

Abstract
Adenosine deaminase acting on RNA (ADAR) 1 is the master editor of the transcriptome, catalyzing the conversion of adenosine to inosine (A-to-I). RNA transcripts fold into a variety of secondary structures including long intramolecular RNA duplexes that are the major substrate of ADAR1. Most A-to-I editing sites occur within RNA duplexes formed by complementary pairing of inverted retrotransposable elements interspersed within noncoding regions of transcripts. This catalytic activity of ADAR1 most likely prevents the abnormal activation of cytosolic nucleic acid sensors by self-dsRNAs. Homozygous disruption of mouse Adar is embryonic lethal due to a toxic type-I interferons response and correspondingly biallelic missense mutations in human ADAR1 cause a severe congenital interferonopathy. Here, we report that Cd19-Cre-mediated Adar gene ablation in the mouse causes a significant defect in the final stages of B cell development with an almost complete absence of newly formed immature and CD23+ mature recirculating B cells in the BM. Adar ablation in pre-B cells induced upregulation of typical interferon-stimulated genes (ISGs) and apoptosis upon further maturation. ADAR1 deficiency also inhibited the in vitro, IL-7-mediated, differentiation of BM-derived B cell precursors. In summary, ADAR1 is required, non-redundantly, for normal B lymphopoiesis in the BM and peripheral maintenance.
Keywords: RNA editing, ADAR1, B cell, lymphopoiesis, epitranscriptomics, Immunology and Microbiology Section, Immune response, Immunity
INTRODUCTION
RNA molecules undergo elaborate post-transcriptional modification, such as editing and methylation, collectively termed epitranscriptomics [1–3]. The most prevalent type of RNA editing in mammalians is the deamination of adenosine into inosine (A-to-I editing) primarily catalyzed by ADAR1. The Adar gene encodes a constitutively expressed p110 isoform and a longer interferon-inducible p150 isoform. The two isoforms have different translation initiation sites and promotors. The p110 isoform is present predominantly in the nucleus whereas the interferon-inducible p150 isoform is usually cytoplasmic [4]. The development of bioinformatics methodologies and tools to map A-to-I editing sites revealed that they are highly abundant in untranslated regions (UTRs) of mRNAs [1, 5]. The majority of editing within UTRs is promiscuous, occurring within long intramolecular RNA duplexes formed by complementary pairing of inverted repeats: e.g., Alu retrotransposons in the human and orthologous short interspersed repetitive elements (SINE) in the mouse [1, 6]. For example, the human transcriptome contains dense clusters of A-to-I editing sites, termed as hyper-edited, localizing mostly in Alu repeats [7].
Genetic ablation of Adar in the mouse is embryonic lethal at midgestation due to defective hematopoiesis and a detrimental type-I interferons response [8–12]. Similarly, infants with mutated ADAR1 develop during early life a severe interferonopathy, the Aicardi-Goutières syndrome (AGS), presenting with encephalopathy and features akin with systemic lupus erythematosus (SLE) and congenital viral infections [13–16]. Recent studies in mouse models show that deletion of genes involved in the innate immune response to double-stranded (ds)RNA rescues ADAR1 deficient mice to birth [8, 17, 18]. Taken as a whole these observations imply that a major biological function of Adar is hyperediting of repetitive elements in the transcriptome to prevent abnormal stimulation of nucleic acid sensing pathways [19, 20]. ADAR1 has been shown to have other conserved biological functions, as follows: (i) infrequent but important modifications of codons within mature mRNAs [21]; (ii) editing-dependent modifications of the alternative splicing of selected pre-mRNAs [22]; (iii) editing-independent and dependent modulation of microRNA biogenesis and/or function [23, 24].
Despite the fact that ADAR1 is essential for embryonal development, normal fetal and adult hematopoiesis, and maintaining innate immune homeostasis in the mouse and human [19, 20], no previous studies have addressed its role in B cell development. In this study, using in vivo Cd19-Cre-mediated conditional ablation of Adar gene, we asked how ADAR1 deficiency affects the late stages of B lymphopoiesis predominantly in the BM.
