

Abstract
Salivary duct carcinoma (SDC) is an aggressive adenocarcinoma of the salivary glands associated with poor clinical outcome. SDCs are known to carry TP53 mutations in about 50%, however, only little is known about alternative pathogenic mechanisms within the p53 regulatory network. Particularly, data on alterations of the oncogenes MDM2 and CDK4 located in the chromosomal region 12q13-15 are limited in SDC, while genomic rearrangements of the adjacent HMGA2 gene locus are well documented in subsets of SDCs. We here analyzed the mutational status of the TP53 gene, genomic amplification of MDM2, CDK4 and HMGA2 rearrangement/amplification as well as protein expression of TP53 (p53), MDM2 and CDK4 in 51 de novo and ex pleomorphic adenoma SDCs.
25 of 51 cases were found to carry TP53 mutations, associated with extreme positive immunohistochemical p53 staining levels in 13 cases. Three out of 51 tumors had an MDM2 amplification, one of them coinciding with a CDK4 amplification and two with a HMGA2 rearrangement/amplification. Two of the MDM2 amplifications occurred in the setting of a TP53 mutation. Two out of 51 cases showed a CDK4 amplification, one synchronously being MDM2 amplified and the other one displaying concurrent low copy number increases of both, MDM2 and HMGA2.
In summary, we here show that subgroups of SDCs display genomic amplifications of MDM2 and/or CDK4, partly in association with TP53 mutations and rearrangement/amplification of HMGA2. Further research is necessary to clarify the role of chromosomal region 12q13-15 alterations in SDC tumorigenesis and their potential prognostic and therapeutic relevance.
Keywords: salivary gland carcinoma, p53, MDM2, CDK4, HMGA2
INTRODUCTION
Salivary duct carcinoma (SDC) is an aggressive adenocarcinoma of the salivary glands, most commonly involving the parotid gland. It is one of the most aggressive salivary gland malignancies, most frequently associated with the occurrence of early distant metastasis and poor prognosis [1]. In the past few years increasing knowledge on recurrent genetic alterations of SDC evolved. In recent studies high percentages of TP53 mutations were detected, involving around 50% of cases of de novo and ex pleomorphic adenoma SDCs [2, 3]. However, there is a large subset of TP53 wildtype (WT) tumors which might harbor alternative alterations in the p53 regulatory network.
For decades, p53 has been a well-known tumor suppressor that is mutated or functionally inactivated in large subsets of human cancers [4]. Physiologically, transcriptional activity and stability of p53 are negatively regulated by the ubiquitin ligase MDM2, involving at least two mechanisms: a) direct blockage of the p53 transactivation domain and b) ubiquitylation-induced proteasomal degradation. Overexpression of MDM2, as found in many human tumors, is therefore capable to functionally impair p53. Inhibition of the MDM2-p53 interaction may therefore restore p53 activity and might offer opportunities for a targeted cancer therapy in tumors characterized by MDM2 overexpression [5]. In this setting, a study performed on MDM2-amplified well-differentiated or dedifferentiated liposarcomas (LS), which are consistently characterized by a high-level genomic amplification (frequently >15-30 copies as clusters) of sequences derived from chromosomal region 12q13-15 comprising the oncogenes MDM2 and CDK4, showed that treatment with the MDM2 antagonist RG7112 activates the p53 pathway and decreases cell proliferation [6]. Most frequently, MDM2 protein deregulation occurs in tumors that retain wildtype TP53, but MDM2 alterations have also been described in subsets of TP53 mutated tumors [4, 6]. It has been shown before that genomic instability affecting the chromosomal region 12q13-15 occurs in subsets of salivary gland carcinomas [7]. Apart from these findings, results from MDM2 transgenic mice developing mammary gland tumors suggest a crucial role for MDM2 in epithelial tumors of glandular differentiation [8].
In LS, the 12q13-15 amplicon usually, but not always, shows a co-amplification of the cell cycle regulator CDK4 together with MDM2 [9]. It has been shown that the small subgroup of MDM2+/CDK4- LS shows favorable prognostic features compared to MDM2+/CDK4+ LS [10]. Knowledge on the CDK4 amplification status therefore provides genomic information on the structural characteristics of the amplicon with MDM2 being located at 12q15 and CDK4 at 12q13.3-12 and it might add further information on an independent oncogenic mechanism apart from p53 dysfunction. Since CDK4 is the key regulator of the G1-S cell-cycle transition and drives cell-cycle progression, CDK4 inhibitors might offer new strategies for a targeted cancer therapy [11, 12].
Another gene in chromosomal region 12q13-15 frequently subject to structural alterations is HMGA2, encoding a high-mobility group protein. Chromosomal breaks of the HMGA2 locus have been described in several benign mesenchymal tumors including lipomas and uterine leiomyomas [13, 14], and HMGA2 amplification was shown in several soft tissue malignancies including liposarcomas where it is almost always co-amplified with MDM2 [9, 15]. In salivary gland tumors, rearrangements/amplifications of HMGA2 are well known in subsets of pleomorphic adenomas (PA) [14, 16]. Carcinomas ex PA have been reported to generally retain HMGA2 rearrangements along with further gene alterations in tumor progression making HMGA2 a potential marker for SDCs arising in PA [16].
Only very little is known about the role of MDM2 or CDK4 in SDC tumorigenesis. The major aim of this study therefore was to systematically evaluate the involvement of MDM2 and CDK4 alterations in SDC and to put them in context with HMGA2 alterations known in SDC. We here report on the rare occurrence of MDM2 and CDK4 amplifications in a large collection of these aggressive salivary neoplasms showing a heterogeneous distribution among TP53 wildtype tumors and those carrying a TP53 mutation.
