

Abstract
The majority of cancer deaths are due to metastases that can occur years or decades after primary tumor diagnosis and treatment. Disseminated tumor cells (DTCs) surviving in a dormant state in target organs appear to explain the timing of this phenomenon. Knowledge on this process is important as it might provide a window of opportunity to prevent recurrences by eradicating dormant DTCs and/or by maintaining DTCs in a dormant state. Importantly, this research might offer markers of dormancy for early monitoring of metastatic relapse. However, our understanding of the mechanisms underlying the regulation of entry into and exit from dormancy is still limited and crippling any therapeutic opportunity. While cancer cell-intrinsic signaling pathways have been linked to dormancy regulation, it is likely that these pathways and the switch controlling reactivation from dormancy are regulated by microenvironmental cues. Here we review and discuss recent findings on how the microenvironment regulates cancer dormancy and raise new questions that may help advance the field.
1. INTRODUCTION
Metastasis formation is responsible for the majority of cancer deaths and is caused by cancer cells disseminated from primary tumors that persist in the host after primary tumor removal. Metastasis formation consists of several steps: local invasion from the primary tumor and intravasation, survival in circulation, extravasation, and proliferation in a target organ microenvironment. Importantly, after extravasation and before proliferation into detectable metastasis, years or even decades can pass. Long time periods where patients present with no evidence of disease (NED) followed by late recurrences are explained by the survival of disseminated tumor cells (DTCs) in a dormant state. The mechanisms that determine the amount of time that can pass between the extravasation of DTCs and their proliferation into metastatic masses are one of the most important questions in cancer biology.
From a cell biology perspective the asymptomatic phase that precedes the reactivation of DTCs to form detectable metastases can be explained by cellular dormancy, where single DTCs survive in a quiescent, reversibly growth-arrested state for long stretches of time, and population-based dormancy, where micrometastases are limited in their growth due to the lack of vascularization and increased cell death that compensates for proliferation or due to immune-mediated responses, which results in a balance between proliferation and DTC death (Aguirre-Ghiso, 2007). While these models of dormancy are not mutually exclusive and might even coexist in the same patient, we will focus on cellular dormancy that is best supported by clinical evidence (Banys, Hartkopf, et al., 2012; Morrissey, Vessella, Lange, & Lam, 2015). Several studies showed that DTCs are frequently found in cancer patients with NED (Braun et al., 2005; Chery et al., 2014; Klein, 2009; Schardt et al., 2005), and the detection of DTCs in the bone marrow is associated with worse clinical outcome in many solid cancers (Braun et al., 2000; Hartkopf et al., 2014, 2015; Thorban, Rosenberg, Busch, & Roder, 2000; Wollenberg et al., 2004). These studies demonstrate that cancer patients with dormant DTCs are at higher risk for metastatic relapses and thus underline the clinical importance of dormancy. The time of NED prior to the reemergence of DTCs from dormancy could be used to for therapeutic intervention but is currently not used to treat patients. Being able to predict if a patient is at risk of recurrence, or not, based on DTC-associated biology would allow more specific therapeutic strategies and avoid overtreatment with antiproliferative therapies that might not address the residual disease biology. Also, dormant DTCs could either be awakened and eradicated or maintained in a dormant state during this time (Aguirre-Ghiso, 2007; Ghajar, 2015; Sosa, Bragado, & Aguirre-Ghiso, 2014). Yet, this window of opportunity is missed, mostly because our understanding of the mechanisms that characterize the dormant state and how DTCs can reemerge from it is cripplingly limited (Aguirre-Ghiso, Bragado, & Sosa, 2013).
Several cancer cell-intrinsic signal pathways that lead to cellular dormancy have been described. Historically, the balance between activated extracellular regulated kinase (ERK1/2) and activated p38α/β was the first signaling mechanism that has been connected reproducibly to DTC dormancy (Adam et al., 2009; Aguirre Ghiso, Kovalski, & Ossowski, 1999; Aguirre-Ghiso, Estrada, Liu, & Ossowski, 2003; Aguirre-Ghiso, Liu, Mignatti, Kovalski, & Ossowski, 2001; Bragado et al., 2013; Kobayashi et al., 2011; Najmi, Korah, Chandra, Abdellatif, & Wieder, 2005; Ruppender et al., 2015; Sosa, Avivar-Valderas, Bragado, Wen, & Aguirre-Ghiso, 2011). Interestingly, early on, a link to the microenvironment was identified in these studies, as the balance between ERK1/2 and p38 signaling was regulated by fibronectin and uPA signaling via the uPA receptor and specific integrins (Aguirre Ghiso et al., 1999; Aguirre-Ghiso et al., 2001). Phosphorylation of p38 leads to the activation of the unfolded protein response pathway, which promotes cell survival and dormancy through ATF6/Rheb/mTOR signaling (Schewe & Aguirre-Ghiso, 2008) and leads to the induction of the dormancy-associated transcription factors DEC2/Sharp1, p27Kip1, p21, and NR2F1 (Bragado et al., 2013; Sosa et al., 2015). These mechanisms are integrated (Fig. 1A) to coordinate a deep but still reversible growth arrest and robust survival pathways. For more in-depth information we would like to refer to reviews previously published by our group (Aguirre-Ghiso, 2007; Sosa et al., 2014). These mechanisms explain how tumor cells regulate specific signal transducers to enter a state of cellular dormancy (G0–G1 arrest). Yet, the fact that tumor cells, which have disseminated from proliferating tumor masses, enter quiescence and stop proliferating but yet maintain reactivating capacity is puzzling. One likely explanation could be the microenvironment partially controlling the switch between DTC proliferation and dormancy.
Fig. 1.

Overview of dormancy-inducing signaling pathways. (A) Overview of dormancy marker expression in DTCs based on known dormancy-signaling pathways. (B) Microenvironment-derived atRA, TGFβ2, and BMP-4 and -7 cooperate to induce a dormant state in DTCs characterized by activating p38 and NR2F1 and inhibiting ERK1/2 signaling. p38 and NR2F1 induce the cell cycle inhibitors p27 and p21, which results in cell cycle arrest (Bragado et al., 2013; Kobayashi et al., 2011; Sosa et al., 2015).
