

Abstract
We selected 19 significantly-mutated genes in AMLs, including FLT3, DNMT3A, NPM1, TET2, RUNX1, CEBPA, WT1, IDH1, IDH2, NRAS, ASXL1, SETD2, PTPN11, TP53, KIT, JAK2, KRAS, BRAF and CBL, and performed massively parallel sequencing for 114 patients with acute myeloid leukemias, mainly including those with normal karyotypes (CN-AML). More than 80% of patients had at least one mutation in the genes tested. DNMT3A mutation was significantly associated with adverse outcome in addition to conventional risk stratification such as the European LeukemiaNet (ELN) classification. We observed clinical usefulness of mutation testing on multiple target genes and the association with disease subgroups, clinical features and prognosis in AMLs.
Keywords: acute myeloid leukemia, mutation, next generation sequencing, DNMT3A
INTRODUCTION
Acute myeloid leukemia (AML) is a biologically heterogeneous disease and its clinical behaviors largely vary among different cases. Prognostic stratification in AML is important to determine treatment strategy. A well-proven prognostic factor is cytogenetic abnormality, according to which patients can be categorized as favorable, intermediate and unfavorable cytogenetic groups [1, 2]. Those with inv(16)/t(16;16), t(8;21) and t(15;17) are classified into favorable cytogenetic risk group while those with complex karyotype, t(6;9), inv(3)/t(3;3), −5/del(5q) and −7/del(7q) are classified into adverse cytogenetic risk group. Cases with intermediate cytogenetic risk group are predominantly with normal cytogenetics (CN-AMLs), constituting about 50% of all AML cases.
Recently, mutations in FLT3, NPM1 and CEBPA have been shown to have significant prognostic impact were suggested to be included in the risk stratification system by European LeukemiaNet (ELN) [2]. In addition, several studies have suggested prognostic implication of mutations in other genes. DNMT3A mutations (about one third of intermediate cytogenetic risk group) were suggested to be associated with adverse outcomes among intermediate-risk cytogenetic group [3]. IDH1/2, which encodes the enzyme isocitrate dehydrogenase, was recently shown to be mutated in about 16% of all AMLs and have significant prognostic impact in CN-AML with mutated NPM1 without FLT3-ITD [4]. In some studies, TET2 mutation was suggested to be associated with poor prognostic impact in CN-AML or intermediate cytogenetic risk group [5, 6]. KIT mutation was suggested to have adverse prognostic impact in AML with t(8;21) or inv(16)/t(16;16) [7]. WT1 mutation in AML was found about 10-15% and associated with adverse prognostic impact in some studies [8, 9]. TP53 mutation is frequently associated with therapy-related myeloid neoplasm with adverse prognostic impact [10, 11]. RUNX1 was found ~20% of de novo CN-AML with short overall survival and relapse free survival [11]. ASXL1, SETD2, JAK2, KRAS, PTPN11, NRAS, BRAF and CBL were found in AML and other myeloproliferative neoplasms such as myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN) with varying frequency and prognostic impact [12–14].
In the present study, we have comprehensively reviewed recent literatures on AML mutations and selected target genes which show relatively high frequencies (NPM1, FLT3, IDH1/2, CEBPA, DNMT3A, WT1), adverse prognostic impact (TET2, ASXL1, NRAS, KRAS, KIT) and associations with secondary/therapy related myeloid neoplasm/cytogenetic abnormalities (JAK2, TP53, PTPN11, BRAF, CBL, SETD2) [5, 12, 14–22]. And we investigated mutation profiles of these 19 genes selected genes [5, 12, 14–22] and their association with clinical parameters and outcomes in Korean patients with AML. Our data suggested a significant impact of DNMT3A mutations over other conventional prognostic factors. Investigation on a relatively homogeneous population from a single institute will provide valuable information to the current knowledge on the impact of gene mutations on AMLs, together with molecular epidemiological insights on this regional population.
RESULTS
Clinical characteristics of 114 AML cases
Demographics and clinical characteristics of enrolled cases are summarized in Table 1 and Supplementary Table S1. Seventy male and 44 female were enrolled. Ninety-eight adult patients (>19 years) and 16 childhood (<20 years) AML patients were enrolled. Ninety-three cases were de novo AMLs and 12 cases were secondary or relapsed AMLs. Two cases were secondary AMLs evolved from primary myelofibrosis and polycythemia vera.
Table 1. Clinical and cytogenetic characteristics of enrolled cases.
| CN-AML (n=77) | CA-AML (n=26) | Unknown* (n=11) | Total | |
|---|---|---|---|---|
| Male/Female | 48/29 | 16/10 | 6/5 | 70/44 |
| Age, years (mean±SD) | 51.4±19.3 | 40.8±25.6 | 61.7±15.5 | 50.0±21.2 |
| Complete remission | 60/63(95.2%) | 20/21(95.2%) | 2/2(100%) | 82/86(95.3%) |
| Relapse | 27/60(45.0%) | 9/20(45.0%) | 1/2(50%) | 37/82(45.1%) |
| Bone marrow transplantation | 35/77(45.5%) | 14/26(53.8%) | 2/2(100%) | 51/105(48.6%) |
| Age group | ||||
| 1-19 years | 7 | 9 | 16 | |
| 20-39 years | 10 | 1 | 1 | 12 |
| 40-59 years | 29 | 8 | 3 | 40 |
| ≥60 | 31 | 8 | 7 | 46 |
| Secondary or relapse | 4 | 8 | 12 | |
| De novo | 73 | 18 | 2 | 93 |
| 2008 WHO classification | ||||
| AML with t(8;21)(q22;q22) | 4 | 4 | ||
| AML with inv(16)(p13q22) | 2 | 2 | ||
| AML with t(15;17)(q22;q21) | 2† | 2 | ||
| AML with myelodysplasia-related changes | 30 | 13 | 43 | |
| AML, not otherwise specified | ||||
| AML with minimal differentiation(M0) | 4 | 1 | 5 | |
| AML without maturation(M1) | 8 | 2 | 10 | |
| AML with maturation(M2) | 20 | 0 | 20 | |
| Acute myelomonocytic leukemia(M4) | 9 | 1 | 10 | |
| Acute monoblastic/monocytic leukemia(M5) | 2 | 1 | 3 | |
| Acute erythroid leukemia(M6) | 2 | 2 | ||
| Acute megakaryoblastic leukemia(M7) | 2 | 0 | 2 | |
| Unknown* | 11 | 11 | ||
| Cytogenetic risk group | ||||
| Favorable | 0 | 8 | 8 | |
| Intermediate | 77 | 7 | 84 | |
| Unfavorable | 11 | 11 | ||
| Unknown* | 11 | 11 |
Cytogenetic results were unavailable or inconclusive
One patient had normal karyotype but fluorescence in situ hybridization and reverse transcript PCR for PML-RARA rearrangement were positive
Categorization and cytogenetic risk group
Of the 114 patients enrolled, 11 had missing or inconclusive cytogenetic results. Among the 103 cases with unequivocal cytogenetic information, 77 were CN-AMLs and 26 were cytogenetically abnormal AMLs (CA-AMLs). As to recurrent cytogenetics, four cases with t(8;21)(q22;q22), 2 cases with inv(16)(p13q22) and 2 cases with t(15;17)(q22;q21) were also enrolled.
