

Abstract
T cells are highly influenced by nutrient uptake from their environment, and changes in overall nutritional status, such as malnutrition or obesity, can result in altered T-cell metabolism and behavior. In states of severe malnutrition or starvation, T-cell survival, proliferation, and inflammatory cytokine production are all decreased, as is T-cell glucose uptake and metabolism. The altered T-cell function and metabolism seen in malnutrition is associated with altered adipokine levels, most particularly decreased leptin. Circulating leptin levels are low in malnutrition, and leptin has been shown to be a key link between nutrition and immunity. The current view is that leptin signaling is required to upregulate activated T-cell glucose metabolism and thereby fuel T-cell activation. In the setting of obesity, T cells have been found to have a key role in promoting the recruitment of inflammatory macrophages to adipose depots along with the production of inflammatory cytokines that promote the development of insulin resistance leading to diabetes. Deletion of T cells, key T-cell transcription factors, or pro-inflammatory T-cell cytokines prevents insulin resistance in obesity and underscores the importance of T cells in obesity-associated inflammation and metabolic disease. Altogether, T cells have a critical role in nutritional immunometabolism.
Keywords: malnutrition, obesity, leptin, T cells, glucose metabolism
Introduction
The ability to use and store nutrients for catabolic and anabolic processes is a fundamental requirement for the life of an organism. Therefore, any imbalance in nutrient consumption and nutritional state can lead to detrimental consequences. Severe under-nutrition or starvation can lead to gradual weight loss, reduction of energy expenditure and metabolic rate, and diminished survival, along with an increased risk of morbidity and mortality from infection. On the other hand, chronic over-nutrition, characterized by a prolonged positive energy imbalance that facilitates obesity, is associated with the development of multiple health problems such as heart disease and diabetes, which contribute directly to increased health cost, morbidity, and mortality.
Over the past few decades, the access to and availability of food sources, education level, and socioeconomic status as well as inherited genetic traits, behavioral, physical, or psychological factors have caused obesity to reach epidemic proportions in the United States. Consistently, the number of obese individuals worldwide has doubled over the past two decades, and prevalence is expected to continue to rise over the coming years [1]. Conversely, while malnutrition is less common in developed countries, the World Health Organization cites malnutrition as the greatest single threat to the world’s public health, and approximately 800 million people are chronically undernourished worldwide [2].
Nutrition and immunity are closely linked. Relative to its size, the immune system is one of the most energy consuming cell systems in the body and, therefore, is strongly affected by nutrient imbalance. One significant player in this context is adipose tissue, which takes on an important intermediate role between nutrient availability and immune cell activation. Under healthy body weight conditions, adipocytes, in concert with a plethora of immune cells, regulate tissue homeostasis and prevent inflammation. However, prolonged nutrient overload which leads to obesity results in accumulation of adiposities that negatively impact the immune function and host defense [3]. Adipose tissue produces many secretory bioactive substances, also known as adipokines (or adipocytokines), which have been found to directly affect immune cells in a pro- or anti-inflammatory manner. Adipokines such as leptin, resistin, and visfatin are increased in obesity and promote inflammation, whereas the adipokine adiponectin is decreased in obesity and suppresses inflammatory response [4–7]. Nutrient shortage or depletion of fat stores as a result of starvation or malnutrition leads to insufficient total energy availability and can also modify the secretion of adipokines, thus impairing immune cell activation [8].
Nutritional status can influence both the innate and the adaptive immune systems. Indeed, numerous studies have identified a link between nutrition and macrophages. Obesity leads to an accumulation of pro-inflammatory macrophages into visceral adipose tissue (VAT), and these inflammatory macrophages comprise 40–60% of adipose tissue-resident immune cells in obesity and produce pro-inflammatory cytokines that drive inflammation [9]. Likewise, pro-inflammatory neutrophils and mast cells are increased in obesity and produce pro-inflammatory cytokines as well [10]. Conversely, Natural Killer T cells (NKT cells), ILC2 cells, and eosinophils have an important role in modulating immune response in adipose tissue by producing anti-inflammatory cytokines, are decreased in obese individuals, and are suggested to have a protective role against diet-induced obesity [11].
In addition to innate immune cells, adaptive immune cells (lymphocytes) also play a role in nutritional immunology, and both T lymphocytes (T cells) and B lymphocytes (B cells) are altered in response to nutritional status [12, 13]. Indeed, several recent studies have uncovered the role of T-cell function and metabolism in response to both under- and over-nutrition. In this review we will focus on the effect of both obesity and malnutrition on T cells. Particularly, we will highlight recent advances in our understanding of T-cell immunometabolism and the mechanisms that link nutrient availability and leptin secretion with T-cell activation and function.
Malnutrition-associated immunodeficiency
Nutrient deprivation caused by severe malnutrition or starvation is a major cause of mortality affecting almost 800 million people worldwide [2]. In addition to chronic malnutrition from food scarcity, malnutrition can also influence immune function in other clinical settings such as in critically-ill hospitalized patients and in patients suffering from cancer or AIDS-related cachexia. In fact, the U.S. National Cancer Institute has estimated that up to 40% of cancer deaths result from infections related to malnutrition. It is therefore clear that malnutrition-induced immune suppression is a major concern in multiple susceptible patient populations.
The physiological response to prolonged starvation can be divided into three stages. The first stage starts after several hours of fasting. It is characterized by hepatic glycogenolysis and adipose lipolysis to maintain blood glucose and fatty acid levels, respectively. The second stage, which can last for several weeks, is caused by depletion of glycogen depots and results in gradual weight loss. In this stage the body switches almost completely to β-oxidation of fatty acids, which generate glycerol as a substrate for gluconeogenesis to sustain energy needs. This process generates high blood levels of ketone bodies such as β-hydroxybutyrate, which can be used directly as a fuel source by many tissues, including the brain. The final stage of starvation starts when fat depots are depleted and skeletal muscle is degraded by proteolysis to gain amino acids as a substrate for gluconeogenesis in the liver. This stage is characterized by a rapid loss of body mass and cannot be sustained for a long period of time. It is accompanied by reduction in energy expenditure, body temperature, and metabolic rate that dramatically reduces locomotor activity, reproduction, and immune cell function and eventually results in death [14].
In a vicious cycle, malnutrition increases susceptibility to infection, while infections simultaneously aggravate malnutrition by decreasing appetite and increasing demand for nutrients. Since immune cells do not possess significant energy stores such as glycogen, and are mostly dependent on uptake of nutrients from their environment to fulfill their energetic needs, these cells are particularly sensitive to the nutrient deprivation seen in malnutrition. Indeed, several studies have demonstrated a significant and rapid decrease in thymocytes, splenocytes, and lymphocytes (particularly T cells) in fasted mice [15–18].
Furthermore, adipose tissue stores have also been shown to affect the size and function of the immune system. In human studies, reduction of fat stores as a result of starvation reduced immune cell function and increased the risk of infectious disease [19]. Adipose tissue-secreted factors (adipokines), which mediate communication between adipose tissue and the immune system, appear to play a key role. Under lean conditions, adipocytes produce high amounts of adiponectin which regulates local inflammation and influences systemic energy homeostasis and insulin sensitivity at a distance. In contrast to adiponectin, leptin is secreted in direct proportion to adipocyte mass [20], and therefore, circulating leptin levels are low in states of malnutrition such as starvation or prolonged fasting. Multiple lines of evidence point to the adipokine leptin as a key link between nutrition and immune cell biology in states of malnutrition. Reduced leptin signaling in the hypothalamus results in adaptations which include decreased body temperature, suppression of reproductive and thyroid function, and stimulation of the hypothalamus-pituitary-adrenal (HPA) axis [21]. These adaptive mechanisms promote survival and limit procreation during malnutrition. Many immune cells are also responsive to leptin, including T cells. In fact, expression of the leptin receptor is upregulated following T-cell activation [22]. Here we will review findings that demonstrate leptin signaling is required to metabolically license pro-inflammatory/effector T cells for activation, and that changes in adipokine levels mediate communication between nutritional status and T-cell immunity.
