

Abstract
Hypoxia is a poor-prognosis microenvironmental hallmark of solid tumours, but it is unclear how it influences the fate of disseminated tumour cells (DTCs) in target organs. Here we report that hypoxic HNSCC and breast primary tumour microenvironments displayed upregulation of key dormancy (NR2F1, DEC2, p27) and hypoxia (GLUT1, HIF1α) genes. Analysis of solitary DTCs in PDX and transgenic mice revealed that post-hypoxic DTCs were frequently NR2F1hi/DEC2hi/p27hi/TGFβ2hi and dormant. NR2F1 and HIF1α were required for p27 induction in post-hypoxic dormant DTCs, but these DTCs did not display GLUT1hi expression. Post-hypoxic DTCs evaded chemotherapy and, unlike ER− breast cancer cells, post-hypoxic ER+ breast cancer cells were more prone to enter NR2F1-dependent dormancy. We propose that primary tumour hypoxic microenvironments give rise to a subpopulation of dormant DTCs that evade therapy. These post-hypoxic dormant DTCs may be the source of disease relapse and poor prognosis associated with hypoxia.
Tumour cell heterogeneity is thought to play an important role in the design of successful therapies1,2. Primary tumours (PTs) contain proliferating, slow-cycling, quiescent or necrotic/apoptotic tumour cells, as well as fast- versus slow-migrating tumour cell populations3–6. Genetic and expression profiling has also revealed genetic heterogeneity in solitary disseminated tumour cells (DTCs) and metastasis7–9.
PT microenvironments are also heterogeneous in composition10,11, but how DTC phenotypic heterogeneity is influenced by PT microenvironments is least understood12. Gene signatures obtained from PTs predict for shorter or longer times to metastasis in patient cohorts13,14. This suggests that PT gene programs, and possibly influenced by the microenvironment, provide information on distinct long-term post-dissemination behaviour (that is, rate of relapse). Some PT gene signatures have been linked to late relapse (>5 years) and these were associated with EMT or dormancy14,15. We found that breast tumours that are enriched for a dormancy signature display longer metastasis-free periods compared with those lacking the signature14. Also DTCs from prostate cancer patients in clinical dormancy for 7–18 years were enriched for the dormancy signature9, which was also present in a subpopulation of prostate cancer DTCs from patients with metastases. Thus, proliferative DTCs and metastases can coexist with dormant DTCs9.
Analysis of the dormancy signature9,14,16 revealed genes differentially regulated by hypoxia10,17–23, an important solid PT microenvironmental feature, which can induce a strong growth arrest24,25. Further, dormant tumour cells were found to upregulate the unfolded protein response26–29, which is also activated by hypoxia30–33. We hypothesized that if hypoxic PT microenvironments spawn subpopulations of DTCs that were imprinted to become dormant, these might escape therapies and fuel incurable metastasis.
Here we show that PT tumour cells exposed to either natural hypoxic microenvironments or those induced using hypoxia-induction nanointravital devices (Hi-NANIVIDs) upregulate both hypoxia and dormancy genes in a reversible manner. Hypoxia-imprinted DTCs that lodged in lungs were more prone to enter an NR2F1-dependent dormant state and evaded chemotherapy. Interestingly, post-hypoxic dormant DTCs did not maintain high expression of hypoxia markers, arguing for a HIF1α-independent maintenance of the dormant phenotype. Further, we found that post-hypoxic ER+ luminal breast cancer cells are more prone to enter dormancy than post-hypoxic TNBC cells. Our work uncovers a previously unrecognized phenotypic diversity initiated in the primary site by hypoxia and carried over by DTCs in target organs. These studies might help explain how hypoxia might contribute to therapy resistance through the unanticipated generation of dormant or slow-cycling DTCs.
RESULTS
Endogenous tumour hypoxia upregulates dormancy markers
We first tested whether dormancy and hypoxia markers were co-expressed in PTs. To measure endogenous hypoxia and HIF1α activity we engineered MDA-MB-231 TNBC cells to express mCherry fused to the oxygen-dependent degradation (ODD) domain of HIF1α under the control of five HIF response elements (HRE) and a constitutively expressed GFP (MDA-MB-231-5HRE-ODD-mCherry/GFP, henceforth MDA-231-HIF reporter cells)34 (Fig. 1a–c). The MDA-231-HIF reporter cells (RED) showed areas where cells had been exposed to hypoxia. In contrast, cells that had not been exposed to significant hypoxia expressed only GFP. ODD-mCherry co-localization with GLUT1 in these microenvironments further supported that MDA-231-HIF reporter cells had sensed hypoxia (Fig. 1a and Supplementary Fig. 1A). Interestingly, DEC2 and NR2F1 (dormancy genes) were also significantly upregulated in hypoxic areas of the MDA-231-HIF tumours (Fig. 1b,c and Supplementary Fig. 1B,C). Quiescent tumour cells were found in naturally occurring hypoxic microenvironments because we found that doxycycline (Dox)-treated Tet-On H2B-GFP T-HEp3 tumours grown in vivo on CAMs for five days displayed accumulation of pimonidazole adducts, which occur only in hypoxic cells35,36 (Fig. 1d–f and Supplementary Fig. 1D). Analysis of 20 human head and neck squamous cell carcinoma (HNSCC) patient samples revealed that within hypoxic, GLUT1high microenvironments NR2F1 was more frequently upregulated (Fig. 1g,h). We conclude that tumour cells in naturally occurring hypoxic microenvironments upregulate dormancy markers and are slow cycling or quiescent.
Figure 1.

Hypoxia induces the expression of dormancy genes in mouse, PDX and human tumours. (a–c) Images and quantification of MDA-231-HIF reporter cell xenografts in nude mice. GFP-tagged (green) cells express mCherry (red) when hypoxic. Sections were stained for GLUT1 (a), DEC2 (b) and NR2F1 (c) (all in blue). Panel I: merged image of a hypoxic area; arrow, hypoxic; arrowhead, normoxic cell. Panels II–IV: separate blue (GLUT1, DEC2, NR2F1), green (GFP) and red (mCherry) channels. Panel V: low-magnification overview. The boxes indicate spontaneous hypoxic (‘H’, purple) and normoxic (‘N’, green) areas; n = 3 tumours were assessed; representative images are shown. The graph shows the percentage of overlap of normoxic or hypoxic areas with the respective staining, mean ± s.d., Mann–Whitney test. n = 45 hypoxic and 45 normoxic high-power fields (HPFs) (×100) pooled from 3 individual tumours; scale bars, 7.5 μm (I–IV) and 100 μm (V). Also see Supplementary Fig. 1A–C for histograms of the different channels. (d) Illustration of the experiment. In vitro-activated Tet-On H2B-GFP-positive T-HEp3 cells were seeded on the CAM and the GFP label was chased in vivo for 5 days. Four hours before tumour collection, 50 μl pimonidazole (40 mg ml−1) was injected intra-amniotically (i.a.). Fixed tumour cells were stained for pimonidazole adducts. (e,f) Images and quantification of pimonidazole (pimo) adduct staining of T-HEp3 TET-On H2B-GFP cytospins. Pimo, red; H2B-GFP, green, DAPI, blue. Graph: mean percentage + s.d. of pimo-positive and -negative H2B-GFP-expressing cells. n = 150 cells pooled from 4 individual tumours; representative images are shown; scale bars, 10 μm; Mann–Whitney test. See also Supplementary Fig. 1D. (g) An HNSCC patient sample stained for GLUT1 and NR2F1. I and II: normoxic and hypoxic tumour area, respectively. The boxes i–iv indicate the areas shown in a–d, respectively. Scale bars, 25 μm (I–II) and 7.5 μm (i–iv). n= 20 tumours were assessed; representative images are shown. (h) Quantification of staining for 20 HNSCC patients, mean ± s.e.m. GLUT1-positive areas showed a higher percentage of nuclear NR2F1-positive staining compared with GLUT1-negative areas. All 20 patients suffered from poorly differentiated HNSCC. n= 20 patients; each data point represents the mean expression of 10 HPFs; Mann–Whitney test.
Nano-device induced hypoxic microenvironments upregulate dormancy markers
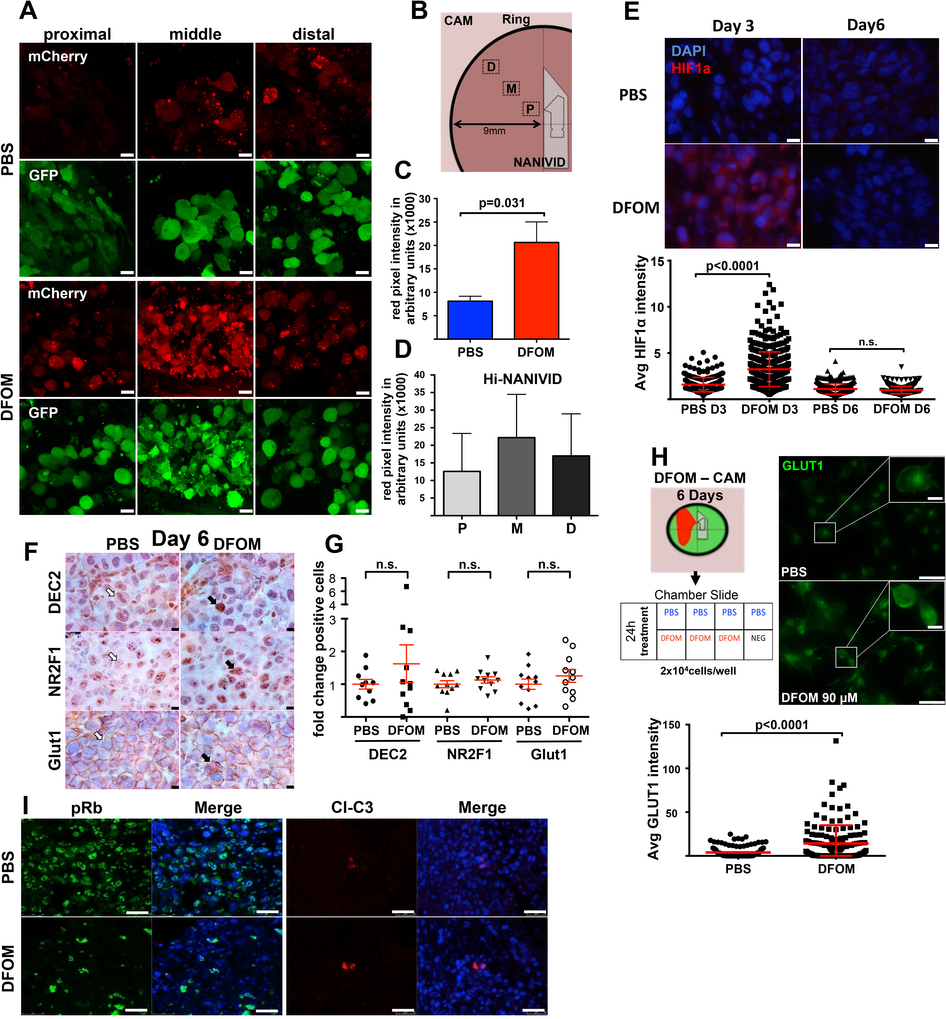
Unlike the in vivo results (Fig. 1), experiments in vitro revealed poor correlation between DEC2 and NR2F1 messenger RNA expression and varying hypoxia levels (Supplementary Fig. 1E). Only NR2F1 expression in breast cancer cell lines showed an upregulation trend in two-dimensional cultures in 1% O2, but not in 5% and 10% O2 versus 21%. Thus, 5–10% O2 levels do not induce hypoxia or dormancy genes suggesting that an in vivo approach is needed to analyse the proposed hypothesis. To this end, we took advantage of the induction NANIVID (iNANIVID) (Fig. 2a,b)37. These microfabricated devices consist of two 3 mm × 1.3 mm glass halves with a lithographically etched chamber that can be loaded with hydrogel solutions containing different cargos37. After insertion into tumours, a 150-μm-wide opening at the side allows a steady flow of compounds from the device, avoiding problems associated with pulsatile systemic or local drug delivery38 (Fig. 2a,b). iNANIVIDs were loaded with Dox, phosphate-buffered saline (PBS) or hypoxia-mimetic agent desferrioxamine (DFOM) and then implanted in T-HEp3 tumours grown on the chicken chorioallantoic membrane (CAM) model37,39 (Fig. 2b).
