

ABSTRACT
Gut homeostasis involves interrelated biological networks that include the immune system, specialized cells of the epithelium, such as Paneth and goblet cells, as well as triggers derived from the microbiota. Disruption of these homeostatic interactions may lead to the pathogenesis of inflammatory bowel diseases (IBD). To develop more targeted and individual treatments in Crohn's disease and ulcerative colitis, it becomes more and more important to link key mechanisms of the disease pathogenesis to distinct IBD subsets. For the first time, our laboratory demonstrated a causal role of the microbiota for the development of Crohn's disease (CD)-like ileitis, supporting the hypothesis that a non-infectious, dysbiotic microbial ecosystem harbors aggressive traits relevant for the induction of chronic inflammation in the disease-susceptible host (i.e. TNFΔARE mouse model). Despite a growing body of evidence claiming a primary role for Paneth cells in the pathogenesis of ileal CD, we showed in the TNFΔARE mouse model that Paneth cell failure or exhaustion is a secondary event to inflammation. Therefore, additional mechanisms may act synergistically to initialize the development of CD-like pathology. Hereby, we propose a novel hypothesis suggesting that individual development of dysbiotic communities is based on stochastic injury and focal inflammation of the epithelial lining that propagate radially, finally leading to an aggressive microbial milieu.
KEYWORDS: Dysbiosis, microbiota, microbiome, inflammation, Crohn's disease, TNFΔARE, IBD, intestinal epithelial cells (IEC), stochastic injuries
Dysbiotic microbiota induces transmissible CD-like ileitis in susceptible host
Our laboratory recently demonstrated that germ-free (GF) TNFΔARE mice do not develop CD-like ileitis, supporting the initial hypothesis that bacterial triggers are essential tassels of disease pathogenesis.1 Of high relevance, only specific (dysbiotic) bacterial communities (so-called responder, “R,” or aggressive microbiota) are associated to a disease phenotype in these mice, capable to transmit disease once transferred into a GF susceptible host (TNFΔARE mice).1 In contrast, microbiota derived from wild-type (WT) as well as from healthy but disease susceptible hosts (so-called non-responder, “NR,” or non-aggressive microbiota) is unable to trigger disease once transferred into GF TNFΔARE mice.1 These results provide functional evidence that dynamic alterations of the microbial ecosystem (dysbiosis) cause transmissible disease in the susceptible host. The use of 16S rRNA gene sequencing demonstrated a clear shift in bacterial community structure of R and NR mice at the age of 8 weeks, culminating at 18 weeks of age and correlating with tissue pathology1 (Fig. 1A and B). In contrast, in early life, during the post weaning phase, R and NR microbiota clustered together, showing no signs of dysbiosis or inflammation (Fig. 1C). This piece of evidence suggests that shifts in microbiota composition develop with gradually increasing inflammation and occur after maternal separation.1 Therefore, the investigation in our laboratory revolves around unraveling the events that trigger changes in the microbiota toward the assembling of dysbiotic bacterial communities. This aspect should be studied by kinetics analysis of the microbiota in different organ systems and of the innate and adaptive immune response (e.g. granulocytes and effector T cells and/or antigen specific T cells) to determine whether the selection of dysbiotic microbiota is based on innate tissue activation or on antigen recognition.
Figure 1.