RESULTS AND DISCUSSION
CD19-Creki-mediated conditional ablation of Adar inhibits B lymphopoiesis
ADAR1 is required for the early stages of fetal and adult hematopoiesis [10, 25]. We now asked whether ADAR1 is also vital for the late phases (from pre-B cell stage and onward) of adult B lymphopoiesis, using a model system of Cd19-Creki-mediated recombination of floxed Adar. The B cell lineage develops initially in the BM from common lymphoid progenitors (CLP) through sequential stages: developing from CLP to pre-pro-B cells (Hardy classification Fr. A), to pro-B cells (Fr. B/C), to pre-B cells (Fr. D), and lastly to immature/new B cells (Fr. E) [26]. The expression of the CD19 molecule is a marker for B lineage-committed cells, and normally its expression starts at the pro-B cell stage [27]. We selected B cell-specific expression of Cre from the Cd19 locus over expression from the mb1 gene, as the efficiency of target gene recombination in the first model system is much lower in early precursors (Fr. B/C), increasing significantly as the B cell mature reaching efficiency of 80-95% in Fr. E to F cells [28]. We used two relevant mouse genotypes Adarfl/flCd19-Creki/+ and Adarfl/flCd19ki/ki to address our question, predicting that the second mutant, homozygous for the Cd19-Creki allele, would display a higher penetrance of biallelic ablation of the floxed Adar gene segment in late B cell precursors.
The FACS analysis of freshly isolated BM cells from Adarfl/flCd19-Creki/+ mice for the major stages of B lymphopoiesis in the BM, revealed a significant reduction of ~70% in the number of immature B cells (Fr. E) compared to littermate Adarfl/+Cd19-Creki/+ control mice (Figure 1A and combined data in Figure 2A). In addition, these mutants also showed a very significant reduction of ~75% in mature recirculating (MR; Fr. F) CD23+CD93− B cells (Figures 1B and combined data in Figure 2B). As predicted, the Adarfl/flCd19-Creki/ki mice displayed a more profound, less variable, reduction of ~95% in the percentage of immature B cells compared to littermate controls, and consequently an almost complete lack of MR B cells (Figures 1A-1B, and combined data in Figures 2A-2B). However, these two mutant mice groups did not show a significant reduction in the development and maintenance of more premature B cells precursors, exhibiting normal numbers of Fr. B to D cells in their BMs (data not shown). The percentages of CD3+ T cells and monocytes in their BM were also normal (data not presented).
Figure 1. Cd19-Cre-mediated Adar disruption causes profound deficiency of immature and recirculating mature B cells in the BM.
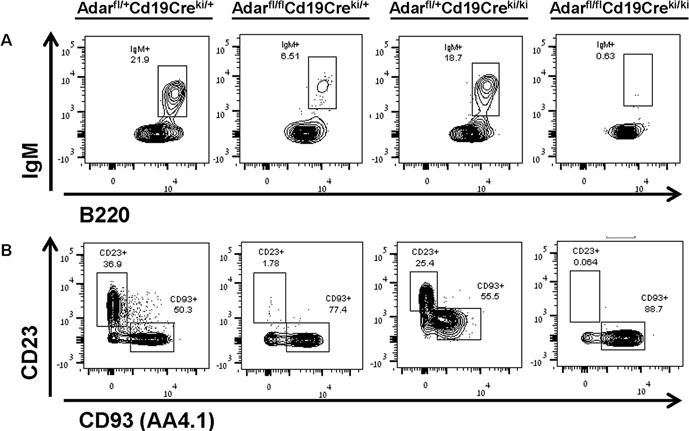
Single cell suspension of BM cells were obtained from 8-16 week-old mice and depleted of erythrocytes. Next, to identify B cell precursors in the last stages of B-lymphopoiesis we immunostained the samples with flourochrome-conjugated mAbs as detailed in Materials and Methods. Samples were acquired by a FACSAria instrument and the data (> 10,000 events per sample) were analyzed/plotted using the FlowJo V.10 software. A. Upper contour plots depict the percentage of immature IgM+ B cells in the indicate group (21.9. 6.51, 18.7 and 0.63, respectively). B. Lower plots show the percentage of MR CD23+CD93− B cells in the indicate group (36.9, 1.78, 25.4, and 0.064; respectively) and other less mature B-lineage cells (50.3, 77.4, 55.5, and 88.7; respectively). The data shown are from a representative mouse from each of the four indicated genotypes.
Figure 2. B lineage specific Adar ablation results in a profound deficiency of immature (naïve) and peripheral (mature) B cell deficiency.

