

Abstract
Genetic alterations of TERT and CTNNB1 have been documented in hepatocellular carcinoma. TERT promoter mutations are the earliest genetic events in the multistep process of hepatocarcinogenesis related to cirrhosis. However, analyses of TERT promoter and CTNNB1 mutations in hepatocellular carcinoma tumor samples have not been performed in the Korean population, where hepatitis B virus-related hepatocellular carcinoma is prevalent. In order to identify the role of TERT promoter and CTNNB1 mutations in the hepatocarcinogenesis and pathogenesis of recurrent hepatocellular carcinoma, we performed the sequence analyses in 140 hepatocellular nodules (including 107 hepatocellular carcinomas), and 8 pairs of matched primary and relapsed hepatocellular carcinomas. TERT promoter and CTNNB1 mutations were only observed in hepatocellular carcinomas but not in precursor lesions. Of 109 patients with hepatocellular carcinoma, 41 (39.0%) and 15 (14.6%) harbored TERT and CTNNB1 mutations, respectively. TERT promotermutations were significantly more frequent in hepatocellular carcinomas related to hepatitis C virus infection (5/6; 83.3%) compared to tumors of other etiologies (P = 0.001). In two cases, discordance in TERT promoter mutation status was observed between the primary and the corresponding recurrent hepatocellular carcinoma. The two patients with discordant cases had early relapses. In conclusion, we identified TERT promoter and CTNNB1 mutations as the most frequent somatic genetic alterations observed in hepatocellular carcinoma, indicating its pivotal role in hepatocarcinogenesis. Furthermore, we suggest the possibility of intratumoral genetic heterogeneity of TERT promoter mutations in hepatocellular carcinoma as indicated by the discordance in TERT promoter mutations between primary and corresponding recurrent hepatocellular carcinoma.
Keywords: TERT, CTNNB1, hepatocellular carcinoma, intratumoral genetic heterogeneity, Pathology Section
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the third leading cause of cancer-related death worldwide. Despite recent advances in therapeutic strategies for the treatment of HCC, it still has a poor prognosis. The recurrence of HCC after resection remains the major cause of death [1, 2]. Patients with HCC have high recurrence rate and intrahepatic recurrence occurs in 70-100% of cases within 5 years after hepatic resection for HCC [3–5].
The majority of HCCs develop in liver cirrhosis related to chronic hepatitis B virus (HBV) infection, chronic hepatitis C virus (HCV) infection, alcohol intake, and obesity. HCC is considered to develop in a multistep process, in which a precursor or premalignant lesion progresses to dysplastic nodules, followed by HCC. Hepatocarcinogenesis has been characterized as the progressive accumulation of a variety of genetic alterations in chronic liver disease. It is well known that HCCs are phenotypically and genetically heterogeneous tumors. A detailed understanding of the genomic alterations in HCC can improve tumor characterization to help identify molecular targets for therapies. In particular, knowledge of the molecular mechanisms of tumor progression could be of potential value for therapeutic decisions.
Recently, genetic alterations of TERT have been documented in HCC [6, 7]. TERT gene alterations identified by exome sequencing in the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA) occur in more than 68% of HCCs and were ancestry-independent [6]. Of note is that somatic mutations in the TERT promoter have been frequently identified in HCC [8]. The frequency of TERT promoter mutations in HCC has been reported to be 44~59% [8–10]. In particular, it has been reported that TERT promoter mutations are the earliest genetic events in the multistep process of hepatocarcinogenesis related to cirrhosis [8]. TERT promoter mutations include 2 hot spots (C228T and C250T) that were identified in various types of tumors. TERT promoter mutations were first reported in melanoma [11], and were subsequently identified in urothelial carcinoma, glioma, and papillary thyroid carcinoma [12–14]. TERT is the catalytic subunit of the telomerase complex, and is a predominant determinant for controlling telomerase activity. Telomerase plays a key role in increasing the longevity of cells by maintaining the length of telomere caps at the end of chromosomes. Telomerase activation is involved in mechanisms of tumorigenesis and telomerase activity is actually upregulated in 85~90% of cancers [15]. The mechanisms of telomerase reactivation in cancer have yet to be fully explored. Activating mutations in the promoter of the TERT gene leads to increased telomerase expression.
A critical role of CTNNB1 (a gene encoding β-catenin) mutations in hepatocarcinogenesis has been established [16–18]. CTNNB1 mutations are among the most frequent genetic alterations in HCC and have been reported in 20~40% of cases. CTNNB1 mutations are more common in HCV-related HCCs compared to HBV-related HCCs [17]. Furthermore, hepatocellular adenomas (HCAs) harboring CTNNB1 mutations are more at risk of malignant transformation leading to the development of HCC [19]. These mutations predominantly occur within exon 3 of the gene, in a region encoding for the protein sequence containing the consensus sites for phosphorylation, and prevent β-catenin from phosphorylation and subsequent degradation. Recently, it has been reported that TERT promoter mutations and CTNNB1 mutations in hepatocellular tumors are significantly associated [8].