The tumor microenvironment is usually defined as the sum of all cellular and extracellular components surrounding cancer cells. In the context of a healthy epithelial tissue, the microenvironment will maintain tissue integrity and is in turn regulated by stromal cells such as fibroblasts and myeloid cells. Several studies support that changes that subvert the tumor microenvironment are required for malignant cells to grow into tumors (Hanahan & Coussens, 2012; Mueller & Fusenig, 2004). Thus, since all adult tissues encode mechanisms to essentially prevent uncontrolled ectopic growth, it is reasonable to hypothesize that a tumor-naïve target organ microenvironment may encode regulatory mechanisms to prevent the expansion of DTCs and this may result in dormancy onset. Similarly, one could propose that changes in the target organ microenvironment might awaken dormant DTCs and allow them to proliferate and thus induce late recurrences. In this chapter we will focus on reviewing recent findings that analyzed the influence of microenvironmental cues and cellular components on dormancy and hypothesize about their influence on dormancy induction and exit from dormancy. The goal is to develop potential answers to persistent questions that need to be addressed to find a solution to the urgent clinical problem of dormancy.
2. MODELS TO STUDY TUMOR CELL DORMANCY
One of the challenges in studying dormancy is that by definition it is undetectable using conventional whole-body imaging tools and takes place over long time periods.
This provides a challenge to drug development, as clinical trials are usually performed with far-progressed patient cohorts. Testing drugs in a metastasis prevention setting with adjuvant therapies would be a radical shift in the standard of clinical trials and requires better insight into dormant disease.
One of the main obstacles to studying dormancy, cited by basic researchers repeatedly, is the lack of model systems. Most basic research relies on fast-growing cancer cell lines and fast transgenic oncogene models. It is also common to use aggressively growing metastasis models where metastases develop without any latency. Moreover, most metastasis assays focus on macrometastases as an end point and rely on the use of clones selected for aggressiveness. Commonly, the presence of solitary DTCs or micrometastases is not investigated and the absence of macrometastases is interpreted as the inability of cancer cells to disseminate without investigating which step of the metastatic cascade was not completed. Therefore, many studies simply miss solitary DTC biology.
Yet, the notion that there is a lack of good models to study dormancy is not correct. Table 1 provides examples of some of the model systems used to study dormancy that will be briefly discussed here.
Table 1.
Models to Study Cancer Dormancy
| Model System | Description | Examples |
|---|---|---|
| In vitro models | Several dormant cancer cells retain quiescence when grown as in 3D in vitro models | Ghajar et al. (2013), Barkan and Green (2011), and Marlow and Dontu (2015) |
| Isolation of latent cancer cell lines | Latent clones can be isolated from spontaneous mouse tumors | 4T07 (Aslakson & Miller, 1992; Gao et al., 2012); D2O.R (Morris, Tuck, Wilson, Percy, & Chambers, 1993) |
| Clonal cell line variants | Cell line variants are established through in vivo selection of clones derived from indolent cell lines that show metastatic growth (e.g., D2.1) or selection of indolent clones from metastatic lines (e.g., MDA-MB231-SCP6) | MDA-MB231-SCP6 (Lu et al., 2011); H2087-LCC; HCC1954-LCC (Malladi et al., 2016) |
| Dormant DTC analysis in PDX tumors | Dormant tumor cells derived from PDX tumors can be analyzed in vivo | Aguirre-Ghiso et al. (2001), Lawson, Bhakta, et al. (2015), and Ossowski and Reich (1980) |
| Spontaneous mouse cancer models | Dormant DTCs and latent metastases can be detected using spontaneous mouse cancer models. Inducible transgenic mouse cancer models allow for investigation of residual dormant DTCs after oncogene withdrawal | Dormant DTCs: MMTV-HER2 (Husemann et al., 2008); indolent lung metastases: MMTV-Wnt1 (Li, Hively, & Varmus, 2000); inducible transgenic mouse models (Abravanel et al., 2015; Felsher & Bishop, 1999; Gunther et al., 2002) |
| Human sample validation | DTCs and CTCs expressing dormancy markers can be isolated from patients | DTCs (Chery et al., 2014; Sosa et al., 2015); CTCs (Vishnoi et al., 2015) |
The in vivo metastasis assay used most commonly is the experimental metastasis assay where cell lines are injected into circulation, either through the tail vein or into the left cardiac ventricle or the iliac artery (Box & Eccles, 2011; Rosol, Tannehill-Gregg, Corn, Schneider, & McCauley, 2004; Wang et al., 2015). Alternatively, cancer cell lines are grown orthotopically or subcutaneously and metastases derived from spontaneously DTCs are monitored. This system allows monitoring multiorgan dormancy by tracking tagged DTCs to monitor solitary cell biology and/or label retention at single-cell resolution, as used in cancer models and studies of hematopoietic stem cell dormancy (Lawson, McDonald, et al., 2015). Even pathway biosensors can be used to monitor dormancy pathways in DTCs to understand their regulation (Aguirre-Ghiso, Ossowski, & Rosenbaum, 2004). Additionally, dormant DTCs can be identified in different target organs by detection of proliferation and dormancy markers (Aguirre-Ghiso et al., 2004; Bragado et al., 2013; Sosa et al., 2015), summarized in Fig. 1A.
Instead of using highly aggressive cancer cell lines (i.e. MDA-MB-231), cell lines that give rise predominantly to dormant DTCs can be used in experimental assays or in in vitro assays (Barkan & Green, 2011). Examples are the dormant breast cancer cell line variants 4T07 variant (Aslakson & Miller, 1992) or D2O.R cells (Morris et al., 1993). Moreover, complementary to the common approach to select aggressively growing clonal cell line variants, several studies have described the selection of latent clonal variants that may be informative (Lu et al., 2011; Malladi et al., 2016). A caveat is that the mechanisms identified may be biased by the clonal selection and not encompass important additional mechanisms of microenvironmental and epigenetic regulation of dormancy regulation. Additionally, dormant DTCs can be identified in murine patient-derived xenograft (PDX) experiments, for example, in head and neck squamous cell carcinoma (Aguirre-Ghiso et al., 2001, 2004; Bragado et al., 2013; Sosa et al., 2015) and breast cancer (Lawson, Bhakta, et al., 2015).