Charts show the percentage of A. immature and B. MR B cells in the BMs from a large number of mutants and littermate control mice. C. Percentage of peripheral blood circulating B cells in the four indicated mice groups. Percentage of D. total B220+ and E. transitional T2/follicular CD23+ splenic B cells. Each point in the graphs represents the percentage of the specified B cell subset for an individual mouse. Mean values are represented with horizontal bars, and error bars represent ±SD. P values were determined by the one-way ANOVA test (compared against Adarfl/+Cd19-Creki/+ controls).
Newly formed conventional B cells (Fr. E) undergo further maturation in the periphery. Therefore, we also determined the effect of Adar ablation on mature splenic and peripheral blood (PB) circulating B cells. The combined FACS data showed, yet again, a significant reduction in the percentage of PB circulating B cells, total splenic B cells, and transitional T2 plus mature CD23+ splenic B cells in the two mutant mice groups compared to respective littermate controls. As expected, the block in B cell peripheral maturation and maintenance was more substantial in Adarfl/flCd19-Creki/ki compared to Adarfl/flCd19ki/+ mutants. The findings that the deleterious biological consequences of Cd19-Creki-mediate Adar gene ablation manifested on or after the immature B cell stage (Fr. E to F) agree with our prediction, based on previous data [28], that the penetrance of Cd19-Cre-mediated Adar recombination should become extensive only in the late phases of B lymphopoiesis.
Previous data from the Rajewsky laboratory on the phenotype of homozygous Cd19-Creki mice, lacking surface CD19 expression, show that conventional B cell development in their BM was undisturbed. However, their peripheral B cells showed a highly reduced capacity to respond to T cell-dependent antigens and for antibody affinity maturation [27]. Moreover, while CD23+ transitional T2 and follicular splenic B cells were present in adequate numbers in CD19 deficient mice, the subset of innate-like CD23− marginal zone B cells was practically absent [29]. We also found grossly normal percentages of Fr. B to E cells in the BM of Adarfl/+Cd19-Creki/ki mice and only a minimal reduction in the percentage of CD23+ splenic B cells (Figures 1–2 and unpresented data). Taken as a whole these observations lead us to conclude: (i) the deficiency in the late stages of B lymphopoiesis in the two mutants is mostly ADAR1-dependent; and (ii) the more profound maturation block in the Adarfl/flCd19-Creki/ki genotype is linked to an almost complete penetrance of Adar ablation in late B cell precursors of these mutant mice.
ADAR1 is required for IL-7-dependent in vitro generation of late B cell precursors
To validate that the significant defect in late B cell precursor maturation in our model is mainly ADAR1-dependent, we cultured BM cells from Adarfl/flCd19-Creki/+ mutants (Cd19 sufficient) with IL-7 for 5 and 7 days. This in vitro culture system permitted us to study the expansion and maturation of IL7R+CD19− B-lineage early precursors into B220+CD19+ late precursors (Fr. D/E). As predicted, we found that the in vitro generation of CD19+ B cell precursors was significantly reduced (p < 0.0001) at both time points in Adarfl/flCd19-Creki/+ mutants compared to control Adarfl/+Cd19-Creki/+ littermate cultures (Figure 3A). In contrast, in vitro generation of late B cell precursors was largely normal in Adarfl/+Creki/ki BM cultures.
Figure 3. B cell precursors from Adarfl/flCd19-Creki/+ display a significantly reduced capacity for IL-7-dependent generation of CD19 B cells in vitro.

A. Bone marrow cells supplemented with rhIL-7 were cultured for 5 or 7 days, as described in methods. Cells were harvested, at the indicated time points, and absolute number of CD19+ B cells per mL was determined by FACS with the 123count eBeads kit. BM cultures were done in triplicates and bar graphs depict mean (±SD) from a representative experiment out of > 3 performed. Absolute CD19+ B cell counts in BM cultures of Adarfl/flCd19-Creki/+ mutants were significantly lower (p < 0.0001 by one-way ANOVA test) compared to the other groups at both time points. B. At the end of BM cultures (day 7), purified DNA from each respective sample was analyzed by PCR to detect relative quantity of either the Adar ∆ allele (lane 1), wt & fl alleles (lane 2), or all the three alleles simultaneously (lane 3). The image was acquired using a Molecular Imager ChemiDoc XRS+ system, and the densitometry analysis of the bands intensity was done using the Image Lab 5.1 software (BioRad Laboratories, Inc.).