The analyses of TERT promoter and CTNNB1 mutations on HCC tumor samples have not been performed in the Korean population, where HBV-related HCC is prevalent. In order to identify the role of TERT promoter mutations and CTNNB1 mutations in hepatocarcinogenesis and the pathogenesis of recurrent HCC, we performed the mutational analyses in full spectrum of precancerous lesions and HCC and in 8 pairs of matched primary and relapsed HCCs.
RESULTS
Prevalence of TERT C228T/C250T and CTNNB1 mutations in hepatocellular nodules
A total of 156 liver nodules were evaluated, including 4 LRNs, 10 LDNs, 1 HDNs, 9 HCAs, 123 HCCs, and 3 combined HCCs and cholangiocarcinomas. Sequence analyses of the TERT promoter and CTNNB1 exon 3 were performed for 123 HCCs, of which 119 and 116 had successful results, respectively. The mutation frequency in each hepatocellular nodule is shown in Table 1. TERT promoter mutations were only observed in HCCs. TERT promoter and CTNNB1 mutations were present in 37.8% (45 of 119) and 13.8% (16 of 116) of HCCs, respectively. All TERT promoter mutations were found at 2 hotspots (C228T and C250T). Among 45 TERT mutant cases, only 2 had C250T, while the rest had C228T. TERT promoter mutations at the 2 hotspots were mutually exclusive.
Table 1. Frequency of TERT promoter and CTNNB1 mutations in hepatocellular nodules.
| TERT C228T & C250T status | CTNNB1 status | ||||
|---|---|---|---|---|---|
| Total | Wild type | Mutant | Wild type | Mutant | |
| (n = 156) | (n = 107) | (n = 45) | (n = 127) | (n = 17) | |
| Hepatic adenoma | 9 | 9 (100%) | 0 | 8 (88.9%) | 1 (11.1%) |
| Regenerating nodule | 4 | 4 (100%) | 0 | 2 (100%) | 0 |
| Low grade dysplastic nodule (Large cell change) | 10 | 10 (100%) | 0 | 8 (100%) | 0 |
| High grade dysplastic nodule | 7 | 7(100%) | 0 | 6 (100%) | 0 |
| Hepatocellular carcinoma | 123 | 74 (60.2%) | 45 (36.6%) | 100 (86.2%) | 16 (13.8%) |
| Combined hepatocellular carcinoma and cholangiocarcinoma | 3 | 3 (100%) | 0 | 3 (100%) | 0 |
In CTNNB1 mutation analysis, 16 out of 116 HCCs (13.8%) had missense point mutations. These mutations were C86T (S29F) in 2 cases, C98G (S33C) in 2 cases, A95C (D32A) in 1 case, G94A (D32N) in 1 case, A95T (D32V) in 1 case, A95G (D32G) in 1 case, C134T (S45F) in 3 cases, T133G (S45A) in 1 case, T109G (S37A) in 1 case, T133C (S45P) in 2 cases, and A121G (T41A) in 1 case. No TERT and CTNNB1 mutations were identified in preneoplastic lesions including 10 LDNs and 7 HDNs, as well as 4 LRNs.
We additionally examined 9 HCAs without malignant transformation including 3 β-catenin-activated HCAs showing nuclear staining of β-catenin. Only 1 case harbored CTNNB1 mutations; however, no TERT promoter mutations were observed in 9 HCAs. There were no mutations in 3 combined HCCs and cholangiocarcinomas.
Clinicopathological demographics of the patients with HCC
A total of 123 HCC samples from 109 patients were analyzed in this study.
The study population consisted of 95 men (87.2%) and 14 women (12.8%), with a median age of 55 years (range, 27-81). Seventy-seven patients (70.6%) were younger than 60 years, and 32 patients (29.4%) were older than 60 years of age. Of these, 61 patients (56.0%) had underlying liver cirrhosis. All patients had chronic liver disease related to HBV infection (80/109, 73.4%), HCV infection (7/109, 6.4%), alcohol consumption (9/1109, 8.3%), and unknown causes (13/109, 11.9%). Of these, 101 patients (92.7%) underwent liver resection and 8 patients (7.3%) underwent liver transplantation. Median follow-up duration was 23 months (range, 0-115 months) after the operation. Of 109 patients, recurrence and death occurred in 37 patients (33.9%) and 12 patients (11.0%), respectively.
Association between TERT / CTNNB1 mutations and clinicopathological features of HCC
Of 109 patients with HCC, 41 (39.0%) and 15 (14.6%) harbored TERT and CTNNB1 mutations, respectively. The association between TERTC228T/CTNNB1 mutations, and clinicopathological factors in HCC patients was evaluated (Table 2).
