Rep. 2012;39:9095–104. doi: 10.1007/s11033-012-1781-y. [DOI] [PubMed] [Google Scholar]
- 38.Rebbeck TR. Molecular epidemiology of the human glutathione S-transferase genotypes GSTM1 and GSTT1 in cancer susceptibility. Cancer Epidemiol Biomarkers Prev. 1997;6:733–43. http://cebp.aacrjournals.org/content/6/9/733 [PubMed] [Google Scholar]
- 39.Lan Q, Chow WH, Lissowska J, Hein DW, Buetow K, Engel LS, Ji B, Zatonski W, Rothman N. Glutathione S-transferase genotypes and stomach cancer in a population-based case-control study in Warsaw, Poland. Pharmacogenetics. 2001;11:655–61. doi: 10.1097/00008571-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 40.Palli D, Saieva C, Gemma S, Masala G, Gomez-Miguel MJ, Luzzi I, D'Errico M, Matullo G, Ozzola G, Manetti R, Nesi G, Sera F, Zanna I, et al. GSTT1 and GSTM1 gene polymorphisms and gastric cancer in a high-risk italian population. Int J Cancer. 2005;115:284–9. doi: 10.1002/ijc.20864. [DOI] [PubMed] [Google Scholar]
- 41.Lao XJ, Peng QL, Lu Y, Li S, Qin X, Chen ZP, Chen JQ. Glutathione S-transferase gene GSTM1, gene-gene interaction, and gastric cancer susceptibility: evidence from an updated meta-analysis. Cancer Cell Int. 2014;14:127. doi: 10.1186/s12935-014-0127-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Futagami S, Hiratsuka T, Shindo T, Horie A, Hamamoto T, Suzuki K, Kusunoki M, Miyake K, Gudis K, Crowe SE, Tsukui T, Sakamoto C. Expression of apurinic/apyrimidinic endonuclease-1 (APE-1) in H. pylori-associated gastritis, gastric adenoma, and gastric cancer. Helicobacter. 2008;13:209–18. doi: 10.1111/j.1523-5378.2008.00605.x. [DOI] [PubMed] [Google Scholar]
- 43.Illuzzi JL, Harris NA, Manvilla BA, Kim D, Li M, Drohat AC, Wilson DM., 3rd Functional assessment of population and tumor-associated APE1 protein variants. PLoS One. 2013;8:e65922. doi: 10.1371/journal.pone.0065922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saadat I, Saadat M. Glutathione S-transferase M1 and T1 null genotypes and the risk of gastric and colorectal cancers. Cancer Lett. 2001;169:21–6. doi: 10.1016/S0304-3835(01)00550-X. [DOI] [PubMed] [Google Scholar]
- 45.Procopciuc LM, Osian G. GSTM1-null genotype as a risk factor for sporadic colorectal cancer in a Romanian population. Association with the NAT2-rapid-acetylator phenotype and exposure to environmental factors. Cancer Invest. 2014;32:53–62. doi: 10.3109/07357907.2013.867972. [DOI] [PubMed] [Google Scholar]
- 46.Hein DW. Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res. 2002;506-507:65–77. doi: 10.1016/S0027-5107(02)00153-7. [DOI] [PubMed] [Google Scholar]
- 47.Kiyohara C, Hirohata T, Inutsuka S. The relationship between aryl hydrocarbon hydroxylase and polymorphisms of the CYP1A1 gene. Jpn J Cancer Res. 1996;87:18–24. doi: 10.1111/j.1349-7006.1996.tb00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhat GA, Shah IA, Makhdoomi MA, Iqbal B, Rafiq R, Nabi S, Masood A, Lone MM, Dar NA. CYP1A1 and CYP2E1 genotypes and risk of esophageal squamous cell carcinoma in a high-incidence region, Kashmir. Tumor Biology. 2014;35:5323–30. doi: 10.1007/s13277-014-1694-6. [DOI] [PubMed] [Google Scholar]
- 49.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–65. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 50.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 51.Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res. 2003;1:1001–8. http://mcr.aacrjournals.org/content/1/14/1001 [PubMed] [Google Scholar]
- 52.Dumont P, Leu JI, Della Pietra AC, 3rd, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet. 2003;33:357–65. doi: 10.1038/ng1093. [DOI] [PubMed] [Google Scholar]