When categorized by the 2008 WHO classification of hematologic malignancies [23], AML-MRC was the most common type (43/114, 37.7 %). AML with maturation (20/52), AML without maturation (10/52) and acute myelomonocytic leukemia (10/52) were most common in AML-NOS.
According to the ELN prognostic stratification [2], eight patients were categorized into favorable, 84 patients were into intermediate, and 11 patients were into unfavorable cytogenetic risk groups. Among the 43 AML-MRC cases, 13 cases had cytogenetic abnormalities. Among these, 12 cases including those with −5/del(5q), −7/del(7q) and complex cytogenetic abnormalities were categorized as poor cytogenetic risk group according to the ELN prognostic stratification.
Information on treatment
Information on treatment regimen was available in 89 adult AML cases. A total of 74 patients received the intensive chemotherapy (high dose cytarabine and idarubicin). Nine patients had received low intensity chemotherapy (hydroxyurea, decitabine, azacitidine etc.), and six patients did not receive chemotherapy due to early death or unstable clinical status. Among 43 AML-MRC patients, 28 (73.6%) had received intensive chemotherapy, and 5 (13.2%) received low intensity chemotherapy. Five AML-MRC patients (13.2%) did not receive chemotherapy.
Among 56 intermediated risk group (i.e. de novo CN-AML with other than mutated NPM1 without FLT3-ITD) patients, 46 (81.2%) had received intensive chemotherapy, and 6 (10.7%) received low intensity chemotherapy. Four AML-MRC patients (7.1%) did not receive chemotherapy.
Mutation spectra, hotspots and co-occurrence
The overall fold coverage (read length/number of reads) was over 1000× (range: 1030-4830×) in all target regions except for some focal regions in KRAS, CEBPA and TET2 (Supplementary Figure S1). Among the 199 variations called and filtered, 166 (83.4%) were confirmed by Sanger sequencing (Supplementary Table S2 and S3). Because 3′-part of CEBPA was sequenced with very low coverage, Sanger sequencing results were integrated as a supplementation. The detection rate of FLT3-ITD by massively parallel sequencing was low (9 of 34 positive cases), especially in those with low mutant allele proportion, so clinical testing results were also integrated.
As a total, ninety-two (80.7%) cases had at least one mutation in the 19 genes investigated, suggesting that this panel could be used effectively as initial workup of AMLs. For all confirmed mutations, we statistically analyzed mutation profiles according to clinical and cytogenetic subgroups (Figure 1 and 2).
Figure 1. Mutational profiles of AML cases analyzed.
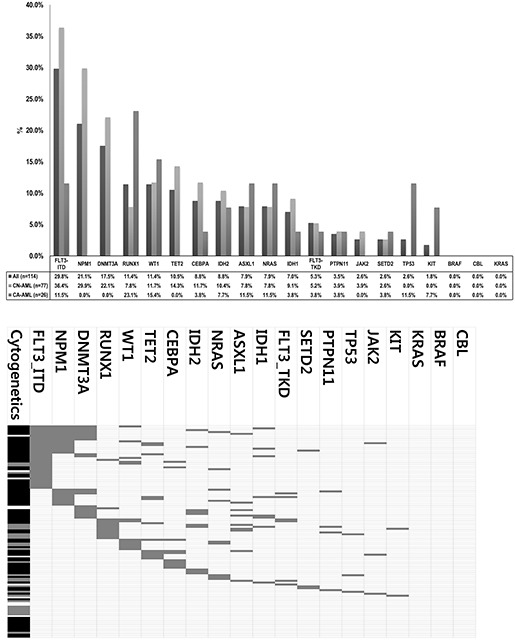
A. Mutation frequencies according to chromosome abnormalities. B. Mutation status in individual cases and co-occurrence among other mutations and chromosome abnormalities. Gray-color box indicates mutated case. For cytogenetic abnormalities, Black-color box indicates CN-AML, Gray-color box indicates CA-AML, White-color box indicates other AML (i.e. unknown cytogenetics).
Figure 2. Mutation spectrums according to the 2008 WHO Classification.
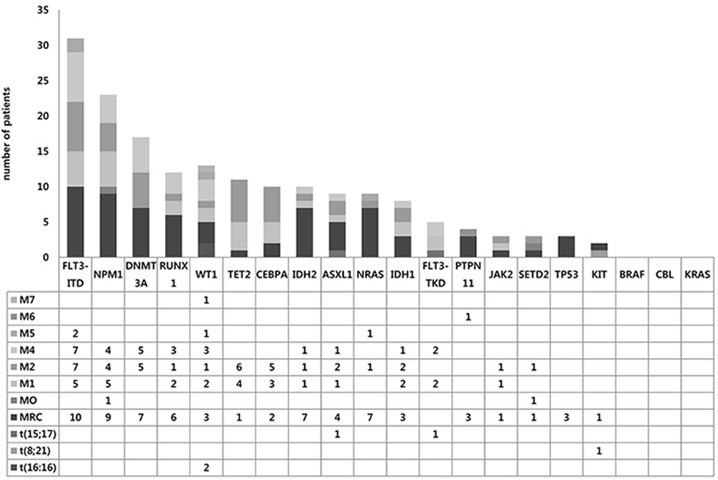
Proportion of mutations in each gene according to AML subgroups.