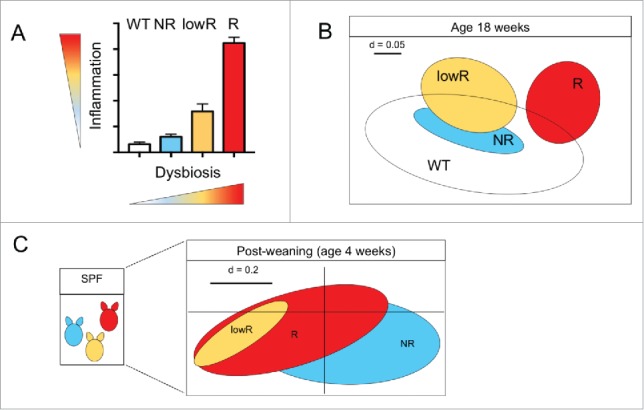
Bacterial profiles and functional dysbiosis mirror ileitis severity in TNFΔARE mice. (A) Independently from the litter of origin, SPF TNFΔARE mice housed in the same cage display a gradient of ileitis severity, ranging from not diseased (i.e., NR), low inflamed (i.e., lowR) and highly inflamed (R) (adapted from1); and inflammation correlates to dysbiosis development in this mouse model. (B) β-diversity analysis shows separation of responder TNFΔARE mice (R) and low-responders (lowR), non-responders (NR) and WT littermate, all mice are 18 weeks of age (adapted from1). (C) Lack of early changes in microbiota phylogeny and dysbiosis in SPF TNFΔARE mice. Non-parametrical multiple dimensional scaling (NMDS) analysis displaying phylogenetic distances between fecal bacterial communities of 4-week old TNFΔARE mice including mice that later in life develop severe (R), intermediate (lowR) and no disease (NR) (adapted from1).
Gut epithelial stochastic injuries generate focal inflammation leading to a dysbiotic microbial ecosystem: A provoking hypothesis
It is very challenging to unravel the mechanisms by which inflammation drives bacterial taxa selection and molds microbiota into a disease-conditioning community. Since cage and litter effects were excluded, our current hypothesis rests on the assumption that morphological modifications of the epithelial interface, previously described as focal lesions,2 occur stochastically in the gut and may alter the local milieu affecting bacterial abundance and fitness.3,4 Actually, while a healthy host is able to seal off the damage, the susceptible individual lacks the ability to restore mucosal integrity, resulting in an altered gut milieu leading to barrier dysfunction and expansion of focal lesions2-4 (Fig. 2A and B). Based on the recently published evidence related to inflammation-mediated changes of bacterial gene expression, we believe that an inflammatory setting generated around focal lesions may up-regulate virulence-associated bacterial genes.5 We speculate that, in such an altered environment, bacterial taxa with aggressive or virulent features may find an ideal niche to propagate and to exert a negative selection on other beneficial commensals. Recent unpublished evidence reports that focal lesion occurrence is mouse strain dependent and that they are enriched in a selected bacterial community.4 Therefore, we suggest the hypothesis that focal lesions may fuel changes in the host microbiota, reprogramming its disease-conditioning nature, and that the transfer of such pre-selected community may results in dysbiosis-associated transmissible disease in a susceptible host (TNFΔARE mice).1-4
Figure 2.
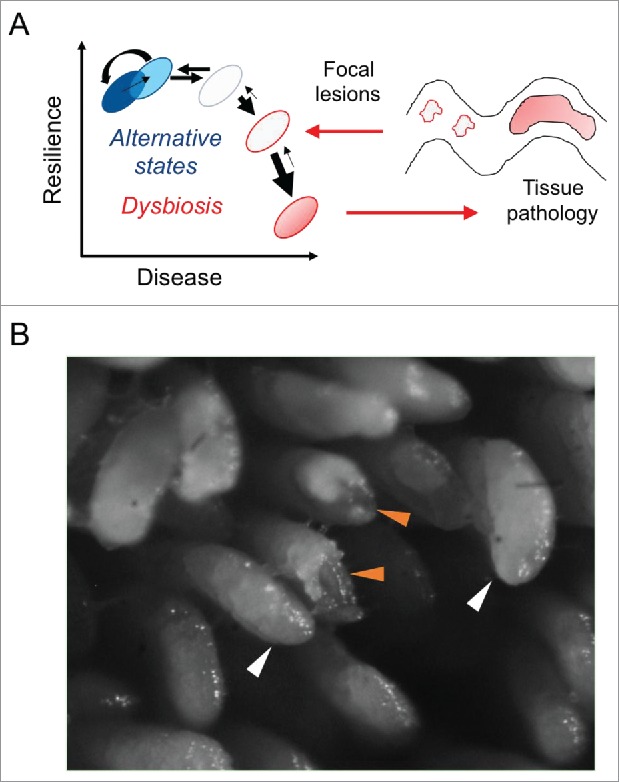
Stochastic occurrence of focal lesions in the gut lining generates local sites of inflammation. (A) The gut microbiota is constantly exposed to intrinsic (i.e., host genetics) and environmental factors (i.e., diet, smoking, antibiotics) that may induce non-permanent alteration in the bacterial community structure, leading to an homeostatic “alternative state.”6 However, the resilience of the gut microbiota may be lost in some pathologies, such as IBD, resulting in dysbiosis. (B) Our current hypothesis focuses on the formation in the epithelial lining of focal lesions, which cannot be sealed off in the genetic susceptible host.3 We speculate that the environmental milieu established in these lesions promotes the selection of specific bacterial taxa with aggressive and virulent features, which fuel inflammatory processes resulting in tissue pathology. Intact villi in GF mice (12 weeks old) are indicated by white arrows, while eroded villi are highlighted by orange arrows.