Additionally, to confirm ongoing Cd19-Cre-mediated Adar gene segment excision in our model, we analyzed the genomic DNA purified from day 5 cultures, by gel electrophoresis for the following PCR products: Adar ∆ allele alone (lane 1), wt & fl alleles (lane 2), or all the three alleles simultaneously (lane 3). We found that loxP site recombination was prominent in vitro in the three mice genotypes (Figure 3B, lane #1 in all samples). Moreover, the densitometry of band intensities of the Adarfl/flCd19-Creki/+ sample (lane #3), revealed that the relative level of ∆ allele was 0.54-fold compared to the fl allele (Figure 3B, see bar graph). This important finding demonstrates, as previously reported [10, 30], that incomplete penetrance of Cre-mediated Adar recombination results in selection against cells with biallelic Adar ablation. Moreover, Adarfl/+Cd19-Creki/ki BM cultures displayed the highest efficiency of Adar recombination: the relative level of the ∆ allele (normalized to fl allele) was 1.51-fold compared to 0.68- and 0.54-fold in the other two littermate groups (Figure 3B, bar graph). Taken together these data confirm that Cd19-Creki homozygosity is coupled with a several-fold higher efficiency of floxed gene segment recombination, supporting the conclusion that the substantial block in B lymphopoiesis in Adarfl/flCd19-Creki/ki mice is mainly due to an almost complete penetrance of Adar ablation and its detrimental biological consequences.
Adar ablation in B cell precursors leads to apoptosis and upregulation of typical ISGs
By Annexin-V staining of BM cells cultured overnight (Figure 4A), we found an increased rate of apoptosis of immature B cells from Adarfl/flCd19-Creki/ki and Adarfl/flCd19-Creki/+ mutants compared to relevant littermate controls (~12- and ~4-fold increase, respectively). Our data agree with previous data showing that the loss of ADAR1 in Hematopoietic stem cells leads to rapid apoptosis [10]. The two major pathways previously linked to the induction of apoptosis in ADAR1 deficient cells include: (i) excessive activation of the dsRNA-dependent eukaryotic translation initiation factor 2-alpha kinase (Eif2ak2/PKR) with subsequent inhibition of protein synthesis; and (ii) dsRNA-dependent RIG-I-like receptors (RLR) mediated activation of IRF3 and a robust type-I interferons response [31, 32].
Figure 4. Cd19-Cre dependent disruption of Adar leads to apoptosis and type-I interferons response.

A. Histograms depict FACS analysis of Annexin V staining of pre-B cells (upper panel) and immature B cells (lower panel) following overnight in vitro incubation. C. Pro-B cells and pre-B cells were isolated from BMs by a FACSAria sorter, and total RNA was purified from the cells as detailed in Methods. Next, we measured by qRTPCR the relative mRNA expression of the indicated ISGs. Bar graphs depict mean of triplicates (± SD), as calculated by the ∆∆CT method, normalized to Gapdh gene expression. Data show a representative experiment out of three performed.
Next, to study whether Adar ablation in B cell precursors was linked to aberrant induction of the PKR and RLR dependent signaling pathways, we first isolated by FACSorting pro-B and pre-B cells from the BMs of Adarfl/flCd19-Creki/ki mice and littermate controls. Total RNA from the FACS-sorted cells was then analyzed by qRTPCR for the relative quantity of a group of selected ISGs downstream of RLR activation (IRF7, CXCL10, RSAD2, and IFIT1). In pro-B cells, we found increased levels of IRF3 and PKR, both linked to the activation of pro-apoptotic pathways following ADAR1 silencing [33, 34]. In pre-B cells, we found increased transcription of all four selected ISGs (Figure 4B). Interestingly, IRF7 that was strongly upregulated in ADAR1 deficient pre-B cells is a major enhancer of the RLR mediated innate immune response [35, 36]; thus implying robust activation of this response in our model. Since the percentage of late B cell precursors (Fr. E/F) was extremely low in the BM of mutant mice, with the majority of these rare cells rapidly undergoing to apoptosis, we could not study the effect of Adar ablation directly in this relevantcell population.