Table 2. Clinicopathologic factors according to TERT promoter and CTNNB1 mutation status.
| Total (n=105) | TERT mutated (n=41, 39.0%) | Wild TERT (n=64, 61.0%) | P-value | Total (n=103) | CTNNB1 mutated (n=15, 14.6%) | Wild CTNNB1 (n=88, 85.4%) | P-value | ||
|---|---|---|---|---|---|---|---|---|---|
| Age | 60 ±10.4 | 54±10.3 | 0.007 | 59 ±10.6 | 56±10.6 | 0.225 | |||
| Sex | Male | 92 (87.6%) | 38 (92.7%) | 54 (84.4%) | 0.207 | 90 (87.4%) | 14 (93.3%) | 76 (86.4%) | 0.686 |
| Female | 13 (12.4%) | 3 (7.3%) | 10 (15.6%) | 13 (12.6%) | 1 (6.7%) | 12 (13.6%) | |||
| Aetiology | HBV | 78 (74.3%) | 23 (56.1%) | 55 (85.9%) | 0.001 | 76 (73.8%) | 10 (66.7%) | 66 (75.0%) | 0.747 |
| HCV | 6 (5.7%) | 5 (12.2%) | 1 (1.6%) | 7 (6.8%) | 1 (6.7%) | 6 (6.8%) | |||
| Alcohol | 8 (7.6%) | 3 (7.3%) | 5 (7.8%) | 7 (6.8%) | 1 (6.7%) | 6 (6.8%) | |||
| Unknown | 13 (12.4%) | 10 (24.4%) | 3 (4.7%) | 13 (6.8%) | 3 (20.0%) | 10 (11.4%) | |||
| LC | Yes | 58 (55.2%) | 24 (58.5%) | 34 (53.1%) | 0.688 | 60 (58.3%) | 10 (66.7%) | 50 (56.8%) | 0.475 |
| No | 47 (44.8%) | 17 (41.5%) | 30 (46.9%) | 43 (41.7%) | 5 (33.3%) | 38 (43.2%) | |||
| Tumor size | <5cm | 63 (60.0%) | 23 (56.1%) | 40 (62.5%) | 0.545 | 64 (62.1%) | 9 (60.0%) | 55 (62.5%) | 0.854 |
| >5cm | 42 (40.0%) | 18 (43.9%) | 24 (37.5%) | 39 (37.9%) | 6 (40.0%) | 33 (37.5%) | |||
| Tumor number | single | 72 (68.6%) | 32 (78.0%) | 40 (62.5%) | 0.131 | 71 (68.9%) | 13 (86.7%) | 58 (65.9%) | 0.138 |
| multiple | 33 (31.4%) | 9 (22.0%) | 24 (37.5%) | 32 (31.1%) | 2 (13.3%) | 30 (34.1%) | |||
| Vascular invasion | No | 76 (72.4%) | 33 (80.5%) | 43 (67.2%) | 0.206 | 74 (71.8%) | 11 (73.3%) | 63 (71.6%) | 0.347 |
| Microvascular | 19 (18.1%) | 4 (9.8%) | 15 (23.4%) | 19 (18.4%) | 4 (26.7%) | 15 (17.0%) | |||
| Macrovascular | 10 (9.5%) | 4 (9.8%) | 6 (9.4%) | 10 (9.7%) | 0 | 10 (11.4%) | |||
| T-stage | 1 | 60 (57.1%) | 28 (68.3%) | 32 (50.0%) | 0.275 | 59 (57.3%) | 10 (66.7%) | 49 (55.7%) | 0.491 |
| 2 | 24 (22.9%) | 6 (14.6%) | 18 (28.1%) | 24 (23.3%) | 2 (13.3%) | 22 (25.0%) | |||
| 3 | 18 (17.1%) | 6 (14.6%) | 12 (18.8%) | 17 (16.5%) | 2 (13.3%) | 15 (17.0%) | |||
| 4 | 3 (2.9%) | 1 (2.4%) | 2 (3.1%) | 3 (2.9%) | 1 (6.7%) | 2 (2.3%) | |||
| Edmondson | I | 4 (3.8%) | 2 (4.9%) | 2 (3.1%) | 0.656 | 5 (4.9%) | 0 | 5 (5.7%) | 0.391 |
| II | 55 (52.4%) | 21 (51.2%) | 34 (53.1%) | 55 (53.4%) | 9 (60.0%) | 46 (52.3%) | |||
| III | 40 (38.1%) | 17 (42.5%) | 23 (35.9%) | 37 (35.9%) | 4 (26.7%) | 33 (37.5%) | |||
| IV | 6 (5.7%) | 1 (2.4%) | 5 (7.8%) | 6 (5.8%) | 2 (13.3%) | 4 (4.5%) | |||
| Recurrence | Yes | 34 (34.0%) | 12 (29.3%) | 23 (35.9%) | 0.479 | 35 (34.0%) | 4 (26.7%) | 31 (35.2%) | 0.518 |
| No | 70 (66.7%) | 29 (70.7%) | 41 (64.1%) | 68 (66.0%) | 11 (73.3%) | 57 (64.8%) | |||
| Death | Yes | 11 (10.5%) | 5 (12.2%) | 6 (9.4%) | 0.747 | 11 (10.7%) | 3 (20.0%) | 8 (9.1%) | 0.199 |
| No | 94 (89.5%) | 36 (87.8%) | 58 (90.6%) | 92 (89.3%) | 12 (80.0%) | 80 (90.9%) |
TERT promoter mutations were significantly more common in HCCs related to HCV infection (5/6; 83.3%) compared to tumors of other etiologies (P = 0.001). The frequency of TERT promoter mutations was 29.5% (23/78) in HCC related to HBV infection. However, there was no significant difference in TERT mutation status regarding sex, age at diagnosis, presence of underlying liver cirrhosis (LC), tumor size, tumor multiplicity, presence of vascular invasion, ES grade, T stage, and AJCC stage. Rates of recurrence and death did not differ based on TERT mutation status.