- 53.Thomas M, Kalita A, Labrecque S, Pim D, Banks L, Matlashewski G. Two polymorphic variants of wild-type p53 differ biochemically and biologically. Mol Cell Biol. 1999;19:1092–100. doi: 10.1128/MCB.19.2.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–87. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 55.Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, Onel K, Yip L, Hwang SJ, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 56.Zhang X, Miao X, Guo Y, Tan W, Zhou Y, Sun T, Wang Y, Lin D. Genetic polymorphisms in cell cycle regulatory genes MDM2 and TP53 are associated with susceptibility to lung cancer. Hum Mutat. 2006;27:110–7. doi: 10.1002/humu.20277. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Y, Liu L, Tang Y, Chen C, Wang Q, Xu J, Yang C, Miao X, Wei S, Chen J, Nie S. Polymorphisms in TP53 and MDM2 contribute to higher risk of colorectal cancer in Chinese population: a hospital-based, case-control study. Mol Biol Rep. 2012;39:9661–8. doi: 10.1007/s11033-012-1831-5. [DOI] [PubMed] [Google Scholar]
- 58.Hong Y, Miao X, Zhang X, Ding F, Luo A, Guo Y, Tan W, Liu Z, Lin D. The role of P53 and MDM2 polymorphisms in the risk of esophageal squamous cell carcinoma. Cancer Res. 2005;65:9582–7. doi: 10.1158/0008-5472.CAN-05-1460. [DOI] [PubMed] [Google Scholar]
- 59.Yang M, Guo Y, Zhang X, Miao X, Tan W, Sun T, Zhao D, Yu D, Liu J, Lin D. Interaction of P53 Arg72Pro and MDM2 T309G polymorphisms and their associations with risk of gastric cardia cancer. Carcinogenesis. 2007;28:1996–2001. doi: 10.1093/carcin/bgm168. [DOI] [PubMed] [Google Scholar]
- 60.Chen CR, Kang Y, Massague J. Defective repression of c-myc in breast cancer cells: a loss at the core of the transforming growth factor beta growth arrest program. Proc Natl Acad Sci U S A. 2001;98:992–9. doi: 10.1073/pnas.98.3.992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA, Jr, Wrana JL, Falb D. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997;89:1165–73. doi: 10.1016/S0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- 63.Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389:631–5. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 64.Houlston RS, Webb E, Broderick P, Pittman AM, Di Bernardo MC, Lubbe S, Chandler I, Vijayakrishnan J, Sullivan K, Penegar S, Colorectal Cancer Association Study C, Carvajal-Carmona L, Howarth K, et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet. 2008;40:1426–35. doi: 10.1038/ng.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Slattery ML, Herrick JS, Lundgreen A, Wolff RK. Genetic variation in the TGF-beta signaling pathway and colon and rectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2011;20:57–69. doi: 10.1158/1055-9965.EPI-10-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tenesa A, Farrington SM, Prendergast JG, Porteous ME, Walker M, Haq N, Barnetson RA, Theodoratou E, Cetnarskyj R, Cartwright N, Semple C, Clark AJ, Reid FJ, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet. 2008;40:631–7. doi: 10.1038/ng.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tomlinson IP, Webb E, Carvajal-Carmona L, Broderick P, Howarth K, Pittman AM, Spain S, Lubbe S, Walther A, Sullivan K, Jaeger E, Fielding S, Rowan A, et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet. 2008;40:623–30. doi: 10.1038/ng.111. [DOI] [PubMed] [Google Scholar]
- 68.Valle L, Serena-Acedo T, Liyanarachchi S, Hampel H, Comeras I, Li Z, Zeng Q, Zhang HT, Pennison MJ, Sadim M, Pasche B, Tanner SM, de la Chapelle A. Germline allele-specific expression of TGFBR1 confers an increased risk of colorectal cancer. Science. 2008;321:1361–5. doi: 10.1126/science.1159397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhong R, Liu L, Zou L, Sheng W, Zhu B, Xiang H, Chen W, Chen J, Rui R, Zheng X, Yin J, Duan S, Yang B, et al. Genetic variations in the TGFbeta signaling pathway, smoking and risk of colorectal cancer in a Chinese population. Carcinogenesis. 2013;34:936–42. doi: 10.1093/carcin/bgs395. [DOI] [PubMed] [Google Scholar]
- 70.Iuliano R, Vismara MF, Dattilo V, Trapasso F, Baudi F, Perrotti N. The role of microRNAs in cancer susceptibility. Biomed Res Int. 2013;2013:591931. doi: 10.1155/2013/591931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nicoloso MS, Sun H, Spizzo R, Kim H, Wickramasinghe P, Shimizu M, Wojcik SE, Ferdin J, Kunej T, Xiao L, Manoukian S, Secreto G, Ravagnani F, et al. Single-nucleotide polymorphisms inside microRNA target sites influence tumor susceptibility. Cancer Res. 2010;70:2789–98. doi: 10.1158/0008-5472.CAN-09-3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cybulski C, Gliniewicz B, Sikorski A, Kladny J, Huzarski T, Gronwald J, Byrski T, Debniak T, Gorski B, Jakubowska A, Wokolorczyk D, Narod SA, Lubinski J. Epistatic relationship between the cancer susceptibility genes CHEK2 and p27. Cancer Epidemiol Biomarkers Prev. 2007;16:572–6. doi: 10.1158/1055-9965.EPI-06-0566. [DOI] [PubMed] [Google Scholar]