In CN-AML, FLT3-ITD was the most common mutation (36.4%), followed by NPM1 and DNMT3A mutations (29.9% and 22.1%, respectively). As previously reported, NPM1 mutations were found only in CN-AML and the majority (19/24, 79.2%) was type A (960insTCTG) mutation, while three Type B (960insCATG) and two Type D (960insCCTG) mutations were identified. DNMT3A mutations were found only in CN-AML, with R882H mutation being most common (9/20, 45%) as previously reported and other mutations being scattered across proline-tryptophan rich, zinc finger and methytransferase domains (Supplementary Figure S2). RUNX1 mutations were found in 13 cases (15 variation) with frameshift being the most predominant mutation type (10/15, 66.6%) (Supplementary Figure S3). WT1 mutations were detected in 13 cases (11.4%) overall, especially in codons 381 and 382 on exon 7 (Supplementary Figure S4). TET2 mutations were distributed throughout coding regions, and loss-of-function mutations including nonsense (9/17) and frameshift (5/17) were commonly observed (Supplementary Figure S5). All IDH1 mutations detected were R132C (100%, 9/9), and IDH2 mutations were detected either as R140Q (60%, 6/10) or R172K (30%, 3/10). NRAS mutations were detected in 10 cases (7.9%) overall and all were missense mutations in codons 12, 13 and 61 (Supplementary Figure S6). ASXL1 mutations were clustered around 5′-part of exon 12 (Supplementary Figure S7). PTPN11 mutations were found in 4 cases (3.5%) overall, with a predominant occurrence in exon 3 (Supplementary Figure S8). KIT mutations were detected only in two cases; AML with t(8;21) and AML-MRC. TP53 mutations were detected in three cases with AML-MRC.JAK2 V617F mutation was detected in secondary AMLs evolved from polycythemia vera and primary myelofibrosis, while one de novo AML (AML without maturation) case also harbored JAK2 V617F mutation. SETD2 mutations were detected in three (2.6%) de novo AMLs. Only one case with double CEBPA mutations was found in this study, whereas 9 cases had only a single CEBPA mutation. No mutation was detected in the CBL, KRAS and BRAF genes.
NPM1 mutations tended to co-occur with DNMT3A (P<0.001) and FLT3-ITD mutations (P<0.001) while DNMT3A mutation was frequently concurrent with IDH1/2 mutation (P=0.002) (Table 2). In line with several previous reports [24–28], high degrees of mutual exclusivity existed between IDH1 and IDH2, WT1 and IDH1/2, and DNMT3A and CEBPA mutations. We also newly observed a high exclusivity between DNMT3A and RUNX1 mutations.
Table 2. Co-occurrence of gene mutations.
| NPM1 | NPM1 | ||||||||
| DNMT3A | wild | mutant | P | FLT3-ITD | wild | mutant | P | ||
| wild | 81 | 13 | <.001 | wild | 71 | 9 | <.001 | ||
| mutant | 9 | 11 | mutant | 19 | 15 | ||||
| FLT3-ITD | WT1 | ||||||||
| DNMT3A | wild | mutant | P | IDH1/2 | wild | mutant | P | ||
| wild | 70 | 24 | .003 | wild | 84 | 13 | .211 | ||
| mutant | 10 | 10 | mutant | 17 | 0 | ||||
| CEBPA | IDH1/2 | ||||||||
| DNMT3A | wild | mutant | P | TET2 | wild | mutant | P | ||
| wild | 84 | 10 | .204 | wild | 86 | 16 | 0.690 | ||
| mutant | 21 | 0 | mutant | 11 | 1 | ||||
| IDH1/2 | RUNX1 | ||||||||
| DNMT3A | wild | mutant | P | ASXL1 | wild | mutant | P | ||
| wild | 85 | 9 | .002 | wild | 95 | 10 | .066 | ||
| mutant | 12 | 8 | mutant | 6 | 3 | ||||
| RUNX1 | IDH1 | ||||||||
| DNMT3A | wild | mutant | P | IDH2 | wild | mutant | P | ||
| wild | 82 | 12 | .459 | wild | 97 | 7 | .532 | ||
| mutant | 19 | 1 | mutant | 9 | 1 | ||||
Different mutation frequencies according to AML subgroups
FLT3-ITD mutations were frequently found in CN-AMLs than CA-AMLs (36.4% vs 11.5%; P=0.017) (Table 3). NPM1 mutations (29.9% vs. 0.0%; P=0.002) and DNMT3A mutations (22.1% vs. 0%; P=0.006) were exclusively found in CN-AML. FLT3-ITD, NPM1, DNMT3A, IDH1/2, TET2, WT1 and RUNX1 mutations were relatively common in de novo AMLs, while RUNX1, NRAS, PTPN11, JAK2, WT1 and IDH1/2 mutations were so in secondary or relapsed AMLs (Table 3). Nonetheless, only JAK2 mutation showed statistical significance.