Transgenic models also provide an opportunity to study dormancy if the right microenvironment and stage of progression are considered. The group of Dr. Klein has shown that in the MMTV-HER2 and MMTV-PyMT breast cancer models, dissemination occurs in parallel to primary tumor development and that these early disseminating tumor cells enter a dormancy period from which they can reemerge (Husemann et al., 2008). This model allows studying how DTCs evolve ectopically and how the tissue microenvironment might shape genetic and epigenetic changes. Thus, using MMTV-HER2 and MMTV-PyMT in early stages allows for analysis of dormant DTCs. The MMTV-WNT1 breast cancer model that is usually considered to be nonmetastatic frequently develops metastasis after prolonged period of indolence that can only be reached when primary tumors are surgically removed (Li et al., 2000). Our lab has described that some target organ microenvironments such as the bone and liver microenvironment foster dormancy in DTCs (Bragado et al., 2013), a finding that we could reproduce in MMTV-HER2 mice (unpublished data). Transgenic models where the oncogene is under the control of an inducible promoter are another way to model dormancy. Several labs have used these models to mimic therapy-induced dormancy (Abravanel et al., 2015; Moody et al., 2002) and how loss of oncogene expression can induce a residual population that allowed studying both intrinsic and microenvironmental regulation of dormancy induced by oncogene inactivation (Felsher, 2008, 2010; Felsher & Bishop, 1999; Giuriato et al., 2006; Rakhra et al., 2010).
Finally, while Kaplan–Meier plots provide some information on how primary tumor signatures affect metastasis-free intervals, they do not inform on the actual gene signatures and mechanisms at play in the residual disease. Thus, it is essential to model dormancy by studying cancer patients beyond the analysis of Kaplan–Meier plots and by isolating CTCs and DTCs and performing single-cell analysis to investigate dormancy markers (Chery et al., 2014; Sosa et al., 2015; Vishnoi et al., 2015). This summary indicates that there are multiplicities of models to extract information related to dormancy that can be validated in human samples.
3. INTRINSIC AND EXTRINSIC SIGNALS CONVERGE TO INDUCE TUMOR CELL DORMANCY
Several studies showed how cues that regulate adult stem cell biology and the interplay between mitogenic and stress-signaling pathways regulate dormancy onset.
These mechanisms are summarized briefly in Fig. 1B and have been reviewed elsewhere recently (Aguirre-Ghiso, 2007; Sosa et al., 2014). However, less work is available on how dormancy and pluripotency pathways, which are commonly associated with aggressive cancer behavior, are integrated. Insight into this problem came from recent work from our lab showing that dormancy of HNSCC DTCs is dependent on NR2F1 signaling, an orphan receptor of the retinoic acid-signaling pathway (Sosa et al., 2015). This study revealed that NR2F1 upregulated both quiescence and pluripotency genes SOX9, OCT4, SOX2, and NANOG. Thus, dormant DTCs coordinate a profound growth arrest with the upregulation of self-renewal genes. This suggests that dormant cancer cells share characteristics of embryonic and adult stem cells. Interestingly, Scognamiglio et al. (2016) showed recently that upon withdrawal of myc signaling, murine embryonic stem cells enter a reversible dormant state where they are quiescent while retaining the expression of NANOG and OCT4. Upon reintroduction of myc signaling, the cells continue to form normal embryos. This myc-dependent dormancy is recapitulated in many mammals, where blastocysts enter a phase of dormancy, called diapause, induced by microenvironmental signals from the uterus. Interestingly, dormancy induced by p38 and NR2F1 signaling is associated with a downregulation of myc transcriptional activity and NR2F1-antagonized myc signaling for proliferation (Sosa et al., 2015). Cancer cell dormancy might therefore resemble an epigenetic state in which cells remain quiescent while retaining their self-renewing capacities. This tumor cell-intrinsic network can be induced by external factors. All-trans retinoic acid (atRA) is abundant in the bone marrow and regulates hematopoietic stem cell renewal (Ghiaur et al., 2013; Purton et al., 2006). Treatment of T-HEp3 cells with atRA induced NR2F1 and TGFβ2 expression in HNSCC DTCs (Sosa et al., 2015). TGFβ2 is a member of the TGFβ family also abundantly present in the bone and has been shown by our group to induce DTC dormancy through p38-dependent signaling, which leads to induction of the dormancy-associated proteins DEC2/Sharp1 and p27Kip1 (Bragado et al., 2013). These findings indicate that in the bone the microenvironment-derived signals TGFβ2 and atRA might cooperate with tumor-intrinsic signals to allow DTCs to enter a dormant state characterized by growth arrest, survival, and pluripotency gene expression. This might lead DTCs to produce an autocrine loop to maintain dormancy but still retain reactivation plasticity. The clinical relevance of these findings was confirmed by a recent study from the group of Dr. Morrisey. They found that DTCs are present in the bone marrow of both prostate cancer patients with late recurrence or with NED, years after treatment of the primary tumor (Chery et al., 2014). While DTCs in the NED group mostly had a dormant and NR2F1/p38-high transcriptome, both dormant and proliferating DTCs were detected in recurring patients (Chery et al., 2014). Additionally, a transcriptome analysis showed considerable heterogeneity in both groups, demonstrating that there may be more than one form of dormancy even within the same patient and organ, and that dormant and slow cycling/proliferating DTCs can be present at the same time within the same organ.
4. COOPERATIVE EXTRINSIC SIGNALS INDUCE DORMANCY WITHIN THE BONE MICROENVIRONMENT
The bone marrow is home to hematopoietic stem cells that undergo tightly controlled steps of differentiation and proliferation during hematopoiesis (Eaves, 2015). DTCs are frequently found in the bone marrow of cancer patients, even in patients with no evidence of metastases (Braun et al., 2005; Pierga et al., 2003). Many of these patients never develop bone metastases and when they do, they usually have long periods of NED before, while many patients with bone DTCs never develop metastases (Sherry, Greco, Johnson, & Hainsworth, 1986). All this indicates that the bone marrow microenvironment therefore seems to be a growth-inhibitory microenvironment and therefore most DTCs enter dormancy. This might seem counterintuitive to the fact that the bone marrow is also the site of hematopoiesis that includes proliferative steps. However, hematopoietic cell expansion is immediately followed by differentiation (not proliferation) and dormancy of adult stem cells coexists with events of proliferation. Therefore, growth events in the bone microenvironment need to be tightly controlled. One way to resolve the paradox of the coexistence of proliferation and quiescence is the formation of niches within the bone microenvironment. Quiescent hematopoietic stem cells are localized in the hematopoietic stem cell niche (Shiozawa et al., 2011). Several studies indicate that DTCs enter the hematopoietic stem cell niche, where they remain dormant. For a detailed summary of these studies, please refer to a recent review by Ghajar (2015). In addition to these hematopoietic stem cell niche-specific effects, atRA and TGFβ2 are both abundant in the bone marrow. Additionally, Kobayashi et al. found that BMP-7, another TGFβ family member secreted by bone stromal cells, was able to induce a reversible dormancy in intra-tibially injected prostate cancer cells through induction of p38 signaling and upregulation of the metastasis suppressor gene NDRG1 (Kobayashi et al., 2011). The selective bone metastatic suppressive function of recombinant BMP-7 in a metastatic prostate cancer xenograft system was previously described by Buijs et al. (2007), who reported a synergistic mechanism in which TGFβ1, amply present in the bone microenvironment, contributes to this BMP-7 function. Thus, in the bone microenvironment, atRA, TGFβ2, and BMP-7 might provide redundant dormancy-inducing cues that might explain why dormancy can be such a stable state, lasting for years or decades (Fig. 1B). Which stromal cells are secreting atRA, TGFβ2, and BMP-7 and whether their expression is uniformly high or enriched in niches within target organs need to be elucidated.