Nucleic acids are the essential carriers of genetic information for all living organisms. Thus, selective identification of foreign nucleic acids is a major undertaking of the mammalian innate immune system [36]. Recent advances imply a role for dysregulation of nucleic acid sensing pathways in human autoimmunity [3, 13, 19]. In fact, mutations in ADAR1 are a genetic cause of the prototypic interferonopathy, AGS, displaying similarities with familial SLE including the production of distinct autoantibodies to nuclear proteins [14, 15]. Moreover, recent studies in mouse models show that genetic deletion of Mavs can rescue Adar−/− mice to birth. Likewise, AdarE861A/E861A knock in mice, expressing only a catalytically inactive ADAR1 and displaying embryonic lethality, could be rescued by concurrent knocking-out of Ifih1, encoding the cytoplasmic dsRNA sensor MDA5 [8, 9]. Activation of MDA5 induces the polymerization of MAVS that recruits and activates selected TRAF E3-ligases. This mediates the activation of the IKK and TBK1 kinases to phosphorylate IκBα and IRF3, respectively, leading to the robust transcription of ISGs and other cytokines [36, 37]. Taken as a whole these observations imply that a major biological function of ADAR1, conserved across mammalians, is the editing of repetitive sequences in transcripts that prevent the abnormal activation of MDA5 by self-dsRNAs. Indeed, our observation that a group of typical ISGs was upregulated in pre-B cells of Adarfl/flCd19-Creki/ki mutant mice is consistent with the latter paradigm.
A previous report from the Nishikura laboratory [38] describes the use of Cd19-Cre mediated ablation of Adar to study its role in the biogenesis of miR-142 by splenic B cells. However, this report did not provide pertinent data regarding the effects of ADAR1 deficiency on B cell development and survival in the BM or spleen, and hence its relevance to our study is small. Pestal et al. have very recently reported that Adar and Mavs double knockout mice, surviving past 1 week of age, show a significant deficiency of mature peripheral B cells, but not of T cells and dendritic cells, within their peripheral lymphoid organs [17]. Unlike our work, the authors did not study the effect of germline Adar deletion on the process of B lymphopoiesis in the BM nor whether ADAR1 was vital, in this model, for the early (Fr. A/B), late (Fr. C-E) and/or final (Fr. F) B development. Importantly, our novel finding that ISGs are significantly upregulated in ADAR1 lacking pre-B cells is consistent with their observation, employing the Adarp150−/− mouse model [39], that the B cell deficiency is ADAR1 p150 isoform dependent [17]. This interferon-inducible isoform, as opposed to the constitutive p110 isoform, performs its catalytic function mainly in the cytosol, playing a key role in the cytosolic regulatory network enabling tolerance to paired RNA components (dsRNAs) of the trascriptome [3, 19, 20].
Pertinently, we have just revealed that hepatocyte-specific ablation of ADAR1 induces massive liver damage with multifocal inflammation and hepatocyte apoptosis. The bioinformatics analysis of the whole transcript expression arrays from these abnormal livers indicated a robust upregulation of ISGs and related immune response genes [30]. Thus, we now communicate multiple data that Adar, is not only vital for stem/progenitor type cells in the fetal and early postnatal period, but also for more differentiated functional cells in adult mice: e.g., hepatocytes and newly formed immature B cells with functional surface IgM receptor.
In conclusion, we show for the first time that ADAR1, the master transcriptome editor, is essential for the late stages of B cell lymphoiesis in the BM and very likely for the survival of newly formed B cells in the periphery. Since the perturbation of nucleic acid sensing pathways and of autoreactive B cells plays a central role in SLE [40, 41] our data imply that further insight into the immunoregulatory role of ADAR1 may be relevant for a subset of autoimmune disorders.
MATERIALS AND METHODS
In vivo B cell-specific Cd19-Cre mediated conditional disruption of Adar
Mice with a loxP-flanked Adar1f7-9 (Adarfl) allele through the germline were previously described [11]. Homozygous Adarfl/fl mice backcrossed into the C57BL/6 background were kindly provided by Stuart H. Orkin (Dana-Farber Cancer Institute, Boston, MA) and Peter H. Seeburg (Max Planck Institute for Medical Research, Heidelberg, Germany). The B6.129P2(C)-Cd19tm1(cre)Cgn/J deleter mice were obtained from The Jackson Laboratory (Bar Harbor, ME). The Cd19-Cre knock-in/knock-out allele (Cd19-Creki) has a Cre gene inserted into the first exon of the Cd19 gene [27].