CTNNB1 mutations were not associated with any clinicopathological factors. CTNNB1 mutations were present in 13.2% (10/76) of HCC related to HBV, and in 14.3% (1/7) HCC related to HCV. Rates of recurrence and death did not differ based on CTNNB1 mutation status.
TERT promoter mutations were found in 46.7% (7/15) and 38.8% (33/85) of CTNNB1 mutant and wild-type CTNNB1 cases, respectively. Conversely, CTNNB1 mutations were found in 17.5% (7/40) and 13.3% (8/60) of TERT promoter mutant and wild-type TERT tumors, respectively. There was no significant association between TERT promoter mutations and CTNNB1 mutations (P = 0.568).
Clinicopathological and molecular details of 8 patients with recurrent HCC
We also analyzed 8 patients for which primary and corresponding recurrent HCC tumor tissues were available. Clinical details of the 8 patients are presented in Table 3. All but 1 case had chronic liver disease related to HBV infection. Two patients died due to HCC. Pathological and molecular details of the 8 patients are presented in Table 4. The clinicopathological and mutational status associated with primary and recurrent HCCs were compared. In 2 cases, discordance in TERT promoter mutation status was observed among primary and corresponding recurrent HCC. The mutation status of the 2 patients was changed from wild to mutant type at recurrence (Figure 1). All patients with discordant cases relapsed within 2 years, which is considered early relapse. Between 2 patients, 1 patient had multiple tumors and microvascular invasion at first presentation. On the other hand, all patients maintained the same CTNNB1 mutational status.
Table 3. Clinical details in 8 patients with recurrent HCC.
| Case No. | Sex | Age at diagnosis | Site (primary/recurrent) | Cause | Underlying cirrhosis | Time to recurrent (month) | F/U duration after relapse | Outcome |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 52 | Liver/Liver | HBV | Yes | 17 | 40 | Death |
| 2 | M | 56 | Liver/Liver | HBV | Yes | 13 | 44 | Alive |
| 3 | M | 50 | Liver/Liver | HBV | No | 17 | 74 | Death |
| 4 | M | 53 | Liver/Liver | HBV | No | 2 | 5 | Alive |
| 5 | M | 74 | Liver/Liver | HBV | No | 13 | 19 | Alive |
| 6 | F | 56 | Liver/Liver | HBV | Yes | 6 | 28 | Alive |
| 7 | M | 79 | Liver/Liver | HCV | Yes | 10 | 4 | Alive |
| 8 | M | 27 | Liver/Liver | HBV | No | 7 | 29 | Alive |
Table 4. Pathological and molecular detains in 8 patients with recurrent HCC.
| Case No. | Stage (primary) | ES grade (primary/recurrent) | Vascular invasion (primary/recurrent) | Tumor multiplicity (primary/recurrent) | TERT mutation (primary/recurrent) | CTNNB1 mutation (primary/recurrent) |
|---|---|---|---|---|---|---|
| 1 | 4 | 3/4 | Micorvascular/Micorvascular invasion | No/No | Wild/Wild | Mutant/Mutant |
| 2 | 2 | 3/2 | No / No invasion | Yes/No | Wild/ND | Wild/Wild |
| 3 | 1 | 3/2 | No / No invasion | Yes/Yes | Wild/Wild | Wild/Wild |
| 4 | 3a | 2/3 | Macrovascular / No invasion | Yes/Yes | Wild/Wild | Wild/ND |
| 5 | 2 | 3/2 | Microvascular / No invasion | Yes/No | Wild/Mutant | Wild/Wild |
| 6 | 2 | 2/2 | No / No invasion | No/No | Wild/Mutant | Wild/Wild |
| 7 | 2 | 3/3 | No / No invasion | Yes/Yes | Mutant/Mutant | Wild/Wild |
| 8 | 1 | 3/3 | No / No invasion | No/No | Wild/Wild | Wild/Wild |
Figure 1. Examples of discordance in TERT mutation status between primary and corresponding recurrent HCC.

A. Histology of primary HCC with ES grade 3 and B. electropherogram of TERT C228T mutation C. Histology of recurrent HCC with ES grade 2 and D. electropherogram of TERT C228T mutation
TERT promoter mutation and CTNNB1 mutation in HCC and preneoplastic lesions
Moreover, we screened 15 patients with primary HCC and matched LRN, LDN, and HDN tissues available. The heat map of the TERT promoter mutations and CTNNB1 mutations are presented in Figure 2. All but 1 case had liver cirrhosis related to HBV infection. The mutation status of preneoplastic cirrhotic lesion to HCC was evaluated. TERT promoter mutations were detected in HCCs, and not observed in the matched LRN and LDN. Similarly, CTNNB1 mutations were detected in HCCs, and not observed in the matched LDN and HDNs.