Table 3. Mutation profile according to cytogenetic abnormality and de novo and secondary/relapsed cases.
| Gene | CN-AML (n=77) | CA-AML(n=26) | P | De novo (n=93) | Secondary or relapsed (n=12) | P |
|---|---|---|---|---|---|---|
| ASXL1 | 6(7.8%) | 3(11.5 %) | .689 | 8(8.6%) | 1(8.3%) | 1.000 |
| BRAF | 0(0.0%) | 0(0.0 %) | - | 0(0.0%) | 0(0.0%) | - |
| CBL | 0(0.0%) | 0(0.0 %) | 0(0.0%) | 0(0.0%) | - | |
| CEBPA | 9(11.7%) | 1(3.8 %) | .445 | 10(10.8%) | 0(0.0%) | .600 |
| DNMT3A | 17(22.1%) | 0(0.0 %) | .006 | 17(18.3%) | 0(0.0%) | .208 |
| FLT3-ITD | 28(36.4%) | 3(11.5 %) | .017 | 31(33.3%) | 1(8.3%) | .101 |
| FLT3-TKD | 4(5.2%) | 1(3.8 %) | 1.000 | 5(5.4%) | 0(0.0%) | 1.000 |
| IDH1 | 7(9.1%) | 1(3.8 %) | .676 | 7(7.5%) | 1(8.3%) | 1.000 |
| IDH2 | 8(10.4%) | 2(7.7 %) | 1.000 | 9(9.7%) | 1(8.3%) | 1.000 |
| JAK2 | 3(3.9%) | 0(0.0 %) | .570 | 1(1.1%) | 2(16.7%) | .034 |
| KIT | 0(0.0%) | 2(7.7 %) | .062 | 1(1.1%) | 1(8.3%) | .216 |
| KRAS | 0(0.0%) | 0(0.0 %) | - | 0(0.0%) | 0(0.0%) | - |
| NPM1 | 23(29.9%) | 0(0.0 %) | .002 | 23(24.7%) | 0(0.0%) | .064 |
| NRAS | 6(7.8%) | 3(11.5 %) | .689 | 6(6.5%) | 3(25.0%) | .065 |
| PTPN11 | 3(3.9%) | 1(3.8 %) | 1.000 | 2(2.2%) | 2(16.7%) | .063 |
| RUNX1 | 6(7.8%) | 6(23.1 %) | .070 | 9(9.7%) | 3(25.0%) | .138 |
| SETD2 | 2(2.6%) | 1(3.8 %) | 1.000 | 3(3.2%) | 0(0.0%) | 1.000 |
| TET2 | 11(14.3%) | 0(0.0 %) | .061 | 11(11.8%) | 1(8.3%) | 1.000 |
| TP53 | 0(0.0%) | 3(11.5 %) | .015 | 2(2.2%) | 1(8.3%) | .308 |
| WT1 | 9(11.7%) | 4(15.4 %) | .734 | 11(11.8%) | 2(16.7%) | .642 |
Among the cases with no recurrent cytogenetic abnormalities according to the 2008 WHO Classification, 92.3% (48/52) of AML, not otherwise specified (AML-NOS) and 81.4% (35/43) of AML-MRC had at least one mutations in the 19 genes examined (Table 4).NRAS mutation was more frequent in AML-MRC (16.3% vs. 3.8%, P=.074), whereas TET2 mutation was less frequent in AML-MRC than AML-NOS (2.3% vs. 19.2%, P=.011). Similar with previous reports [10, 29], we observed a relatively high frequency of TP53 mutations in AML-MRC (0.0% vs. 7.1%, P=.089).
Table 4. Comparison of mutation profiles and clinical characteristics between AML-MRC and AML-NOS.
| AML-NOS(n=52) | AML-MRC(n=43) | P | |
|---|---|---|---|
| Age | 46.9±21.2 | 52.9±20.6 | .112 |
| Sex(male/female) | 33/19 | 28/15 | .867 |
| Complete remission | 44/46(95.7%) | 28/30(94.7%) | .645 |
| Relapse | 15/44(34.1%) | 19/28(57.6%) | .005 |
| Bone marrow transplantation | 29/52(55.8%) | 15/43(34.9%) | .042 |
| Gene mutation | |||
| ASXL1 | 4(7.7%) | 4(9.3%) | 1.000 |
| CEBPA | 8(15.4%) | 2(4.7%) | .107 |
| DNMT3A | 10(19.2%) | 7(16.3%) | .709 |
| FLT3-ITD | 21(40.3%) | 10(23.3%) | .076 |
| FLT3-TKD | 4(7.7%) | 0(0.0%) | .124 |
| IDH1 | 5(9.6%) | 3(7.0%) | .725 |
| IDH2 | 3(5.8%) | 6(16.3%) | .177 |
| NPM1 | 14(26.9%) | 9(20.9%) | .497 |
| NRAS | 2(3.8%) | 7(16.3%) | .074 |
| PTPN11 | 1(1.9%) | 3(9.3%) | .325 |
| RUNX1 | 6(11.5%) | 6(14.0%) | .764 |
| SETD2 | 2(3.8%) | 1(2.3%) | 1.000 |
| TET2 | 10(19.2%) | 1(2.3%) | .011 |
| TP53 | 0(0.0%) | 3(7.0%) | .089 |
| WT1 | 8(15.4%) | 3(7.0%) | .335 |
| JAK2 | 2(3.8%) | 1(2.3%) | 1.000 |
Complete remission and relapse rates according to mutation status
Complete remission (CR) rate was 95.3% (82/86) and relapse rate was 45.1% (37/82). RUNX1 mutation was associated with low complete remission rate (57.1% vs. 100%, P=.001). We observed that NRAS and DNMT3A mutations were associated with high relapse rates (41.7% vs. 100%, P=.037 for NRAS; 38.9%, vs. 80.0%, P=.034, for DNMT3A) in adult de novo AML (Supplementary Table S4).
Overall survival according to mutation status
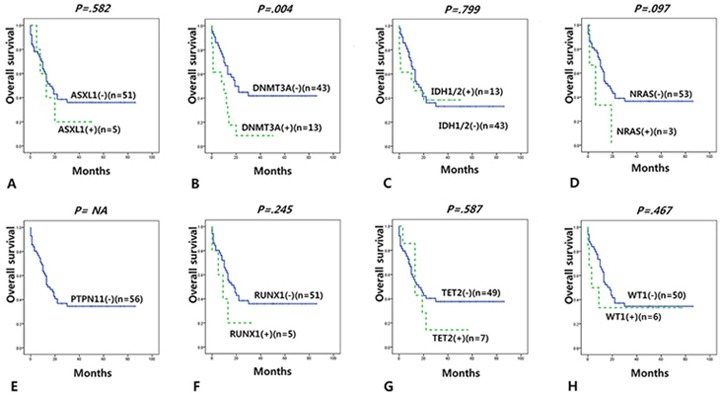
We investigated overall survival (OS) according to mutation status in adult (>19 years) de novo AMLs with normal karyotype (n=66) or in intermediate cytogenetics risk group (n=68) and found that DNMT3A mutation was significantly associated with short OS in a multivariate cox regression model (Supplementary Table S5).