RESULTS
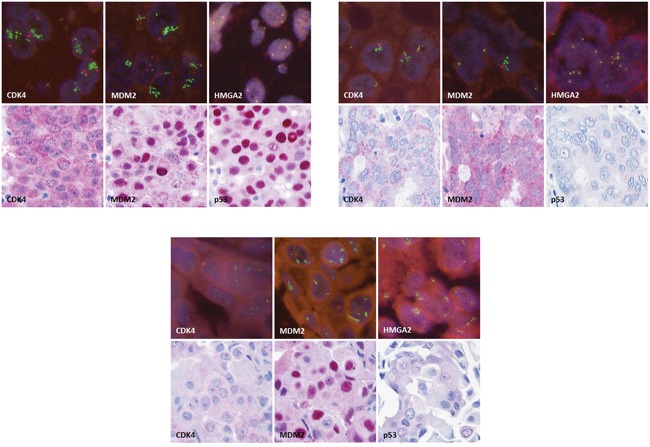
51 SDC cases were analyzed for TP53 mutational status, MDM2, CDK4 and HMGA2 genomic amplification as well as HMGA2 rearrangement and p53, MDM2 and CDK4 protein expression. The clinicopathological characteristics of these 51 patients are summarized in Table 1, and the results of the mutation screen, FISH and immunohistochemical analyses are displayed in Figure 1. In Figure 2, images of immunohistochemical stainings and FISH analyses are shown exemplarily for cases M117 (Figure 2A), K210 (Figure 2B) and M52 (Figure 2C).
Table 1. Clinical data of the patients included in the study.
| Patients' characteristics | N (%) |
|---|---|
| Patients | 51 |
| Male | 38 (74.5%) |
| Female | 13 (25.5%) |
| Age (years) | |
| Mean ± SD | 66.2 ± 13.0 |
| Median | 68 |
| Minimum/Maximum | 36/90 |
| Histology | |
| SDC de novo | 39 (76.5%) |
| SDC ex pleomorphic adenoma | 12 (23.5%) |
| Resection margins | |
| R0 | 25 (49.0%) |
| R1 | 17 (33.3%) |
| R2 | 1 (2.0%) |
| Rx | 8 (15.7%) |
| pT-stage | |
| pTx | 1 (2.0%) |
| pT1 | 12 (23.5%) |
| pT2 | 5 (9.8%) |
| pT3 | 16 (31.4%) |
| pT4a | 16 (31.4%) |
| pT4b | 1 (2.0%) |
| pN-stage | |
| pNx | 2 (3.9%) |
| pN0 | 10 (19.6%) |
| pN1 | 8 (15.7%) |
| pN2 | 30 (58.8%) |
| pN3 | 1 (2.0%) |
| Extracapsular spread | |
| Unknown | 23 (45.1%) |
| Yes | 15 (29.4%) |
| No | 13 (25.5%) |
| cM-stage | |
| cMx | 12 (23.6%) |
| cM0 | 30 (58.8%) |
| cM1 | 9 (17.6%) |
| Lymphangiosis | |
| Unknown | 15 (29.4%) |
| Yes | 22 (43.1%) |
| No | 14 (27.5%) |
| Hemangiosis | |
| Unknown | 18 (35.3%) |
| Yes | 14 (27.5%) |
| No | 19 (37.3%) |
| Perineural invasion | |
| Unknown | 19 (37.3%) |
| Yes | 22 (43.1%) |
| No | 10 (19.6%) |
| Type of parotidectomy | |
| Unknown | 1 (2.0%) |
| Lateral | 4 (7.8%) |
| Total | 24 (47.1%) |
| Radical | 20 (39.2%) |
| Subtotal | 2 (3.9%) |
| Neck dissection | |
| Yes | 49 (96.1%) |
| No | 2 (3.9%) |
Figure 1. Results of immunohistochemical stainings for p53, MDM2 and CDK4 and fluorescence in-situ hybridization analyses for MDM2, CDK4 and HMGA2, displayed for each case (PA: pleomorphic adenoma; n.a.: not available; EP: extreme positive; EN: extreme negative; NE: non-extreme, RA: rearrangement; ampl.: amplification).