T-cell dysfunction in malnutrition
Malnutrition has been linked to immune dysfunction in settings of starvation and cachexia in both human and animal studies [19, 23–25]. Numerous groups have specifically examined the effect of malnutrition on T cells. Severe malnutrition or starvation has been found to alter T-cell number by influencing both T-cell survival and proliferation. For example, mice fasted for 48 hours have significantly decreased thymocyte and splenocyte counts compared to ad libitum-fed control mice [16–18]. In an alternative model of malnutrition, mice fed a protein-deficient diet had atrophic spleens and decreased T-cell numbers compared to control fed mice [26, 27]. Similar findings were seen in studies of malnourished humans. For example, both CD4+ and CD8+ T-cell counts were decreased in blood samples from malnourished children in comparison to T-cell counts from normal weight controls [28].
Many studies have also described the effect of malnutrition on decreased T-cell cytokine production as well as decreased capacity to respond appropriately to stimulation or cytokines [29, 30]. In a rat model, protein energy malnutrition impaired the ability of T cells to proliferate and produce the Type 1 T helper cell (Th1)-associated cytokine interferon (IFN)-γ [31]. Similarly, activated T cells from fasted mice had greatly decreased production of the pro-inflammatory cytokines interleukin (IL)-2 and IFN-γ compared to T cells from ad libitum fed control mice [18]. Similar results have been observed in humans studies: malnourished children were found to have decreased levels of cytokines which promote differentiation of CD4+ T helper cells into pro-inflammatory Th1 cells: IL-12, IL-18, and IL-21; decreased levels of cytokines produced by Th1 cells: IFN-γ and IL-2; as well as impaired Th1-cell proliferation [32, 33]. The same group also observed that malnourished children showed an increase in the relative expression of the Type 2 T helper cell (Th2) cytokine IL-4 and the anti-inflammatory cytokine IL-10 [33]. More recent studies have compared the effect of malnutrition/fasting on effector CD4+ T cells (Teff, such as Th17 cells) and regulatory T cells (Treg) and found that fasted mice had a more significant drop in Teff, particularly Th17, cell number than in Treg [34]. Overall, these studies demonstrate that malnutrition leads to a shift in the balance of cytokines in favor of an anti-inflammatory response and immune tolerance. Consequently, malnutrition can increase susceptibility to infection; however, these cytokine shifts also offer protection against autoimmune disease. This has been demonstrated in mouse models of systemic lupus erythematosus (SLE) [35, 36] and experimental autoimmune encephalomyelitis (EAE) [34, 37–40], among others. The results of these studies suggest that calorie-restriction and fasting prevent autoimmunity by decreasing T-cell responses and inflammatory cytokine production.
As mentioned previously, adipokines have been shown to influence immunity and infection response in states of malnutrition, and many studies have demonstrated that adiponectin and leptin play opposite roles in inflammation and immunity [41]. Low levels of leptin are associated with high rates of death from infectious diseases [16, 21, 42]. In a 2014 study of malnourished Ugandan infants, low circulating leptin levels were found to be the single most important biomarker to predict mortality [43]. It is now clear that both genetic leptin deficiency and starvation/fasting-induced hypoleptinemia have dramatic effects on T cells including reduced T-cell numbers, decreased Teff cells, increased proportion of Treg cells, and protection against certain forms of autoimmune disease [15, 17, 18, 44–47]. Initial studies of exogenous administration of leptin during starvation demonstrated that leptin-treated mice were protected from lymphoid atrophy, indicating a role for leptin in the immune dysfunction of starvation [16]. Treatment of fasted animals with leptin also rescued impaired T-cell proliferation and inflammatory cytokine production, and did so when leptin was delivered either in vivo to fasted animals or in vitro to T cells isolated from fasted mice [18, 34].
Leptin deficiency in both mouse (ob−/− leptin mutant mice) and human resulted in decreased total T-cell number, decreased CD4+ T helper cell number, decreased pro-inflammatory cytokine production, and reduced sensitivity of T cells to activating stimuli, resulting in increased susceptibility to intracellular infections as well as protection against several forms of autoimmunity [16, 48–51]. Importantly, these immune defects were reversed following treatment with recombinant leptin protein [52–55]. Similar immune defects were also seen in studies of leptin receptor mutant mice (db−/− mice): activated CD4+ T cells from db−/− mice had decreased T-cell cytokine production and decreased proliferation compared to T cells from wildtype controls [18, 56]. And consistent with findings reported in malnourished animals and humans, either neutralization of leptin in wildtype mice or genetic leptin deficiency increased susceptibility to intracellular infections and decreased susceptibility to multiple autoimmune diseases: EAE, glomerulonephritis, colitis, and hepatitis [57].
It must be noted, however, that leptin is a pleiotropic hormone, and as such, there has been some debate as to whether the effects of leptin on T-cell immunity are mediated through direct effects on T cells or through indirect, cell-extrinsic mechanisms. At present, there is evidence for both. Many studies have demonstrated a direct effect of leptin on T cells in vitro [15, 58–60], whereas others have shown that leptin can influence T cells indirectly [61–63]. Additionally, some reports suggest leptin may also mediate its effects on immune cell populations via central signaling; for example, intracerebroventricular injections of leptin were sufficient to rescue lymphocyte numbers in fasted mice [64]. Recent studies have confirmed a direct role for leptin in Teff-cell function and differentiation. T cell-specific leptin receptor knockout mice have now been shown by two groups to be critical for T-cell polarization into pro-inflammatory Th1 and Th17 subsets which secrete inflammatory cytokines IFN-γ and IL-17, respectively [34, 45]. Indeed, Gerriets et al. demonstrated that both malnutrition-associated hypoleptinemia and T cell-specific leptin receptor knockout suppressed Th1 or Th17-cell number and function [34]. Moreover, both Reis et al. and Gerriets et al. demonstrated the importance of T-cell leptin-signaling in autoimmune disease, and found leptin signaling critical for Teff function in mouse models of EAE and colitis [34, 45].
Adiponectin also plays a critical role in controlling inflammation and immunity. While less than 10% of human peripheral blood T cells express the adiponectin receptor on their surface, most T cells store adiponectin receptors intracellularly and upregulate expression following stimulation. Adiponectin signaling in T cells leads to apoptosis, decreased proliferation, and decreased cytokine production in response to antigen stimulation [65]. Specifically, adiponectin signaling has been shown to upregulate sirtuin 1 (SIRT1) and peroxisome proliferator-activated receptor γ (PPARγ) to inhibit expression of the Th17-associated transcription factor, RORγt [66]. Additionally, adiponectin has been found to be required to promote Treg in a mouse model of EAE [67]. Collectively these studies show that adiponectin plays a protective role in inflammatory and autoimmune diseases by negatively regulating antigen-specific T cells, inhibiting Teff-cell differentiation, and modulating Treg-cell homeostasis.
Altered T-cell metabolism in malnutrition
Numerous studies have shown that the cellular metabolism of T cells varies under conditions of rest, activation, and memory generation [68–70], and we have reviewed this topic previously [71]. Non-proliferating quiescent lymphocytes (such as naïve and memory T cells) utilize a mixed fuel metabolism of glucose, lipids, and amino acids via oxidative phosphorylation to generate energy for immune surveillance and maintenance of memory. In contrast, Teff-cell activation results in a metabolic switch to anabolic metabolism and glycolysis as the primary metabolic program [72, 73]. This switch to anabolic metabolism enables the use of nutrients not only for generating energy but also for the construction of the molecular building blocks that are incorporated into cellular biomass to support activated T-cell growth, proliferation, and cytokine production [74]. Moreover, metabolic pathways also have a role in the differentiation of CD4+ T cells. Teff cells such as Th1, Th2, and Th17 cells express high amounts of the glucose transporter Glut1, and utilize a largely glycolytic metabolism; Treg, on the other hand, which suppress T-cell responses and maintain homeostasis, express low concentrations of Glut1 and require oxidative metabolism to generate energy for their survival and suppressive function [68, 75, 76], but when cultured in vitro, Treg cells were shown to engage in both glycolysis and fatty acid oxidation to enhance proliferation [77]. Moreover, conditions such as acute infection or inflammation provided signals that increased glycolysis and expression of Glut1 levels in Treg [78]. These metabolic changes directly modified Treg function to down-regulate the transcription factor Foxp3 and to become more proliferative and less suppressive. As inflammatory signals resolved, Foxp3 levels were increased and downregulated Glut1, thereby favoring mitochondrial oxidative pathways and Treg-cell suppressive capacity [78].