- 73.Slattery ML, Lundgreen A, Herrick JS, Wolff RK. Genetic variation in RPS6KA1, RPS6KA2, RPS6KB1, RPS6KB2, and PDK1 and risk of colon or rectal cancer. Mutat Res. 2011;706:13–20. doi: 10.1016/j.mrfmmm.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 75.Slattery ML, Herrick JS, Lundgreen A, Fitzpatrick FA, Curtin K, Wolff RK. Genetic variation in a metabolic signaling pathway and colon and rectal cancer risk: mTOR, PTEN, STK11, RPKAA1, PRKAG2, TSC1, TSC2, PI3K and Akt1. Carcinogenesis. 2010;31:1604–11. doi: 10.1093/carcin/bgq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Monig SP, Baldus SE, Hennecken JK, Spiecker DB, Grass G, Schneider PM, Thiele J, Dienes HP, Holscher AH. Expression of MMP-2 is associated with progression and lymph node metastasis of gastric carcinoma. Histopathology. 2001;39:597–602. doi: 10.1046/j.1365-2559.2001.01306.x. [DOI] [PubMed] [Google Scholar]
- 78.Price SJ, Greaves DR, Watkins H. Identification of novel, functional genetic variants in the human matrix metalloproteinase-2 gene: role of Sp1 in allele-specific transcriptional regulation. J Biol Chem. 2001;276:7549–58. doi: 10.1074/jbc.M010242200. [DOI] [PubMed] [Google Scholar]
- 79.Kim J, Pyun JA, Cho SW, Lee K, Kwack K. Lymph node metastasis of gastric cancer is associated with the interaction between poly (ADP-ribose) polymerase 1 and matrix metallopeptidase 2. DNA Cell Biol. 2011;30:1011–7. doi: 10.1089/dna.2011.1250. [DOI] [PubMed] [Google Scholar]
- 80.Jiao S, Hsu L, Berndt S, Bezieau S, Brenner H, Buchanan D, Caan BJ, Campbell PT, Carlson CS, Casey G, Chan AT, Chang-Claude J, Chanock S, et al. Genome-wide search for gene-gene interactions in colorectal cancer. PLoS One. 2012;7:e52535. doi: 10.1371/journal.pone.0052535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng X, Wang L, Zhu Y, Guan Q, Li H, Xiong Z, Deng L, Lu J, Miao X, Cheng L. The SNP rs961253 in 20p12. 3 is associated with colorectal cancer risk: a case-control study and a meta-analysis of the published literature. PLoS One. 2012;7:e34625. doi: 10.1371/journal.pone.0034625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nocito A, Dahm F, Jochum W, Jang JH, Georgiev P, Bader M, Graf R, Clavien PA. Serotonin regulates macrophage-mediated angiogenesis in a mouse model of colon cancer allografts. Cancer Res. 2008;68:5152–8. doi: 10.1158/0008-5472.can-08-0202. [DOI] [PubMed] [Google Scholar]
- 83.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–41. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 84.Suzuki H, Maruyama R, Yamamoto E, Kai M. Epigenetic alteration and microRNA dysregulation in cancer. Front Genet. 2013;4:258. doi: 10.3389/fgene.2013.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kang C, Song JJ, Lee J, Kim MY. Epigenetics: An emerging player in gastric cancer. World J Gastroenterol. 2014;20:6433–47. doi: 10.3748/wjg.v20.i21.6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li J, Jin H, Wang X. Epigenetic Biomarkers: Potential Applications in Gastrointestinal Cancers. ISRN Gastroenterol. 2014;2014:464015. doi: 10.1155/2014/464015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu WK, Law PT, Lee CW, Cho CH, Fan D, Wu K, Yu J, Sung JJ. MicroRNA in colorectal cancer: from benchtop to bedside. Carcinogenesis. 2011;32:247–53. doi: 10.1093/carcin/bgq243. [DOI] [PubMed] [Google Scholar]
- 88.Wu WK, Lee CW, Cho CH, Fan D, Wu K, Yu J, Sung JJ. MicroRNA dysregulation in gastric cancer: a new player enters the game. Oncogene. 2010;29:5761–71. doi: 10.1038/onc.2010.352. [DOI] [PubMed] [Google Scholar]
- 89.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 90.Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov. 2013;12:847–65. doi: 10.1038/nrd4140. [DOI] [PMC free article] [PubMed] [Google Scholar]