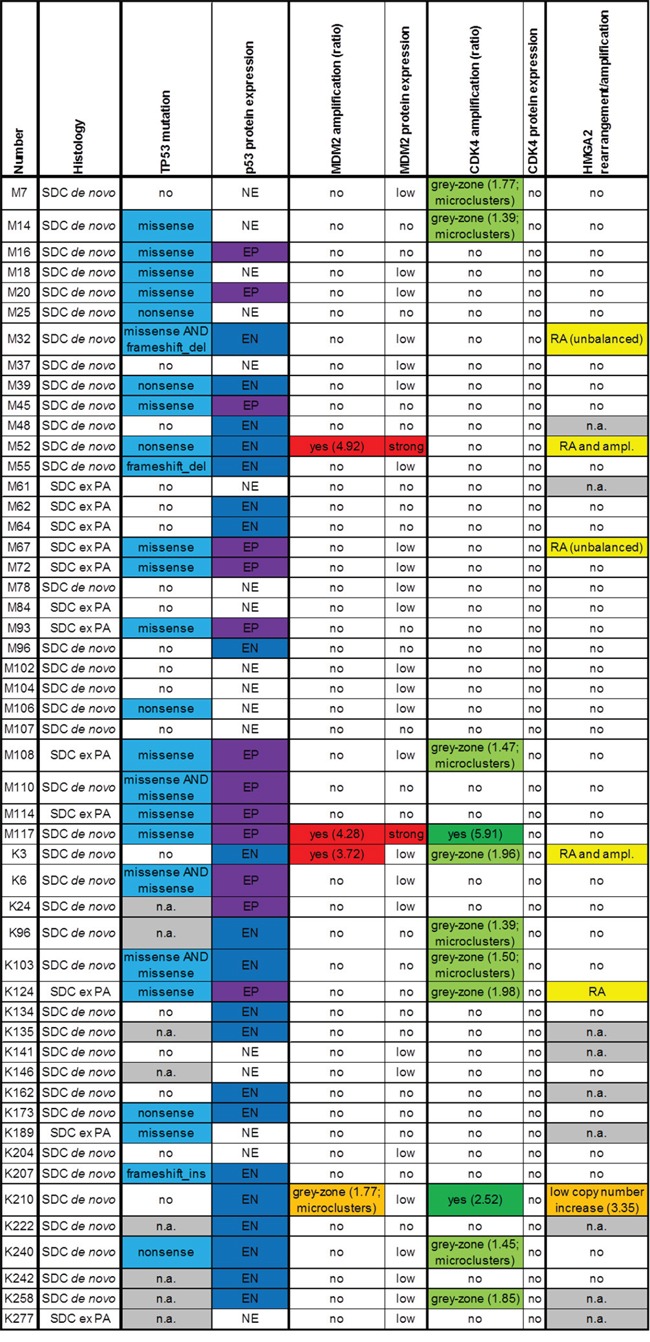
Figure 2. Immunohistochemical stainings for CDK4, MDM2 and p53 (original magnification 250x) and fluorescence in-situ hybridization for CDK4, MDM2 and HMGA2 (original magnification 630x), exemplarily for case M117 A., case K210 B. and case M52 C.
TP53 mutations were detected in 25 of these 51 cases (Table 2). In 8 cases, the TP53 mutational status could not be evaluated due to insufficient DNA content. Immunohistochemistry for p53 showed an extreme positive (EP) staining result in 13 cases, all of these cases displaying a TP53 missense point mutation except for one case in which the TP53 mutational status could not be evaluated due to minor DNA quality. In 8 cases with various TP53 mutations, an extreme negative (EN) p53 immunohistochemical staining was detected and 5 cases with TP53 mutations showed a non-extreme (NE) staining level for p53. An extreme negative (EN) p53 staining was detected in 8 cases without TP53 mutation and in 5 further cases in which TP53 mutational status could not be evaluated.
Table 2. List of TP53 mutations detected in the analysis of 51 SDCs.
| No. | TP53 mutation |
|---|---|
| M14 | Exon 8 p.Pro278Arg (c.833C>G) |
| M16 | Exon 5 p.Tyr163His (c.487T>C) |
| M18 | Exon 4 p.Pro89Leu (c.266C>T) |
| M20 | Exon 7 p.Gly245Ser (c.733G>A) |
| M25 | Exon 10 p.Arg342* (c.1024C>T) |
| M32 | Exon 4 p.Pro75Leu (c.224C>T); Exon 6 p.Arg209fs (c.626_627delGA) |
| M39 | Exon 6 p.Gln192* (c.574C>T) |
| M45 | Exon 5 p.Asn131Tyr (c.391A>T) |
| M52 | Exon 8 p.Arg306* (c.916C>T) |
| M55 | Exon 6 p.Arg209fs (c.626_627delGA) |
| M67 | Exon 8 p.Phe270Cys (c.809T>G) |
| M72 | Exon 5 p.Lys132Glu (c.394A>G) |
| M93 | Exon 6 p.Tyr220Cys (c.659A>G) |
| M106 | Exon 9 p.Gln331* (c.991C>T) |
| M108 | Exon 5 p.Arg175His (c.524G>A) |
| M110 | Exon 7 p.Ile232Met (c.696C>G); p.Ser241Phe (c722C>T) |
| M114 | Exon 5 p.His168Arg (c.503A>G) |
| M117 | Exon 5 p.Ser127Phe (c.380C>T) |
| K6 | Exon 7 p.Gly245Val (c.734G>T), Exon 10 p.Gly360Ala (c.1079G>C) |
| K103 | Exon 8 p.Arg273Leu (c.818G>T), p.Phe270Ser (c.809T>C) |
| K124 | Exon 7 p.Asn239Ser (c.716A>G) |
| K173 | Exon 4 p.Tyr103* (c.309C>A) |
| K189 | Exon 6 p.His214Arg (c.641A>G) |
| K207 | Exon 8 p.Val274fs (c.819_820insT) |
| K240 | Exon 4 p.Gln100* (c.298C>T) |
Three out of the total 51 cases displayed an MDM2 amplification, one case showed an MDM2 grey-zone amplification in FISH analyses. Two cases with MDM2 amplification strongly stained for MDM2, one MDM2 amplified case and the grey-zone amplified case showed low MDM2 immunohistochemical staining levels. Two of the MDM2 amplified cases synchronously carried TP53 mutations. All MDM2 amplifications and the MDM2 grey-zone amplification occurred in de novo SDCs, none was detected in an SDC ex pleomorphic adenoma.
In two cases, we detected a CDK4 amplification, one coinciding with an MDM2 amplification and one in the setting of an MDM2 grey-zone amplification. Nine cases showed a CDK4 grey-zone amplification, one of these cases in the setting of an MDM2 amplification. However, the immunohistochemical CDK4 staining was negative in all cases.