We now appreciate that T-cell function and metabolism are intimately linked, and alterations in the cellular metabolic state can lead to changes in T-cell function [71]. Because activated Teff cells are largely dependent on glycolysis to fulfill their energy requirements for proliferation and function, these cells are particularly sensitive to low nutrient levels observed during starvation or malnutrition. Indeed, low glucose levels found in starved or fasted mice may contribute to the differentiation of T cells into Treg over Teff cells [79]. In a study by Saucillo et al., activated CD4+ T cells from fasted animals had decreased glucose uptake and glycolysis [18]. However, when the fasted animals were treated with leptin, T-cell glucose metabolism returned to levels seen in the ad libitum fed control group [18]. Using the db−/− model, this study went on to show that leptin signal was required for normal T-cell Glut1 expression and glucose metabolism following T-cell activation, and that db−/− mice had decreased production of lactate and decreased mitochondrial oxidation, as measured by extracellular flux analysis [18]. These findings were only observed in activated cells, as naïve/resting T cells, which are less glycolytic at baseline, did not have altered glucose metabolism in db−/− mice.
The same group used a T cell-specific leptin receptor conditional knockout model to confirm that these effects of leptin on T-cell metabolism were direct and mediated through the leptin receptor. Indeed, activated T cells from leptin receptor conditional knockout mice were found to have decreased inflammatory cytokine production and decreased glucose metabolism, similar to activated T cells from db−/− mice [18]. Moreover, comparison of cellular metabolism of in vitro differentiated Teff (Th1 and Th17) and Treg cells from leptin receptor conditional knockout mice versus littermate controls found that the differentiated Th1 and Th17 cells from T cell-specific leptin receptor conditional knockout mice had decreased glucose uptake, glycolysis, lactate production, and mitochondrial respiration compared to Teff cells from control mice. However, Treg glucose metabolism, which is lower at baseline, was unchanged by the presence or absence of T cell-specific leptin signaling [34]. Altogether, these reports suggest that leptin is a central regulator of the pro-inflammatory response and can link nutritional status and immunity by directly promoting T-cell glucose metabolism and thereby enhancing the differentiation, proliferation and functionality of Teff cells (Fig. 1).
Figure 1. The role of leptin in Teff-mediated inflammation.
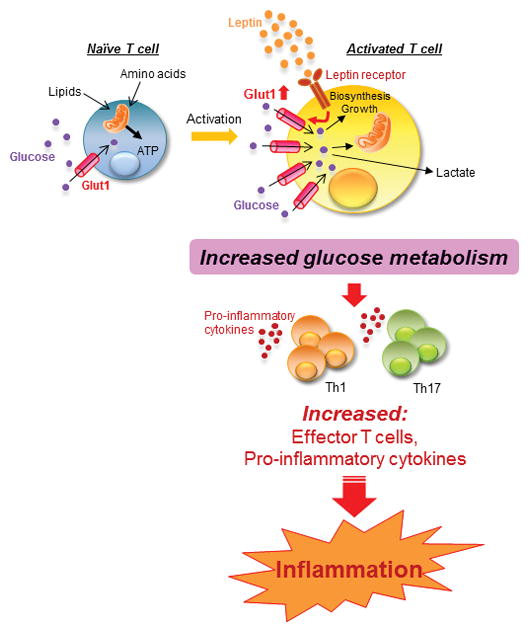
Leptin receptor expression is upregulated following T cell activation, and T cell leptin signaling leads to increased expression of the glucose transporter Glut1. Increased Glut1 expression, in turn, promotes glucose uptake and glycolysis, and thereby fuels Teff (Th1 and Th17) differentiation and pro-inflammatory cytokine production, leading to inflammation.
Key signaling molecules that mediate nutritional effects on T-cell immunity
Nutrient deficiencies may influence T-cell metabolism via cytoplasmic nutrient sensors including AMP-activated protein kinase (AMPK) and the mammalian target of rapamycin (mTOR) [80]. AMPK is activated during fasting and starvation and has a role in restoring energy homeostasis. Cellular stress, such as that caused by nutrient starvation, leads to an increased AMP/ATP ratio, which in turn promotes the phosphorylation and activation of AMPK. Activation of AMPK promotes glucose uptake and glycolysis, fatty acid uptake and fatty acid oxidation, as well as autophagy [81]. In addition, and in order to maintain energy, AMPK inhibits large molecule synthesis and blocks anabolic pathways, such as gluconeogenesis and synthesis of glycogen, fatty acids and triglycerides [81]. In T cells, AMPK is required for activation of lipid oxidation and Treg function [68].
mTOR directly impacts T-cell differentiation and proliferation by integrating environmental signals (nutrients, energy stores, and growth factors) and coordinating the metabolism of the cell according to its need to proliferate or functionally differentiate [82, 83]. However, when nutrient availability is limited such as in starvation, mTOR activity is impaired, resulting in inhibition in the proliferation of Teff cells [17]. Reduced mTOR activation has been found to be partly mediated through the activation of AMPK [84]. Moreover, the mTOR pathway behaves differently in Teff versus Treg cells; in Treg, mTOR inhibition following starvation leads to elevation in the expression of Foxp3 and results in preferential differentiation and proliferation [85, 86]. Recent studies have further defined the role of mTOR signaling in Treg: Treg have been found to have relatively high mTORC1 activity [87], and deletion of tuberous sclerosis 1 (TSC1), a negative regulator of mTOR, in Treg impaired expression of Foxp3 and Treg suppressive capacity [88]. Likewise, activation of mTOR signaling by Toll-like receptor (TLR) signals also impaired Treg suppressive capacity [78].
mTOR activity and leptin influence one another in both the hypothalamus and the peripheral immune system. In the hypothalamus, mTOR is activated in response to leptin and the effect of leptin on food intake is mTOR-dependent. In addition, mice treated with the mTOR inhibitor rapamycin had reduced body weight and decreased epididymal fat pads (visceral adipose depots). Those mice also reduced daily food efficiency and had lower serum leptin and insulin levels compared to control mice fed a high-fat diet [89]. In lymphocytes, the leptin-mTOR axis has been shown to control the survival and proliferation of autoreactive T cells and the responsiveness of Treg cells in ob−/− and db−/− mice [86], although these results may be a consequence of indirect effects, as T cells from T cell-specific leptin receptor conditional knockout mice did not display any changes in mTOR activation [34], suggesting that other downstream signaling pathways may be involved in leptin-mediated metabolic changes in T cells. One such candidate signaling protein is hypoxia-inducible factor-1α (HIF-1α) protein. HIF-1α is a transcription factor that is upregulated in settings of decreased oxygen tension. It has also been shown to have a critical role in promoting both Th17 differentiation and T-cell glycolytic metabolism [90]. Upon activation, HIF-1α promotes the expression of several glycolysis-related genes, such as Glut1 and 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase3 (PFKFB3) [91]. HIF-1α can also increase glucose uptake and glycolysis, and reduce oxidative phosphorylation and oxygen consumption in T cells [92]. Both Reis et al. and Gerriets et al. have demonstrated a direct role for leptin in promoting HIF-1α expression in Teff cells [34, 45].
Immune adaptation to obesity
On the other end of the nutritional spectrum, obesity is a growing epidemic in the United States with over 30% of Americans currently classified as obese, and another one third overweight [1]. A similar trend has been observed in many developed countries worldwide. The obesity epidemic is accompanied by numerous associated health risks, including insulin resistance leading to type 2 diabetes mellitus, and hyperlipidemia and hypertension leading to increased cardiovascular and renal disease [93]. At the same time, obesity increases the risk of multiple forms of autoimmunity such as multiple sclerosis, thyroid autoimmunity, and type 1 diabetes [94–96], as well as increases susceptibility to select infections due to impaired host defense [97]. Indeed, obesity is now known to be an independent risk factor for both seasonal flu and pandemic H1N1 influenza infections [98–100]. Moreover, obese patients are at increased risk of developing complications such as sepsis, pneumonia and bacteremia following surgical procedures [101], are more prone to Helicobacter pylori infection [102], and obese children were found to have three times greater risk of being asymptomatic carriers of Neisseria meningitides [103].