Figure 2.

Hypoxia induction using iNANIVID. (a) Magnified image (×10) of an iNANIVID. (b) Illustration depicting the iNANIVID in a tumour on the chicken CAM. The tumour is contained in a Teflon ring for support. The device opening is in the upper left quadrant. The area of influence is shown in green. (c) A three-day-old Tet-On H2B-GFP T-HEp3 tumour with an implanted iNANIVID (10 mM doxycycline or PBS (see Supplementary Fig. 1F)). First panel (top row, left): stereoscopic image of the tumour in situ (×4). Boxes i, ii and iii indicate the areas of the following images. i: tip and opening of the iNANIVID in situ. ii: merged image of a tumour area proximal to the iNANIVID showing GFP-positive cells. The dashed line indicates the Teflon ring. iii: merged image of a tumour area distal to the iNANIVID showing GFP-positive cells. The dashed line indicates the iNANIVID. Boxes I+II: details of the proximal (I) and distal (II) tumour areas. Scale bars, 250 μm; n = 3 tumours were assessed; representative images are shown. See also Supplementary Figs 1F and 2A–D for the DFOM diffusion gradient. (d) Relative mRNA expression levels in T-HEp3 CAM tumours treated in vivo for 3 or 6 days with Hi-NANIVID or PBS-iNANIVID. GAPDH was used as a housekeeping gene. n= 3 tumours per group, PCR in triplicate, mean + s.e.m.; *P < 0.001, two-tailed Student’s t-test. (e) IHC staining for DEC2, NR2F1 and GLUT1 in 3-day-old PBS- or Hi-NANIVID (DFOM) T-HEp3 CAM tumours. White arrows, negative; black arrows, positive cells. Scale bars, 10 μm; n= 3 tumours were assessed; representative images are shown. (f) Quantification of IHC staining for DEC2, NR2F1 and GLUT1 in 3-day-old PBS- or Hi-NANIVID T-HEp3 CAM tumours. Horizontal bars indicate median ± s.e.m., n = 23 HPFs pooled from 3 tumours; Mann–Whitney test. See Supplementary Fig. 2E,H for day 6. (g,h) Quantification of IF staining for phospho-Rb and cleaved caspase-3 of T-HEp3 CAM tumours treated for 3 days with PBS- or Hi-NANIVID. Mean + s.d.; n = 500 cells pooled from 3 tumours; two-tailed Student’s t-test. See Supplementary Fig. 2I for staining.
We implanted the devices within Tet-On-H2B-GFP-expressing T-HEp3 HNSCC tumours, where the H2B-GFP fusion protein is induced by Dox16. This construct provides a marker for quiescence due to the extremely slow turnover of the H2B-GFP fusion protein in non-proliferative cells40. After three days in vivo, only the Dox-iNANIVID tumours showed H2B-GFP induction proximal (micrometre range Fig. 2cb) and distal (millimetre range Fig. 2cc) to the iNANIVID opening (Fig. 2ca and Supplementary Fig. 1F)37. Thus, the device supports the efficient release of small molecules and diffusion into the tumour tissue in the millimetre range (Fig. 2cc).
We used iNANIVIDs37 loaded with DFOM (1 mM) (refs 41–43) (henceforth Hi-NANIVID) to mimic hypoxic microenvironments. Within 4.5 h a gradient is established when using EGF or hypoxia-mimetic drugs and chemoattractants37. When T-HEp3 and MDA-231-HIF reporter tumours were implanted with Hi- or PBS-NANVIDs (control) (Supplementary Fig. 2A–D) we found that after three days the gradient is stabilized and that Hi-NANIVID but not PBS-NANIVID regions strongly induced the hypoxia biosensor (Fig. 1 and Supplementary Fig. 2A–D). Further, the dormancy markers NR2F1 and p27 (ref. 16) and the hypoxia genes HIF1α and GLUT1 (ref. 44) were induced in tumours with Hi-NANIVID microenvironments (Fig. 2d). Because the device empties within the first 4.5 h (ref. 37), gene and protein upregulation were reversible (Fig. 2d) and by day 6 they returned to basal levels (Supplementary Fig. 2E–G). While DEC2 mRNA was not elevated in Hi-NANIVID microenvironments (Fig. 2d), immunohistochemistry (IHC) or immunofluorescence (IF) revealed a significant upregulation of DEC2, NR2F1, HIF1α and GLUT1 protein at three days (Fig. 2e,f and Supplementary Fig. 2E). GLUT1 showed membrane localization, while HIF1α, NR2F1 and DEC2 showed nucleocytoplasmic distribution (Fig. 2e,f and Supplementary Fig. 2E). After six days, no differences in DEC2, NR2F1, HIF1α and GLUT1 protein expression were obvious between the DFOM and PBS groups (Supplementary Fig. 2D–G), which is probably due to the absence of DFOM and an extinction of the hypoxic response. Supplementary Fig. 2H supports this conclusion because cells obtained from DFOM Hi-NANIVID microenvironments after six days had downregulated the hypoxic response (Fig. 2d and Supplementary Fig. 2E–G), but restored hypoxia gene expression if re-exposed to DFOM (Supplementary Fig. 2H).
Consistent with the induction of the dormancy genes NR2F1, DEC2 and p27, tumour cells in Hi-NANIVID microenvironments showed reduced phosphorylation of retinoblastoma protein (phospho-Rb, pRb) indicating a G0–G1 arrest16 (Supplementary Fig. 2I). As reported previously37,45, the devices and/or their content were not toxic because histologic analysis and cleaved caspase-3 (CCL-3—apoptosis marker) levels were very low and similar in both PBS and Hi-NANIVID microenvironments (Fig. 2h and Supplementary Figs 2I and 3A).
Hypoxia and dormancy markers are co-expressed in quiescent tumour cells
We found no upregulation of p27, NR2F1, DEC2 or GLUT1 expression in PBS-NANIVID microenvironments (Fig. 3a–c). However, Hi-NANIVID microenvironments displayed tumour cells with upregulation of these markers (Figs 2f and 3a–d and Supplementary Fig. 3B–E). These were also co-expressed in the same tumour cells as significant correlation (r2 0.70–0.98) was found for upregulation of HIF1α and p27 (Supplementary Fig. 3B–E), supporting also that GLUT1high/HIF1αhigh cells are growth arrested. Tumour cells in Hi-NANIVID microenvironments were also pRblow compared with PBS-NANIVID microenvironments (Fig. 3a). Regardless of the microenvironment, DEC2high tumour cells were negative for pRb (Fig. 3a,b). Thus, the dormancy markers NR2F1, DEC2 and p27 are more commonly upregulated in GLUT1high/HIF1αhigh tumour cells. NR2F1 was shown to induce a repressive chromatin state where H3K27me3 marks accumulated in the nucleus of dormant NR2F1high cells16. We found that H3K27me3 levels were significantly enriched in hypoxic, GLUT1high, but not in normoxic, GLUT1low microenvironments (Fig. 3e,f). H3K4me3 levels did not change between GLUT1-positive or -negative cells (Supplementary Fig. 3F). Thus, the hypoxic microenvironment induces changes in specific genes and global chromatin reorganization. Upregulation of p27 was dependent on NR2F1 and HIF1α expression because it was significantly reduced following NR2F1 and HIF1α knockdown (Fig. 3g–i and Supplementary Fig. 3G–J). Thus, both NR2F1 and HIF1α may contribute to the induction of p27 expression and growth arrest of tumour cells in hypoxic microenvironments.
Figure 3.

Primary tumour hypoxia induces the expression of quiescence and dormancy genes. (a) Double IF stainings for DEC2 and pRb, NR2F1 and p27, and p27 and GLUT1 of 3-day-old PBS- or Hi-NANIVID (DFOM)-treated T-HEp3 CAM tumours; scale bars, 7.5 μm; 3 individual tumours were assessed, representative images are shown. (b–d) Quantification of the double IF stainings; bars indicate mean ± s.d., n = 16, 22 and 21 HPFs (b, c and d, respectively) pooled from 3 tumours per treatment; Mann–Whitney test. NS, not significant. (e) A T-HEp3 CAM tumour stained for H3K27me3 (green), GLUT1 (red) and DAPI (blue). Scale bars, 25μm; n = 3 tumours were assessed; representative images are shown. (f) Quantification of H3K27me3 staining in normoxic (GLUT1-negative) and hypoxic (GLUT1-positive) areas. Mean + s.e.m.; n = 1,500 cells pooled from 3 tumours; two-tailed Student’s t-test. See Supplementary Fig. 3F for H3K4me3. (g) A T-HEp3 tumour cytospin stained for p27 and human vimentin. Cells were transfected with NR2F1, HIF1α or control siRNA for 24 h while in normoxia (21% O2) or hypoxia (1% O2). Cells (3 × 105) were seeded on CAMs for 48 h. Scale bars, 10 μm; n = 10 tumours were assessed; representative images are shown. (h,i) Quantification of p27 staining for NR2F1 and HIF1α knockdown, respectively. See Supplementary Fig. 3G,H for knockdown efficiency before seeding. The graph shows the percentage of p27-positive T-HEp3 cells per tumour. Bars indicate mean + s.d. n = 4 tumours (siNR2F1), n = 6 tumours (siHIF1α) per treatment, each having >100 cells scored; two-tailed Student’s t-test.