HMGA2 FISH could be analyzed in 42 out of 51 cases. 5 cases showed rearrangement and/or amplification of HMGA2, among these 3 de novo and 2 ex PA SDCs. Two of the 5 cases displayed HMGA2 rearrangement and amplification, both occurring in association with an MDM2 amplification; the third MDM2 (and CDK4) amplified case displayed no HMGA2 alteration. One of the HMGA2 rearrangements occurred in the setting of a CDK4 grey-zone amplification, the two further HMGA2 rearrangements were detected in cases without MDM2 and/or CDK4 amplification. One CDK4 amplified tumor with grey-zone MDM2 amplification showed only a low HMGA2 copy number increase.
The 5-year overall survival (OS) in our collection of SDCs was 39.6% (Figure 3A). TP53 mutated cases showed a tendency to a worse 5-year OS (20.5% with TP53 mutation vs. 53.3% without TP53 mutation) but statistical significance was not reached (p=0.267) (Figure 3B). No statistically significant differences in 5-year OS were detected for MDM2 and/or CDK4 amplified/grey-zone amplified subgroups.
Figure 3.
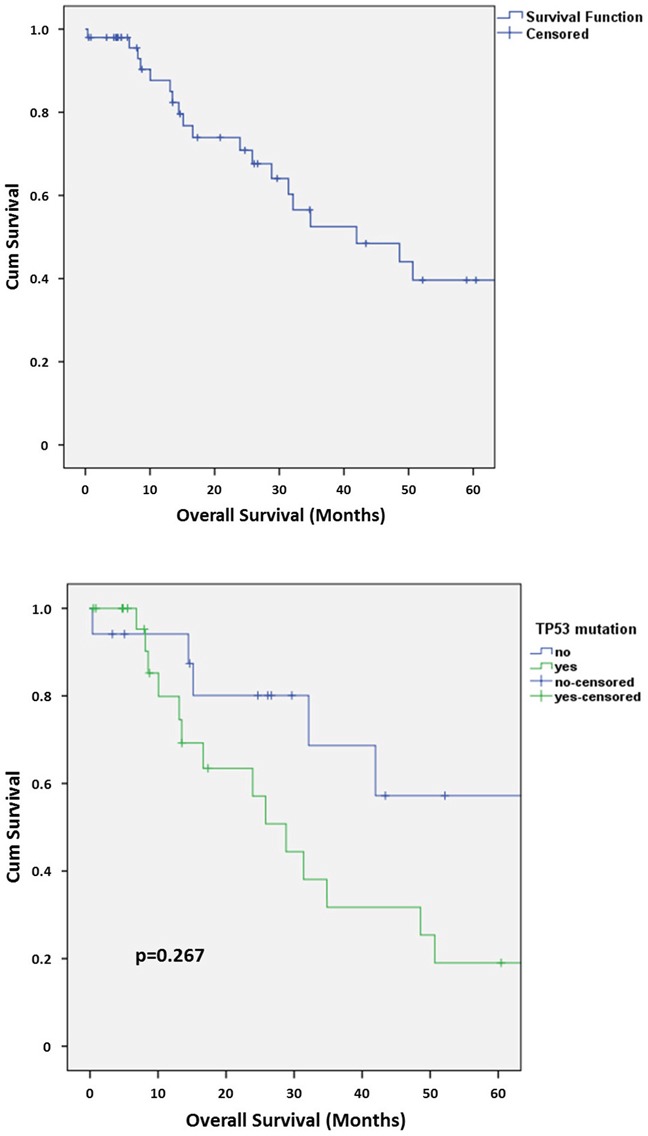
A. Kaplan-Meier chart of overall survival in the collection of SDCs. B. Kaplan-Meier chart of overall survival in the collection of SDCs depending on the presence or absence of a TP53 mutation.
DISCUSSION
Recent genomic profiling studies confirmed a high frequency of TP53 mutations in de novo and ex PA SDCs [2, 3], affecting about 50% of the tumors, pointing to an outstanding relevance of the p53 tumor suppressing network in the pathogenesis of these rare but highly aggressive tumors. With particular regard to the remaining 50% of SDCs harboring wildtype sequences for TP53, the major aim of this study was to elucidate the role of another key player within the p53 regulatory network, i.e. the ubiquitin ligase MDM2, in SDC. Due to the co-localization of MDM2 with the cell cycle regulator CDK4 in the chromosomal region 12q13-15 and a documented proneness of this region to genomic alterations, these regulators of cell growth and fate are frequently co-amplified in some human malignancies including a subset of soft tissue tumors [9]. Apart from providing details on the characteristics of the amplicon, knowledge of the CDK4 amplification status might add further information on an independent oncogenic mechanism apart from p53 dysfunction, opening therapeutic options. Involvement of the chromosomal region 12q13-15 in salivary gland tumorigenesis has been documented before with the HMGA2 gene showing rearrangements and genomic amplification in subsets of PA and SDCs ex PA [16]. Since HMGA2 furthermore represents an almost constant partner of MDM2 in the 12q13-15 amplicon known in other tumors, we complemented our analysis with the analysis of HMGA2 to get further insight into the structure of the amplicon [9].