5. ENDOSTEAL AND PERIVASCULAR NICHES SUPPORT CANCER CELL DORMANCY
In addition to cooperative microenvironment-derived dormancy-inducing cues, the tendency of DTCs to enter dormancy depends on their specific localization within the organ. A recent review by Ghajar summarizes most of the available data on the dormant niche (Ghajar, 2015). Below, we will therefore only briefly cover the most recent publications and raise questions associated with this topic. Lawson et al. found that dormant myeloma bone marrow DTCs were engaging with osteoblasts in the endosteum, whereas proliferating DTCs were not (Lawson, McDonald, et al., 2015). Remarkably, proliferating cells introduced to an experimental endosteal niche entered dormancy. Dormant DTCs can be released from the endosteum and activated through enhanced osteoclast activity induced by sRANKL (Lawson, McDonald, et al., 2015). This shows that dormancy is a reversible state crucially regulated by the microenvironment. It further indicates that while osteoblasts might be involved in inducing dormancy, osteoclast activity might be involved in the escape from dormancy by triggering a vicious cycle that characterizes osteolytic bone metastases (Fig. 2A). The group of Dr. Taichman accordingly showed the dormancy-inducing effect of osteoblasts, showing that osteoblast-derived growth arrest-specific 6 (GAS6) protein signaling through its receptor Axl induces dormancy in prostate cancer DTCs (Shiozawa et al., 2010; Taichman et al., 2013). Interestingly, under hypoxic conditions a negative feedback loop of GAS6/Axl is inhibited, leading to increased Axl expression and possibly maintenance of dormancy in hypoxic microenvironments (Mishra et al., 2012). Additionally, another study had shown that osteoclasts might mediate escape from dormancy (Lu et al., 2011). They found that osteoclast progenitors are recruited to VCAM1+ DTCs through integrin α4β1 signaling. This triggered osteoclast formation in vicinity of DTCs and entry into the vicious cycle (Lu et al., 2011). In contrast, in absence of VCAM1 expression, DTCs failed to recruit osteoclast progenitors and entered prolonged periods of dormancy before the formation of osteolytic macrometastases. While this indicates that osteoclasts drive metastasis formation, the mechanisms of dormancy and whether DTCs entered cellular or population-based dormancy were not addressed (Lu et al., 2011). Additionally, this study compared cell lines with different bone metastasis properties, suggesting that whether DTCs enter dormancy depends on their expression of VCAM1 upon arrival in the bone (Fig. 2B). However, clinical evidence implies that DTCs in the bone commonly go through a phase of dormancy from which they can reemerge spontaneously (Braun et al., 2005; Chery et al., 2014; Pierga et al., 2003). Thus, whether the spontaneous exit from dormancy might be due to spontaneous upregulation of VCAM1 or due to different mechanisms leading to the recruitment of osteoclasts remains to be elucidated.
Fig. 2.

Extrinsic signals inducing dormancy in disseminated tumor cells (DTCs) in different microenvironments. (A) Dormancy induction in the bone microenvironment is in part mediated by osteoblasts through GAS6/AXL signaling (Shiozawa et al., 2010; Taichman et al., 2013). DTCs can escape osteoblast-induced dormancy through activation of TYRO or through activation of osteoclasts. Osteoclasts can be activated through RANKL (Lawson, McDonald, et al., 2015) or through recruitment of osteoclast progenitors via VCAM1/integrin α4β1 signaling (Lu et al., 2011). How these dormancy escape mechanisms occur spontaneously in patients and whether they resemble alternative dormancy pathways or cooperate remain to be established. (B) DTCs in the perivascular niche frequently enter dormancy due to thrombospondin (TSP-1) signaling (Ghajar et al., 2013). Activated endothelium on the other hand releases periostin (POSTN) and TGFβ1 that induce DTC proliferation and possibly escape from dormancy. (C) BMP-4, a TGFβ family member enriched in the lung microenvironment, induces dormancy (Gao et al., 2012). Overexpression of CoCo, a BMP inhibitor, allows DTCs to escape dormancy.
In addition to the endosteal niche, Ghajar et al. found a link between the proliferative state of endothelial cells and the dormancy of DTCs associated with the vessels (Fig. 2B). They identified that resting endothelium produces thrombospondin-1 (TSP-1) that induces quiescence in DTCs (Ghajar et al., 2013). In contrast, DTCs in contact with neovascular tips showed enhanced proliferation and this was mediated by secretion of TGFβ1 and periostin (POSTN) by endothelial tip cells. Since all DTCs by default have to pass through the perivascular niche upon dissemination, the question arises whether dormancy might be the default program of extravasating DTCs and only DTCs entering through activated endothelial tip cell niches are the ones who immediately start to proliferate.
Overall, these studies indicate that DTC dormancy within one target organ microenvironment might be regulated by the specific localization of DTCs in the endosteal or the perivascular niche. It will be interesting to understand whether DTCs might move incidentally into a niche where they enter dormancy or whether they are actively recruited. Furthermore, it remains to be elucidated whether the dormant niche is identical with the adult stem cell niche.
6. REACTIVATION FROM DORMANCY
The studies discussed above show that the induction and maintenance of dormancy could be influenced by microenvironment-derived factors such as atRA, TGFβ2, BMP-7, and TSP-1 and by interaction with resting endothelial cells and osteoblasts. Yet, it is a hallmark of dormancy that it is a reversible growth arrest and the exit from dormancy is the clinically most relevant phase. Several studies describe how DTCs escape dormancy, but not how these dormancy escape mechanisms are triggered spontaneously. However, dormancy can last for years and decades, indicating that dormancy-inducing cues can be very stable. This implies that relapses include the switch of DTCs from a dormant into a proliferative state, most likely due to changes in their microenvironment that disrupted the stable dormant state. However, most of the dormancy escape mechanisms described to this date are dormancy exit signals, but do not address how these exit signals are activated spontaneously.