We further reclassified adult cases according to the ELN classification which incorporates FLT3-ITD, NPM1 and CEBPA mutation status. DNMT3A and PTPN11 mutation were associated with short OS in the favorable risk ELN group (i.e. de novo CN-AML with mutated NPM1 without FLT3-ITD or de novo CN-AML with doubly-mutated CEBPA, n=10) (Supplementary Figure S9). DNMT3A mutations were associated with short OS in intermediate risk group (i.e. de novo CN-AML with other than mutated NPM1 without FLT3-ITD, n=56) (Figure 3).
Figure 3. Overall survival according to gene mutations in intermediate group (adult de novo CN-AML with other than mutated NPM1 without FLT3-ITD) according to the ELN classification.
Relapse free survival according to mutation status
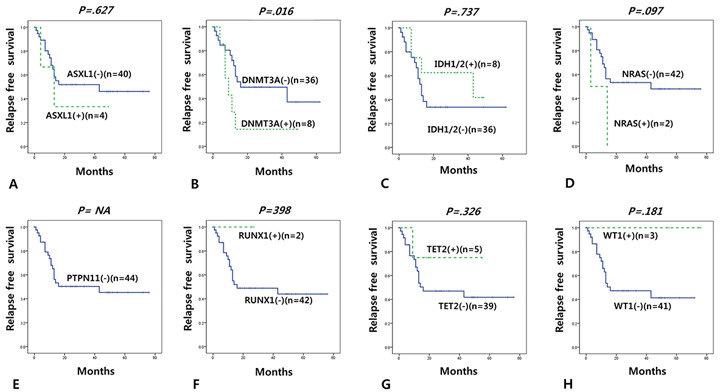
We also investigated relapse free survival (RFS) according to mutation status in 53 adult (>19 years) de novo CN-AML cases. In a multivariable cox regression model, only DNMT3A mutation was associated with short RFS (Supplementary Table S6). We observed the similar results in analysis of RFS in 55 adult (>19 years) de novo intermediate cytogenetic risk group AML cases (Supplementary Table S6). When re-grouped by the ELN classification, DNMT3A was associated with short RFS in the intermediate risk ELN group (Figure 4 and Supplementary Figure S10).
Figure 4. Relapse free survival according to gene mutations in intermediate group (adult de novo CN-AML with other than mutated NPM1 without FLT3-ITD) according to the ELN classification.
Clinical outcomes of adult de novo AML-MRC and AML-NOS
AML-MRC had significantly shorter RFS than AML-NOS, while OS was not significantly different (Supplementary Figure S11).
DISCUSSION
Considering the high prevalence of gene mutations in AML their prognostic impact and disease monitoring, mutation profiling at initial diagnosis may be necessary in addition to the conventional workups [4, 6, 30]. Recent high throughput sequencing technologies may be useful for detecting mutations in a number of target genes [19, 31, 32]. Compared to the whole genome or exome sequencing with highly sophiscated, time-consuming and intensive efforts, targeted gene sequencing may be a practical and ideal method for clinical testing in AML, with low cost, short turnaround time and less burden for bioinformatics [31, 33, 34]. Through a custom targeted panel, we could detect most of the target sites with high fold coverages (>1,000) and more than 80% of cases had at least one mutation in the genes investigated, suggesting the targeted sequencing strategy could be useful for initial workup. However, we had suboptimal coverages for CEBPA, where guanine-cytosine (GC) content is high and PCR amplification is problematic, and FLT3-ITD region, where large tandem duplication is common and short amplicon sequencing can miss some mutations. So for such mutations, conventional Sanger sequencing or PCR-fragment length analysis may be needed as a supplement [35].
As a whole, we observed mutation frequencies similar to previous studies in Western countries [3, 6, 10, 25, 27, 30] as well as in Asian countries. Wang et al [36] reported high mutation rates (>10%) in CEBPA, NPM1, DNMT3A, FLT3-ITD, NRAS, IDH2 and WT1. Kihara et al [37] reported high mutation rates of FLT3, NPM1, CEBPA, DNMT3A and KIT mutations in Japanese patients with AML. Some studies focusing on a single or a few genes have been reported in Korean patients with AML. DNMT3A R882 was detected 7.5% [38] and 18.3% [39] in Korean patients with CN-AML. Ahn et al reported that mutation rates for IDH1/2, TET2, NRAS and WT1 were 16.5%, 8.7%, 6.1% and 14.8%, respectively, in 115 Korean patients with CN-AML [39].
The impact of FLT3-ITD, NPM1 and CEBPA mutations on clinical outcome are well-established now, as implemented in the ELN classification [2, 28]. However, we observed particularly significant impact of DNMT3A mutations in the favorable or intermediate risk ELN group. As well, relapse rate of DNMT3A-mutated cases was significantly higher than unmutated cases (80.0% vs. 38.9%, P=0.034). Ahn et al [39] also reported poor prognostic impact (short OS, short RFS and high relapse rate) of DNMT3A R882 mutations in Korean patients with CN-AML after hematopoietic cell transplantation. Other previous studies also support that DNMT3A mutation can be an important prognostic factor [25, 26, 40–42] although some studies failed to find such associations [38, 43]. DNMT3A mutation was known to be more frequent in CN-AMLs, and we surprisingly observed a high exclusivity of the mutations in CN-AMLs compared CA-AMLs (22.1% vs. 0%). This might be due to the small number of CA-AMLs analyzed but should be further investigated in an extended set of patients. Considering the high positive rates and possible clinical implications, we may suggest that DNMT3A can be used in routine diagnostics and as an additional factor to the ELN risk stratification. We also observed possible prognostic implication of some genes with lower frequencies, including WT1, PTPN11, NRAS and ASXL1. Comprehensive mutation profiling may be needed when clinical implication of these rare mutations is established by future large studies and meta-analyses.(35)
In conclusion, we observed the recurrent mutation profile and their clinical significance using Korean AML patients. Although relatively small number of enrolled cases and heterogeneous population, it is first comprehensive mutation analysis using Korean patients. Considering the wide heterogeneity in mutational spectrums and clinical characteristics, it will be essentially needed to accumulate more data on the mutational profile and clinical associations in AMLs from different centers and working groups.