Hypoxia does not affect intravasation or extravasation
We tested the ability of hypoxic cells to intravasate in vivo, using the CAM assay (Fig. 4a–c)46,47. MDA-231-HIF reporter cells were incubated in 1% or 21% O2 for 72 h prior to inoculation on CAMs48 and T-HEp3-GFP cells cultured in 21% O2 served as a positive intravasation control39,46 (see Methods). Intravasated T-HEp3-GFP and MDA-231-HIF reporter cells were readily detected in the lower CAM (Fig. 4c). Exposure to 1% O2 for three days did not significantly change the ability of these cells to intravasate (Fig. 4c). To test extravasation potential, T-HEp3-GFP cells were exposed to 21% or 1% O2 for three days and then injected i.v. into nude mice. After 24 h lungs were perfused to eliminate the blood and circulating tumour cells that did not extravasate and the lung-lodged GFP-positive cells were quantified (see Methods for details). The T-HEp3-GFP DTCs overall extravasation efficiency was ~0.4% and extravasation efficiency did not differ between post-hypoxic or -normoxic cells (Fig. 4d).
Figure 4.

Hypoxia does not significantly influence intravasation and extravasation. (a) Illustration of the CAM system. The tumour cells are seeded on the chorionic epithelium and penetrate this monolayer to reach the mesodermal vasculature. The CAM perpendicular opposite the implantation site, the lower CAM, is harvested 48 h after seeding. Following digestion of the CAM, the number of intravasated cells is counted. (b) Lower CAM cell suspension with MDA-231-HIF reporter cells pretreated for 72 h with 21% O2 (left) or 1% O2 (right). Scale bars, 25 μm; n = 3 CAMs per treatment were assessed; representative images are shown. (c) Quantification of cells counted in lower CAM. T-HEp3 GFP cells are used as a positive control. n = 3 CAMs with T-HEp3 GFP, n = 7 CAMs with MDA-MB-231-HIF reporter cells; bars indicate mean; Mann–Whitney test. (d) Post-hypoxia extravasation test. T-HEp3-GFP cells (3 × 105) were cultured in normoxia (21% O2) or hypoxia (1% O2) for 72 h and then tail vein injected in nude mice. Twenty-four hours later, the lungs were retrieved, collagenased and GFP+ cells were counted. n= 7 mice (normoxia), n= 6 mice (hypoxia); bars indicate mean; Mann–Whitney test.
PT hypoxia increases the frequency of dormant DTCs
We next tested the fate of post-hypoxic DTCs in lungs by using tumour cells isolated from Tet-On H2B-GFP T-HEp3 PTs influenced by PBS-or Hi-NANIVID microenvironments and treated with Dox to generate H2B-GFP+ nucleosomes. Three-day-old tumours were digested and cells were tail vein injected into nude mice that did not receive any Dox, allowing us to track the H2B-GFP label-retaining (quiescent) DTCs (Fig. 5a).
Figure 5.

Post-hypoxic DTCs become quiescent in metastatic sites. (a) PBS-or Hi-NANIVID Tet-On H2B-GFP T-HEp3 CAM tumours were treated with doxycycline (Dox) for 3 days to activate H2B-GFP. T-HEp3-H2B-GFP cells (5 × 105) were injected i.v. in nude mice. Five and ten days post-injection, GFP+ cells were quantified in digested lungs. For total number of cells, H2B-GFP was re-induced (Dox) for 16 h in vitro. (b) Lung cell suspension at day 5 and 10. Inset: T-HEp3-H2B-GFP-positive cell. Scale bars, 25 μm and 5 μm; n = 4 (day 5) and 3 (day 10) mice were assessed; representative images are shown. (c) Total cells per mouse after re-induction of H2B-GFP. n = 4 mice (day 5), n= 3 mice (day 10). Bars indicate mean. Mann–Whitney test. (d) LRCs (H2B-GFP+) as a percentage of the total cell number (after reinduction, triple count, see Fig. 5c). Bars indicate mean. n = 4 mice (day 5), n = 3 mice (day 10). Mann–Whitney test. (e) 231-H2B-Dendra2 cells were cultured in 21% O2 or 1% O2 for 3 days and photoconverted (green to red). Cells (1 × 106) were injected i.v. in nude mice. At 8 and 16 days later, the lungs were digested. The total cell number (Dendra2 green) and number of LRCs (Dendra2 green and red) was quantified. (f) 21% or 1% O2-pretreated cells in situ at day 8 and 16. Inset: LRCs. Scale bars, 15 μm and 5 μm; n= 3 mice were assessed; representative images are shown. (g,h) Total cell number at day 8 and day 16 (green) and red-label-retaining cells at the indicated time points (only green and red). n= 3 mice each group; each point is the average of 3 counts per mouse. Bars indicate mean; ‘N’ normoxia, ‘H’ hypoxia; Mann–Whitney test. (i) MDA-MB-231 cells were cultured for 3 days in 21% O2 (normoxia, ‘N’ in j) or 1% O2 (hypoxia, ‘H’ in j) and then tail vein injected. After 30 days, lungs were co-stained for p27 and human vimentin (see Supplementary Fig. 4A). Positive: double positive cells for vimentin (red) and p27 (green); negative: vimentin positive cells that were negative for p27. DAPI was used to detect nuclear DNA in all cells. Scale bars, 10 μm, n= 3 mice were assessed, representative images are shown. (j) Quantification of 30-day MDA-MB-231 DTCs stained for p27 and human vimentin (see Fig. 5i). Bars show mean ± s.e.m.; n= 3 mice; >100 cells scored per mouse; Mann–Whitney test.
Enzymatically digested lungs were used to quantify the number of label-retaining T-HEp3-H2B-GFP cells (LRCs) (Fig. 5a,b) and then the cell suspensions were cultured with Dox to re-induce H2B-GFP and quantify total tumour cells (Fig. 5a–c). The total number of T-HEp3 cells in lungs five days after injection was similar, regardless of whether they originated from a PBS- or Hi-NANIVID microenvironment (Fig. 5c). Post-hypoxic DTCs displayed a non-statistically significant trend to increased quiescence at five days post-injection, versus DTCs from PBS-NANIVID microenvironments (Fig. 5d). At ten days, cells from PBS-NANIVID tumours had diluted the label entirely, while label retention in Hi-NANIVID DTCs persisted at levels similar to five days (Fig. 5d), suggesting a dormant-like state.
The total number of post-hypoxic lung DTCs was greater at ten days than in the post-normoxic group (Fig. 5c). However, the number of total post-normoxic cells decreased compared with day 5, suggesting some overall attrition of tumour cells that does not occur if cells were post-hypoxic (Fig. 5c). That more DTCs were counted in mice that were injected with post-hypoxic tumour cells, suggests that the cells that did not enter dormancy, survive, colonize more efficiently and/or grow more aggressively (Fig. 5c). The tools we use allowed detection of LRCs, but the dynamics of different DTC subpopulations is difficult to estimate. Nevertheless, these data suggest that PT hypoxia might induce a heterogeneous population of both dormant-like cells and also more efficiently surviving and growing cells.
To further test the connection between hypoxia and DTC quiescence, we used MDA-MB-231 cells stably expressing an H2B-Dendra2 fluorescent protein (231-H2B-Dendra2). The excitation emission of Dendra2 is similar to that of GFP, but following irradiation with 405 nm light, Dendra2 is stably photoconverted to emit in the red fluorescence wavelength. The fusion to H2B allowed for long-term tracking of LRCs. 231-H2B-Dendra2 cells cultured in hypoxic and normoxic conditions for three days were photoconverted generating >95% of the cells H2B-Dendra2 red (Fig. 5e). These cells were tail vein injected and later the lungs were retrieved, digested and the number of green/red or only green LRCs was counted (Fig. 5f). The total number of 231-H2B-Dendra2 cells at day 8 post-injection was not significantly different between the two groups, although post-hypoxic DTCs trended towards less proliferation (Fig. 5g). Nevertheless, already at this time point it was evident that hypoxia had induced LRCs, as evidenced by ~80% of post-hypoxic 231-H2B-Dendra2 cells retaining red H2B-Dendra2 (Fig. 5h). At day 16, we observed a significantly lower number of post-hypoxic 231-H2B-Dendra2 cells, compared with post-normoxic DTCs (Fig. 5g) and ~70% of the former cells at day 16 retained varying levels of H2B-Dendra2-red protein (Fig. 5h). Further, post-hypoxic solitary DTCs in lungs one month after i.v. injection were more frequently high for p27 expression than post-normoxic DTCs (Fig. 5i,j), suggesting that post-hypoxic DTCs are able to maintain a long-lived quiescent phenotype.
Post-hypoxic T-HEp3 cells displayed increased plating efficiency followed by re-oxygenation, while the MDA-MB-231 cells displayed a slightly lower plating efficiency (Supplementary Fig. 3I). While this assay does not account for quiescence, it reliably informs on survival and proliferation phenotypes. Our results suggest that hypoxia can induce dormancy in a subpopulation of tumour cells that after dissemination may become quiescent in target organs (Fig. 5a–j). This correlated with their reduced efficiency in vitro to form colonies after hypoxia (Supplementary Fig. 3I). In contrast, T-HEp3 cells displayed a heterogeneous response in vivo with a mixture of faster-growing cells and quiescent cells in the same target organ (Fig. 5a–d). The enhanced metastatic capacity of post-hypoxic T-HEp3 cells compared with post-normoxic cells (Fig. 5a–d) may be due to enhanced survival detected in plating efficiency experiments in vitro (Supplementary Fig. 3I).