Figure 2. Heat map of TERT promoter and CTNNB1 mutations.
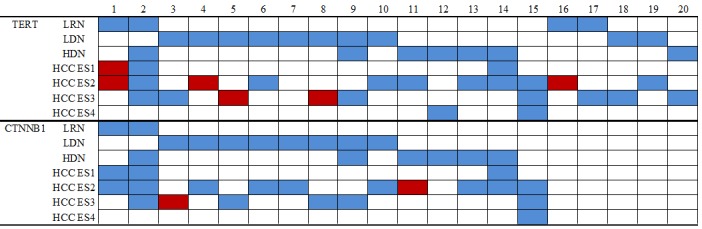
Each case is represented by a single column.
In cases with multiple HCCs, TERT promoter and CTNNB1 mutation status did not differ based on the ES grade. The mutation status also did not differ in single HCC showing different ES.
DISCUSSION
We evaluated the frequency of TERT promoter and CTNNB1 mutations in 146 hepatocellular nodules including HCAs, LRNs, LDNs, HDNs, HCCs, and combined HCCs and cholangiocarcinomas. Among the 121 HCC cases (including recurrent tumors in a patient and multiple tumors showing a different ES grade in a patient), 37.8% (45 of 119) and 13.8% (16 of 116) harbored TERT promoter mutations and CTNNB1 mutations, respectively. Of 109 patients with HCC, 41 (39.0%) and 15 (14.6%) harbored TERT and CTNNB1 mutations, respectively. In HCC, previously published data show that the frequency of TERT promoter and CTNNB1 mutations were 44~59% and 20~40%, respectively. The frequencies of these mutations in the present study were slightly lower compared to those reported in previous studies. These may have been due to the higher prevalence of HBV-related HCC in Korea. In the present study, the frequency of HCCs related to HBV infection was 74.3% (78/109), which is much higher compared to those reported by previous studies upon analyses of these mutations. Nault et al. [8] identified TERT promoter mutations in HCC to be more common in non-related HBV. It is also known that the insertion of HBV DNA into the TERT promoter induces telomerase transcription [20, 21]. Several studies reported that CTNNB1 mutations were identified at higher frequencies in HCCs related to HCV infection [22–24]. In our study, TERT promoter mutations were significantly more common in HCC related to HCV infection (5/6; 83.3%) compared to tumors of other etiologies; however, CTNNB1 mutations did not differ between HBV-related HCC and other etiologies-related HCC.
We did not identify a significant association between TERT promoter mutations and CTNNB1 mutations. However, the limited number of HCV-related HCC cases analyzed, restrains the conclusion that TERT promoter mutations might be significantly associated with WNT pathway gene alterations, such as CTNNB1 in HCV-related HCC and non-viral HCCs [6, 8].
The absence of TERT promoter mutations in our series of 15 HCAs without malignant transformation is consistent with a previous study, which reported that TERT promoter mutations are common in HCAs with malignant transformation [8].
In this study, TERT promoter and CTNNB1 mutations were not identified in LRN, but were identified in matched HCC, indicating that they were somatic mutations.
Recently, TERT promoter mutations were identified in 6% of LDNs, 19% of HDNs, and 61% of early HCCs [25]. The prevalence of mutations gradually increased with the degree of dysplasia, indicating TERT promoter mutations were the earliest genetic events in the multistep process of hepatocarcinogenesis [8, 25]. However, we never identified any mutations in the precursor lesions of HCC. One reason for the observed discrepancy could be attributed to the small number of dysplastic nodules studied. A further reason this discrepancy could be diagnostic confusion concerning equivocal nodular lesions in the cirrhotic liver. Regarding the pathological and clinical diagnosis of equivocal nodular lesions found in cirrhotic liver, there is a discrepancy in interpretation [26]. Although the majority of nodular lesions are HCC in cirrhotic background, equivocal nodules in which it is difficult to distinguish between well-differentiated HCC and high-grade dysplastic nodules. Therefore, a large study with precancerous lesions accurately diagnosed is warranted to resolve an issue. Another reason could be the differences in the ethnic population; a cohort enriched for HBV-related HCC patients.
Meanwhile, Park [27] et al. described that CTNNB1 mutations were not found in precursor lesions of HCC and were not uniformly present in all tumor lesions, indicating that these mutations are late events in hepatocarcinogenesis.