We detected MDM2 amplifications in 3 cases, interestingly all classified as de novo SDCs. Two of these MDM2 amplified cases showed a concurrent rearrangement/co-amplification of HMGA2. According to Bahrami et al. [16], who proposed alterations of HMGA2 as a marker for SDCs ex PA, it could be hypothesized if these two cases might also have arisen in totally obscured PA, however, there was no evidence for a pre-existing PA, neither by histopathology nor with respect to clinical history. One of the MDM2 amplified SDCs harbored no HMGA2 alteration while 3 SDCs with HMGA2 rearrangement did not show an MDM2 amplification, implying that alterations of these two genes are obviously not strictly connected in SDCs. Thus, MDM2 amplification appears to be a genomic alteration at least partly independent from HMGA2 status and particularly from pathogenesis (de novo vs. ex PA) in SDC. Since only two of the cases harboring an MDM2 amplification revealed a strong immunohistochemical staining for MDM2, immunohistochemical staining alone appears not to be a reliable marker of MDM2 amplification in SDC. Interestingly, two of the MDM2 amplified cases synchronously carried TP53 mutations, while one MDM2 amplification occurred in a TP53-wildtype setting. In another TP53-wildtype tumor a grey-zone MDM2 amplification was detected. A very recent study on 37 SDCs revealed one MDM2 amplified case, also in the setting of a TP53 mutation [2]. When focusing on the MDM2-p53 regulatory connection, mechanistically, tumors with functionally inactivating mutations in TP53 cannot be expected to substantially gain in oncogenic potential due to a concomitant amplification of MDM2 since it usually acts as an oncogenic factor by downregulation of wildtype p53 protein. In contrast, in tumors harboring particular TP53 mutations and retaining their wildtype TP53 allele, MDM2 amplification might contribute to oncogenesis by additionally disrupting the remaining functional p53 levels. Additionally, several reports argue in favor of an oncogenic role for MDM2 independently from the p53 status [8, 17, 18]. In any case, based on our findings, in a small subset of SDCs, MDM2 amplification obviously may serve as an alternative mechanism leading to dysregulation of the p53 network, thereby offering an option for a targeted MDM2-directed therapy [6]. With respect to documented activation steps of MDM2 through AKT1 in salivary acinar cells and mammary epithelium [19, 20], MDM2 amplification might represent an alternative or additional mechanism in deregulating the p53 network besides well-known and pathogenetically relevant genetic alterations of the PI3K/AKT signaling pathway in SDC [3].
The immunohistochemical staining for p53 partly correlated with the TP53 mutational status: With 48% of TP53 mutated cases and none of the TP53 wildtype cases showing an extreme positive (EP) staining for p53 protein expression, EP p53 staining reliably went along with a missense TP53 mutation. 32% of TP53 mutated cases showed an extreme negative (EN) p53 staining and 20% of TP53 mutated cases a non-extreme (NE) p53 staining. In contrast, cases without TP53 mutation displayed an EN p53 staining result in 44% and a NE staining in 56%. So, in summary, in case of an EN or a NE p53 staining, a reliable prediction of TP53 mutational status is not possible in SDCs whereas an EP p53 expression is strongly suggestive of an underlying TP53 missense mutation.
Interestingly, TP53 mutated cases showed a tendency to a worse 5-year OS (Figure 3B), pointing to a prognostic relevance of a TP53 mutation in SDC.
Out of the MDM2 amplified SDCs identified in this study, two tumors showed a concomitant CDK4 amplification, one a grey-zone CDK4 amplification and one lacked an amplification of CDK4. This finding is in agreement with genomic data from a subset of liposarcomas, which frequently show a variable amplification of sequences of the chromosomal region 12q13-15. Independently from a copy number change of MDM2, eight tumors showed a CDK4 grey-zone amplification (one coinciding with a HMGA2 rearrangement), pointing to a CDK4-mediated cell-cycle dysregulation in SDC tumorigenesis. CDK4 inhibitors might therefore offer new strategies for targeted therapeutic approaches in SDCs as recently discussed for several entities [11, 12]. Interestingly, none of the CDK4 amplified or grey-zone amplified cases showed a positive immunohistochemical staining for CDK4 (with solid staining result of the positive control), so immunohistochemical CDK4 staining obviously is not reliably suitable for the detection of CDK4 amplified SDC. Anyway, lack of immunohistochemical CDK4 protein detection does not exclude a pathogenic role of the CDK4 oncogene and may be due to tight protein regulation beyond the threshold of staining sensitivity. Contrasting with our immunohistochemical results and potentially pointing to different characteristics of diagnostically employed antibodies, a previously reported study on five SDC cases demonstrated CDK4 protein expression in four of five cases, accompanied by MDM2 protein expression in two cases; however, CDK4 and MDM2 amplification status was not determined in that study [21].
As described above, in a subset of liposarcomas the small subgroup of MDM2+/CDK4- tumors shows favorable prognostic features compared to MDM2+/CDK4+ tumors [10]. In our collection of SDCs the 5-year OS was 39.6%. We could not detect any statistically significant differences of the 5-year OS in the MDM2 and/or CDK4 amplified/grey-zone amplified subgroups, which is probably due to the limited case numbers contained in the smaller subgroups. Larger cohorts of these aggressive tumors should be analyzed for a better understanding of a potential prognostic impact of MDM2 and/or CDK4 alterations.
In this study, we investigated for the first time systematically the involvement of MDM2 and CDK4 in the carcinogenesis of SDCs by analyzing the TP53 mutational status, MDM2 and CDK4 amplification and HMGA2 rearrangement/amplification as well as protein expression of p53, MDM2 and CDK4. We showed that in subgroups of SDCs, MDM2 and/or CDK4 amplification might play a pathogenic role, in part apparently in association with other genetic alterations. Further work is mandatory to clarify the role of MDM2 and CDK4 alterations in the tumorigenesis of SDCs (especially in the TP53 mutated subgroup), the potential prognostic relevance of these alterations in SDCs and the feasibility of MDM2- and/or CDK4-directed therapeutic strategies in SDCs.