Paneth cell dysfunction does not precede inflammation and ileal pathology in TNFΔARE mouse model of CD-like ileitis
Abnormalities in Paneth cells structure and function have been increasingly recognized in IBD, and a growing body of evidence on Paneth cell lineage ablation supports their role in inflammation.7 Emerging data from genome-wide association studies (GWAS) in human cohorts also suggest that Paneth cells may play a critical role in the disease pathogenesis of IBD, specifically targeting CD. Key genes involved in Paneth cell function include XBP1, ORMDL3, NOD2, ATG16L1 and IRGM.6,8-11 The genetic deletion of Nod2 gene in mice results in Paneth cell dysfunction with a substantial reduction in the expression of a specific class of antimicrobial peptides (i.e., α-defensins) in the crypts of these mice, mirroring Paneth cell malfunction found in CD patients carrying the NOD2 mutation.12,13 Mice bearing mutations in genes associated with autophagy (ATG16L1 and IRGM genes) or in a gene involved in the endoplasmic reticulum stress response (XBP1) develop exacerbated intestinal inflammation and abnormalities in Paneth cell morphology and function, including diminished antimicrobial peptide secretion.11,14-16
It is necessary to acknowledge that not all studies support the finding that the deficiency of α-defensins expression is a predisposing factor for CD. For instance, Simms and coworkers showed that reduced levels of α-defensins are seen in CD, however this reduction is correlated with inflammation rather than with the NOD2 genotype of patients.17 This observation is in agreement with our findings, demonstrating that Paneth cell dysfunction is the consequence of inflammation rather than its cause.1 Of note, our transfer experiments of microbiota from R mice into GF TNFΔARE mice provided clear evidence that transcriptional activation of TNF and ileal pathology preceded Paneth cell dysfunction1 (Fig. 3). Failure of Paneth cell functionality, determined by the reduced protein expression of lysozyme and cryptdin-2 using immunofluorescence analysis, was an event subsequent to inflammation, and that was not caused by Paneth cell apoptosis in TNFΔARE mice.1 One tempting hypothesis is that the generation of reacting oxygen species (ROS) in response to inflammatory processes may lead to the functional exhaustion of Paneth cells.18 This is speculative and requires validation in TNFΔARE mice. It seems plausible to assume that the various IBD-related mouse models represent a distinct subset of patients and thus one subset of the disease pathogenesis.
Figure 3.
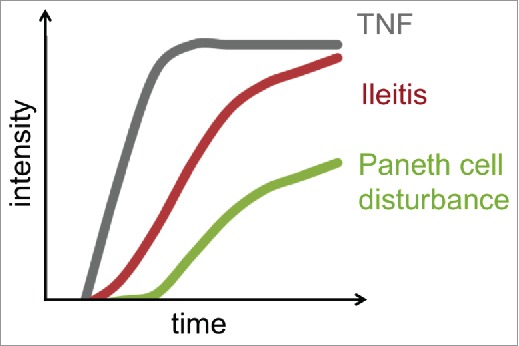
Inflammation (TNF levels in total tissue samples) and tissue pathology (CD-like ileitis H&E score) precede loss of Paneth cells function in TNFΔARE mouse model.