Obesity leads to immune cell changes characterized by a low-grade chronic inflammatory state, with increased circulating pro-inflammatory cytokines such as tumor necrosis factor (TNF), IL-6 and C-reactive protein (CRP), the expression of which are mediated by signaling molecules such as nuclear factor-kappa B (NF-κB) and phosphoinositide 3-kinase (PI3K), by glucocorticoids, and by catecholamines [104]. This change in cytokine milieu is associated with both a change in the number and proportion of circulating immune cells as well as an influx of pro-inflammatory immune cells into the visceral adipose tissue. Many studies have implicated macrophages as a key player in the development of obesity-associated metabolic disease, and inhibition or deletion of macrophages from VAT has been found to inhibit the development of insulin resistance in mouse models of obesity [105]. Likewise, neutrophils and mast cells are also increased in obesity and produce pro-inflammatory cytokines [10]. Conversely, Natural Killer T cells (NKT cells), ILC2 cells, and eosinophils, which have a protective anti-inflammatory effect, are decreased in obese individuals [11].
More recently, lymphocytes have also been highlighted as key players in the development of obesity-associated inflammation. In obese VAT, there is an influx of effector B and T cells (particularly Th1 and CD8+ T cells) along with a decreased proportion of Treg [79, 106–108]. The increase in pro-inflammatory T cells leads to increased secretion of cytokines that can directly impair insulin action in target tissues. In this regard, secretion of IFN-γ by VAT Th1 cells and CD8+ T cells was shown to promote insulin resistance through direct effects on adipocyte metabolism and by polarizing, activating and stimulating VAT pro-inflammatory (classically activated/M1) macrophages [109]. In addition, TNF secretion by activated T cells directly impaired insulin-mediated glucose uptake via stimulation of inhibitor of NF-κB kinase subunit beta (IKKβ) and c-Jun amino-terminal kinase (JNK) [110].
These changes are critical in obesity-associated pathologies, as animal models that knockout or disable T or B-cell responses in obesity both prevent the accumulation of inflammatory cytokines and improve insulin sensitivity [111–116]. Immunotherapy aimed at blunting Th1 cells using CD3-specific antibody (or its F(ab′) fragment) or by depleting adipose tissue of T cells was shown to control adipose inflammation and insulin sensitivity in mice [107]. Similarly, depletion of CD8+ T cells with an anti-CD8 antibody significantly reduced local VAT inflammation, glucose intolerance, and insulin resistance, whereas transfer of CD8+ T cells into CD8-deficient mice increased pro-inflammatory gene expression, glucose intolerance, and insulin resistance [106].
T cells can also play a role in modulating insulin resistance. Treg numbers are reduced in obesity [117], and Treg have been shown to alter macrophage polarization and improve insulin sensitivity [79, 105]. Consistently, depletion of Treg cells resulted in an increase in systemic insulin resistance [118], whereas expansion of Treg cells led to improvement in glucose tolerance and insulin sensitivity in high fat diet-induced obese mice [79, 118]. There may also be a role for insulin in regulating Treg function directly [119]. A study by Han et al. showed that insulin receptors are expressed on Treg, and that insulin signaling directly influenced Treg function by decreasing IL-10 production via AKT/mTOR [119]. Moreover, Treg from obese VAT produced less IL-10 and more IFN-γ than Treg from lean animals [119]. These findings suggest that obesity-associated hyperinsulinemia can promote inflammation by directly reducing Treg suppression and thereby driving inflammation.
T cell metabolism and adipokines in obesity
Given the broad list of obesity-associated complications that may result from impaired immunity, it is critically important that we work to understand the mechanisms by which obesity causes immune dysregulation to promote disease. As discussed previously, the regulation of nutrient uptake and utilization is critically important for the control of lymphocyte differentiation and function [71], and the pathways that control lymphocyte function and metabolism are intimately linked. Lymphocyte metabolism in effector, regulatory, or memory cells may be altered in obesity and/or obesity-associated type 2 diabetes in which the metabolic environment is altered. As immune cells require extracellular nutrient uptake for their survival, they are extremely sensitive to these changes in the lymphocyte microenvironment. It is therefore likely that an excessive level of nutrients in obesity and/or obesity-associated type 2 diabetes is likely to have a significant impact on T-cell immunometabolism in addition to function.
Similar to glucose, fatty acids are important in fueling an immune response, as they are a readily available source of abundant energy. The impact of excess circulating non-essential fatty acids, a hallmark of obesity [120], on immune cells has not been fully characterized. However, it is clear that elevated intake of saturated fatty acids (such as palmitate and stearate) is associated with inflammation and increased metabolic disease, whereas unsaturated fatty acids may prevent or decrease inflammation [121]. One recent study showed that adipocyte-released fatty acids increased T-cell proliferation towards Th17 and led to increased secretion of the pro-inflammatory cytokines IFN-γ and IL-17 [122]. Additionally, increasing levels of the saturated fatty acid palmitate, but not of unsaturated fatty acids, were shown to be important for the activation of T cells and led to dose-dependent increases in cytokine production, increased generation of reactive oxygen species, and increased lipid peroxidation in vitro [123]. Similarly, short chain fatty acids have been shown to enhance Teff differentiation and cytokine production, via activation of mTOR, in a context dependent manner [124]. Conversely, the polyunsaturated fatty acids docosahexaenoic acid (DHA) and alpha-linoleic acid have been shown to increase secretion of the anti-inflammatory T-cell cytokine IL-10, decrease pro-inflammatory cytokine production, and impair T-cell proliferation [121]. The exact mechanism by which fatty acids regulate T-cell metabolism in obesity requires further investigation.
Another mechanism by which obesity may influence immune response is through adipocyte-secreted hormones (adipokines), such as the hormones leptin, resistin, and visfatin. As we already discussed, circulating leptin concentrations increase in proportion to adipose mass, and leptin has a key role in the activation of T cells. Leptin has been shown in both mouse and human to promote effector Th1 and Th17-cell number, and leptin is required to upregulate the glucose transporter Glut1 to allow metabolic reprograming of activated effector T cells and fuel T-cell proliferation and cytokine production [18]. This suggests a pathway by which obesity-associated hyperleptinemia alters lymphocyte metabolism, thereby promoting the chronic low-grade T-cell inflammation seen in obesity. Similar to leptin, resistin levels are increased markedly in animal models of both genetic and diet-induced obesity and reduced by fasting [125]. In human studies the correlation of resistin and nutritional status is less clear, but increased resistin expression has been correlated with inflammatory markers, coronary artery disease, cardiovascular disease, asthma, and rheumatoid arthritis [126, 127]. Numerous studies have demonstrated that resistin is a pathogenic factor with a role both in inflammation and metabolism.
Lastly, nutritional signals in obesity are integrated by T cells via AMPK and mTOR, which may be altered in the obese state and appear to play a role in obesity-associated inflammation. In the case of AMPK signaling, Gauthier at el. have shown that AMPK levels are low in adipose tissue from obese/insulin resistant subjects compared to obese/insulin sensitive subjects [128]. This finding suggests that AMPK keeps inflammation and metabolic disease restrained in obesity. On the other hand, in response to nutrient availability, T cells may become activated and trigger the mTOR pathway to up-regulate Glut1 surface expression and shift these cells toward the use of glycolysis and glutaminolysis to fulfil energetic and anabolic needs. The mTOR pathway also contributes to the differentiation of CD4+ T cells into Teff cells (such as Th1 and Th17) and inhibits Treg-cell differentiation [83, 85]. Moreover, T-cell metabolism is also mediated by AKT signaling which increases utilization of glucose and amino acids and promotes the mTOR pathway and glycolysis, thus supporting effector T-cell differentiation, growth, and function [17].
Conclusions
Overall, there is growing evidence that adipocytes and immune cells coordinate nutritional status and immunity (Fig. 2). One mechanism for this interaction is through secretion of adipokines, such as leptin, that can directly convey the state of nutrient availability to peripheral lymphocytes and other immune cells. This communication of nutritional status to immune cells is critical to maintain energy balance in immunological defense, as activated effector lymphocytes have a very high metabolic demand which is essential for growth, proliferation, and the production of proteins required for a successful immune response. In states of limited nutrients, such as starvation or fasting, T-cell number is low and activated T cells are unable to upregulate glucose metabolism to fuel effector T-cell function, i.e. proliferation and inflammatory cytokine production. This malnutrition-associated immunodeficiency can lead to increased susceptibility to infection while simultaneously offering protection against several forms of autoimmune disease. Conversely, immunometabolic changes in obesity may promote effector T-cell differentiation and inflammatory cytokine production and thereby increase the risk of obesity-associated metabolic diseases. Understanding the relevant cytokines, hormones, signaling pathways, and mechanisms by which nutritional status and adipose tissue communicate to immune cells will hopefully inform treatment of both malnutrition-associated immunodeficiency and obesity-associated inflammation.