For example, as mentioned above, engagement with osteoclasts in the bone environment can allow exit of DTCs from dormancy (Lawson, McDonald, et al., 2015; Lu et al., 2011). What leads to the spontaneous recruitment of osteoclasts toward DTCs? Lu et al. showed that NFkB signaling leads to activation of VCAM1 expression (Lu et al., 2011). Which factors derived from the microenvironment might be able to activate NFkB and subsequent VCAM1 upregulation in dormant DTCs? Alternatively, can osteoclasts be activated systemically? Could comorbidities such as bone injury or osteoporosis be triggering osteoclast activation? Can dormancy escape be a matter of chance, induced by coincidental colocalization of osteoclasts and dormant DTCs? Does aging affect the expression of dormancy inducers allowing for a slow reactivation process?
Ghajar et al. showed that activation of the endothelium can lead to reactivation of dormant DTCs residing in the perivascular niche (Ghajar et al., 2013). Thus, in the case of perivascular dormancy, is the reemergence of DTCs a matter of chance when the endothelium in their vicinity becomes activated, as shown by Ghajar et al. in vitro (Ghajar et al., 2013)? Alternatively, could DTCs be activated by interaction with stromal cells such as macrophages and therefore move out of the perivascular niche that maintained their dormancy?
Investigating the mechanisms of breast cancer dormancy in the lung, the group of Dr. Giancotti found that the secreted BMP-4 inhibitor protein CoCo reactivates dormant breast cancer cells in the lung through blockade of BMP signaling (Gao et al., 2012). CoCo is secreted by cancer cells and accumulates specifically in the pericellular matrix, where it can neutralize BMP-4 secreted by the stroma, and thus enable DTCs to exit dormancy. The blockade of BMP/p-Smad-driven inhibition of self-renewal led to proliferation of single breast cancer DTCs in the lungs of mice. In contrast, breast cancer cells with low expression of CoCo remained dormant (Fig. 2C). This effect was specific to BMP-rich target organs, as CoCo expression predicted for lung, a BMP-rich organ, but not bone or brain metastasis in a large patient cohort (Gao et al., 2012). However, Gao et al. used two clonal variants that either entered dormancy due to low expression of CoCo or were able to escape due to high CoCo expression (4T1/4T07 breast cancer cell lines). While these identified CoCo as a mechanism to evade dormancy, it remains to be elucidated whether dormant DTCs can upregulate CoCo spontaneously.
A recent preclinical study by Sansone et al. (2016) found that chronic hormone therapy of mice with ER-positive luminal breast cancer led to induction of a dormant, CD133high/ERlow/mitochondrialow population of cells, which produced IL-6 in an autocrine manner. These cells showed an increase in pluripotency genes and were able to exit dormancy by utilizing IL-6/Stat3/Notch3-driven, ER-independent self-renewal and mitochondria reactivation. The use of an anti-IL6R antibody restored ER expression in HT-resistant cells and double treatment with HT/anti-IL6R antibodies in vivo was effective even in HT-resistant tumors (Sansone et al., 2016). These results demonstrate that dormancy may be induced by therapy and that microenvironment-derived cytokines such as IL-6 may be able to reawaken dormant cells. Yet, the cellular source of IL-6 and how its secretion is regulated were not addressed.
In summary, while the phase of dormancy exit is clinically relevant, our understanding how spontaneous exit from dormancy occurs is still very limited. Part of the reason for this is that this process is hard to study. Many commonly used metastasis models use highly aggressive clonal cell line variants that do not undergo dormancy. Only a few cell lines enter dormancy in vivo and while these models do allow identification and study of dormant cells, following the spontaneous escape of dormant cells from dormancy in situ remains a technical challenge. Possibly, recent advances in live imaging might allow to follow the fate of dormant tumor cells and thereby allow us to gain insight into the microenvironmental changes that facilitate dormancy escape. This could be achieved by the use of fluorescently tagged spontaneous mouse models where the fate of single DTCs and their switch from single cells into proliferating metastases in target organs could be followed by live imaging. Such studies could be combined with systemic effects on DTC behavior such as therapy or activation of bone marrow progenitor cells.
7. THE IMMUNE SYSTEM AND DORMANCY
The ability of the immune system to affect tumor progression has been established over the last decades. Interestingly, the immune system can have both tumor-promoting and tumor-inhibitory functions. Inflammatory reactions and association of tumor cells with myeloid cells have been implicated with tumor-promoting functions (for reviews, see Baxter & Hodgkin, 2002; Grivennikov, Greten, & Karin, 2010; Mantovani, Allavena, Sica, & Balkwill, 2008). On the other hand, it has been shown that the adaptive immune system is able to recognize tumor-specific antigens and inhibit tumor growth (Baxter & Hodgkin, 2002; Gajewski, Schreiber, & Fu, 2013).
Metastasis-promoting effects of myeloid cells have recently been reviewed elsewhere (Quail & Joyce, 2013). Yet, the majority of these studies focus on mechanisms how myeloid cells facilitate macrometastatic growth but do not address the interaction of solitary DTCs with myeloid cells and whether this might provide a dormancy/growth switch. Work by Pollard and colleagues revealed that inflammatory Ly6C+ monocytes are required for breast cancer cell extravasation in the lung, a process mediated by chemokine C-C-motif ligand 2 (CCL2) and vascular endothelial growth factor (VEGF) (Qian et al., 2009, 2011; Qian & Pollard, 2010) (Fig. 3A). In addition, a study from the lab of Dr. Massague showed that macrophages are involved in the survival of breast cancer cells in the lung after their extravasation (Chen, Zhang, & Massague, 2011). This is mediated by binding of VCAM1 expressed on cancer cells to β-1-integrin-positive macrophages which leads to induction of Akt signaling in DTCs and allows them to escape TRAIL-induced apoptosis (Chen et al., 2011). These studies imply that monocytes and monocyte-derived macrophages are crucial for extravasation and survival of DTCs.
Fig. 3.

Effects of macrophages and NK cells on dormancy. (A) Monocytes are recruited to DTCs by CCL2 and assist their extravasation in the lung through VEGF secretion (Qian et al., 2009, 2011). In the lung tissue, these monocytes differentiate into metastasis-associated macrophages that promote DTC proliferation (Kitamura et al., 2015; Qian et al., 2015). (B) The role of monocytes and macrophages in dormancy has not been established. If DTCs require monocytes to extravasate, why do some DTCs enter dormancy instead of entering a cycle of macrophage-assisted proliferation? Can niche-derived factors such as TSP-1 in the endothelial niche override the growth-promoting effect of macrophages? Are there macrophage subtypes that induce dormancy? Do some DTCs fail to retain growth-promoting macrophages? Can DTCs extravasate independent of monocytes and therefore enter dormancy? (C) Activation of WNT signaling allows DTCs to enter a proliferative state in which they are more susceptible to cytotoxic signals from NK cells. Once cells enter a dormant state through activation of DKK1 and subsequent WNT inhibition, they are able to escape NK cytotoxic signals (Malladi et al., 2016).