Notably, we first described 2 cases of discordant TERT promoter mutations between primary and corresponding recurrent HCCs. Two patients with discordant cases had early relapses. In HCC, early recurrence might be largely related to metastasis from the primary tumor, while late recurrence might be due to de novo tumors from non-tumoral lesions on a cirrhotic background that are independent of resected primary tumors. From our findings, we could speculate two hypotheses. TERT promoter mutations in recurrent HCCs could be from minor subclones, which were not detected at the initial presentation by Sanger sequencing technology. Conventional sequencing might have missed intratumoral heterogeneity by representing only the dominant clone. TERT promoter mutations are not acquired in proportion to cancer progression; however, are already encoded in the primary tumors. Even though we identified that there was no heterogeneous distribution of TERT promoter and CTNNB1 mutations from multiple regions in a single HCC, intratumoral genetic heterogeneity could not be fully ruled out. It is known that HCC shows morphological and immunophenotypical heterogeneity, indicating that HCC can display intratumoral genetic heterogeneity. Intratumoral genetic heterogeneity may indicate tumor evolution, adaptation to environmental stress, and response to treatment. Furthermore, intratumoral genetic heterogeneity is a practical challenge with clinical implications in the era of targeted therapy. Therefore, understanding of intratumoral genetic heterogeneity is crucial in clinical management. Recently, Friemel [28] et al. identified that heterogeneous intratumoral mutational status of TP53 and CTNNB1 mutations was present in 22% of HCCs. However, there are no data regarding intratumoral heterogeneity of other somatic mutations.
It could also be assumed that the development of a tumor cell clone showing TERT promoter mutations in recurrence was not present in the primary HCC. It is thought that TERT promoter mutations were acquired during their progression, not as early events in HCC. However, this is in contrast to the current view that TERT promoter mutations are among the earliest genetic alterations involved in malignant transformation [25].
We are fully aware that the number of cases in our study is too small to reach a definitive conclusion. Furthermore, these speculations could be supported by deep sequencing to detect mutations with low frequencies and by multiregional sequencing, which reveals cancer genome obtained from multiple regions in a single tumor. The role of TERT promoter mutations in hepatocarcinogenesis, and the pathogenesis of tumor progression remain to be functionally investigated.
In conclusion, we identified TERT promoter mutations to be the most frequent somatic genetic alterations in HCC, indicating that they play a pivotal role in hepatocarcinogenesis. Furthermore, we suggest the possibility of intratumoral genetic heterogeneity of TERT promoter mutations in HCC through the discordance in TERT promoter mutations between primary and corresponding recurrent HCCs. Further studies in a large cohort are needed to support our observations.
MATERIALS AND METHODS
Patient selection and characteristics
Study approval was obtained from the Institutional Review Board of Konkuk University Medical Center (KUH 1210050). This study included 111 patients who underwent hepatectomy for HCC (n = 109)/HCA (n = 2), and 7 patients who underwent liver needle biopsy for HCA between 2006 and 2016 at the Konkuk University Medical Center in Seoul, Korea. A total of 156 liver nodules from 113 patients were evaluated, including 4 large regenerating nodules (LRNs), 10 low grade dysplastic nodules (LDNs), 7 high-grade dysplastic nodules (HDNs), 9 HCAs, 123 HCCs, and 3 combined HCAs and cholangiocarcinomas. Tumor stage was determined according to the American Joint Committee on Cancer (AJCC) staging system. The histologic grade of differentiation was determined on the basis of Edmondson and Steiner (ES) classification [29]. Histologic grades of worst differentiation were recorded. All of the 156 hematoxylin and eosin-stained slides were reviewed; representative tumor tissue samples were selected from each case.
DNA extraction
DNA was extracted from formaldehyde fixed-paraffin embedded (FFPE) tumor tissue. Briefly, 100 μL of the MultiTech DNA extraction solution containing 16 mM (NH4)2SO4, 50 mM Tris-HCl, pH 8.5, 1 mM EDTA pH 8.0, and 0.5% Tween-20 was added to dissected cells in a 1.5-mL microcentrifuge tube. Next, proteinase K (Takara Bio. Inc., Shiga, Japan) was added to a final concentration of 200 μg/mL, and the tissue was digested at 56°C for 1 h. Following digestion, Chelex-100 (Bio-Rad, CA, USA) was added to a final concentration of 10% in the PCR tubes, and the tubes were heated to 100°C for 10 min in a dry bath incubator (Major Science, New Taipei City, Taiwan). After gentle shaking, the tubes were centrifuged at 12,000 ×g for 10 min for DNA elution [30].
Identification of TERT promoter mutations by PCR amplification and direct sequencing
Standard PCR was carried out for genetic sequencing to identify TERT promoter mutations. Briefly, a fragment of the TERT promoter was amplified by PCR on genomic DNA using primers 5′-AGTGGATTCGCGGGCACAGA-3′ (sense) and 5′-CAGCGCTGCCTGAAACTC-3′ (antisense). About 40-50 ng of genomic DNA was used in the PCR, which was carried out with an initial denaturation step at 95°C for 3 min, followed by ten cycles of 95°C denaturation for 30 sec, 58~62°C annealing for 30 sec, and 68°C elongation for 1 min. This was followed by 30 cycles under the same settings, except for the elongation step that was modified to continue for an additional 5 sec in each cycle. The PCR was completed with a final elongation step at 68°C for 7 min. The PCR products were subsequently subjected to a sequencing reaction using the BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City CA, USA). Sequencing was performed using the following PCR reaction settings: 35 cycles of denaturation for 60 sec at 96°C, annealing for 5 sec at 50°C, and elongation for 4 min at 60°C. PCR was performed in a final volume of 10 μL, containing 2.5 μL of 70 ng/μL PCR product, 0.5 μL of sequencing primer (3 picomoles), 5X buffer, and AmpliTaq DNA polymerase. DNA sequence was then read on an ABI 3730XL DNA Analyzer (Applied Biosystems). Sequences were aligned using the ContigExpress alignment program (InforMax, Frederick MD, USA).