Figure 2. Nutritional status influences T cell response.
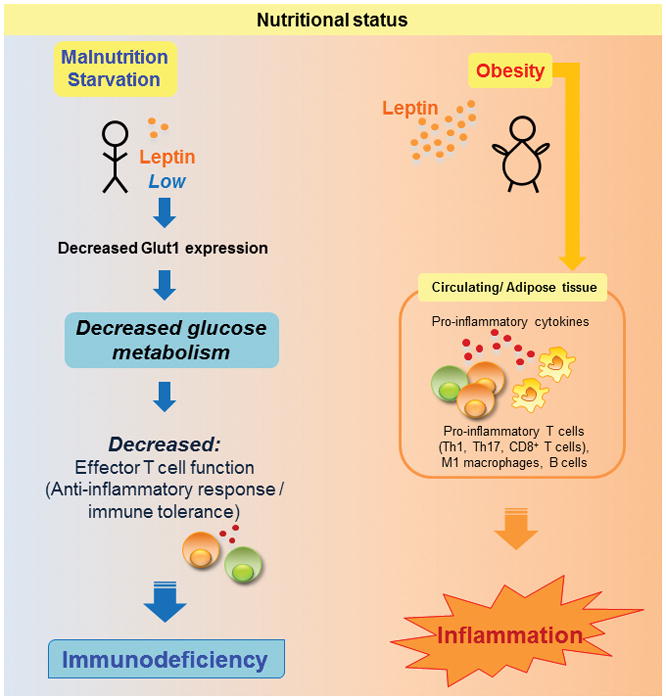
Malnutrition and starvation are associated with hypoleptinemia and decreased T cell Glut1 expression, leading to decreased glucose metabolism and impaired Teff function. Conversely, obesity, which is associated with elevated circulating leptin, is associated with increased numbers of Th1, Th17, CD8+ T cells, B cells, and M1 (classically activated) macrophages, leading to increased production of pro-inflammatory cytokines in both circulation and in adipose tissue, thereby accelerating inflammation.
Acknowledgments
This work was supported by the National Institutes of Health Grant R01-DK106090 (N.J.M.), the National Multiple Sclerosis Society Research Grant RG-5333 (K.D. and N.J.M.), and the Alliance for Lupus Research (S.C.)
Abbreviations
- Th1
Type 1 T helper cell
- Th2
Type 2 T helper cell
- Th17
Type 17 T helper cell
- Teff
effector T cell
- Treg
regulatory T cell
- IFN-γ
interferon-γ
- EAE
experimental autoimmune encephalomyelitis
- AMPK
AMP-activated protein kinase
- mTOR
mammalian target of rapamycin
- HIF-1α
hypoxia-inducible factor-1α
- VAT
visceral adipose tissue
Footnotes
Conflict of Interest Statement
The authors declare no commercial or financial conflict of interest.
References
- 1.Ogden CL, Carroll MD, Fryar CD, Flegal KM. Prevalence of Obesity Among Adults and Youth: United States, 2011–2014; NCHS Data Brief. 2015:1–8. [PubMed] [Google Scholar]
- 2.McGuire S FAO IFAD WFP. The State of Food Insecurity in the World 2015: Meeting the 2015 International Hunger Targets: Taking Stock of Uneven Progress; Rome: FAO; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]; Adv Nutr. 2015;6:623–624. doi: 10.3945/an.115.009936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milner JJ, Beck MA. The impact of obesity on the immune response to infection. Proc Nutr Soc. 2012;71:298–306. doi: 10.1017/S0029665112000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naylor C, Petri WA., Jr Leptin Regulation of Immune Responses. Trends Mol Med. 2016;22:88–98. doi: 10.1016/j.molmed.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, Pedersen AA, Kalthoff C, Tullin S, Sams A, Summer R, Walsh K. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem. 2010;285:6153–6160. doi: 10.1074/jbc.M109.088708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silswal N, Singh AK, Aruna B, Mukhopadhyay S, Ghosh S, Ehtesham NZ. Human resistin stimulates the pro-inflammatory cytokines TNF-alpha and IL-12 in macrophages by NF-kappaB-dependent pathway. Biochem Biophys Res Commun. 2005;334:1092–1101. doi: 10.1016/j.bbrc.2005.06.202. [DOI] [PubMed] [Google Scholar]
- 7.Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, Tilg H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. 2007;178:1748–1758. doi: 10.4049/jimmunol.178.3.1748. [DOI] [PubMed] [Google Scholar]
- 8.Wing EJ, Magee DM, Barczynski LK. Acute starvation in mice reduces the number of T cells and suppresses the development of T-cell-mediated immunity. Immunology. 1988;63:677–682. [PMC free article] [PubMed] [Google Scholar]
- 9.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huh JY, Park YJ, Ham M, Kim JB. Crosstalk between Adipocytes and Immune Cells in Adipose Tissue Inflammation and Metabolic Dysregulation in Obesity. Mol Cells. 2014 doi: 10.14348/molcells.2014.0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, Balk SP, O’Shea D, O’Farrelly C, Exley MA. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity. 2012;37:574–587. doi: 10.1016/j.immuni.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerriets VA, MacIver NJ. Role of T cells in malnutrition and obesity. Front Immunol. 2014;5:379. doi: 10.3389/fimmu.2014.00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winer DA, Winer S, Chng MH, Shen L, Engleman EG. B Lymphocytes in obesity-related adipose tissue inflammation and insulin resistance. Cell Mol Life Sci. 2014;71:1033–1043. doi: 10.1007/s00018-013-1486-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wensveen FM, Valentic S, Sestan M, Wensveen TT, Polic B. Interactions between adipose tissue and the immune system in health and malnutrition. Semin Immunol. 2015;27:322–333. doi: 10.1016/j.smim.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 15.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 16.Howard JK, Lord GM, Matarese G, Vendetti S, Ghatei MA, Ritter MA, Lechler RI, Bloom SR. Leptin protects mice from starvation-induced lymphoid atrophy and increases thymic cellularity in ob/ob mice. J Clin Invest. 1999;104:1051–1059. doi: 10.1172/JCI6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Procaccini C, De Rosa V, Galgani M, Carbone F, Cassano S, Greco D, Qian K, Auvinen P, Cali G, Stallone G, Formisano L, La Cava A, Matarese G. Leptin-induced mTOR activation defines a specific molecular and transcriptional signature controlling CD4+ effector T cell responses. J Immunol. 2012;189:2941–2953. doi: 10.4049/jimmunol.1200935. [DOI] [PubMed] [Google Scholar]
- 18.Saucillo DC, Gerriets VA, Sheng J, Rathmell JC, Maciver NJ. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol. 2014;192:136–144. doi: 10.4049/jimmunol.1301158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caulfield LE, de Onis M, Blossner M, Black RE. Undernutrition as an underlying cause of child deaths associated with diarrhea, pneumonia, malaria, and measles. Am J Clin Nutr. 2004;80:193–198. doi: 10.1093/ajcn/80.1.193. [DOI] [PubMed] [Google Scholar]
- 20.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 21.Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- 22.Papathanassoglou E, El-Haschimi K, Li XC, Matarese G, Strom T, Mantzoros C. Leptin receptor expression and signaling in lymphocytes: kinetics during lymphocyte activation, role in lymphocyte survival, and response to high fat diet in mice. J Immunol. 2006;176:7745–7752. doi: 10.4049/jimmunol.176.12.7745. [DOI] [PubMed] [Google Scholar]
- 23.Schlaudecker EP, Steinhoff MC, Moore SR. Interactions of diarrhea, pneumonia, and malnutrition in childhood: recent evidence from developing countries. Curr Opin Infect Dis. 2011;24:496–502. doi: 10.1097/QCO.0b013e328349287d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antwi A. Assessment and management of severe malnutrition in children. West Afr J Med. 2011;30:11–18. doi: 10.4314/wajm.v30i1.69878. [DOI] [PubMed] [Google Scholar]
- 25.Dewys WD, Begg C, Lavin PT, Band PR, Bennett JM, Bertino JR, Cohen MH, Douglass HO, Jr, Engstrom PF, Ezdinli EZ, Horton J, Johnson GJ, Moertel CG, Oken MM, Perlia C, Rosenbaum C, Silverstein MN, Skeel RT, Sponzo RW, Tormey DC. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am J Med. 1980;69:491–497. doi: 10.1016/s0149-2918(05)80001-3. [DOI] [PubMed] [Google Scholar]