MATERIALS AND METHODS
Patient data and specimens
The investigation was conducted according to the Declaration of Helsinki on biomedical research involving human subjects. The retrospective study included 51 patients with newly diagnosed salivary duct carcinoma. Among these, 30 cases were derived from the University Hospital of Muenster and 21 cases from the University Hospital of Cologne (the latter having partly been included in a previous study [3]). Patients from Cologne have been treated at the Department of Otorhinolaryngology, Head and Neck Surgery at the University Hospital of Cologne between 1998 and 2011, patients from Muenster have been treated at the Department of Otorhinolaryngology, Head and Neck Surgery at the University Hospital of Muenster between 2000 and 2014. All patients were subjected to primary definitive surgery and potential adjuvant radiation according to patients' cancer stage. Tumor staging was adapted to the 7th edition of the UICC TNM classification for carcinomas of the salivary glands. Patients were followed up at the outpatients department of Cologne or Muenster, respectively, at periodic visits in 3 to 6 months. At the time of analysis, 22 patients had died and 11 patients had developed a histologically confirmed relapse. Mean follow-up time was 22.2 months (range 0 to 161). The study was approved by the local Ethics Committees.
Formalin-fixed and paraffin-embedded (FFPE) material of the patients was obtained from the archives of the Departments of Pathology at the University Hospitals of Cologne and Muenster, respectively. All tumors were re-evaluated microscopically and by means of immunohistochemistry by two experienced pathologists with regard to histopathological diagnosis in accordance with WHO 2005 classification of tumors of salivary glands.
From FFPE material of all included cases, two core biopsies out of the tumor area were taken to assemble tissue microarrays (TMA).
For statistical analysis, the IBM SPSS Statistics 22 software (SPSS Inc., Chicago, IL, USA) was applied and Kaplan-Meier survival analysis and Log rank test were performed. The significance level was set at p<0.05.
FISH analyses and immunohistochemistry
Fluorescence in-situ hybridization (FISH) analyses and immunohistochemical stainings were conducted on slides from TMAs. FISH analyses were performed as described previously [22, 23] using the ZytoLight® SPEC MDM2/CEN 12 Dual Color Probe for assessment of MDM2 amplification and the ZytoLight® SPEC CDK4/CEN 12 Dual Color Probe for assessment of CDK4 amplification (ZytoVision GmbH, Bremerhaven, Germany). HMGA2 FISH analysis was performed according to a previously published assay [16, 24] using BACs RP11-662G15 and RP11-1025D9 (Life Technologies by Thermo Fisher Scientific Inc., Waltham, USA). At least 40-60 tumor cell nuclei of each tumor sample were analyzed. Amplification of MDM2 and CDK4 was defined as an MDM2/centromer 12 (CEN12) or CDK4/centromer 12 (CEN12) ratio ≥2.0 or an average number of MDM2 or CDK4 signals per tumor cell nucleus ≥6 or large clusters of MDM2 or CDK4 signals ≥10%, respectively. Grey-zone amplification was defined as an MDM2/centromer 12 (CEN12) or CDK4/centromer 12 (CEN12) ratio ≥1.8 or as microclusters of ≥5 MDM2 or CDK4 signals in ≥15% of tumor cell nuclei, respectively, based on modified scoring algorithms for HER2 and FGFR1 as published before [23, 25]. For HMGA2 rearrangement and amplification were evaluated as published before [16].
Immunohistochemical staining was conducted on a Dako Autostainer (Dako Deutschland GmbH, Hamburg, Germany) following the manufacturer's instructions. Three μm sections were cut from the TMAs, followed by heat induced antigen retrieval in low (for p53) or high (for MDM2 and CDK4) pH buffer. For visualization LSAB method with AP/RED was used. Following antibodies and concentrations were used: p53 (1:3000, clone DO-7, Dako), MDM2 (1:100, clone IF2, Invitrogen by Thermo Fisher Scientific Inc., Waltham, USA) and CDK4 (1:100, clone DCS-31, Invitrogen). Sections were counterstained with hematoxylin and tiled with Cytoseal (Thermo Fisher Scientific Inc.). For scoring of the immunohistochemical stainings for p53, MDM2 and CDK4 only nuclear staining was rated. The staining intensity was evaluated semiquantitavely into 0, 1, 2 or 3 by comparing within the different tumor samples. To determine percentage labelling indices, all tumor cells within the cores were analyzed using high-power (400x) magnification. A sum score was calculated out of the staining intensity and the percentage labelling index, and for MDM2 and CDK4 value ranges for staining level were defined as follows: 0: no staining; >0 to 50: low staining level; >50 to 100: intermediate staining level; >100: strong staining. Sum score of p53 staining was interpreted modified according to Boyle et al. [26] as follows: 0: extreme negative (EN); >50: extreme positive (EP); >0 to 50: non-extreme (NE, intermediate patterns).
Assessment of TP53 mutational status
To complete the data on TP53 mutational status as previously described for a smaller subset of samples [3] for the whole cohort, assessment of TP53 mutational status was carried out as follows:
Tumor macrodissection, DNA extraction and quantification
Sections were prepared from FFPE material and stained with hematoxylin & eosin (H&E). Six additional sections of 6 μm thickness were cut, mounted onto glass slides and used for macrodissection. In total, 1 cm2 tumor area corresponding to the tumor area of H&E-stained section was scraped off with a scalpel and collected into plastic tubes. Subsequently, the DNA was automatically extracted using the Maxwell DNA FFPE isolation kit on a Maxwell platform (Promega GmbH, Mannheim, Germany). Fluorometric DNA quantification was performed according to the Qubit dsDNA HS assay (Qubit 2.0, Life Technologies, Carlsbad, CA, USA).