Dormancy phenotypes in post-hypoxic ER+ and ER− breast cancer cells
ER+ luminal breast cancer shows late metastatic relapse14,49. Thus, we tested whether post-hypoxic ZR-75-1 ER+ luminal breast cancer cells would be more prone to enter a dormancy-like phenotype than TNBC MDA-MB-231 cells by virtue of carrying a ‘pre-encoded’ dormancy signature not observed in MDA-MB-231 cells14. 231-H2B-Dendra2 or ZR-75-1-H2B-Dendra2 (75-1-H2B-Dendra2) cells grown in either 21% O2 or 1% O2 for three days were photoconverted and embedded in 3D Matrigel at low density (150 75-1-H2B-Dendra2 and 15 231-H2B-Dendra2 cells mm−2) to mimic solitary DTC behaviour and cultured in 21% O2 for up to 21 days (Fig. 6a). Unlike in vivo, 231-H2B-Dendra2 cells exposed to hypoxia or normoxia in vitro were indistinguishable in their ability to generate LRCs (Fig. 6b). Thus, the in vivo lung microenvironment contributes factors that make this tissue more restrictive for post-hypoxic DTCs and reveals a phenotype not detected in vitro (Figs 5f–j and 6b). In contrast, post-hypoxic 75-1-H2B-Dendra2 cells showed a significantly higher number of LRCs from day 12 that persisted for at least 21 days (Fig. 6c). Following NR2F1 knockdown, post-hypoxic 75-1-H2B-Dendra2 cells showed a significant loss in label retention at day 10, compared with controls (Fig. 6m and Supplementary Fig. 3J). Plating efficiency of ZR-75-1 cells after hypoxia was also increased (Supplementary Fig. 3I) suggesting that hypoxia-induced quiescence might be coupled to enhanced survival in ER+ cells. These data suggest that post-hypoxic ER+ luminal cells might be more prone than ER− cells to enter quiescence in an NR2F1-dependent manner. However, in vivo, post-hypoxic MDA-MB-231 cells can also enter a dormant-like phenotype in the lungs.
Figure 6.

Post-hypoxic breast cancer cells become quiescent in vitro. (a) Illustration of the experiment. Triple-negative 231-H2B-Dendra2 and ER+ 75-1-H2B-Dendra2 cells were grown as a monolayer for 72 h in normoxic or hypoxic conditions. Cells were photoconverted (PC), seeded in 8-well chamber slides coated with Matrigel at low densities and cultured in normoxia. The green/red label was chased for 21 days. 75-1-H2B-Dendra2 were also transfected with 50 nM NR2F1 or control siRNA for 24 h during the hypoxia or normoxia incubation and followed for 10 days in Matrigel. See Supplementary Fig. 3J for knockdown efficiency at 72 h. (b,c) Quantification of red-label-retaining 231-H2B-Dendra2 and 75-1-H2B-Dendra2 cells, respectively. Bars show mean + s.d. at the indicated days after seeding. n = 8 (231-H2B-Dendra2), n = 6 (75-1-H2B-Dendra2), n= 4 (75-1-H2B-Dendra2 knockdown NR2F1) independent experiments; one-tailed Student’s t-test.
Post-hypoxic DTCs express dormancy but not hypoxia markers and resist chemotherapy
We wondered whether the post-hypoxic dormant-like phenotype depended on active hypoxic and/or dormancy programs. T-HEp3 cells originating from PBS or Hi-NANIVID microenvironments were delivered i.v. into nude mice. Later the lungs were excised and prepared for IF analysis (Fig. 7a). Using IF for human-specific vimentin and dormancy markers we tested the expression of NR2F1, DEC2, p27 or GLUT1 in T-HEp3 DTCs in the lung (Fig. 7a–f and Supplementary Fig. 4A). Scoring of ~2,000 lung DTCs per antigen combination revealed that: solitary post-normoxic DTCs contained a higher proportion of NR2F1-, DEC2- and p27-positive cells compared with the PBS-NANIVID PTs (Fig. 7c–f); solitary post-hypoxic DTCs and Hi-NANIVID PTs showed a similar percentage of marker-positive cells (Figs 1f and 7c–f); post-hypoxic DTCs at day 5 contained a large percentage of NR2F1-, p27- and DEC2-positive DTCs compared with post-normoxic DTCs (Fig. 7c–e); this difference persisted for NR2F1 and p27 at day 10, but was only statistically significant for p27 (Fig. 7c,d). The post-normoxic DTCs maintained the same level of GLUT1 expression as the PTs, while the percentage of GLUT1-positive Hi-NANIVID-primed DTCs was slightly reduced compared with the PT levels (Figs 2f and 7f). GLUT1 expression was low and not different between Hi- or PBS-NANIVID-primed DTCs, arguing for an attenuated hypoxic response.
Figure 7.

Post-hypoxic DTCs upregulate a dormancy signature. (a) Schematic of the experiment. PBS- or Hi-NANIVID T-HEp3 CAM tumours were collagenased after 72 h. Cells (5 × 105) were injected in the tail vein of nude mice. After 5 and 10 days, the lungs were retrieved and snap frozen for IF staining. (b) Positive and negative single lung DTCs and micrometastatic cells stained for human vimentin (red) and NR2F1, p27, DEC2 or GLUT1 (all: green), DAPI (blue). Scale bars, 10 μm; n= 12 mice were assessed; representative images are shown. (c–f) Quantification of the NR2F1, p27, DEC2 or GLUT1 staining of single DTCs. Blue bars indicate cells pretreated with PBS-iNANIVID on the CAM, red bars indicate Hi-NANIVID-treated cells. Brown bar: micrometastases were present only in the Hi-iNANIVID day 10 group. Grey bar: cells in PBS-iNANIVID PTs (see Fig. 2e,f). Whiskers: 5th–95th percentile; bars: median. n = 2,000 cells pooled form 3 mice per treatment and time point; Mann–Whitney and Kruskal–Wallis test. (g) TGFβ2-positive and -negative DTCs and periphery and centre of a lung micrometastasis. Asterisk: extracellular TGFβ2; arrow: TGFβ2 positive DTC. The experimental conditions were the same as in a. Scale bars, 10 μm and 25 μm, respectively; n = 12 mice were assessed; representative images are shown. (h) Quantification of TGFβ2 staining. Whiskers: 5th–95th percentile; bars show mean. n = 2,000 cells pooled from 3 mice per treatment and time point. Kruskal–Wallis and two-tailed Student’s t-test.
Interestingly, micrometastases (>5-cell clusters) were detectable only in the post-hypoxic group at 10 days after injection (Fig. 7b). Micrometastases had a lower percentage of NR2F1-positive cells than post-hypoxic DTCs in the same lung, while not differing significantly from post-normoxic DTCs (Fig. 7c). The micrometastases were also less positive for p27, except when comparing with post-normoxic DTCs at five days (Fig. 7d). The most striking difference was GLUT1 expression (Fig. 7b,f). Only ~30% of solitary DTCs expressed GLUT1, independent of their post-normoxic or -hypoxic origin and time in lungs, while ~85% of the cells in the micrometastases were GLUT1hi (Fig. 7f), a percentage that is even higher than in Hi-NANIVID PTs (Fig. 2f). Only DEC2 expression was equally low in percentage of micrometastatic cells and DTCs, both in the PBS- and DFOM-treated groups at day 10 (Fig. 7e).
When we tested the levels of the dormancy-inducing cytokine TGFβ2 (ref. 50) in the T-HEp3 lung DTCs and metastases, we found that DTCs, regardless of their hypoxic state, were more frequently positive for intracellular TGFβ2 versus metastases that dramatically reduced all detectable TGFβ2 in tumour cells or stroma (Fig. 7g,h). Importantly, Hi-NANIVID DTCs were more frequently positive for TGFβ2 (Fig. 7g,h).
We next asked whether solitary dormant DTCs could survive chemotherapy. The experiment was conducted as in Fig. 5a for ten days post-injection, when we find mostly dormancy-marker-expressing solitary DTCs in lungs (Figs 5 and 7). However, after seeding in the lung mice were treated with vehicle (NaCl 0.9%) or cisplatin at 3.5 mg kg−1 (standard chemotherapy for HNSCC) for the remainder of the experiment (Fig. 8a–d). In agreement with their non-proliferative state (Figs 5 and 7) we found more single DTCs when these were post-hypoxic (Fig. 8b). These DTCs were p27hi (Fig. 7b,d) and non-proliferative, as confirmed by the lower frequency of pRb+ DTCs (Supplementary Fig. 4B). Cisplatin caused, over this short treatment period, a moderate but still significant reduction in the numbers of post-normoxic single DTCs (Fig. 8b), probably because a subpopulation of these cells (45–50%, see Fig. 7d and Supplementary Fig. 4B) are proliferating. This decrease in single DTC numbers was correlated with increased apoptosis (TUNEL staining) after cisplatin treatment (Fig. 8c,d). In contrast, post-hypoxic DTCs (from Hi-NANIVID tumours) were unaffected by cisplatin (Fig. 8d) and this correlated with lower frequency of pRb+ cells (Supplementary Fig. 4B), more p27+ cells (~80%, see Fig. 7d) and no changes in apoptosis (Fig. 8c,d). We conclude that non-proliferative DTCs can evade chemotherapy and this escape is more pronounced for post-hypoxic DTCs.
Figure 8.

Evasion of chemotherapy by post-hypoxic DTCs and NR2F1 induction in spontaneously seeded PyMT DTCs. (a) T-HEp3 tumours grew for 3 days on the CAM with PBS- or Hi-NANIVIDs, as in Fig. 7. Five days after tail vein injection, mice were i.p. injected every other day with 3.5 mg kg−1 cisplatin or 0.9% NaCl solution. At ten days post-injection, the lungs were collected. (b) Quantification of single DTCs positive for human vimentin per HPF (×40) in the lungs of cisplatin- and control-treated nude mice at day 10. Bars: mean + s.e.m.; n = 21 HPFs pooled from 3 (PBS ctrl) or 4 (all other) mice; one-tailed Student’s t-test. (c) T-HEp3 DTCs in cisplatin-treated mouse lungs at day 10, stained for TUNEL (green), human vimentin (red) and DAPI (blue). Arrows: negative and positive cell for TUNEL. Scale bars, 10 μm, n= 12 mice were assessed; representative images are shown. (d) Quantification of TUNEL staining. Bars: mean + s.e.m., n = 3 mice per treatment, 50 DTCs scored per mouse; one-tailed Student’s t-test. See Supplementary Fig. 4B for pRb staining of single DTCs. (e) Non-tumour bearing FVB mice were injected in the mammary fat with cells from spontaneous MMTV-PyMT-Dendra2 transgenic primary tumours. Tumours were photoconverted in vivo for 5 min. Lungs were retrieved 7 days later. See also Supplementary Fig. 4C,D. (f) Dendra2-green signal identified spontaneous DTCs in the lungs. Fifty-five per cent of the DTCs were label-retaining cells (LRCs) and 37% were positive for NR2F1. n = 187 spontaneous DTCs pooled from 3 mice. Bars, mean + s.d. (g) NR2F1 staining of PyMT-Dendra2 DTCs. NR2F1, blue. Scale bars, 10 μm; n = 3 mice were assessed; representative images are shown. (h) Images of LRC and NLRC PyMT-Dendra2 DTCs before and after additional 30 s photoconversion on a slide. Scale bars, 10 μm; n = 3 mice were assessed; representative images are shown. See Supplementary Fig. 4E for fold change in intensity. (i) Sixty-seven per cent of the LRCs and <1% of the non-label-retaining cells (NLRCs) were positive for NR2F1. Bars: mean + s.d.; n = 187 DTCs pooled from 3 mice; two-tailed Student’s t-test. See Supplementary Fig. 4F for NR2F1 in MMTV-PyMT-Dendra2 primary tumour.