Work from the group of Dr. Pollard also showed that in the lung tissue, monocytes differentiate into metastasis-associated macrophages (MAMs) where they support metastasis growth (Kitamura et al., 2015; Qian et al., 2015) (Fig. 3A). Blocking of MAM reduced metastasis burden even when metastases had established already. These studies were performed in highly aggressive mouse models where metastases form rapidly after extravasation. In these models, monocytes are required for extravasation, thereafter differentiate into MAMs, and as such mediate metastatic growth. However, these models do not show a dormancy phase, precluding the formation of metastases. They therefore do not contribute to our understanding how the fate of solitary DTCs that enter a dormant state prior to their entry into proliferation is affected by MAMs or other immune cells. Clinical evidence shows that in the majority of patients, there is a significant lag between extravasation (that likely already occurred at the time of surgery) and proliferation into detectable metastasis (Hartkopf et al., 2014; Lilleby, Stensvold, Mills, & Nesland, 2013). Therefore, it seems that extravasation and activation of proliferation can be significantly uncoupled processes. This raises several questions (depicted in Fig. 3B): Can other microenvironment-derived signals, such as TSP-1 in the perivascular niche, induce dormancy even when DTCs are associated with macrophages? Can monocytes differentiate into a growth-suppressive macrophage phenotype? Do some DTCs fail to retain macrophages and therefore enter dormancy? Could it be a matter of stoichiometry where monocytes supporting extravasation interact with several DTCs so that there are not enough macrophages to support subsequent growth? Is there a macrophage-independent extravasation step in some cases and some DTCs may thus not interact with macrophages at all? Can DTCs be irresponsive to MAM-derived growth signals? Live imaging analysis might provide the answers to the exact stoichiometry of the macrophage–DTC interaction during extravasation and subsequent growth promotion. However, all will depend on what models are used to test this hypothesis and it is likely that the use of clonal variants selected for aggressive growth will reveal the same mechanisms.
While macrophages are mostly associated with tumor-promoting features, a recent study by the lab of Dr. Massague showed that NK cells are involved in dormancy of DTCs after reactivation (Malladi et al., 2016). Using a model of latency competent cancer cells where dormant clones were selected from an experimental metastasis assay with a HER2+ breast cancer and a lung adenocarcinoma cell line, the authors could confirm previous findings that dormant cancer cells activate the p38 and self-renewal pathways through Sox2 and Sox9 (Sosa et al., 2015). Malladi et al. found that Sox2 also induced a growth arrest by inhibition of Wnt signaling and downstream proliferative pathways through activation of DKK1 (Fig. 3C). Once DKK1 induced a dormant state, DTCs were able to avoid NK cell-mediated cell death, whereas DKK1 low-proliferating DTCs were susceptible to NK cell cytotoxicity. This suggests that immune evasion is a result of dormancy induced by different microenvironmental signals rather than dormancy being a result of NK cells on DTCs.
Adaptive immune cells have also been shown to negatively affect tumor growth. This has recently been confirmed in clinical practice when a new generation of immune therapies targeting immune checkpoint molecules such as CTLA4, PD-1, and PDL-1 has led to great success (Pardoll, 2012). Yet again, our understanding of the role of adaptive immunity in dormancy is surprisingly limited. We have known for a long time that immune suppression after organ transplants is linked to an enhanced risk to develop cancer (Ross, 2007), indicating that the immune system can suppress tumor growth. Cases where melanoma was transferred through a kidney transplant from organ donors with NED for up to 16 years (MacKie, Reid, & Junor, 2003) indicate that dormancy of kidney DTCs can be mediated through adaptive immunity and that these DTCs exit dormancy upon immune suppression after organ transplantation. However, whether this phenomenon is functionally linked to a loss of CD8 cell cytotoxic activity or NK cell activity is still unclear.
Only few studies investigated the role of CD4 and CD8 T-cells in maintaining dormancy. It has been reported that dormant tumor cells seem to be less susceptible to adaptive immune cell responses and show reduced tumor antigen expression (Matsuzawa, Takeda, Narita, & Ozawa, 1991; Weinhold, Miller, & Wheelock, 1979). Additionally, it has been shown that dormant leukemia cells express PDL1, which allows them to inhibit T-cells (Saudemont & Quesnel, 2004). These studies indicate tumor cells escape immune-mediated cell killing and are therefore able to survive in a dormant state (Fig. 4A). Other studies indicate that T-cell-derived factors might induce dormancy in DTCs (Fig. 4B). For example, CD8 T-cell-derived interferon γ (IFNγ) has been shown to induce dormancy in a murine lymphoma model (Farrar et al., 1999). In the Rip-Tag2 mouse model for pancreatic cancer, CD4 T-cells induced angiogenic population-based dormancy through IFNγ and tumor necrosis factor α (TNFα) signaling (Muller-Hermelink et al., 2008). It remains to be elucidated whether evasion of T-cell-induced cell death and T-cell-induced dormancy are exclusive processes or whether they might be different mechanisms, depending on the tumor type and the specific immune response mechanisms. Furthermore, it remains to be elucidated whether immune-mediated dormancy always resembles population-based dormancy or whether T-cell-derived factors such as IFNγ might also be able to induce cellular dormancy in DTCs (Fig. 4C).
Fig. 4.
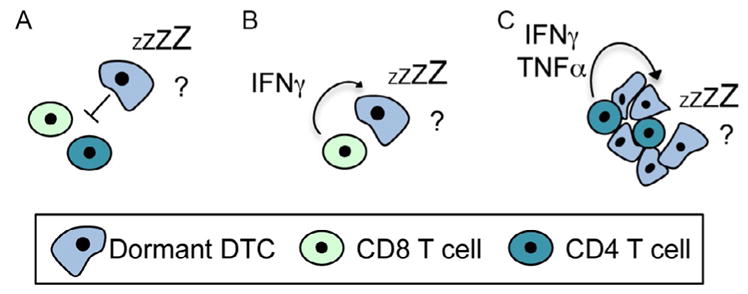
Effects of T-cells on dormancy. (A) Some studies suggest that dormant DTCs survive by escaping adaptive immune responses (Matsuzawa et al., 1991; Saudemont & Quesnel, 2004; Weinhold et al., 1979). (B) Other studies indicate that T-cell-derived IFNγ and TNFR signaling induce dormancy (bottom) (Farrar et al., 1999; Muller-Hermelink et al., 2008). (C) Whether immune-mediated dormancy resembles cellular dormancy or population-based dormancy, where tumor cell killing and proliferation are in equilibrium, is not clear either.