NGS library construction
Pre-verified multiplex PCR primer sets (summarized in Supporting Information Supplemental Table 1 A) were used to amplify the exonic region of TP53 (customized GeneRead DNAseq Mix-n-Match V2 panel, Qiagen GmbH, Hilden, Germany). Target enrichment was processed by means of the GeneRead DNAseq Panel PCR V2 Kit (Qiagen), following the manufacturer's instructions. All purification and size selection steps were performed utilizing Agencourt AMPure XP magnetic beads (Beckman Coulter, Inc., Brea, CA, USA). End repair, A-addition and ligation to NEXTflex-96 DNA barcodes (Bioo Scientific, Austin, Texas, USA) was carried out using the GeneRead DNA Library I Core Kit (Qiagen). Amplification of adapter-ligated DNA was conducted using NEXTflex primers (Bioo Scientific) and the HiFi PCR Master Mix (GeneRead DNA I Amp Kit, Qiagen). Next generation sequencing was performed applying 12.5 pM library pools (2% PhiX V3 control) and the MiSeq Reagent v2 chemistry (Illumina, Inc., San Diego, Ca, USA).
NGS data analysis
Fastq files were generated by the MiSeq Reporter software (Illumina) and further analyzed by means of the CLC Biomedical Genomics Workbench software (CLC bio, Qiagen). The total batch of identified TP53 variants was filtered according to following criteria and then validated by Sanger sequencing: Hotspot artefacts and reading errors were filtered, which are recognized by high occurrence and constant frequency within the cohort. In addition, synonymous variants, germline single nucleotide polymorphisms (SNPs) and all variants below 4% allelic frequency were filtered, ending up with variants listed in Supporting Information Supplemental Table 2. Additional information on the functional impact of detected TP53 mutations is provided in Supporting Information Supplemental Table 3.
Sanger sequencing
Conventional Sanger sequencing was conducted according to standard procedures using the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific Inc.) and various TP53 primer sets (summarized in Supporting Information Supplemental Table 1 B).
SUPPLEMENTARY TABLES
Acknowledgments
There are no financial disclosures from any authors.
Footnotes
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest.
REFERENCES
- 1.Eveson JW, Auclair P, Gnepp DR, El-Naggar AK. Tumours of the Salivary glands. In: Barnes L, Eveson JW, Reichart P, Sidransky D, editors. World health organization classification of tumours Pathology and genetics Head and neck tumours. Lyon: IARC Press; 2005. pp. 209–81. [Google Scholar]
- 2.Chiosea SI, Williams L, Griffith CC, Thompson LD, Weinreb I, Bauman JE, Luvison A, Roy S, Seethala RR, Nikiforova MN. Molecular characterization of apocrine salivary duct carcinoma. The American Journal of Surgical Pathology. 2015;39:744–52. doi: 10.1097/PAS.0000000000000410. [DOI] [PubMed] [Google Scholar]
- 3.Grunewald I, Vollbrecht C, Meinrath J, Meyer MF, Heukamp LC, Drebber U, Quaas A, Beutner D, Huttenbrink KB, Wardelmann E, Hartmann W, Buttner R, Odenthal M, et al. Targeted next generation sequencing of parotid gland cancer uncovers genetic heterogeneity. Oncotarget. 2015;6:18224–37. doi: 10.18632/oncotarget.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nature reviews Cancer. 2013;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science (New York, NY) 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 6.Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, Geho D, Middleton SA, Vassilev LT, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. The Lancet Oncology. 2012;13:1133–40. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- 7.Persson F, Andren Y, Winnes M, Wedell B, Nordkvist A, Gudnadottir G, Dahlenfors R, Sjogren H, Mark J, Stenman G. High-resolution genomic profiling of adenomas and carcinomas of the salivary glands reveals amplification, rearrangement, and fusion of HMGA2. Genes, chromosomes & cancer. 2009;48:69–82. doi: 10.1002/gcc.20619. [DOI] [PubMed] [Google Scholar]
- 8.Lundgren K, Montes de Oca Luna R, McNeill YB, Emerick EP, Spencer B, Barfield CR, Lozano G, Rosenberg MP, Finlay CA. Targeted expression of MDM2 uncouples S phase from mitosis and inhibits mammary gland development independent of p53. Genes & development. 1997;11:714–25. doi: 10.1101/gad.11.6.714. [DOI] [PubMed] [Google Scholar]
- 9.Italiano A, Bianchini L, Keslair F, Bonnafous S, Cardot-Leccia N, Coindre JM, Dumollard JM, Hofman P, Leroux A, Mainguene C, Peyrottes I, Ranchere-Vince D, Terrier P, et al. HMGA2 is the partner of MDM2 in well-differentiated and dedifferentiated liposarcomas whereas CDK4 belongs to a distinct inconsistent amplicon. International journal of cancer. 2008;122:2233–41. doi: 10.1002/ijc.23380. [DOI] [PubMed] [Google Scholar]