Spontaneous non-proliferative lung DTCs display an NR2F1high profile
We next tested whether spontaneously occurring DTCs could upregulate dormancy markers with the same kinetics as i.v.-delivered cells. We used the MMTV-PyMT-Dendra2 immune-competent model (Supplementary Fig. 4C) where tumours developed by injecting PyMT-Dendra2 cells in the mammary fat pad were photoconverted in vivo (Fig. 8e). This strategy, which allowed testing the fate of spontaneously spread tumour cells, revealed that after one week the DTCs (CD45negative ruling out phagocytosis of DTCs) showed Dendra2-green protein in large intracellular cytosolic bodies (Fig. 8g,h and Supplementary Fig. 4D). We confirmed the identity of Dendra2 green or doubly labelled green/red-label-retaining PyMT-Dendra2 DTCs by photoconversion and ~90% of the DTCs were photoconvertible (Supplementary Fig. 4E); in 10% of DTCs, Dendra2 levels were very low hampering photoconversion. We found that seven days after photoconversion in the PT, 55% of the lung DTCs were red at different degrees, suggesting heterogeneous cycling rates (Fig. 8f,g). The dormancy regulator NR2F1 (ref. 16) was downregulated in the PyMT PTs (<1% NR2F1-positive cells, Supplementary Fig. 4F). However, NR2F1 detection via IF was positive in 37% of the spontaneous DTCs (Fig. 8f) and in 67% of PyMT-Dendra2 red DTCs, while 33% of label-retaining cells were negative for NR2F1 (Fig. 8i). A small proportion of DTCs (<1%) did not retain Dendra2 red label but were NR2F1+ and majority of green-only DTCs were NR2F1negative (Fig. 8i). NR2F1 expression was more frequent (37%) in DTCs than in PT cells (<1%) arguing that NR2F1 is upregulated after dissemination. We conclude that spontaneous DTCs can remain in, or quickly enter, an NR2F1high non-proliferative state as DTCs in lungs.
DISCUSSION
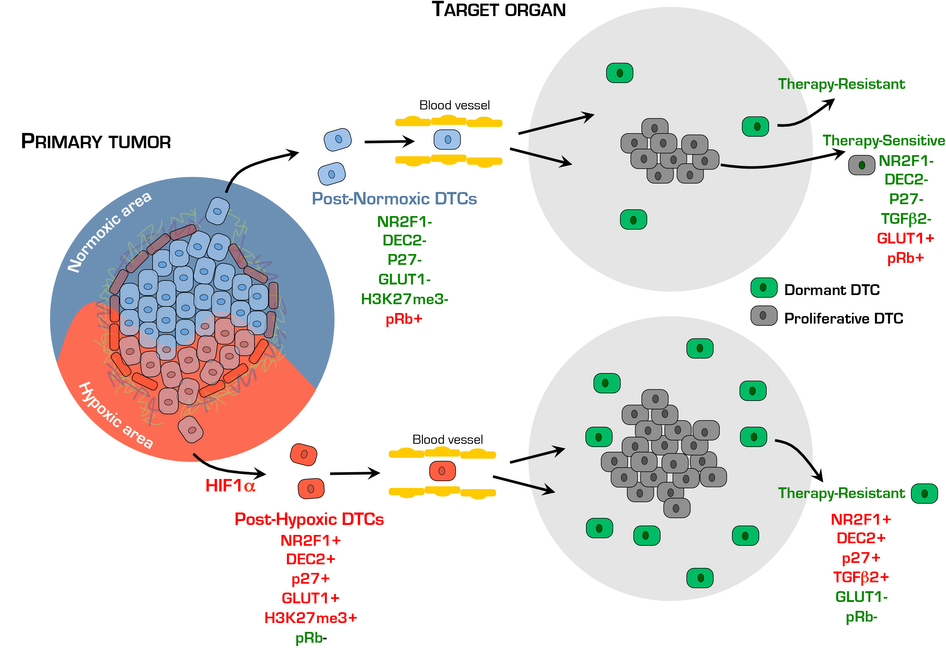
Our work reveals that hypoxic microenvironments in primary lesions contain subpopulations of tumour cells that not only activate a hypoxic response, but also a long-term dormancy-like program29 (Supplementary Fig. 5). Once cells disseminated, the expression of dormancy markers persisted, but the hypoxic response did not (Supplementary Fig. 5). This suggests that the dormancy-like response is more long-lived than the hypoxic program or that another hypoxia-responsive pathway not linked to GLUT1 expression is activated concomitantly with the dormancy program. The identity of this pathway is unknown, but hypoxia-activated p38 (ref. 51) is a likely candidate, as p38 can induce NR2F1 (ref. 16) and HIF1α (ref. 51). The dormancy markers NR2F1 and p27 had the longest half-life and p27 was detected as long as one month in post-hypoxic DTCs, the equivalent of ~3 years in humans. DEC2 was also induced by hypoxia, possibly post-transcriptionally18, but it was marginally expressed or absent in proliferative DTCs or tumours, suggesting that it may be an early executor of hypoxia-induced dormancy. We also confirmed in immune-competent PyMT-Dendra2 mice52 that NR2F1+ quiescent DTCs emerge rapidly in lungs. Thus, both PT stress microenvironments and the target organ might cooperate to induce a dormant phenotype, as previously proposed11 (Supplementary Fig. 5).
Post-extravasation DTC dormancy, rather than intravasation and extravasation, was significantly induced by hypoxia. We also found post-hypoxic T-HEp3 cells to be more frequently TGFβ2high, while growing metastases silenced the TGFβ2 signal (Supplementary Fig. 5). TGFβ2 expression is detectable in the lung, a site permissive for metastasis, but that also harbours dormant DTCs50. Here, we found TGFβ2 upregulated in specific lung niches harbouring dormant DTCs. While additional testing of TGFβ2high niches’ function in the lung is needed, awakening of numerous dormant DTCs (TGFβ2high/NR2F1high/p27high) might explain why p38α/β (activated by TGFβ2) and TGFβRI inhibitors enhance lung metastasis50. It is also possible that dormant DTCs orchestrated TGFβ2hi niches that may help drive and maintain dormancy, or that post-hypoxic DTCs may actively home to TGFβ2hi niches or fail to disrupt these niches.
NR2F1 holds DTCs in lungs in a dormant state16 and we now show that hypoxia in PTs might initiate an NR2F1-, HIF1α- and p27-induced dormancy program. This was also true for ER+ breast cancer cells, which show an enrichment for a dormancy signature strongly associated with longer time to metastasis14. In malignant cells upregulation of NR2F1 can induce remodelling of chromatin into a repressive state16. Our data show that a similar reprogramming might be occurring in PT hypoxic microenvironments, as cells that were GLUT1high also accumulated abundant H3K27me3 repressive marks in areas of growth arrest where HIF1α and p27 were usually co-upregulated (Supplementary Fig. 5). Thus, the PT hypoxia-induced reprogramming might precede and allow for the long-lived nature of the dormancy program in DTCs. Our data further suggest that PT hypoxic microenvironments induce, even in metastasis-permissive sites such as the lung, a dormancy program16,50. The duration of dormancy may be more variable in the lung, given that proliferative micro- and macrometastasis lesions appear to coexist with dormant DTCs. These data are important because hypoxia-induced dormant-like DTCs survive chemotherapy by evading apoptosis, potentially allowing dormant DTCs to reactivate and contribute to relapse. Our data are not in disagreement with the findings that hypoxia fuels metastasis10,53–55, but rather reveal that hypoxia spawns an additional phenotype where a large subpopulation of DTCs may enter dormancy and fuel recurrences by evading therapy.
Using previously unavailable technological advancements, namely the Hi-NANIVID37,38 and a hypoxia biosensor34, we could induce and monitor spatially defined hypoxic stress microenvironments in vivo. The potential of the NANIVID is not limited only to its applications in this study. For example, it can be used for direct intravital imaging while the tumour area is influenced37. A caveat is that once the device is implanted the experiment starts and pulse-chase-like experiments cannot be performed in the same tumour area. However, this device is being upgraded to include regulated release systems and its direct integration in imaging windows, which will allow monitoring single-cell biology events in real time in a reversible manner in the same tumour area37.
Our study shows how hypoxia in PT microenvironments can have a long-lasting influence on the fate of DTCs. This suggests that therapies that target such dormancy mechanisms might be useful to eliminate quiescent tumour cells during minimal residual disease56, or in combination with anti-proliferative therapies, to concomitantly target proliferative and quiescent or slow-cycling cells and thus therapeutically address the full heterogeneity of disseminated disease.
METHODS
Cell lines
The tumorigenic HEp3 (T-HEp3) head and neck squamous cell carcinoma (HNSCC) PDX line was derived form a lymph node metastasis of a HNSCC patient, and maintained as described previously57. The triple-negative, basal-like MDA-MB-231 and the ER+ luminal-like ZR-75-1 breast cancer cell lines were described previously58,59 and acquired from ATCC. Cell were routinely checked to exclude mycoplasma contamination. No cell lines used in this study were found in the database of commonly misidentified cell lines that is maintained by ICLAC and NCBI Biosample. The cell lines were not further authenticated after purchase from ATCC. Using Lipofectamine transfection reagent (Invitrogen) according to the manufacturer’s instructions, these cell lines were stably transfected with different reporter proteins (see Supplementary Table 1).