Identification of CTNNB1 mutations by PCR amplification and direct sequencing
In order to amplify exon 3 of the CTNNB1 gene, 2 PCR methods were used with catenin F (CCT GGC TAT CAT TCT GCT TTT C), catenin R (TCA AAA CTG CAT TCT GAC TTT CA), Beta GF (CCA ATC TAC TAA TGC TAA TAC TG), and Beta GR (CTG CAT TCT GAC TTT CAG TAA GG) primers. The first PCR method using catenin F and catenin R primers was programmed as follows: initial denaturation at 94°C for 2 min, 35 cycles of denaturation at 94°C for 30 sec, annealing at 58°C for 30 sec, and elongation at 72°C for 1 min, followed by a final extension cycle at 72°C for 10 min. The second method used was Touchdown PCR (TD-PCR) with Beta GF and Beta GR primers. The PCR was programmed as follows: initial step of denaturation at 95°C for 2 min, 20 cycles of denaturation at 95°C for 30 sec, annealing temperature starting at 69°C for 30 sec (decreasing by 1°C /2 cycles), and elongation at 72°C for 1 minute. This was followed by 30 cycles of denaturation at 95°C for 30 sec, annealing at 59°C for 30 sec, elongation at 72°C for 1 min, and a final elongation cycle at 72°C for 10 min. Finally, the PCR products were purified and sequenced (Macrogen, Korea). Sequencing reactions were performed in the DNA Engine Tetrad 2 Peltier Thermal Cycler (BIO-RAD) using the ABI BigDye (R) Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems), following the protocols supplied by the manufacturer. Single pass sequencing was performed on each template using F primer. The fluorescently labeled fragments were purified using the method recommended by Applied Biosystems, as it removed the unincorporated terminators and dNTPs. The samples were subjected to electrophoresis in an ABI 3730xl DNA Analyzer (Applied Biosystems).
Statistical analysis
For the analysis of the relationship between clinicopathological factors and presence of TERT C228T/CTNNB1 mutations, Pearson chi-squared test and Fisher's exact test were used. Linear-by-linear testing was used to examine the associations between T stage, AJCC stage, and TERT C228T/CTNNB1 mutations. The time to recurrence-free survival was defined from the day of first surgery until recurrence. All tests were 2-sided, with P < 0.05 considered as statistically significant. All statistical analyses were performed using the SPSS software (SPSS Inc., Chicago IL, USA).
Footnotes
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
- 1.Cha C, Fong Y, Jarnagin WR, Blumgart LH, DeMatteo RP. Predictors and patterns of recurrence after resection of hepatocellular carcinoma. J Am Coll Surg. 2003;197:753–758. doi: 10.1016/j.jamcollsurg.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 2.Shah SA, Greig PD, Gallinger S, Cattral MS, Dixon E, Kim RD, Taylor BR, Grant DR, Vollmer CM. Factors associated with early recurrence after resection for hepatocellular carcinoma and outcomes. J Am Coll Surg. 2006;202:275–283. doi: 10.1016/j.jamcollsurg.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Belghiti J, Panis Y, Farges O, Benhamou JP, Fekete F. Intrahepatic recurrence after resection of hepatocellular carcinoma complicating cirrhosis. Ann Surg. 1991;214:114–117. doi: 10.1097/00000658-199108000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen MF, Hwang TL, Jeng LB, Wang CS, Jan YY, Chen SC. Postoperative recurrence of hepatocellular carcinoma. Two hundred five consecutive patients who underwent hepatic resection in 15 years. Arch Surg. 1994;129:738–742. doi: 10.1001/archsurg.1994.01420310070012. [DOI] [PubMed] [Google Scholar]
- 5.Lise M, Bacchetti S, Da Pian P, Nitti D, Pilati PL, Pigato P. Prognostic factors affecting long term outcome after liver resection for hepatocellular carcinoma: results in a series of 100 Italian patients. Cancer. 1998;82:1028–1036. doi: 10.1002/(sici)1097-0142(19980315)82:6<1028::aid-cncr4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 6.Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H, Yamamoto S, Shinbrot E, Hama N, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–1273. doi: 10.1038/ng.3126. [DOI] [PubMed] [Google Scholar]
- 7.Jhunjhunwala S, Jiang Z, Stawiski EW, Gnad F, Liu J, Mayba O, Du P, Diao J, Johnson S, Wong KF, Gao Z, Li Y, Wu TD, et al. Diverse modes of genomic alteration in hepatocellular carcinoma. Genome Biol. 2014;15:436. doi: 10.1186/s13059-014-0436-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, Laurent A, Cherqui D, Balabaud C, Zucman-Rossi J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:2218. doi: 10.1038/ncomms3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC, Grollman AP, He TC, He Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110:6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quaas A, Oldopp T, Tharun L, Klingenfeld C, Krech T, Sauter G, Grob TJ. Frequency of TERT promoter mutations in primary tumors of the liver. Virchows Arch. 2014;465:673–677. doi: 10.1007/s00428-014-1658-7. [DOI] [PubMed] [Google Scholar]