- 26.Pena-Cruz V, Reiss CS, McIntosh K. Sendai virus infection of mice with protein malnutrition. J Virol. 1989;63:3541–3544. doi: 10.1128/jvi.63.8.3541-3544.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor AK, Cao W, Vora KP, De La Cruz J, Shieh WJ, Zaki SR, Katz JM, Sambhara S, Gangappa S. Protein energy malnutrition decreases immunity and increases susceptibility to influenza infection in mice. J Infect Dis. 2013;207:501–510. doi: 10.1093/infdis/jis527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Najera O, Gonzalez C, Toledo G, Lopez L, Ortiz R. Flow cytometry study of lymphocyte subsets in malnourished and well-nourished children with bacterial infections. Clin Diagn Lab Immunol. 2004;11:577–580. doi: 10.1128/CDLI.11.3.577-580.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez L, Gonzalez C, Flores L, Jimenez-Zamudio L, Graniel J, Ortiz R. Assessment by flow cytometry of cytokine production in malnourished children. Clin Diagn Lab Immunol. 2005;12:502–507. doi: 10.1128/CDLI.12.4.502-507.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pelletier DL, Frongillo EA, Jr, Habicht JP. Epidemiologic evidence for a potentiating effect of malnutrition on child mortality. Am J Public Health. 1993;83:1130–1133. doi: 10.2105/ajph.83.8.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mengheri E, Nobili F, Crocchioni G, Lewis JA. Protein starvation impairs the ability of activated lymphocytes to produce interferon-gamma. J Interferon Res. 1992;12:17–21. doi: 10.1089/jir.1992.12.17. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez-Torres C, Gonzalez-Martinez H, Miliar A, Najera O, Graniel J, Firo V, Alvarez C, Bonilla E, Rodriguez L. Effect of malnutrition on the expression of cytokines involved in Th1 cell differentiation. Nutrients. 2013;5:579–593. doi: 10.3390/nu5020579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez-Martinez H, Rodriguez L, Najera O, Cruz D, Miliar A, Dominguez A, Sanchez F, Graniel J, Gonzalez-Torres MC. Expression of cytokine mRNA in lymphocytes of malnourished children. J Clin Immunol. 2008;28:593–599. doi: 10.1007/s10875-008-9204-5. [DOI] [PubMed] [Google Scholar]
- 34.Gerriets VA, Danzaki K, Kishton RJ, Eisner W, Nichols AG, Saucillo DC, Shinohara ML, MacIver NJ. Leptin directly promotes T-cell glycolytic metabolism to drive effector T-cell differentiation in a mouse model of autoimmunity. Eur J Immunol. 2016;46:1970–1983. doi: 10.1002/eji.201545861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy ED, Roths JB. A Y chromosome associated factor in strain BXSB producing accelerated autoimmunity and lymphoproliferation. Arthritis Rheum. 1979;22:1188–1194. doi: 10.1002/art.1780221105. [DOI] [PubMed] [Google Scholar]
- 36.Kubo C, Gajar A, Johnson BC, Good RA. The effects of dietary restriction on immune function and development of autoimmune disease in BXSB mice. Proc Natl Acad Sci U S A. 1992;89:3145–3149. doi: 10.1073/pnas.89.7.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piccio L, Stark JL, Cross AH. Chronic calorie restriction attenuates experimental autoimmune encephalomyelitis. J Leukoc Biol. 2008;84:940–948. doi: 10.1189/jlb.0208133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanna V, Di Giacomo A, La Cava A, Lechler RI, Fontana S, Zappacosta S, Matarese G. Leptin surge precedes onset of autoimmune encephalomyelitis and correlates with development of pathogenic T cell responses. J Clin Invest. 2003;111:241–250. doi: 10.1172/JCI16721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esquifino AI, Cano P, Jimenez V, Cutrera RA, Cardinali DP. Experimental allergic encephalomyelitis in male Lewis rats subjected to calorie restriction. J Physiol Biochem. 2004;60:245–252. doi: 10.1007/BF03167069. [DOI] [PubMed] [Google Scholar]
- 40.Kafami L, Raza M, Razavi A, Mirshafiey A, Movahedian M, Khorramizadeh MR. Intermittent feeding attenuates clinical course of experimental autoimmune encephalomyelitis in C57BL/6 mice. Avicenna J Med Biotechnol. 2010;2:47–52. [PMC free article] [PubMed] [Google Scholar]
- 41.Stofkova A. Leptin and adiponectin: from energy and metabolic dysbalance to inflammation and autoimmunity. Endocr Regul. 2009;43:157–168. [PubMed] [Google Scholar]
- 42.Boden G, Chen X, Mozzoli M, Ryan I. Effect of fasting on serum leptin in normal human subjects. J Clin Endocrinol Metab. 1996;81:3419–3423. doi: 10.1210/jcem.81.9.8784108. [DOI] [PubMed] [Google Scholar]
- 43.Bartz S, Mody A, Hornik C, Bain J, Muehlbauer M, Kiyimba T, Kiboneka E, Stevens R, Bartlett J, St Peter JV, Newgard CB, Freemark M. Severe acute malnutrition in childhood: hormonal and metabolic status at presentation, response to treatment, and predictors of mortality. J Clin Endocrinol Metab. 2014;99:2128–2137. doi: 10.1210/jc.2013-4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y, Yu Y, Matarese G, La Cava A. Cutting edge: fasting-induced hypoleptinemia expands functional regulatory T cells in systemic lupus erythematosus. J Immunol. 2012;188:2070–2073. doi: 10.4049/jimmunol.1102835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reis BS, Lee K, Fanok MH, Mascaraque C, Amoury M, Cohn LB, Rogoz A, Dallner OS, Moraes-Vieira PM, Domingos AI, Mucida D. Leptin receptor signaling in T cells is required for Th17 differentiation. J Immunol. 2015;194:5253–5260. doi: 10.4049/jimmunol.1402996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu Y, Liu Y, Shi FD, Zou H, Matarese G, La Cava A. Cutting edge: Leptin-induced RORgammat expression in CD4+ T cells promotes Th17 responses in systemic lupus erythematosus. J Immunol. 2013;190:3054–3058. doi: 10.4049/jimmunol.1203275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amarilyo G, Iikuni N, Shi FD, Liu A, Matarese G, La Cava A. Leptin promotes lupus T-cell autoimmunity. Clin Immunol. 2013;149:530–533. doi: 10.1016/j.clim.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 48.Ozata M, Ozdemir IC, Licinio J. Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J Clin Endocrinol Metab. 1999;84:3686–3695. doi: 10.1210/jcem.84.10.5999. [DOI] [PubMed] [Google Scholar]
- 49.Farooqi IS, Wangensteen T, Collins S, Kimber W, Matarese G, Keogh JM, Lank E, Bottomley B, Lopez-Fernandez J, Ferraz-Amaro I, Dattani MT, Ercan O, Myhre AG, Retterstol L, Stanhope R, Edge JA, McKenzie S, Lessan N, Ghodsi M, De Rosa V, Perna F, Fontana S, Barroso I, Undlien DE, O’Rahilly S. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. 2007;356:237–247. doi: 10.1056/NEJMoa063988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chandra RK. Cell-mediated immunity in genetically obese C57BL/6J ob/ob) mice. Am J Clin Nutr. 1980;33:13–16. doi: 10.1093/ajcn/33.1.13. [DOI] [PubMed] [Google Scholar]
- 51.Faggioni R, Moser A, Feingold KR, Grunfeld C. Reduced leptin levels in starvation increase susceptibility to endotoxic shock. Am J Pathol. 2000;156:1781–1787. doi: 10.1016/S0002-9440(10)65049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paz-Filho GJ, Delibasi T, Erol HK, Wong ML, Licinio J. Cellular immunity before and after leptin replacement therapy. J Pediatr Endocrinol Metab. 2009;22:1069–1074. doi: 10.1515/jpem.2009.22.11.1069. [DOI] [PubMed] [Google Scholar]
- 53.Paz-Filho G, Mastronardi C, Delibasi T, Wong ML, Licinio J. Congenital leptin deficiency: diagnosis and effects of leptin replacement therapy. Arq Bras Endocrinol Metabol. 2010;54:690–697. doi: 10.1590/s0004-27302010000800005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, Lechler RI, DePaoli AM, O’Rahilly S. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Faggioni R, Jones-Carson J, Reed DA, Dinarello CA, Feingold KR, Grunfeld C, Fantuzzi G. Leptin-deficient (ob/ob) mice are protected from T cell-mediated hepatotoxicity: role of tumor necrosis factor alpha and IL-18. Proc Natl Acad Sci U S A. 2000;97:2367–2372. doi: 10.1073/pnas.040561297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mandel MA, Mahmoud AA. Impairment of cell-mediated immunity in mutation diabetic mice (db/db) J Immunol. 1978;120:1375–1377. [PubMed] [Google Scholar]