MATERIALS AND METHODS
Study population
We obtained bone marrow samples from 114 patients with AML diagnosed at Samsung Medical Center from 2008 to 2012, as well as peripheral blood samples from 4 healthy controls. Genomic DNA was isolated using the Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA). Diagnosis are based on 2008 WHO classification of tumors of haematopoietic and lymphoid tissues [23]. AML-MRC was defined as AML with morphological features of myelodysplasia or a prior history of a myelodysplastic syndrome (MDS) or myelodysplastic/myeloproliferative neoplasm (MDS.MPN), or MDS-related cytogenetic abnormalities, and absence of the specific genetic abnormalities. AML-NOS were classified according to morphology, cytochemical statin and/or immunophenotypic results. Chromosome study was performed with a standard protocol and karyotypes were described according to the International System for Human Cytogenetic Nomenclature (ISCN) 2009. The Institional Review Board at Samsung Medical Center approved the current study (IRB No. 2014-05-016-002).
Targeted high-throughput sequencing
From a literature review, we selected 19 genes significantly mutated in AML [5, 12, 15, 16]]; they included FLT3-ITD, FLT3-TKD, DNMT3A, NPM1, TET2, RUNX1, CEBPA, WT1, IDH1, IDH2, NRAS, ASXL1, SETD2, PTPN11, TP53, KIT, JAK2, KRAS, BRAF and CBL (Supplementary Table S7). Custom target enrichment and amplicon library preparation was done using the Access Array System (Fluidigm, South San Francisco, CA, USA),[18] followed by massively parallel sequencing on the Miseq System (Illumina, San Diego, CA, USA) [44].
Analysis of high-throughput sequencing data
A schematic overview of bioinformatics analysis is presented in Supplementary Figure S12. After quality control by the SAMtool and fastQC softwares,(46) raw files were aligned to the human reference genome (build GRCh37) using the Burrows-Wheeler Aligner (BWA) with default parameters.(47) Local alignment was done with the Genome Analysis Toolkit (GATK), followed by variant calling by the GATK-Haplotype caller and Varscan algorithms.(48) Read depth and coverage were estimated using the BEDTools.(49) Called variants were annoatated using the SnpEff software(50) and filtered according to the following strategy; i) variation calls detected in normal healthy control were excluded, ii) only missense, nonsense, framshift, start gain and splice site mutations were included, iii) variations found over 0.1% of general population in public databases (NCBI dbSNP and ESP6000)(51) were excluded, iv) only variations with allele frequecy >20% in GATK-Haplotype caller and/or Varscan were included, and v) variants were further filtered by visual inspection on Inegrative Genomics Viwer (IGV) [45]. SIFT and PolyPhen softwares [46, 47] were used for in-silico analysis of damaging effects of missense variations.
Crosscheck and validation by Sanger sequencing
Variants filtered by the strategy mentioned above were further confirmed by Sanger sequecing. For FLT3-ITD, FLT3-TKD, JAK2 V617F, KIT, CEBPA and NPM1 mutations, we integrated clinical testing results wherever available; all these were tested by Sanger sequencing and FLT3-ITD was double-checked by PCR and fluorescent fragment length analysis. Primers used for Sanger sequecing are summarized in Supplementary Table S8. Sequencing was performed using the BigDye Terminator Cycle Sequencing Ready Reaction Kit on an ABI Prism 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The sequence data was analyzed using the Sequencher software (Gene Codes Corp., Ann Arbor, MI, USA).
Statistical analysis and clinical parameters
Statistical analysis was performed using the PASW Statistics 20.0 software (IBM, Armonk, NY, USA). Significance between categorical data was calculated by Chi-square or Fisher's exact tests. Differences in survival between mutation groups were analyzed by Kaplan–Meier estimates. Multivariable Cox regression analysis was performed to examine the impact of mutations along with other clinical variables. OS was measured from the time from diagnosis to death or last follow-up. RFS was defined only for patients achieving CR, measured from the date of achievement of a remission until the date of relapse or death from CR, death or any cause. A P-value of <0.05 was considered as statistically significant.
SUPPLEMENTARY FIGURES AND TABLES
Acknowledgments
We gratefully acknowledge contributions of participants enrolled in this study. This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2012R1A1A2043879).