We generated T-HEp3 cells expressing a Histone 2B (H2B)-GFP fusion protein under the control of the tetracycline-controlled transactivator (Tet-On) system (Tet-On H2B-GFP T-HEp3). These cells produce the nuclear H2B-GFP tag when in the presence of doxycycline, a more stable tetracycline analogue, and are unable to produce this tagged fusion protein in the absence of it. Due to the slow turnover of the Histone 2B protein, the GFP tag stays with cells for extended time, even if doxycycline is withdrawn; yet any cell division will quickly dilute the label down.
We also generated MDA-MB-231 cells expressing both a permanently active GFP and, under the control of five hypoxic response elements and an oxygen-dependent degradation domain (5HRE-ODD), a red mCherry fluorescent protein (MDA-MB-231-5HRE-ODD-mCherry-GFP)34. These cells always express the GFP and only in hypoxic conditions also express the red mCherry protein, which allows for detection of different oxygenation status.
The MDA-MB-231-H2B-Dendra2 and ZR-75-1-H2B-Dendra2 cells express a fusion protein of Histone 2B and the photoswitchable fluorescent protein Dendra2, which can be irreversibly switched from green to a highly photostable red emission by near-UV light (400–405 nm). We also utilized cell suspensions from the Dendra2 tag in the MMTV polyoma middle T (MMTV-PyMT)-driven spontaneous breast cancer model in immunocompetent mice. These MMTV-PyMT-Dendra2 tumours express the soluble Dendra2 tag in the cytoplasm52. All plasmids also included the KanMX cassette that confers kanamycin resistance for clonal selection. Selection of successfully transfected clones through addition of G418 to culture medium was started 48 h after transfection.
iNANIVID
The novel induction nano-intravital device (iNANIVID) consists of two microfabricated glass halves with an etched chamber that can be loaded with hydrogel solutions containing different chemical compounds, which are released over time37. The iNANIVIDs can be used in the in vivo chicken chorioallantoic membrane (CAM) model to influence the tumour microenvironment. We used iNANIVIDs loaded with 10 mM doxycycline (Dox), 1 mM desferrioxamine (DFOM, Hi-NANIVID) and phosphate-buffered saline (PBS, PBS-NANIVID). DFOM is an iron chelator and has previously been used extensively to mimic hypoxia. Iron is needed in the PHD-mediated selective hydroxylation of HIF1α proline residues, which initiates proteosomal degradation. By blocking the degradation, DFOM leads to HIF1α accumulation within normoxic cells.
Photoconversion
A custom-built 405 nm LED-Array (3A/20V) was constructed for photoconversion of the Dendra2-expressing cells in vitro and in vivo. Photoconversion of plated cells was carried out for 90–120 s, depending on the cell density and visually controlled for >95% photoconversion (Figs 5e–h and 6a–c). For the spontaneously disseminated PyMT-Dendra2 experiment, the skin covering the tumour was incised and temporarily retracted to expose the tumour, which was photoconverted for 5 min in vivo (Fig. 8e). On the lung slides, the Dendra2-green signal was used to identify the DTCs. Using a Leica DM 5500 B microscope (Leica), the cells were imaged before and after 60 s irradiation with the DAPI channel (405 nm), to photoconvert additional Dendra2-green protein to Dendra2 red (Fig. 8h and Supplementary Fig. 4C–E). The change in pixel intensity was measured using the Metamorph software (Supplementary Fig. 4C). For red-label-retaining cells, the red pixel intensity in arbitrary units after photoconversion was normalized to the value before photoconversion. For non-label-retaining cells the absolute value for red pixel intensity in arbitrary units is used.
Chicken chorioallantoic membrane (CAM) model
The chicken chorioallantoic membrane (CAM) model and the serial inoculation of T-HEp3 tumours have been previously described57. For the CAM xenografts we used premium specific pathogen-free (SPF), fertile, 8-day-incubated embryonated chicken eggs supplied by Charles River Laboratories. For the Tet-On H2B-GFP T-HEp3 label chase experiment (Fig. 1d–f), the cells were incubated with 1 μg ml−1 Dox in normal tissue culture conditions. Cells (5 × 105) were seeded on CAMs and tumours were grown for five days, without Dox treatment. At day 5, 50 μl of pimonidazole (40 mg ml−1, HP1-100Kit, Hypoxyprobe) was injected into the embryos and incubated for 4 h. The tumours were excised and immediately collagenased. The cells were fixed in 4% formaldehyde and cytospins were prepared and stained for pimonidazole adducts (see Supplementary Table 2).
For all iNANIVID inoculation experiments, a layer of minced T-HEp3 tumour was seeded inside a sterile Teflon ring on the chorioallantoic membrane. In this ring, on the first layer of tumour cells, the iNANIVID was inoculated, as shown in Fig. 2b. Then a second layer of minced T-HEp3 tumour was added to cover the iNANIVID completely. The ring kept the mass of minced tumour together. Following tumour implantation, the CAMs were incubated for 3 and 6 days at 37 °C before the tumour was recovered and the upper left quadrant was harvested for further analysis (area of influence, Fig. 2b). The stereoscopic images in Fig. 2c and Supplementary Fig. 1F were taken with a Zeiss Lumar V12.0. The intravasation experiment has been described previously46. For the experiment (Fig. 4b,c), 106 T-HEp3 Tet-On-H2B-GFP or MDA-MB-231-5HRE-ODD-mCherry-GFP cells were inoculated on the CAM. The CAMs carrying T-HEp3 Tet-On-H2B-GFP cells were treated with 5 μg ml−1 doxycycline daily to sustain the H2B-GFP expression. Prior to inoculation the MDA-MB-231-HIF reporter cells were incubated for 72 h in either normoxic (only GFP expressed) or hypoxic conditions (to activate the 5HRE-ODD-mCherry red fluorescing protein). After 48 h, the egg membranes perpendicular opposite to the implantation site (lower CAM) were recovered and intravasated cells in the vasculature were detected and quantified. For the DFOM diffusion experiments (Supplementary Fig. 2A–D), 5 × 105 MDA-231-HIF reporter cells were seeded on recipient CAMs and resulting tumour nodules were harvested 7 days later, minced and re-seeded in Teflon rings and implanted with NANIVIDs. After 3 days, whole CAMs with tumours were removed from the eggs and mCherry and GFP signals were imaged using a LSM 880 Zeiss confocal microscope.
Mouse xenograft models
For tail vein and mammary fat pad injections we used female, 8–10-week-old athymic Foxn1 nu/nu mice acquired from Harlan Sprague Dawley. The MMTV-PyMT immune-competent transgenic mice were female, 10–12 week old and the mouse model as a robust model for breast cancer was described previously52. All mouse experiments were conducted according to institutionally approved animal protocols (IACUC MSSM: LA11-00017, 11-0032; Einstein: 20130909). No statistical method was used to predetermine sample size; we used empirical methods from past studies. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
In the MDA-MB-231-5HRE-ODD-mCherry-GFP xenograft experiments (Fig. 1a–c and Supplementary Fig. 1A–C), 2 × 106 cells were subcutaneously injected in the inguinal mammary fat pad of athymic Foxn1 nu/nu mice and tumours were harvested at 1.5–2 cm diameter and immediately snap frozen for IF staining. For the in vivo extravasation experiment (Fig. 4d), stably GFP-expressing T-HEp3 cells were grown in monolayer in normoxia (21%O2) or hypoxia (1%O2) for 72 h. Then, 3 × 105 cells in 50 μl PBS were tail vein injected in Foxn1 nu/nu mice. Twenty-four hours later mice were euthanized and the lungs were flushed with 3 ml ice-cold PBS to exclude blood from the analysis and dissected from the animals. Then, one lung of each animal was minced and collagenased for 30 min at 37 °C. The total lung cell suspension (1 ml) was manually screened for GFP+ cells using a direct fluorescence microscope (Nikon Eclipse Ti-S equipped with an EXFO X-Cite 120 fluorescence illumination system) and the total cell number per animal was calculated. For all tail vein injection experiments, the indicated amounts of pretreated cells suspended in 50 μl PBS were injected in Foxn1 nu/nu mice and the animals were euthanized using CO2 at the designated time points (Figs 5, 7 and 8).
For the in vivo label-retention assays (Fig. 5), the lungs of the mice were flushed with 3 ml PBS and one lung was immediately mounted in Tissue-Tek O.C.T. Compound (Sakura Finetek USA), snap frozen and stored it −80 °C until further use. The other lung was minced and digested using collagenase-IA (Sigma-Aldrich) for 30 min at 37 °C.
In case of the TET-ON-H2B-GFP T-HEp3 label-retention experiment (Fig. 5a–d), the resulting cell suspension was diluted to 10 ml volume and the amount of tagged tumour cells (H2B-GFP, Dendra2 green/red) in three 100 μl samples of each animal was manually counted using a fluorescence microscope (see above). Using the total number of lung-lodged T-HEp3 cells (obtained by re-inducing the TET-ON-H2B-GFP by culturing the total lung cell suspension with doxycycline for 16 h) (Fig. 5c), the percentage of label-retaining cells was calculated (Fig. 5d).
For the 231-H2B-Dendra2 tail vein experiment (Fig. 5e–h), the lungs were treated as above. Following digestion of the lungs, the amount of Dendra2-green cells was counted manually as above, followed by assessment of Dendra2-red expression. Using these results, the total number of lung-lodged 231-H2B-Dendra2 cells (Dendra2 green, Fig. 5g) and the percentage of red-label-retaining cells (Dendra2 green and red, Fig. 5h) were calculated per mouse.
In the chemotherapy experiment (Fig. 8a–d and Supplementary Fig. 4B) we used 3.5 mg kg−1 body weight cisplatin (NDC 63323-103-65, Fresenius Kabi) or an equal volume of sterile, injection-grade 0.9% NaCl solution for i.p. injection. Mice were weighed every other day before injection to account for possible weight loss due to therapy.
For the spontaneous lung DTC experiments we used MMTV-PyMT-Dendra2 mice (Fig. 8e–i and Supplementary Fig. 4C–E). The spontaneous, luminal-like mammary tumours carry the photoswitchable Dendra2 protein. When the tumours were palpable in the fat pad, they were excised, minced and injected in the inguinal mammary fat pad of non-tumour-bearing FVB mice, to eliminate the effect of multiple simultaneous tumours. After about 8 weeks, when the tumours were 1.5–2 cm in diameter, the whole tumour was photoconverted in vivo for 5 min using a custom-built 405 nm light-emitting, low-heat diode. This method caused no thermal damage to the tissue and the wound was closed over the tumour. Seven days later the animals were euthanized and the lungs were snap frozen in O.C.T. for further analysis.