According to the immune editing hypothesis, tumors go through a phase of elimination, followed by a phase of equilibrium and a phase of escape where the evolutionary pressure of the immune system has resulted in immune escape mechanisms, through intrinsic changes such as reduced tumor antigen expression, through extrinsic changes by creating an immune-suppressive microenvironment, or through a combination of both (Schreiber, Old, & Smyth, 2011). The immune editing hypothesis refers to phases during primary tumor development. However, we now have strong evidence that primary tumors and metastases develop in parallel (Klein, 2009). This is based on large cohort patient studies (Banys, Gruber, et al., 2012; Braun et al., 2005; Sanger et al., 2011; Schardt et al., 2005; Turajlic & Swanton, 2016) and studies with spontaneous murine tumor models for breast cancer (Husemann et al., 2008) and pancreatic cancer (Rhim et al., 2012), showing that tumor cells can disseminate during pre-malignant and preinvasive asymptomatic disease stages. Thus, while primary tumors undergo phases of immune editing, tumor cells already disseminate and enter tumor-naïve target organs. This raises several interesting questions posed below.
The immune editing hypothesis further postulates an equilibrium phase characterized by a balance between tumor cell killing through immune cells and tumor cell proliferation. However, there is only little tumor cell apoptosis and tumor cell proliferation during this phase (Koebel et al., 2007). This implies that limited cancer growth during the equilibrium phase might rather be due to growth-inhibitory effects of the immune system with some contribution of tumor cell killing. This is supported by a recent study showing that in a murine pancreatic cancer model, inhibition of focal adhesion kinase led to disease stabilization and reduced proliferation and augmented immunity (Jiang et al., 2016). Further, if DTCs disseminate during the equilibrium phase, could they be more susceptible to enter immune-induced dormancy in target organs? Another interesting question is how DTCs manage the immune escape phase in target organs. Are those DTCs that have retained high tumor antigen expression and are derived from primary tumors with immune-suppressive microenvironments more prone to be eradicated by the immune system when they enter a tumor-naïve organ microenvironment or do they enter immune-mediated dormancy? Interestingly, a recent study by Payne et al. found that therapy-induced dormant mammary carcinoma cells were unable to use immune editing to escape immunotherapy in a mouse model (Payne et al., 2016). Even slowly cycling, Ki-67low cells were able to escape immunotherapy, indicating that cellular but not immune or vascular dormancy is the driving mechanism behind this biology. This observation might lead to combination therapies where immunotherapy targets dormant cells, while established antiproliferative therapies target the proliferating or slow cycling populations.
Another interesting unanswered question is whether DTCs are able to escape the immune system in target organs by entering immune-protected sites. As mentioned above and reviewed elsewhere (Ghajar, 2015), DTCs can become dormant by entering stem cell niches. Adult stem cell niches have been shown to be immune-protected sites. The hematopoietic stem cell niche, for example, is rich in CD4+CD25+ regulatory T-cells (Zou et al., 2004) that mediate immune protection of hematopoietic stem cells (Fujisaki et al., 2011). It will be interesting to address which DTC niches provide immune-privileged sites that protect DTCs from eradication throughout the immune system and how these DTCs can exit their dormant niche and form metastases without being eradicated by the immune system. Would systemic immune suppression allow DTCs to exit immune-protected sites and form metastases—a phenomenon observed in immune-suppressed organ transplant recipients?
Novel immune therapies against CTLA-4 and PD-1 achieved great immediate success in certain patient populations (Pardoll, 2012). However, it is unknown whether the success of these immune therapies is based on eradication of cancer cells or whether they also contribute to a switch into immune-mediated dormancy. Data about long-term disease progression are not available yet since these new therapies have only been applied in the clinic for less than 5 years. It therefore remains to be elucidated whether novel immune therapies might in fact contribute to dormant minimal residual disease and whether late relapses might occur.
In addition to a limited understanding of long-term effects of immune therapies on minimal residual disease and late relapses due to dormant DTCs, we also do not fully understand whether there might be an association of dormancy and nonresponders to immune therapy. A recent study has shown that the total load of nonsynonymous mutations seems to correlate with a patient’s response to CTLA4 therapy (Van Allen et al., 2015). Thus, the more mutation a tumor has acquired, the better the response to immune therapy, possibly because the developed escape mechanisms rely on inhibition of costimulatory factors such as CTLA4 and PD-1. However, not all DTCs are necessarily derived from a highly proliferative invasive tumor with high mutator phenotypes. As mentioned above, it is now accepted that dissemination occurs in parallel to primary tumor development and results in increased heterogeneity and discordance of mutations in primary tumors and metastases (Klein, 2009; Turajlic & Swanton, 2016). DTCs that disseminated early will have a lower mutational burden than the primary tumor at the time of diagnosis. Could those early DTCs therefore be resistant to immune therapy?
In summary, it is important to highlight that our understanding of immune-mediated dormancy currently limits our understanding of long-term effects of immune therapies. More insight into this subject might hold the key to more targeted immune therapies and possibly combined immune and dormancy therapies.