- 57.Procaccini C, Pucino V, Mantzoros CS, Matarese G. Leptin in autoimmune diseases. Metabolism. 2015;64:92–104. doi: 10.1016/j.metabol.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 58.Rodriguez L, Graniel J, Ortiz R. Effect of leptin on activation and cytokine synthesis in peripheral blood lymphocytes of malnourished infected children. Clin Exp Immunol. 2007;148:478–485. doi: 10.1111/j.1365-2249.2007.03361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin-Romero C, Santos-Alvarez J, Goberna R, Sanchez-Margalet V. Human leptin enhances activation and proliferation of human circulating T lymphocytes. Cell Immunol. 2000;199:15–24. doi: 10.1006/cimm.1999.1594. [DOI] [PubMed] [Google Scholar]
- 60.Fujita Y, Murakami M, Ogawa Y, Masuzaki H, Tanaka M, Ozaki S, Nakao K, Mimori T. Leptin inhibits stress-induced apoptosis of T lymphocytes. Clin Exp Immunol. 2002;128:21–26. doi: 10.1046/j.1365-2249.2002.01797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palmer G, Aurrand-Lions M, Contassot E, Talabot-Ayer D, Ducrest-Gay D, Vesin C, Chobaz-Peclat V, Busso N, Gabay C. Indirect effects of leptin receptor deficiency on lymphocyte populations and immune response in db/db mice. J Immunol. 2006;177:2899–2907. doi: 10.4049/jimmunol.177.5.2899. [DOI] [PubMed] [Google Scholar]
- 62.Gove ME, Sherry CL, Pini M, Fantuzzi G. Generation of leptin receptor bone marrow chimeras: recovery from irradiation, immune cellularity, cytokine expression, and metabolic parameters. Obesity (Silver Spring) 2010;18:2274–2281. doi: 10.1038/oby.2010.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gruver AL, Ventevogel MS, Sempowski GD. Leptin receptor is expressed in thymus medulla and leptin protects against thymic remodeling during endotoxemia-induced thymus involution. J Endocrinol. 2009;203:75–85. doi: 10.1677/JOE-09-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanaka M, Suganami T, Kim-Saijo M, Toda C, Tsuiji M, Ochi K, Kamei Y, Minokoshi Y, Ogawa Y. Role of central leptin signaling in the starvation-induced alteration of B-cell development. J Neurosci. 2011;31:8373–8380. doi: 10.1523/JNEUROSCI.6562-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilk S, Scheibenbogen C, Bauer S, Jenke A, Rother M, Guerreiro M, Kudernatsch R, Goerner N, Poller W, Elligsen-Merkel D, Utku N, Magrane J, Volk HD, Skurk C. Adiponectin is a negative regulator of antigen-activated T cells. Eur J Immunol. 2011;41:2323–2332. doi: 10.1002/eji.201041349. [DOI] [PubMed] [Google Scholar]
- 66.Zhang K, Guo Y, Ge Z, Zhang Z, Da Y, Li W, Zhang Z, Xue Z, Li Y, Ren Y, Jia L, Chan KH, Yang F, Yan J, Yao Z, Xu A, Zhang R. Adiponectin Suppresses T Helper 17 Cell Differentiation and Limits Autoimmune CNS Inflammation via the SIRT1/PPARgamma/RORgammat Pathway. Mol Neurobiol. 2016 doi: 10.1007/s12035-016-0036-7. [DOI] [PubMed] [Google Scholar]
- 67.Piccio L, Cantoni C, Henderson JG, Hawiger D, Ramsbottom M, Mikesell R, Ryu J, Hsieh CS, Cremasco V, Haynes W, Dong LQ, Chan L, Galimberti D, Cross AH. Lack of adiponectin leads to increased lymphocyte activation and increased disease severity in a mouse model of multiple sclerosis. Eur J Immunol. 2013;43:2089–2100. doi: 10.1002/eji.201242836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, Rathmell JC. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180:4476–4486. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38:2438–2450. doi: 10.1002/eji.200838289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–852. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- 75.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, Rathmell JC. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20:61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, Haeberli L, Huck C, Turka LA, Wood KC, Hale LP, Smith PA, Schneider MA, MacIver NJ, Locasale JW, Newgard CB, Shinohara ML, Rathmell JC. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, Faicchia D, Marone G, Tramontano D, Corona M, Alviggi C, Porcellini A, La Cava A, Mauri P, Matarese G. The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements. Immunity. 2016;44:406–421. doi: 10.1016/j.immuni.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, Warmoes MO, de Cubas AA, MacIver NJ, Locasale JW, Turka LA, Wells AD, Rathmell JC. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol. 2016;17:1459–1466. doi: 10.1038/ni.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bourke CD, Berkley JA, Prendergast AJ. Immune Dysfunction as a Cause and Consequence of Malnutrition. Trends Immunol. 2016 doi: 10.1016/j.it.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 83.Peter C, Waldmann H, Cobbold SP. mTOR signalling and metabolic regulation of T cell differentiation. Curr Opin Immunol. 2010;22:655–661. doi: 10.1016/j.coi.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 84.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Procaccini C, De Rosa V, Galgani M, Abanni L, Cali G, Porcellini A, Carbone F, Fontana S, Horvath TL, La Cava A, Matarese G. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33:929–941. doi: 10.1016/j.immuni.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. 2013;499:485–490. doi: 10.1038/nature12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Park Y, Jin HS, Lopez J, Elly C, Kim G, Murai M, Kronenberg M, Liu YC. TSC1 regulates the balance between effector and regulatory T cells. J Clin Invest. 2013;123:5165–5178. doi: 10.1172/JCI69751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chang GR, Chiu YS, Wu YY, Chen WY, Liao JW, Chao TH, Mao FC. Rapamycin protects against high fat diet-induced obesity in C57BL/6J mice. J Pharmacol Sci. 2009;109:496–503. doi: 10.1254/jphs.08215fp. [DOI] [PubMed] [Google Scholar]
- 90.Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, Luo W, Zeller K, Shimoda L, Topalian SL, Semenza GL, Dang CV, Pardoll DM, Pan F. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 92.Molon B, Cali B, Viola A. T Cells and Cancer: How Metabolism Shapes Immunity. Front Immunol. 2016;7:20. doi: 10.3389/fimmu.2016.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li Z, Bowerman S, Heber D. Health ramifications of the obesity epidemic. Surg Clin North Am. 2005;85:681–701. v. doi: 10.1016/j.suc.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 94.Harpsoe MC, Basit S, Andersson M, Nielsen NM, Frisch M, Wohlfahrt J, Nohr EA, Linneberg A, Jess T. Body mass index and risk of autoimmune diseases: a study within the Danish National Birth Cohort. Int J Epidemiol. 2014 doi: 10.1093/ije/dyu045. [DOI] [PubMed] [Google Scholar]
- 95.Duntas LH, Biondi B. The interconnections between obesity, thyroid function, and autoimmunity: the multifold role of leptin. Thyroid. 2013;23:646–653. doi: 10.1089/thy.2011.0499. [DOI] [PubMed] [Google Scholar]
- 96.Lukens JR, Dixit VD, Kanneganti TD. Inflammasome activation in obesity-related inflammatory diseases and autoimmunity. Discov Med. 2011;12:65–74. [PMC free article] [PubMed] [Google Scholar]
- 97.Falagas ME, Kompoti M. Obesity and infection. Lancet Infect Dis. 2006;6:438–446. doi: 10.1016/S1473-3099(06)70523-0. [DOI] [PubMed] [Google Scholar]