To generate mice with B cell lineage-specific disruption of Adar we first bred Adarfl/fl mice to Cd19-Creki/ki deleter mice to produce Adarfl/+Cd19-Creki/+offspring that were further interbred to generate four different genotypes. Two relevant mutant genotypes (Adarfl/flCd19ki/ki / Adarfl/flCd19-Creki/+) with high and low efficiency of biallelic B cell-specific AdarΔ7-9 somatic gene segment excision, respectively, and two relevant littermate controls (Adarfl/+Cd19-Creki/ki / Adarfl/+Cd19-Creki/+). Genomic DNA samples isolated from the tail tips of 3wk mice pups were analyzed by multiplex PCR that simultaneously detects Adar the wt(+), floxed(fl), and excised (Δ7-9) alleles, as previously described [11]. In paralleled, the presence of the Cd19-Creki and/or the wt (+) allele was determined by a different multiplex PCR assay [27]. Additionally, to verify actual Cd19-Creki-dependent Adar gene disruption, selected DNA samples from BM cells of 8-16 week old mutant and littermate control mice were analyzed by multiplex PCR for the presence of Adar +, fl, or excised Δ7-9 alleles. The mice were bred and housed in a SPF facility, and did not display any gross abnormalities as mature adults (3-6 months of age). The Institutional Animal Care and Use Committee of the Weizmann Institute of Science (Rehovot, Israel) approved all animal experiments.
FACS analysis of B cell developmental stages
Single-cell suspensions of BM, Spleen, and of blood cells were obtained from 8-16 week old mice. Depletion of erythrocytes from the various sample was achieved using Red Blood Cell Lysis Buffer (Sigma-Aldrich Chemie GmbH, Steinheim, Germany). Next, the cells were immunostained with a relevant mixture of flourochrome conjugated mAbs against any of the following selected mouse B cell surface antigens B220/CD45R, CD19, CD43, HSA/CD24, CD25, AA4.1/CD93, CD23, IgM, and IgD; all obtained from eBioscience Inc. (San Diego, CA). Next, by polychromatic flow cytometry we analyzed the samples for relevant B cell developmental stages in the BM and periphery according to the Hardy classification and the Philadelphia nomenclature [26, 42]. The following B cells stages were identified: pro-B/Fr. B-C (B220+ CD25− CD43+ CD24++ CD19+/++ CD93+); pre-B/Fr. D (B220++ CD25+ CD43− CD24++ CD19+++ CD93+ IgM−); immature/Fr. E (B220+++ CD43+ CD24hi CD19+++ CD93+ CD25−/IgM+ IgD−); mature recirculating/Fr. F (B220+++ CD19+++ CD23++/hi CD93−); and splenic transitional T2 plus follicular (B220+++ CD19+++ CD23++/hi). Data were acquired on a FACSAria instrument (BD Biosciences, San Jose, CA), and analyzed/plotted using the FlowJo V.10 software (FlowJo, Ashland, OR).
Isolation of pro- and pre-B cells from BM
Single-cell suspensions of BM derived cells from 8-16 week old mutant and littermate controls were depleted of red blood cells and immunostained for relevant cell surface markers, as detailed above. Subsequently, we sorted out pro-B cells and pre B-cells using a FACSAria instrument to high purity (~95%), as previously described [43]. Next, the purified cell were lysed with TRIzol® Reagent (Invitrogen, Carlsbad, CA) and total RNA was purified using Direct-zol™ RNA Kits (Zymo Research Corporation, Irvine, CA) for subsequent downstream analysis. The quantification of selected gene transcripts was performed by quantitative PCR (qRTPCR) using specific predesigned pair of unlabeled PCR primers and a TaqMan® probe (TaqMan® Gene Expression Assays) and analyzed on an ABI PRISM 7300 Sequence Detection system (all reagents and the analyzer were from Applied Biosystems, Thermo Fisher Scientific Inc.). Selected DNA samples from pre-B cells of Adarfl/flCd19-Creki/ki mutants and controls were also analyzed by multiplex PCR to verify enrichment for the AdarΔ7-9 allele in the mutant mice samples.