Footnotes
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
- 1.Slovak ML, Kopecky KJ, Cassileth PA, Harrington DH, Theil KS, Mohamed A, Paietta E, Willman CL, Head DR, Rowe JM, Forman SJ, Appelbaum FR. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000;96:4075–4083. [PubMed] [Google Scholar]
- 2.Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 3.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, Dicker F, Fasan A, Haferlach C, Haferlach T, Kern W, Schnittger S, Kohlmann A. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. 2012;26:934–942. doi: 10.1038/leu.2011.326. [DOI] [PubMed] [Google Scholar]
- 6.Metzeler KH, Maharry K, Radmacher MD, Mrozek K, Margeson D, Becker H, Curfman J, Holland KB, Schwind S, Whitman SP, Wu YZ, Blum W, Powell BL, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29:1373–1381. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim HJ, Ahn HK, Jung CW, Moon JH, Park CH, Lee KO, Kim SH, Kim YK, Kim HJ, Sohn SK, Kim SH, Lee WS, Kim KH, et al. KIT D816 mutation associates with adverse outcomes in core binding factor acute myeloid leukemia, especially in the subgroup with RUNX1/RUNX1T1 rearrangement. Ann Hematol. 2013;92:163–171. doi: 10.1007/s00277-012-1580-5. [DOI] [PubMed] [Google Scholar]
- 8.Owen C, Fitzgibbon J, Paschka P. The clinical relevance of Wilms Tumour 1 (WT1) gene mutations in acute leukaemia. Hematol Oncol. 2010;28:13–19. doi: 10.1002/hon.931. [DOI] [PubMed] [Google Scholar]
- 9.King-Underwood L, Pritchard-Jones K. Wilms' tumor (WT1) gene mutations occur mainly in acute myeloid leukemia and may confer drug resistance. Blood. 1998;91:2961–2968. [PubMed] [Google Scholar]
- 10.Devillier R, Mansat-De Mas V, Gelsi-Boyer V, Demur C, Murati A, Corre J, Prebet T, Bertoli S, Brecqueville M, Arnoulet C, Recher C, Vey N, Mozziconacci MJ, Delabesse E, Birnbaum D. Role of ASXL1 and TP53 mutations in the molecular classification and prognosis of acute myeloid leukemias with myelodysplasia-related changes. Oncotarget. 2015;6:8388–8396. doi: 10.18632/oncotarget.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Peng J, Tang G, Goswami M, Young KH, Singh R, Medeiros LJ, Kantarjian HM, Luthra R, Wang SA. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J Hematol Oncol. 2015;8:45. doi: 10.1186/s13045-015-0139-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rocquain J, Carbuccia N, Trouplin V, Raynaud S, Murati A, Nezri M, Tadrist Z, Olschwang S, Vey N, Birnbaum D, Gelsi-Boyer V, Mozziconacci MJ. Combined mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in myelodysplastic syndromes and acute myeloid leukemias. BMC Cancer. 2010;10:401. doi: 10.1186/1471-2407-10-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohgami RS, Ma L, Merker JD, Gotlib JR, Schrijver I, Zehnder JL, Arber DA. Next-generation sequencing of acute myeloid leukemia identifies the significance of TP53, U2AF1, ASXL1, and TET2 mutations. Mod Pathol. 2015;28:706–714. doi: 10.1038/modpathol.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aranaz P, Hurtado C, Erquiaga I, Migueliz I, Ormazabal C, Cristobal I, Garcia-Delgado M, Novo FJ, Vizmanos JL. CBL mutations in myeloproliferative neoplasms are also found in the gene's proline-rich domain and in patients with the V617FJAK2. Haematologica. 2012;97:1234–1241. doi: 10.3324/haematol.2011.052605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez-Mercado M, Yip BH, Pellagatti A, Davies C, Larrayoz MJ, Kondo T, Perez C, Killick S, McDonald EJ, Odero MD, Agirre X, Prosper F, Calasanz MJ, Wainscoat JS, Boultwood J. Mutation patterns of 16 genes in primary and secondary acute myeloid leukemia (AML) with normal cytogenetics. PLoS One. 2012;7:e42334. doi: 10.1371/journal.pone.0042334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, Habdank M, Spath D, Morgan M, Benner A, Schlegelberger B, Heil G, Ganser A, Dohner H. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 17.Duncavage EJ, Abel HJ, Szankasi P, Kelley TW, Pfeifer JD. Targeted next generation sequencing of clinically significant gene mutations and translocations in leukemia. Mod Pathol. 2012;25:795–804. doi: 10.1038/modpathol.2012.29. [DOI] [PubMed] [Google Scholar]
- 18.Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohgami RS, Ma L, Merker JD, Gotlib JR, Schrijver I, Zehnder JL, Arber DA. Next-generation sequencing of acute myeloid leukemia identifies the significance of TP53, U2AF1, ASXL1, and TET2 mutations. Mod Pathol. 2014 doi: 10.1038/modpathol.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christiansen DH, Desta F, Andersen MK, Pedersen-Bjergaard J. Mutations of the PTPN11 gene in therapy-related MDS and AML with rare balanced chromosome translocations. Genes Chromosomes Cancer. 2007;46:517–521. doi: 10.1002/gcc.20426. [DOI] [PubMed] [Google Scholar]
- 21.Christiansen DH, Andersen MK, Desta F, Pedersen-Bjergaard J. Mutations of genes in the receptor tyrosine kinase (RTK)/RAS-BRAF signal transduction pathway in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2005;19:2232–2240. doi: 10.1038/sj.leu.2404009. [DOI] [PubMed] [Google Scholar]
- 22.Side LE, Curtiss NP, Teel K, Kratz C, Wang PW, Larson RA, Le Beau MM, Shannon KM. RAS, FLT3, and TP53 mutations in therapy-related myeloid malignancies with abnormalities of chromosomes 5 and 7. Genes Chromosomes Cancer. 2004;39:217–223. doi: 10.1002/gcc.10320. [DOI] [PubMed] [Google Scholar]
- 23.Swerdlow SH, Campo E, Nancy LH, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. 2008. [Google Scholar]
- 24.Ilyas A, Ahmad S, Faheem M, Naseer M, Kumosani TA, Al-Qahtani M, Gari M, Ahmed F. Next generation sequencing of acute myeloid leukemia: influencing prognosis. BMC Genomics. 2015;16:S5. doi: 10.1186/1471-2164-16-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Renneville A, Boissel N, Nibourel O, Berthon C, Helevaut N, Gardin C, Cayuela JM, Hayette S, Reman O, Contentin N, Bordessoule D, Pautas C, Botton S, et al. Prognostic significance of DNA methyltransferase 3A mutations in cytogenetically normal acute myeloid leukemia: a study by the Acute Leukemia French Association. Leukemia. 2012;26:1247–1254. doi: 10.1038/leu.2011.382. [DOI] [PubMed] [Google Scholar]