- 10.Italiano A, Bianchini L, Gjernes E, Keslair F, Ranchere-Vince D, Dumollard JM, Haudebourg J, Leroux A, Mainguene C, Terrier P, Chibon F, Coindre JM, Pedeutour F. Clinical and biological significance of CDK4 amplification in well-differentiated and dedifferentiated liposarcomas. Clinical cancer research. 2009;15:5696–703. doi: 10.1158/1078-0432.CCR-08-3185. [DOI] [PubMed] [Google Scholar]
- 11.Dickson MA. Molecular pathways: CDK4 inhibitors for cancer therapy. Clinical cancer research. 2014;20:3379–83. doi: 10.1158/1078-0432.CCR-13-1551. [DOI] [PubMed] [Google Scholar]
- 12.Leontieva OV, Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle. 2013;12:3063–9. doi: 10.4161/cc.26130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashar HR, Fejzo MS, Tkachenko A, Zhou X, Fletcher JA, Weremowicz S, Morton CC, Chada K. Disruption of the architectural factor HMGI-C: DNA-binding AT hook motifs fused in lipomas to distinct transcriptional regulatory domains. Cell. 1995;82:57–65. doi: 10.1016/0092-8674(95)90052-7. [DOI] [PubMed] [Google Scholar]
- 14.Schoenmakers EF, Wanschura S, Mols R, Bullerdiek J, Van den Berghe H, Van de Ven WJ. Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nat Genet. 1995;10:436–44. doi: 10.1038/ng0895-436. [DOI] [PubMed] [Google Scholar]
- 15.Berner JM, Meza-Zepeda LA, Kools PF, Forus A, Schoenmakers EF, Van de Ven WJ, Fodstad O, Myklebost O. HMGIC, the gene for an architectural transcription factor, is amplified and rearranged in a subset of human sarcomas. Oncogene. 1997;14:2935–41. doi: 10.1038/sj.onc.1201135. [DOI] [PubMed] [Google Scholar]
- 16.Bahrami A, Perez-Ordonez B, Dalton JD, Weinreb I. An analysis of PLAG1 and HMGA2 rearrangements in salivary duct carcinoma and examination of the role of precursor lesions. Histopathology. 2013;63:250–62. doi: 10.1111/his.12152. [DOI] [PubMed] [Google Scholar]
- 17.Alkhalaf M, Ganguli G, Messaddeq N, Le Meur M, Wasylyk B. MDM2 overexpression generates a skin phenotype in both wild type and p53 null mice. Oncogene. 1999;18:1419–34. doi: 10.1038/sj.onc.1202448. [DOI] [PubMed] [Google Scholar]
- 18.Jones SN, Hancock AR, Vogel H, Donehower LA, Bradley A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:15608–12. doi: 10.1073/pnas.95.26.15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng X, Xia W, Yang JY, Hsu JL, Lang JY, Chou CK, Du Y, Sun HL, Wyszomierski SL, Mills GB, Muller WJ, Yu D, Hung MC. Activation of murine double minute 2 by Akt in mammary epithelium delays mammary involution and accelerates mammary tumorigenesis. Cancer research. 2010;70:7684–9. doi: 10.1158/0008-5472.CAN-09-3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Limesand KH, Schwertfeger KL, Anderson SM. MDM2 is required for suppression of apoptosis by activated Akt1 in salivary acinar cells. Molecular and cellular biology. 2006;26:8840–56. doi: 10.1128/MCB.01846-05. doi: MCB.01846-05 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Etges A, Pinto DS, Jr, Kowalski LP, Soares FA, Araujo VC. Salivary duct carcinoma: immunohistochemical profile of an aggressive salivary gland tumour. Journal of clinical pathology. 2003;56:914–8. doi: 10.1136/jcp.56.12.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedrichs N, Kriegl L, Poremba C, Schaefer KL, Gabbert HE, Shimomura A, Paggen E, Merkelbach-Bruse S, Buettner R. Pitfalls in the detection of t(11;22) translocation by fluorescence in situ hybridization and RT-PCR: a single-blinded study. Diagnostic molecular pathology: the American journal of surgical pathology, part B. 2006;15:83–9. doi: 10.1097/00019606-200606000-00004. doi: 00019606-200606000-00004 [pii] [DOI] [PubMed] [Google Scholar]
- 23.Schildhaus HU, Heukamp LC, Merkelbach-Bruse S, Riesner K, Schmitz K, Binot E, Paggen E, Albus K, Schulte W, Ko YD, Schlesinger A, Ansen S, Engel-Riedel W, et al. Definition of a fluorescence in-situ hybridization score identifies high- and low-level FGFR1 amplification types in squamous cell lung cancer. Modern pathology. 2012;25:1473–80. doi: 10.1038/modpathol.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bahrami A, Dalton JD, Shivakumar B, Krane JF. PLAG1 alteration in carcinoma ex pleomorphic adenoma: immunohistochemical and fluorescence in situ hybridization studies of 22 cases. Head Neck Pathol. 2012;6:328–35. doi: 10.1007/s12105-012-0353-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, McShane LM, Paik S, Pegram MD, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Journal of clinical oncology. 2007;25:118–45. doi: 10.1200/JCO.2006.09.2775. doi: JCO.2006.09.2775 [pii] [DOI] [PubMed] [Google Scholar]
- 26.Boyle DP, McArt DG, Irwin G, Wilhelm-Benartzi CS, Lioe TF, Sebastian E, McQuaid S, Hamilton PW, James JA, Mullan PB, Catherwood MA, Harkin DP, Salto-Tellez M. The prognostic significance of the aberrant extremes of p53 immunophenotypes in breast cancer. Histopathology. 2014;65:340–52. doi: 10.1111/his.12398. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.