BM cell cultures
To induce the IL7-dependent in vitro expansion and differentiation of early IL7R+ lymphoid progenitors, BM cultures were prepared as previously described [43]. Briefly, single cell suspension of BM cells obtained from the femurs of 8-16 week-old mice were first depleted of erythrocytes using Red Blood Cell Lysis Buffer (Sigma-Aldrich Chemie GmbH), per manufacturer's instructions. Then, BM cells were cultured, at 5 × 105 cells/ml in a 6-well plate, for 5 and 7 days in Iscove modified Dulbecco medium supplemented with 100 IU/mL of recombinant human IL-7 (PeproTech, Rocky Hill, NJ). At the end of the experiment, we performed absolute counting of B220+ CD19+ cells by flow cytometry employing the 123count eBeads kit (eBioscience Inc.), according to manufacturer's instructions. As expected, usually at the end of culturing most of the cells were in vitro formed B220intCD19+ B cell precursors.
Statistical analysis
Statistically significant differences between group means were determined by one-way ANOVA test with Dunnett's post hoc multiple comparison method, using Prism win V.5.02 (GraphPad Software, Inc. La Jolla, CA).
Acknowledgments
The authors thank Shirley Oren Ben-Shoshan for technical assistance.
G.R. is the incumbent of the Djerassi Chair in Oncology at the Sackler School of Medicine, Tel-Aviv University, Tel-Aviv 6997801, Israel.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
This study was supported by The Varda and Boaz Dotan Research Center in Hemato-oncology affiliated with the CBRC of Tel Aviv University (I.G. and G.R.); The Israeli Centers for Research Excellence I-CORE Program (ISF grants no. 41/11 and no. 1796/12 to G.R.); and The Flight Attendant Medical Research Institute (G.R.).
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors' response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
- 1.Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, Olshansky M, Rechavi G, Jantsch MF. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 2.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 3.O'Connell MA, Mannion NM, Keegan LP. The Epitranscriptome and Innate Immunity. PLoS Genet. 2015;11:e1005687. doi: 10.1371/journal.pgen.1005687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.George CX, Wagner MV, Samuel CE. Expression of interferon-inducible RNA adenosine deaminase ADAR1 during pathogen infection and mouse embryo development involves tissue-selective promoter utilization and alternative splicing. J Biol Chem. 2005;280:15020–15028. doi: 10.1074/jbc.M500476200. [DOI] [PubMed] [Google Scholar]
- 5.Eisenberg E, Adamsky K, Cohen L, Amariglio N, Hirshberg A, Rechavi G, Levanon EY. Identification of RNA editing sites in the SNP database. Nucleic Acids Res. 2005;33:4612–4617. doi: 10.1093/nar/gki771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neeman Y, Levanon EY, Jantsch MF, Eisenberg E. RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA. 2006;12:1802–1809. doi: 10.1261/rna.165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porath HT, Carmi S, Levanon EY. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat Commun. 2014:5. doi: 10.1038/ncomms5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349:1115–1120. doi: 10.1126/science.aac7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, Jantsch MF, Dorin J, Adams IR, Scadden AD, Ohman M, Keegan LP, et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014;9:1482–1494. doi: 10.1016/j.celrep.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10:109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. Liver Disintegration in the Mouse Embryo Caused by Deficiency in the RNA-editing Enzyme ADAR1. Journal of Biological Chemistry. 2004;279:4894–4902. doi: 10.1074/jbc.M311347200. [DOI] [PubMed] [Google Scholar]
- 12.Wang Q, Khillan J, Gadue P, Nishikura K. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science. 2000;290:1765–1768. doi: 10.1126/science.290.5497.1765. [DOI] [PubMed] [Google Scholar]
- 13.Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- 14.Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, Baxter P, Benko WS, Bergmann C, Bertini E, Biancheri R, Blair EM, et al. Clinical and Molecular Phenotype of Aicardi-Goutières Syndrome. The American Journal of Human Genetics. 2007;81:713–725. doi: 10.1086/521373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goutieres F. Aicardi-Goutieres syndrome. Brain Dev. 2005;27:201–206. doi: 10.1016/j.braindev.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 17.Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity. 2015;43:933–944. doi: 10.1016/j.immuni.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Funabiki M, Kato H, Miyachi Y, Toki H, Motegi H, Inoue M, Minowa O, Yoshida A, Deguchi K, Sato H, Ito S, Shiroishi T, Takeyasu K, Noda T, Fujita T. Autoimmune Disorders Associated with Gain of Function of the Intracellular Sensor MDA5. Immunity. 2014;40:199–212. doi: 10.1016/j.immuni.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 19.Liddicoat BJ, Chalk AM, Walkley CR. ADAR1, inosine and the immune sensing system: distinguishing self from non-self. Wiley Interdisciplinary Reviews: RNA. 2016;7:157–172. doi: 10.1002/wrna.1322. [DOI] [PubMed] [Google Scholar]