- 26.Ribeiro AF, Pratcorona M, Erpelinck-Verschueren C, Rockova V, Sanders M, Abbas S, Figueroa ME, Zeilemaker A, Melnick A, Lowenberg B, Valk PJ, Delwel R. Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood. 2012;119:5824–5831. doi: 10.1182/blood-2011-07-367961. [DOI] [PubMed] [Google Scholar]
- 27.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyer SC, Levine RL. Translational implications of somatic genomics in acute myeloid leukaemia. Lancet Oncol. 2014;15:e382–394. doi: 10.1016/S1470-2045(14)70008-7. [DOI] [PubMed] [Google Scholar]
- 29.Pedersen-Bjergaard J, Christiansen DH, Desta F, Andersen MK. Alternative genetic pathways and cooperating genetic abnormalities in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2006;20:1943–1949. doi: 10.1038/sj.leu.2404381. [DOI] [PubMed] [Google Scholar]
- 30.Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrozek K, Nicolet D, Whitman SP, Wu YZ, Schwind S, Powell BL, Carter TH, Wetzler M, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011;118:6920–6929. doi: 10.1182/blood-2011-08-368225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spencer DH, Abel HJ, Lockwood CM, Payton JE, Szankasi P, Kelley TW, Kulkarni S, Pfeifer JD, Duncavage EJ. Detection of FLT3 internal tandem duplication in targeted, short-read-length, next-generation sequencing data. J Mol Diagn. 2013;15:81–93. doi: 10.1016/j.jmoldx.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 32.Mori A, Deola S, Xumerle L, Mijatovic V, Malerba G, Monsurro V. Next generation sequencing: new tools in immunology and hematology. Blood Res. 2013;48:242–249. doi: 10.5045/br.2013.48.4.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Myllykangas S, Ji HP. Targeted deep resequencing of the human cancer genome using next-generation technologies. Biotechnol Genet Eng Rev. 2010;27:135–158. doi: 10.1080/02648725.2010.10648148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rao AV, Smith BD. Are results of targeted gene sequencing ready to be used for clinical decision making for patients with acute myelogenous leukemia? Curr Hematol Malig Rep. 2013;8:149–155. doi: 10.1007/s11899-013-0161-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grossmann V, Schnittger S, Schindela S, Klein HU, Eder C, Dugas M, Kern W, Haferlach T, Haferlach C, Kohlmann A. Strategy for robust detection of insertions, deletions, and point mutations in CEBPA, a GC-rich content gene, using 454 next-generation deep-sequencing technology. J Mol Diagn. 2011;13:129–136. doi: 10.1016/j.jmoldx.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang B, Liu Y, Hou G, Wang L, Lv N, Xu Y, Xu Y, Wang X, Xuan Z, Jing Y, Li H, Jin X, Deng A, et al. Mutational spectrum and risk stratification of intermediate-risk acute myeloid leukemia patients based on next-generation sequencing. Oncotarget. 2016;7:32065–78. doi: 10.18632/oncotarget.7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, Chen F, Asou N, Ohtake S, Miyawaki S, Miyazaki Y, Sakura T, Ozawa Y, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014;28:1586–1595. doi: 10.1038/leu.2014.55. [DOI] [PubMed] [Google Scholar]
- 38.Park SH, Choi JC, Kim SY, Yi J, Oh SH, Kim IS, Kim HH, Chang CL, Lee EY, Song MK, Shin HJ, Chung JS. Incidence and prognostic impact of DNMT3A mutations in Korean normal karyotype acute myeloid leukemia patients. Biomed Res Int. 2015;2015:723682. doi: 10.1155/2015/723682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahn JS, Kim HJ, Kim YK, Lee SS, Jung SH, Yang DH, Lee JJ, Kim NY, Choi SH, Jung CW, Jang JH, Kim HJ, Moon JH, et al. DNMT3A R882 Mutation with FLT3-ITD Positivity Is an Extremely Poor Prognostic Factor in Patients with Normal-Karyotype Acute Myeloid Leukemia after Allogeneic Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant. 2016;22:61–70. doi: 10.1016/j.bbmt.2015.07.030. [DOI] [PubMed] [Google Scholar]
- 40.Ostronoff F, Othus M, Ho PA, Kutny M, Geraghty DE, Petersdorf SH, Godwin JE, Willman CL, Radich JP, Appelbaum FR, Stirewalt DL, Meshinchi S. Mutations in the DNMT3A exon 23 independently predict poor outcome in older patients with acute myeloid leukemia: a SWOG report. Leukemia. 2013;27:238–241. doi: 10.1038/leu.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marcucci G, Metzeler KH, Schwind S, Becker H, Maharry K, Mrozek K, Radmacher MD, Kohlschmidt J, Nicolet D, Whitman SP, Wu YZ, Powell BL, Carter TH, et al. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J Clin Oncol. 2012;30:742–750. doi: 10.1200/JCO.2011.39.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thol F, Damm F, Ludeking A, Winschel C, Wagner K, Morgan M, Yun H, Gohring G, Schlegelberger B, Hoelzer D, Lubbert M, Kanz L, Fiedler W, et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol. 2011;29:2889–2896. doi: 10.1200/JCO.2011.35.4894. [DOI] [PubMed] [Google Scholar]
- 43.Roller A, Grossmann V, Bacher U, Poetzinger F, Weissmann S, Nadarajah N, Boeck L, Kern W, Haferlach C, Schnittger S, Haferlach T, Kohlmann A. Landmark analysis of DNMT3A mutations in hematological malignancies. Leukemia. 2013;27:1573–1578. doi: 10.1038/leu.2013.65. [DOI] [PubMed] [Google Scholar]
- 44.Moonsamy PV, Williams T, Bonella P, Holcomb CL, Hoglund BN, Hillman G, Goodridge D, Turenchalk GS, Blake LA, Daigle DA, Simen BB, Hamilton A, May AP, Erlich HA. High throughput HLA genotyping using 454 sequencing and the Fluidigm Access Array System for simplified amplicon library preparation. Tissue Antigens. 2013;81:141–149. doi: 10.1111/tan.12071. [DOI] [PubMed] [Google Scholar]
- 45.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 47.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013 doi: 10.1002/0471142905.hg0720s76. Chapter 7:Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.