Conclusions and key open questions
Compositional and functional shifts in the intestinal microbial ecosystem are considered to represent key mechanisms in the pathogenesis of IBD. High-throughput sequencing analysis of microbial communities from patients with Crohn's disease and ulcerative colitis identified risk patterns associated with these distinct IBD phenotypes, however functional evidence for the existence of disease-conditioning ecosystems (dysbiosis) or the selection of single taxa (pathobionts) as underlying cause of chronic inflammation is still lacking.6 Until now, mechanistic studies about the role of bacteria in the pathogenesis of CD with predominant ileal inflammation were hampered by the lack of adequate GF mouse models. GF animal models susceptible to inflammatory pathologies are excellent tools to study the causal role of microbial ecosystems in disease initiation. However, when using a GF mouse model approach, it is essential to investigate bacterial colonization efficiency, which may be affected by several factors. In fact, a specific time window is required to develop a stable microbiota composition and a normal mucus system.19-21 Moreover, host mechanisms are important for the selection of a host-specific microbiota, e.g., host specificity,22 host immune system maturation,23 host gut selective pressure24 and host glycan repertoire.19,25 The use of antibiotics in place of GF mouse models should be carefully examined owing to the specific effects of antibiotics on different bacterial taxa, possibly resulting in selection of aggressive microbiota, and on the host epithelium, such as mucus thickening processes.19
For the first time, our laboratory showed that inflammation-associated changes in the microbiota caused transmissible disease in the genetically susceptible host (TNFΔARE mice) but not in WT recipients.1 Schaubeck and coworkers demonstrated that microbial communities, although developed under the same experimental conditions, may undergo progressive changes in their structures, leading to a more aggressive phenotype over time.1 The current goal is to identify the trigger that drives microbiota modification from neutral to aggressive nature. A growing body of evidence reports that several IBD susceptibility genes are associated with Paneth cell dysfunction. Nevertheless, in the TNFΔARE mouse model, Paneth cell loss was a secondary event to inflammation and tissue pathology.1 Given that TNF reduces epithelial barrier function by promoting sharp increases in tight junction permeability,26 it can be speculated that specific levels of TNF, such as those detected in lowR and R mice, are necessary to induce intestinal epithelial cells impairment, resulting in Paneth cell failure. To explain different grade of disease onset in our TNFΔARE mouse model, we propose that stochastic injuries in the gut mucosa generate focal inflammation leading to a dysbiotic microbial ecosystem with an individual selection of bacteria. This may explain the so far unmet effort to identify a unified microbial profile or single bacterial taxa responsible for the pathogenesis of IBD. Inflamed tissue areas in the gut are often enriched or depleted of selective bacterial taxa (e.g., Proteobacteria or Clostridia), most likely highlighting the fact that those bacteria quickly adapt to an altered milieu rather than being responsible for disease development.
Despite these findings,1 we still have no understanding about the characteristic features of disease-conditioning (dysbiotic) microbial communities. Therefore, our long-term aim is to better identify the functional plasticity of complex microbial ecosystems toward changes in the intestinal milieu, such as those triggered by focal injuries and inflammation. Mechanistically, the most important question is centered on a systematic understanding of microbe-host interactions leading to the onset of disease. Most importantly, will it be possible to predict disease onset or relapse based on individual microbiota composition and function? Although fecal material transplantation (FMT) is a quite promising option for IBD treatment, its therapeutic efficacy is still poor, supporting the hypothesis that the selection patient-specific donor microbiota is not rationalized.27,28
In conclusion, a systematic investigation of the molecular mechanisms underpinning the development of dysbiosis in the context of stochastically appearing focal injuries in the gut mucosa is likely to offer new insights into the pathophysiology of chronic inflammatory diseases.