- 11.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 12.Landa I, Ganly I, Chan TA, Mitsutake N, Matsuse M, Ibrahimpasic T, Ghossein RA, Fagin JA. Frequent somatic TERT promoter mutations in thyroid cancer: higher prevalence in advanced forms of the disease. J Clin Endocrinol Metab. 2013;98:E1562–1566. doi: 10.1210/jc.2013-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rachakonda PS, Hosen I, de Verdier PJ, Fallah M, Heidenreich B, Ryk C, Wiklund NP, Steineck G, Schadendorf D, Hemminki K, Kumar R. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc Natl Acad Sci U S A. 2013;110:17426–17431. doi: 10.1073/pnas.1310522110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simon M, Hosen I, Gousias K, Rachakonda S, Heidenreich B, Gessi M, Schramm J, Hemminki K, Waha A, Kumar R. TERT promoter mutations: a novel independent prognostic factor in primary glioblastomas. Neuro Oncol. 2015;17:45–52. doi: 10.1093/neuonc/nou158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 16.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, Perret C. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang H, Fujii H, Sankila A, Mahler-Araujo BM, Matsuda M, Cathomas G, Ohgaki H. Beta-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am J Pathol. 1999;155:1795–1801. doi: 10.1016/s0002-9440(10)65496-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zucman-Rossi J, Benhamouche S, Godard C, Boyault S, Grimber G, Balabaud C, Cunha AS, Bioulac-Sage P, Perret C. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene. 2007;26:774–780. doi: 10.1038/sj.onc.1209824. [DOI] [PubMed] [Google Scholar]
- 19.Nault JC, Bioulac-Sage P, Zucman-Rossi J. Hepatocellular benign tumors-from molecular classification to personalized clinical care. Gastroenterology. 2013;144:888–902. doi: 10.1053/j.gastro.2013.02.032. [DOI] [PubMed] [Google Scholar]
- 20.Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, Mulawadi FH, Wong KF, Liu AM, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44:765–769. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- 21.Paterlini-Brechot P, Saigo K, Murakami Y, Chami M, Gozuacik D, Mugnier C, Lagorce D, Brechot C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22:3911–3916. doi: 10.1038/sj.onc.1206492. [DOI] [PubMed] [Google Scholar]
- 22.Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA, Pawlik TM, Daniel HD, Kannangai R, Offerhaus GJ, Velculescu VE, Wang L, Zhou S, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43:828–829. doi: 10.1038/ng.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clement B, Balabaud C, Chevet E, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, Arai Y, Takahashi H, Shirakihara T, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 25.Nault JC, Calderaro J, Di Tommaso L, Balabaud C, Zafrani ES, Bioulac-Sage P, Roncalli M, Zucman-Rossi J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology. 2014;60:1983–1992. doi: 10.1002/hep.27372. [DOI] [PubMed] [Google Scholar]
- 26.Kojiro M. Focus on dysplastic nodules and early hepatocellular carcinoma: an Eastern point of view. Liver Transpl. 2004;10:S3–8. doi: 10.1002/lt.20042. [DOI] [PubMed] [Google Scholar]
- 27.Park JY, Park WS, Nam SW, Kim SY, Lee SH, Yoo NJ, Lee JY, Park CK. Mutations of beta-catenin and AXIN I genes are a late event in human hepatocellular carcinogenesis. Liver Int. 2005;25:70–76. doi: 10.1111/j.1478-3231.2004.0995.x. [DOI] [PubMed] [Google Scholar]
- 28.Friemel J, Rechsteiner M, Frick L, Bohm F, Struckmann K, Egger M, Moch H, Heikenwalder M, Weber A. Intratumor heterogeneity in hepatocellular carcinoma. Clin Cancer Res. 2015;21:1951–1961. doi: 10.1158/1078-0432.CCR-14-0122. [DOI] [PubMed] [Google Scholar]
- 29.Edmondson HA, Steiner PE. Primary carcinoma of the liver: a study of 100 cases among 48,900 necropsies. Cancer. 1954;7:462–503. doi: 10.1002/1097-0142(195405)7:3<462::aid-cncr2820070308>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 30.Oh SY, Lee HT. Efficiency of EGFR mutation analysis for small microdissected cytological specimens using multitech DNA extraction solution. Cancer Cytopathol. 2015;123:401–412. doi: 10.1002/cncy.21550. [DOI] [PubMed] [Google Scholar]