- 98.Paich HA, Sheridan PA, Handy J, Karlsson EA, Schultz-Cherry S, Hudgens MG, Noah TL, Weir SS, Beck MA. Overweight and obese adult humans have a defective cellular immune response to pandemic H1N1 influenza A virus. Obesity (Silver Spring) 2013;21:2377–2386. doi: 10.1002/oby.20383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morgan OW, Bramley A, Fowlkes A, Freedman DS, Taylor TH, Gargiullo P, Belay B, Jain S, Cox C, Kamimoto L, Fiore A, Finelli L, Olsen SJ, Fry AM. Morbid obesity as a risk factor for hospitalization and death due to 2009 pandemic influenza A(H1N1) disease. PLoS One. 2010;5:e9694. doi: 10.1371/journal.pone.0009694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Louie JK, Acosta M, Samuel MC, Schechter R, Vugia DJ, Harriman K, Matyas BT California Pandemic Working, G. A novel risk factor for a novel virus: obesity and 2009 pandemic influenza A (H1N1) Clin Infect Dis. 2011;52:301–312. doi: 10.1093/cid/ciq152. [DOI] [PubMed] [Google Scholar]
- 101.Choban PS, Flancbaum L. The impact of obesity on surgical outcomes: a review. J Am Coll Surg. 1997;185:593–603. doi: 10.1016/s1072-7515(97)00109-9. [DOI] [PubMed] [Google Scholar]
- 102.Arslan E, Atilgan H, Yavasoglu I. The prevalence of Helicobacter pylori in obese subjects. Eur J Intern Med. 2009;20:695–697. doi: 10.1016/j.ejim.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 103.Uberos J, Molina-Carballo A, Fernandez-Puentes V, Rodriguez-Belmonte R, Munoz-Hoyos A. Overweight and obesity as risk factors for the asymptomatic carrier state of Neisseria meningitidis among a paediatric population. Eur J Clin Microbiol Infect Dis. 2010;29:333–334. doi: 10.1007/s10096-009-0849-7. [DOI] [PubMed] [Google Scholar]
- 104.Gil A, Maria Aguilera C, Gil-Campos M, Canete R. Altered signalling and gene expression associated with the immune system and the inflammatory response in obesity. Br J Nutr. 2007;98(Suppl 1):S121–126. doi: 10.1017/S0007114507838050. [DOI] [PubMed] [Google Scholar]
- 105.McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. 2014;41:36–48. doi: 10.1016/j.immuni.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 106.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T, Nagai R. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 107.Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, Dorfman R, Wang Y, Zielenski J, Mastronardi F, Maezawa Y, Drucker DJ, Engleman E, Winer D, Dosch HM. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15:921–929. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang H, Youm YH, Vandanmagsar B, Ravussin A, Gimble JM, Greenway F, Stephens JM, Mynatt RL, Dixit VD. Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: implications for systemic inflammation and insulin resistance. J Immunol. 2010;185:1836–1845. doi: 10.4049/jimmunol.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. 1983;158:670–689. doi: 10.1084/jem.158.3.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 111.Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, Libby P. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res. 2008;103:467–476. doi: 10.1161/CIRCRESAHA.108.177105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.O’Rourke RW, White AE, Metcalf MD, Winters BR, Diggs BS, Zhu X, Marks DL. Systemic inflammation and insulin sensitivity in obese IFN-gamma knockout mice. Metabolism. 2012;61:1152–1161. doi: 10.1016/j.metabol.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Khan IM, Dai Perrard XY, Perrard JL, Mansoori A, Smith CW, Wu H, Ballantyne CM. Attenuated adipose tissue and skeletal muscle inflammation in obese mice with combined CD4+ and CD8+ T cell deficiency. Atherosclerosis. 2014;233:419–428. doi: 10.1016/j.atherosclerosis.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stolarczyk E, Vong CT, Perucha E, Jackson I, Cawthorne MA, Wargent ET, Powell N, Canavan JB, Lord GM, Howard JK. Improved insulin sensitivity despite increased visceral adiposity in mice deficient for the immune cell transcription factor T-bet. Cell Metab. 2013;17:520–533. doi: 10.1016/j.cmet.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, Leong HX, Glassford A, Caimol M, Kenkel JA, Tedder TF, McLaughlin T, Miklos DB, Dosch HM, Engleman EG. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17:610–617. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.DeFuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, Markham D, Strissel KJ, Watkins AA, Zhu M, Allen J, Bouchard J, Toraldo G, Jasuja R, Obin MS, McDonnell ME, Apovian C, Denis GV, Nikolajczyk BS. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci U S A. 2013;110:5133–5138. doi: 10.1073/pnas.1215840110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wagner NM, Brandhorst G, Czepluch F, Lankeit M, Eberle C, Herzberg S, Faustin V, Riggert J, Oellerich M, Hasenfuss G, Konstantinides S, Schafer K. Circulating regulatory T cells are reduced in obesity and may identify subjects at increased metabolic and cardiovascular risk. Obesity (Silver Spring) 2013;21:461–468. doi: 10.1002/oby.20087. [DOI] [PubMed] [Google Scholar]
- 118.Bapat SP, Myoung Suh J, Fang S, Liu S, Zhang Y, Cheng A, Zhou C, Liang Y, LeBlanc M, Liddle C, Atkins AR, Yu RT, Downes M, Evans RM, Zheng Y. Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature. 2015;528:137–141. doi: 10.1038/nature16151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Han JM, Patterson SJ, Speck M, Ehses JA, Levings MK. Insulin inhibits IL-10-mediated regulatory T cell function: implications for obesity. J Immunol. 2014;192:623–629. doi: 10.4049/jimmunol.1302181. [DOI] [PubMed] [Google Scholar]
- 120.Bergman RN, Ader M. Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol Metab. 2000;11:351–356. doi: 10.1016/s1043-2760(00)00323-4. [DOI] [PubMed] [Google Scholar]
- 121.Hubler MJ, Kennedy AJ. Role of lipids in the metabolism and activation of immune cells. J Nutr Biochem. 2016;34:1–7. doi: 10.1016/j.jnutbio.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ioan-Facsinay A, Kwekkeboom JC, Westhoff S, Giera M, Rombouts Y, van Harmelen V, Huizinga TW, Deelder A, Kloppenburg M, Toes RE. Adipocyte-derived lipids modulate CD4+ T-cell function. Eur J Immunol. 2013;43:1578–1587. doi: 10.1002/eji.201243096. [DOI] [PubMed] [Google Scholar]
- 123.Stentz FB, Kitabchi AE. Palmitic acid-induced activation of human T-lymphocytes and aortic endothelial cells with production of insulin receptors, reactive oxygen species, cytokines, and lipid peroxidation. Biochem Biophys Res Commun. 2006;346:721–726. doi: 10.1016/j.bbrc.2006.05.159. [DOI] [PubMed] [Google Scholar]
- 124.Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, Kim CH. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 2015;8:80–93. doi: 10.1038/mi.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 126.Ohmori R, Momiyama Y, Kato R, Taniguchi H, Ogura M, Ayaori M, Nakamura H, Ohsuzu F. Associations between serum resistin levels and insulin resistance, inflammation, and coronary artery disease. J Am Coll Cardiol. 2005;46:379–380. doi: 10.1016/j.jacc.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 127.Senolt L, Housa D, Vernerova Z, Jirasek T, Svobodova R, Veigl D, Anderlova K, Muller-Ladner U, Pavelka K, Haluzik M. Resistin in rheumatoid arthritis synovial tissue, synovial fluid and serum. Ann Rheum Dis. 2007;66:458–463. doi: 10.1136/ard.2006.054734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gauthier MS, O’Brien EL, Bigornia S, Mott M, Cacicedo JM, Xu XJ, Gokce N, Apovian C, Ruderman N. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun. 2011;404:382–387. doi: 10.1016/j.bbrc.2010.11.127. [DOI] [PMC free article] [PubMed] [Google Scholar]