- 20.Mannion N, Arieti F, Gallo A, Keegan LP, O'Connell MA. New Insights into the Biological Role of Mammalian ADARs; the RNA Editing Proteins. Biomolecules. 2015;5:2338–2362. doi: 10.3390/biom5042338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solomon O, Oren S, Safran M, Deshet-Unger N, Akiva P, Jacob-Hirsch J, Cesarkas K, Kabesa R, Amariglio N, Unger R, Rechavi G, Eyal E. Global regulation of alternative splicing by adenosine deaminase acting on RNA (ADAR) RNA. 2013;19:591–604. doi: 10.1261/rna.038042.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishikura K. Functions and Regulation of RNA Editing by ADAR Deaminases. Annu Rev Biochem. 2009;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Iizasa H, Davuluri RV, Nishikura K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell. 2013;153:575–589. doi: 10.1016/j.cell.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.XuFeng R, Boyer MJ, Shen H, Li Y, Yu H, Gao Y, Yang Q, Wang Q, Cheng T. ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proc Natl Acad Sci U S A. 2009;106:17763–17768. doi: 10.1073/pnas.0903324106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- 27.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-l cell development in CD19-deficient mice. Nature. 1995;376:352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 28.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Brooks SR, Li X, Anzelon AN, Rickert RC, Carter RH. The Physiologic Role of CD19 Cytoplasmic Tyrosines. Immunity. 2002;17:501–514. doi: 10.1016/s1074-7613(02)00426-0. [DOI] [PubMed] [Google Scholar]
- 30.Ben-Shoshan SO, Kagan P, Sultan M, Barabash Z, Dor C, Jacob-Hirsch J, Harmelin A, Pappo O, Marcu-Malina V, Ben-Ari Z, Amariglio N, Rechavi G, Goldstein I, Safran M. ADAR1 deletion induces NFkappaB and interferon signaling dependent liver inflammation and fibrosis. RNA Biol. 2016 doi: 10.1080/15476286.2016.1203501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McAllister CS, Samuel CE. The RNA-activated protein kinase enhances the induction of interferon-beta and apoptosis mediated by cytoplasmic RNA sensors. J Biol Chem. 2009;284:1644–1651. doi: 10.1074/jbc.M807888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vitali P, Scadden ADJ. dsRNAs containing multiple IU pairs are sufficient to suppress interferon induction and apoptosis. Nature structural & molecular biology. 2010;17:1043–1050. doi: 10.1038/nsmb.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toth AM, Li Z, Cattaneo R, Samuel CE. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J Biol Chem. 2009;284:29350–29356. doi: 10.1074/jbc.M109.045146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfaller CK, Li Z, George CX, Samuel CE. Protein kinase PKR and RNA adenosine deaminase ADAR1: new roles for old players as modulators of the interferon response. Curr Opin Immunol. 2011;23:573–582. doi: 10.1016/j.coi.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 36.Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 37.Seth RB, Sun L, Ea C-K, Chen ZJ. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-B and IRF3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 38.Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward SV, George CX, Welch MJ, Liou L-Y, Hahm B, Lewicki H, de la Torre JC, Samuel CE, Oldstone MB. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:331–336. doi: 10.1073/pnas.1017241108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banchereau J, Pascual V. Type I Interferon in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Immunity. 2006;25:383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 41.Kirou KA, Lee C, George S, Louca K, Peterson MGE, Crow MK. Activation of the interferon- pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis & Rheumatism. 2005;52:1491–1503. doi: 10.1002/art.21031. [DOI] [PubMed] [Google Scholar]
- 42.Osmond DG, Rolink A, Melchers F. Murine B lymphopoiesis: towards a unified model. Immunol Today. 1998;19:65–68. doi: 10.1016/s0167-5699(97)01203-6. [DOI] [PubMed] [Google Scholar]
- 43.Oliver PM, Wang M, Zhu Y, White J, Kappler J, Marrack P. Loss of Bim Allows Precursor B Cell Survival But Not Precursor B Cell Differentiation in the Absence of Interleukin 7. The Journal of Experimental Medicine. 2004;200:1179–1187. doi: 10.1084/jem.20041129. [DOI] [PMC free article] [PubMed] [Google Scholar]