Human samples
All human tumour samples were de-identified and written informed consent was obtained from all patients prior to operation. Approval was granted under IRB number HSM no. 12-00145/GCO no. 12-0366.
Tissue culture experiments
All cells, if not indicated otherwise, were cultured in normoxic (21%O2) or hypoxic (1%O2) cell culture conditions. The T-HEp3 and MDA-MB-231 cells were cultured in DMEM medium (Corning Cellgro) with 10% FBS (Sigma, for T-HEp3 and Atlanta Biologicals, for MDA-MB-231) and 1% Pen/Strep (Gibco). ZR-75-1 cells were grown in RPMI medium (Corning Cellgro) with 10% FBS and 1% Pen/Strep (both the same as for MDA-MB-231). The medium of Dendra2-transfected cells was supplemented with G418 (InvivoGen) for selection maintenance (MDA-MB-231: 1.0 mg ml−1; ZR-75-1: 0.5 mg ml−1; T-HEp3: 0.4 mg ml−1).
For the re-exposure to DFOM in vitro (Supplementary Fig. 2H), T-HEp3 cells derived from the Hi-NANIVID CAM tumours at day 6 were plated on 8-well chamber slides (20,000 cells per well), allowed to attach for 24 h and then treated with PBS or DFOM (90 μM, Sigma-Aldrich) for 24 h before fixation and staining for GLUT1.
For the plating efficiency assay, MDA-MB-231, ZR-75-1 and T-HEp3 cells were cultured for 24 h in normoxia or for 24 h, 48 h or 72 h in hypoxia, followed by plating (1,000 cells per well) and culture in standard TC conditions. Colonies (clusters of >5 cells) were counted at day 8 (see Supplementary Fig. 3I).
Knockdown experiments
The knockdown experiments (Figs 3h,i and 6c and Supplementary Fig. 3G,H,J) were conducted in antibiotic-free medium for 24 h, using iPORT NeoFX Transfection Agent (Thermo Fisher) according to the manufacturer’s instructions. NR2F1 (50 nM) and HIF1α (20 nM) siRNA, as well as equal amounts of scrambled control siRNA were used, as in our previous publication16 and in this paper. See Supplementary Table 4 for further information on siRNA.
Matrigel assay
231-H2B-Dendra2, ZR-75-1 H2B-Dendra2 and ZR-75-1 H2B-Dendra2 NR2F1 knockdown cells were seeded in Matrigel-coated (50 μl per well) 8-well chamber slides (Fig. 6a–c). Medium containing 5% FBS and 2% Matrigel was changed every other day. The fluorescent cells in the Matrigel were imaged and scored using a Nikon Eclipse Ti-S microscope on the indicated days.
TUNEL assay
Paraffin-embedded tissue sections were stained using the In Situ Cell Death Detection kit (Roche) according to the manufacturer’s instructions. Briefly, following slide dewaxation and rehydration in xylene and a graded alcohol series, slides were microwaved in 10 mM citrate buffer, pH 6.0 for 12 min. TUNEL reaction was performed at 37 °C for 30 min and stopped by washing the slides with 0.3 M NaCl 0.03M Na2C6H6O7. Vimentin staining was performed as described above.
Quantitative PCR (qPCR)
Whole RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. Following reverse transcription using MMuLV RT (NEB), quantitative qPCR for GLUT1, DEC2, NR2F1, p27 and HIF1α was performed as described previously16 using Taq DNA polymerase (NEB) in the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). GAPDH was used as a housekeeping gene. All primers were purchased from IDT. For the specific primer sequences see Supplementary Table 3.
Immunohistochemistry (IHC) and immunofluorescence (IF) assays
Paraffin-embedded tissue sections were stained using the Vectastain ABC-kit (Vector Laboratories) according to the manufacturer’s instructions. See Supplementary Table 2 for complete list of antibodies. Briefly, following slide hydration in xylene and a graded alcohol series, slides were microwaved in 10 mM citrate buffer for 12 min for antigen retrieval. Endogenous peroxidase activity was quenched by 3% H2O2. Binding of the primary antibody was carried out at 4 ° C overnight, with detection by secondary antibodies at room temperature for 1 h. Liquid DAB was used as chromogen (Vector Laboratories).
For IF stainings, paraffin-embedded sections were first de-paraffinized and rehydrated and antigen retrieval was conducted as described above; frozen sections were thawed to room temperature while covered with PBS. Cell membranes were permeabilized with 0.5% Triton X in PBS for 5 min at room temperature. Sections were blocked with 3% normal goat serum (NGS) in PBS for 60 min at room temperature prior to the first antibody incubation. Antibody binding was carried out at 4 °C overnight, followed by washing with PBS (3 × 5 min) and blocking for 60 min at room temperature. A secondary, fluorescent antibody reacting to the primary was applied for 1 h in the dark at room temperature (all: Invitrogen). Slides were mounted with Pro-Long Gold with DAPI (Life Technologies). The IF and IHC stainings were evaluated using a Leica DM 5500 B microscope with DAPI, GFP, Cy-3 and Y5 filter cubes or a Leica TCS SP5 II confocal Laser-microscope (Leica). If not otherwise indicated, slides were scored manually by bright-field (IHC) or fluorescence microscopy (IF). Total cell numbers per high-power field (40×, 100×, see legend) were counted and the percentages of positive or negative cells were calculated.
Scoring using ImageJ and MetaMorph software
The positive area in the MDA-MB-231-HIF reporter xenografts was evaluated using the open-source processing software Fiji (ImageJ) (Fig. 1a–c and Supplementary Figs 1A–C, 2A–D and 3B). The pictures were duplicated; one picture was thresholded to the staining appropriate intensities and a binary image was created. The area covered by positive staining was quantified in the original file using the binary image as a template. In the xenografts, the area covered by the specific antigen was normalized to the expression of mCherry for hypoxic areas and GFP for normoxic areas. The histograms were created using Fiji (ImageJ). For the diffusion analysis (Supplementary Fig. 2A–D), pixel intensity of the red channel in >60 cell clusters was measured at indicated distances from the iNANIVID, as indicated above. For the xenografts, 20 high-power fields were scored.
For the scoring of nuclear HIF1α and nuclear p27 in CAM tumour sections (Supplementary Fig. 3C–E), the nuclei of T-HEp3 cells in 5–6 different fields per tumour were outlined and the integrated fluorescence intensities of the red channel (HIF1α) and from the green channel (p27) were quantified using MetaMorph software (Molecular Devices). For HIF1αhigh and p27high nuclear levels we considered values >1.5× the negative control. The GLUT1 integrated fluorescence intensity in individual cells on stained CAM tumour sections or in tissue culture (Supplementary Fig. 2E,H) was measured using MetaMorph software.
Statistics and reproducibility
All statistical analysis was carried out using the GraphPad Prism software version 5.0d. Non-parametric Mann–Whitney test or Student’s t-test was used to calculate the significance in differences between two groups, Kruskal–Wallis test was used to calculate the difference between more than three groups, and Pearson’s correlation was used to correlate staining in single cells, as indicated in the figure legends; a P value of <0.05 was considered significant. All in vivo (both mouse and CAM) and in vitro experiments were independently repeated and included at least three biologically independent samples, as indicated in the legends. All IF or IHC stainings were performed in duplicate and accompanied by appropriate isotype and negative controls.
Data availability
Source data for Figs 2d and 5c,d,g,h and Supplementary Figs 1E and 3F,G,I have been provided as Supplementary Table 5. All other data supporting the findings of this study are available from the corresponding author on request.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank the Aguirre-Ghiso, Condeelis and Castracane laboratories for useful discussions. We thank N. Linde and M. S. Sosa for help and advice during initial phases of this study on the detection of TGFβ2 and NR2F1. This study was supported by: the Samuel Waxman Cancer Research Foundation Tumor Dormancy Program to J.A.A.-G.; the NIH/NCI TMEN U54CA163131 to J. Condeelis, P.J.K., J. Castracane and J.A.A.-G.; NIH/NCI grants CA109182 and CA191430 to J.A.A.-G.; DoD-BCRP Breakthrough Award (BC132674) to J.A.A.-G. and J. Condeelis; NCI Cancer Center P30 grant CA196521 to J.A.A.-G.; the TCI Young Scientist Cancer Research Award JJR Fund and NCI K22CA196750 grants to J.J.B.-C.; and the German Research Foundation (DFG) Fellowship (FL 865/1-1) and University Hospital Duesseldorf, Department of General and Visceral Surgery to G.F. Special optical devices were constructed and validated in the Gruss Lippoer Biophotonic Center and Integrated Imaging Program at Einstein. All imaging was performed in the Microscopy CORE at the Icahn School of Medicine at Mount Sinai. We thank N. Tzavaras (Microscopy CORE) for his technical help.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization: J.A.A.-G., J. Condeelis, P.J.K., A.A.-V., G.F.; methodology: J. Condeelis, J. Castracane, D.E., A.R.N., J.F.C., V.C.; investigation: G.F., A.A.-V., A.R.N., J.F.C., V.C.; resources: Y.W., M.R.P., J.K.W., V.V., J.F.C., J.J.B.-C.; writing: G.F., J.A.A.-G.; visualization: G.F., A.A.-V.; funding acquisition: J. Condeelis, J. Castracane, P.J.K., J.A.A.-G., J.J.B.-C., G.F.; supervision: J.A.A.-G., J. Condeelis.
COMPETING FINANCIAL INTERESTS
J.A.A.-G. receives funding from E. Lilly and co. J. Condeelis is a consultant for, and has equity in MetaStat, Inc.; J. Condeelis is also a consultant for Deciphera Pharmaceuticals.