Disclosure of potential conflicts of interest
No conflicts of interest were disclosed.
References
- [1].Schaubeck M, Clavel T, Calasan J, Lagkouvardos I, Haange SB, Jehmlich N, Basic M, Dupont A, Hornef M, von Bergen M, et al.. Dysbiotic gut microbiota causes transmissible Crohn's disease-like ileitis independent of failure in antimicrobial defence. Gut 2016; 65:225–237; PMID:25887379; http:dx.doi.org/ 10.1136/gutjnl-2015-309333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rodriguez-Palacios A, Kodani T, Kaydo L, Pietropaoli D, Corridoni D, Howell S, Katz J, Xin W, Pizarro TT, Cominelli F, et al.. Stereomicroscopic 3D-pattern profiling of murine and human intestinal inflammation reveals unique structural phenotypes. Nat Commun 2015; 6:7577; PMID:26154811; http://dx.doi.org/ 10.1038/ncomms8577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Butto LF, Haller D. Dysbiosis in intestinal inflammation: Cause or consequence. Int J Med Microbiol 2016; 306(5):302-9; PMID:27012594 [DOI] [PubMed] [Google Scholar]
- [4].Rodriguez-Palacios A, Buttó LF, Bederman IR, Haller D, Cominelli F. Su1879 Spatial 3D-Stereomicroscopic, microbial and metabolic characterization of intestinal villous erosions and ulcerations in mice. Gastroenterology 2016; 150:S578; http://dx.doi.org/ 10.1016/S0016-5085(16)31978-3 [DOI] [Google Scholar]
- [5].Ocvirk S, Sava IG, Lengfelder I, Lagkouvardos I, Steck N, Roh JH, et al.. Surface-Associated Lipoproteins Link Enterococcus faecalis Virulence to Colitogenic Activity in IL-10-Deficient Mice Independent of Their Expression Levels. PLoS Pathog 2015; 11:e1004911; PMID:26067254; http://dx.doi.org/ 10.1371/journal.ppat.1004911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Buttó LF, Schaubeck M, Haller D. Mechanisms of microbe-host-interaction in Crohn's Disease: Dysbiosis vs. Pathobiont selection. Frontiers Immunol 2015; 6:555; http://dx.doi.org/ 10.3389/mmu.2015.00555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wehkamp J, Stange EF. Paneth's disease J Crohns Colitis 2010; 4:523-31; PMID:21122555; http://dx.doi.org/ 10.1016/j.crohns.2010.05.010 [DOI] [PubMed] [Google Scholar]
- [8].Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al.. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491:119-24; PMID:23128233; http://dx.doi.org/ 10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al.. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015; 47:979-86; PMID:26192919; http://dx.doi.org/ 10.1038/ng.3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kaser A, Adolph TE, Blumberg RS. The unfolded protein response and gastrointestinal disease. Semin Immunopathol 2013; 35:307-19; PMID:23588234; http://dx.doi.org/ 10.1007/s00281-013-0377-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, Glickman JN, Tschurtschenthaler M, Hartwig J, Hosomi S, et al.. Paneth cells as a site of origin for intestinal inflammation. Nature 2013; 503:272-6; PMID:24089213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, et al.. Reduced Paneth cell alpha-defensins in ileal Crohn's disease. Proc Natl Acad Sci U S A 2005; 102:18129-34; PMID:16330776; http://dx.doi.org/ 10.1073/pnas.0505256102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005; 307:731-4; PMID:15692051; http://dx.doi.org/ 10.1126/science.1104911 [DOI] [PubMed] [Google Scholar]