Note: Supplementary Information is available in the online version of the paper
References
- 1.Polyak K, Shipitsin M, Campbell-Marrotta L, Bloushtain-Qimron N, Park SY. Breast tumor heterogeneity: causes and consequences. Breast Cancer Res. 2009;11:S18. [Google Scholar]
- 2.Sharma SV, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischer CA, et al. Co-overexpression of p21 and Ki-67 in head and neck squamous cell carcinoma relative to a significantly poor prognosis. Head Neck. 2011;33:267–273. doi: 10.1002/hed.21440. [DOI] [PubMed] [Google Scholar]
- 4.Gronroos TJ, et al. Hypoxia, blood flow and metabolism in squamous-cell carcinoma of the head and neck: correlations between multiple immunohistochemical parameters and PET. BMC Cancer. 2014;14:876–887. doi: 10.1186/1471-2407-14-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 6.Gligorijevic B, Bergman A, Condeelis J. Multiparametric classification links tumor microenvironments with tumor cell phenotype. PLoS Biol. 2014;12:e1001995. doi: 10.1371/journal.pbio.1001995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein CA. Selection and adaptation during metastatic cancer progression. Nature. 2013;501:365–372. doi: 10.1038/nature12628. [DOI] [PubMed] [Google Scholar]
- 8.Klein CA, et al. Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet. 2002;360:683–689. doi: 10.1016/S0140-6736(02)09838-0. [DOI] [PubMed] [Google Scholar]
- 9.Chery L, et al. Characterization of single disseminated prostate cancer cells reveals tumor cell heterogeneity and identifies dormancy associated pathways. Oncotarget. 2014;5:9939–9951. doi: 10.18632/oncotarget.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer. 2014;14:430–439. doi: 10.1038/nrc3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bragado P, Sosa MS, Keely P, Condeelis J, Aguirre-Ghiso JA. Microenvironments dictating tumor cell dormancy. Recent Results Cancer Res. 2013;195:25–39. doi: 10.1007/978-3-642-28160-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sosa MS, Bragado P, Debnath J, Aguirre-Ghiso JA. Regulation of tumor cell dormancy by tissue microenvironments and autophagy. Adv Exp Med Biol. 2013;734:73–89. doi: 10.1007/978-1-4614-1445-2_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alvarez JV, et al. Par-4 downregulation promotes breast cancer recurrence by preventing multinucleation following targeted therapy. Cancer Cell. 2013;24:30–44. doi: 10.1016/j.ccr.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim RS, et al. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS ONE. 2012;7:e35569. doi: 10.1371/journal.pone.0035569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng Q, et al. A signature of epithelial-mesenchymal plasticity and stromal activation in primary tumor modulates late recurrence in breast cancer independent of disease subtype. Breast Cancer Res. 2014;16:407–420. doi: 10.1186/s13058-014-0407-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sosa MS, et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat Commun. 2015;6:6170. doi: 10.1038/ncomms7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alarcon R, Koumenis C, Geyer RK, Maki CG, Giaccia AJ. Hypoxia induces p53 accumulation through MDM2 down-regulation and inhibition of E6-mediated degradation. Cancer Res. 1999;59:6046–6051. [PubMed] [Google Scholar]
- 18.Piccolo S, Enzo E, Montagner M. p63, Sharp1, and HIFs: master regulators of metastasis in triple-negative breast cancer. Cancer Res. 2013;73:4978–4981. doi: 10.1158/0008-5472.CAN-13-0962. [DOI] [PubMed] [Google Scholar]
- 19.Montagner M, et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature. 2012;487:380–384. doi: 10.1038/nature11207. [DOI] [PubMed] [Google Scholar]
- 20.Goda N, et al. Hypoxia-inducible factor 1α is essential for cell cycle arrest during hypoxia. Mol Cell Biol. 2003;23:359–369. doi: 10.1128/MCB.23.1.359-369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galson DL, et al. The orphan receptor hepatic nuclear factor 4 functions as a transcriptional activator for tissue-specific and hypoxia-specific erythropoietin gene expression and is antagonized by EAR3/COUP-TF1. Mol Cell Biol. 1995;15:2135–2144. doi: 10.1128/mcb.15.4.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yadav V, Matsakas A, Lorca S, Narkar VA. PGC1β activates an antiangiogenic program to repress neoangiogenesis in muscle ischemia. Cell Rep. 2014;8:783–797. doi: 10.1016/j.celrep.2014.06.040. [DOI] [PubMed] [Google Scholar]
- 23.Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem. 2013;288:10819–10829. doi: 10.1074/jbc.M112.442939. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 25.Liu L, et al. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chéry L, et al. Characterization of single disseminated prostate cancer cells reveals tumor cell heterogeneity and identifies dormancy associated pathways. Oncotarget. 2014;5:9939–9951. doi: 10.18632/oncotarget.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ranganathan AC, Ojha S, Kourtidis A, Conklin DS, Aguirre-Ghiso JA. Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Res. 2008;68:3260–3268. doi: 10.1158/0008-5472.CAN-07-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006;66:1702–1711. doi: 10.1158/0008-5472.CAN-05-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adam AP, et al. Computational identification of a p38SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer Res. 2009;69:5664–5672. doi: 10.1158/0008-5472.CAN-08-3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koumenis C, Wouters BG. “Translating” tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol Cancer Res. 2006;4:423–436. doi: 10.1158/1541-7786.MCR-06-0150. [DOI] [PubMed] [Google Scholar]
- 31.Bi M, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005;24:3470–3481. doi: 10.1038/sj.emboj.7600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romero-Ramirez L, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64:5943–5947. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
- 33.Koumenis C, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol Cell Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, et al. Direct visualization of the phenotype of hypoxic tumor cells at single cell resolution in vivo using a new hypoxia probe. Intravital. 2016;5:e1187803. doi: 10.1080/21659087.2016.1187803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azuma C, Raleigh JA, Thrall DE. Longevity of pimonidazole adducts in spontaneous canine tumors as an estimate of hypoxic cell lifetime. Radiat Res. 1997;148:35–42. [PubMed] [Google Scholar]
- 36.Chou SC, Flood PM, Raleigh JA. Marking hypoxic cells for complement and cytotoxic T lymphocyte-mediated lysis: using pimonidazole. Br J Cancer Suppl. 1996;27:S213–S216. [PMC free article] [PubMed] [Google Scholar]
- 37.Williams JK, et al. Validation of a device for the active manipulation of the tumor microenvironment during intravital imaging. IntraVital. 2016;5:e1182271. doi: 10.1080/21659087.2016.1182271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raja WK, et al. Development path and current status of the NANIVID: a new device for cancer cell studies. J Micro Nanolithogr. 2012;11:013013–013023. doi: 10.1117/1.JMM.11.1.013013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ossowski L. Plasminogen activator dependent pathways in the dissemination of human tumor cells in the chick embryo. Cell. 1988;52:321–328. doi: 10.1016/s0092-8674(88)80025-4. [DOI] [PubMed] [Google Scholar]
- 40.Foudi A, et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol. 2009;27:84–90. doi: 10.1038/nbt.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo M, et al. Hypoxia-mimetic agents desferrioxamine and cobalt chloride induce leukemic cell apoptosis through different hypoxia-inducible factor-1α independent mechanisms. Apoptosis. 2006;11:67–77. doi: 10.1007/s10495-005-3085-3. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez-Elsner T, et al. Synergistic cooperation between hypoxia and transforming growth factor-β pathways on human vascular endothelial growth factor gene expression. J Biol Chem. 2001;276:38527–38535. doi: 10.1074/jbc.M104536200. [DOI] [PubMed] [Google Scholar]
- 43.Triantafyllou A, et al. Cobalt induces hypoxia-inducible factor-1α (HIF-1α) in HeLa cells by an iron-independent, but ROS-, PI-3K- and MAPK-dependent mechanism. Free Radic Res. 2006;40:847–856. doi: 10.1080/10715760600730810. [DOI] [PubMed] [Google Scholar]
- 44.Marotta D, et al. In vivo profiling of hypoxic gene expression in gliomas using the hypoxia marker EF5 and laser-capture microdissection. Cancer Res. 2011;71:779–789. doi: 10.1158/0008-5472.CAN-10-3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aguirre Ghiso JA. Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene. 2002;21:2513–2524. doi: 10.1038/sj.onc.1205342. [DOI] [PubMed] [Google Scholar]
- 46.Kim J, Yu W, Kovalski K, Ossowski L. Requirement for specific proteases in cancer cell intravasation as revealed by a novel semiquantitative PCR-based assay. Cell. 1998;94:353–362. doi: 10.1016/s0092-8674(00)81478-6. [DOI] [PubMed] [Google Scholar]
- 47.Zijlstra A, Lewis J, Degryse B, Stuhlmann H, Quigley JP. The inhibition of tumor cell intravasation and subsequent metastasis via regulation of in vivo tumor cell motility by the tetraspanin CD151. Cancer Cell. 2008;13:221–234. doi: 10.1016/j.ccr.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ossowski L. In vivo invasion of modified chorioallantoic membrane by tumor cells: the role of cell surface-bound urokinase. J Cell Biol. 1988;107:2437–2445. doi: 10.1083/jcb.107.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klein CA. Framework models of tumor dormancy from patient-derived observations. Curr Opin Genet Dev. 2011;21:42–49. doi: 10.1016/j.gde.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 50.Bragado P, et al. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat Cell Biol. 2013;15:1351–1361. doi: 10.1038/ncb2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shemirani B, Crowe DL. Hypoxic induction of HIF-1α and VEGF expression in head and neck squamous cell carcinoma lines is mediated by stress activated protein kinases. Oral Oncol. 2002;38:251–257. doi: 10.1016/s1368-8375(01)00052-5. [DOI] [PubMed] [Google Scholar]
- 52.Gligorijevic B, Kedrin D, Segall JE, Condeelis J, van Rheenen J. Dendra2 photoswitching through the Mammary Imaging Window. J Vis Exp. 2009 doi: 10.3791/1278. http://dx.doi.org/10.3791/1278. [DOI] [PMC free article] [PubMed]
- 53.Erler JT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 54.Finger EC, et al. Hypoxic induction of AKAP12 variant 2 shifts PKA-mediated protein phosphorylation to enhance migration and metastasis of melanoma cells. Proc Natl Acad Sci USA. 2015;112:4441–4446. doi: 10.1073/pnas.1418164112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chaturvedi P, et al. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J Clin Invest. 2013;123:189–205. doi: 10.1172/JCI64993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14:611–622. doi: 10.1038/nrc3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ossowski L, Reich E. Changes in malignant phenotype of a human carcinoma conditioned by growth environment. Cell. 1983;33:323–333. doi: 10.1016/0092-8674(83)90414-2. [DOI] [PubMed] [Google Scholar]
- 58.Cailleau R, Olive M, Cruciger QV. Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro. 1978;14:911–915. doi: 10.1007/BF02616120. [DOI] [PubMed] [Google Scholar]
- 59.Engel LW, et al. Establishment and characterization of three new continuous cell lines derived from human breast carcinomas. Cancer Res. 1978;38:3352–3364. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for Figs 2d and 5c,d,g,h and Supplementary Figs 1E and 3F,G,I have been provided as Supplementary Table 5. All other data supporting the findings of this study are available from the corresponding author on request.