- [14].Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al.. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008; 456:259-63; PMID:18849966; http://dx.doi.org/ 10.1038/nature07416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu B, Gulati AS, Cantillana V, Henry SC, Schmidt EA, Daniell X, Grossniklaus E, Schoenborn AA, Sartor RB, Taylor GA. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am J Physiol Gastrointest Liver Physiol 2013; 305:G573-84; PMID:23989005; http://dx.doi.org/ 10.1152/ajpgi.00071.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, et al.. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008; 134:743-56; PMID:18775308; http://dx.doi.org/ 10.1016/j.cell.2008.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Simms LA, Doecke JD, Walsh MD, Huang N, Fowler EV, Radford-Smith GL. Reduced alpha-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn's disease. Gut 2008; 57:903-10; PMID:18305068; http://dx.doi.org/ 10.1136/gut.2007.142588 [DOI] [PubMed] [Google Scholar]
- [18].Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y, et al.. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 2006; 12:446-51; PMID:16565722; http://dx.doi.org/ 10.1038/nm1388 [DOI] [PubMed] [Google Scholar]
- [19].Johansson ME, Hansson GC. Immunological aspects of intestinal mucus and mucins. Nat Rev Immunol 2016; 16(10):639-49; PMID:27498766; http://dx.doi.org/ 10.1038/nri.2016.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Johansson ME, Jakobsson HE, Holmen-Larsson J, Schutte A, Ermund A, Rodriguez-Pineiro AM, Arike L, Wising C, Svensson F, Bäckhed F, et al.. Normalization of Host Intestinal Mucus Layers Requires Long-Term Microbial Colonization. Cell Host & Microbe 2015; 18:582-92; PMID:26526499; http://dx.doi.org/ 10.1016/j.chom.2015.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hansen CH, Nielsen DS, Kverka M, Zakostelska Z, Klimesova K, Hudcovic T, Tlaskalova-Hogenova H, Hansen AK. Patterns of early gut colonization shape future immune responses of the host. PLoS One 2012; 7:e34043; PMID:22479515; http://dx.doi.org/ 10.1371/journal.pone.0034043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Arrieta MC, Walter J, Finlay BB. Human microbiota-associated mice: A model with challenges. Cell Host Microbe 2016; 19:575-8; PMID:27173924; http://dx.doi.org/ 10.1016/j.chom.2016.04.014 [DOI] [PubMed] [Google Scholar]
- [23].Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, et al.. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 2012; 149:1578-93; PMID:22726443; http://dx.doi.org/ 10.1016/j.cell.2012.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rawls JF, Mahowald MA, Ley RE, Gordon JI. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 2006; 127:423-33; PMID:17055441; http://dx.doi.org/ 10.1016/j.cell.2006.08.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li H, Limenitakis JP, Fuhrer T, Geuking MB, Lawson MA, Wyss M, Brugiroux S, Keller I, Macpherson JA, Rupp S, et al.. The outer mucus layer hosts a distinct intestinal microbial niche. Nat Commun 2015; 6:8292; PMID:26392213; http://dx.doi.org/ 10.1038/ncomms9292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mashukova A, Wald FA, Salas PJ. Tumor necrosis factor alpha and inflammation disrupt the polarity complex in intestinal epithelial cells by a posttranslational mechanism. Mol Cell Biol 2011; 31:756-65; PMID:21135124; http://dx.doi.org/ 10.1128/MCB.00811-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sha S, Liang J, Chen M, Xu B, Liang C, Wei N, Wu K. Systematic review: faecal microbiota transplantation therapy for digestive and nondigestive disorders in adults and children. Aliment Pharmacol Ther 2014; 39:1003-32; PMID:24641570; http://dx.doi.org/ 10.1111/apt.12699 [DOI] [PubMed] [Google Scholar]
- [28].Paramsothy S, Kamm M, Walsh A, van den Boaerde J, Samuel D, Leong R, et al.. Multi donor intense faecal microbiota transplantation is an effective treatment for resistant ulcerative colitis: A randomised Placebo-Controlled trial. Gastroenterology 2016; 150, Issue 4, Supplement 1; S122-S123; http://dx.doi.org/ 10.1016/S0016-5085(16)30517-0 [DOI] [Google Scholar]