8. SUMMARY AND OUTLOOK
Two alternative strategies to use the therapeutic window of dormancy have been discussed. Since dormancy provides a mechanism by which DTCs evade current therapies, one could devise ways to reawaken dormant DTCs prior to therapy. Forcing DTCs into proliferation and using currently available antiproliferative therapies might kill the majority of the cells. Yet, from the treatment of primary cancers we know that this might also induce the selection of resistant clones (Pisco et al., 2013), leading to a potentially worsened outcome. The other alternative would be to induce a perpetual dormancy. By evaluating which signals keep DTCs dormant, one could artificially keep the cells in a dormant state, thus preventing overt metastasis. Both possibilities have been reviewed in depth elsewhere (Aguirre-Ghiso et al., 2013; Ghajar, 2015; Sosa et al., 2011, 2014). For the latter strategy, we need to find out whether dormancy therapies need to be administered continuously or can be given in pulses which would be more tolerable for NED patients. Can we identify markers that indicate a dormant vs a reawakening phenotype of DTCs (Aguirre-Ghiso et al., 2013)? One possibility might be to measure levels of dormancy-inducing factors in the blood. A few studies indicate that this might be possible. High TGFβ2 levels in the blood seem to correlate with therapy success (Kopp, Jonat, Schmahl, & Knabbe, 1995; Lucia, Sporn, Roberts, Stewart, & Danielpour, 1998). In addition, analysis of the dormancy status of DTCs isolated from bone marrow of patients with NED might allow predicting relapses. In fact, recent work (Chery et al., 2014; Sosa et al., 2015) indicated for the first time that analysis of bone marrow DTCs can be predictive of late recurrences and that markers derived from understanding the basic mechanisms of dormancy are expressed in DTCs from NED patients arguing they could serve as dormancy markers. These studies indicate that understanding the mechanism of dormancy might allow to follow patients with NED more closely in order to predict their metastatic relapse before it occurs. Development of dormancy therapies might allow treating those patients with reduced levels of dormancy markers. This could change the life expectancy of patients with disseminated cancer drastically since current therapies administered to patients with overt metastases often fail. Moreover, once we better understand the mechanisms of dormancy and immune responses, combined dormancy and immune therapy might be an exciting new avenue in cancer therapy.
References
- Abravanel DL, Belka GK, Pan TC, Pant DK, Collins MA, Sterner CJ, et al. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. The Journal of Clinical Investigation. 2015;125(6):2484–2496. doi: 10.1172/JCI74883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam AP, George A, Schewe D, Bragado P, Iglesias BV, Ranganathan AC, et al. Computational identification of a p38SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer Research. 2009;69(14):5664–5672. doi: 10.1158/0008-5472.CAN-08-3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. The Journal of Cell Biology. 1999;147(1):89–104. doi: 10.1083/jcb.147.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nature Reviews Cancer. 2007;7(11):834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA, Bragado P, Sosa MS. Metastasis awakening: Targeting dormant cancer. Nature Medicine. 2013;19(3):276–277. doi: 10.1038/nm.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK) Cancer Research. 2003;63(7):1684–1695. [PubMed] [Google Scholar]
- Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Molecular Biology of the Cell. 2001;12(4):863–879. doi: 10.1091/mbc.12.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA, Ossowski L, Rosenbaum SK. Green fluorescent protein tagging of extracellular signal-regulated kinase and p38 pathways reveals novel dynamics of pathway activation during primary and metastatic growth. Cancer Research. 2004;64(20):7336–7345. doi: 10.1158/0008-5472.CAN-04-0113. [DOI] [PubMed] [Google Scholar]
- Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Research. 1992;52(6):1399–1405. [PubMed] [Google Scholar]
- Banys M, Gruber I, Krawczyk N, Becker S, Kurth R, Wallwiener D, et al. Hematogenous and lymphatic tumor cell dissemination may be detected in patients diagnosed with ductal carcinoma in situ of the breast. Breast Cancer Research and Treatment. 2012;131(3):801–808. doi: 10.1007/s10549-011-1478-2. [DOI] [PubMed] [Google Scholar]
- Banys M, Hartkopf AD, Krawczyk N, Kaiser T, Meier-Stiegen F, Fehm T, et al. Dormancy in breast cancer. Breast Cancer (Dove Medical Press) 2012;4:183–191. doi: 10.2147/BCTT.S26431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan D, Green JE. An in vitro system to study tumor dormancy and the switch to metastatic growth. Journal of Visualized Experiments. 2011:54. doi: 10.3791/2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter AG, Hodgkin PD. Activation rules: The two-signal theories of immune activation. Nature Reviews Immunology. 2002;2(6):439–446. doi: 10.1038/nri823. [DOI] [PubMed] [Google Scholar]
- Box GM, Eccles SA. Simple experimental and spontaneous metastasis assays in mice. Methods in Molecular Biology. 2011;769:311–329. doi: 10.1007/978-1-61779-207-6_21. [DOI] [PubMed] [Google Scholar]
- Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, et al. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nature Cell Biology. 2013;15(11):1351–1361. doi: 10.1038/ncb2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S, Pantel K, Muller P, Janni W, Hepp F, Kentenich CR, et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. The New England Journal of Medicine. 2000;342(8):525–533. doi: 10.1056/NEJM200002243420801. [DOI] [PubMed] [Google Scholar]
- Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, et al. A pooled analysis of bone marrow micrometastasis in breast cancer. The New England Journal of Medicine. 2005;353(8):793–802. doi: 10.1056/NEJMoa050434. [DOI] [PubMed] [Google Scholar]
- Buijs JT, Rentsch CA, van der Horst G, van Overveld PG, Wetterwald A, Schwaninger R, et al. BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. The American Journal of Pathology. 2007;171(3):1047–1057. doi: 10.2353/ajpath.2007.070168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhang XH, Massague J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell. 2011;20(4):538–549. doi: 10.1016/j.ccr.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chery L, Lam HM, Coleman I, Lakely B, Coleman R, Larson S, et al. Characterization of single disseminated prostate cancer cells reveals tumor cell heterogeneity and identifies dormancy associated pathways. Oncotarget. 2014;5(20):9939–9951. doi: 10.18632/oncotarget.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaves CJ. Hematopoietic stem cells: Concepts, definitions, and the new reality. Blood. 2015;125(17):2605–2613. doi: 10.1182/blood-2014-12-570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar JD, Katz KH, Windsor J, Thrush G, Scheuermann RH, Uhr JW, et al. Cancer dormancy. VII. A regulatory role for CD8+ T cells and IFN-gamma in establishing and maintaining the tumor-dormant state. Journal of Immunology. 1999;162(5):2842–2849. [PubMed] [Google Scholar]
- Felsher DW. Oncogene addiction versus oncogene amnesia: Perhaps more than just a bad habit? Cancer Research. 2008;68(9):3081–3086. doi: 10.1158/0008-5472.CAN-07-5832. discussion 3086. [DOI] [PubMed] [Google Scholar]
- Felsher DW. MYC inactivation elicits oncogene addiction through both tumor cell-intrinsic and host-dependent mechanisms. Genes & Cancer. 2010;1(6):597–604. doi: 10.1177/1947601910377798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Molecular Cell. 1999;4(2):199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 2011;474(7350):216–219. doi: 10.1038/nature10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nature Immunology. 2013;14(10):1014–1022. [Google Scholar]
- Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A, et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell. 2012;150(4):764–779. doi: 10.1016/j.cell.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghajar CM. Metastasis prevention by targeting the dormant niche. Nature Reviews. Cancer. 2015;15(4):238–247. doi: 10.1038/nrc3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, et al. The perivascular niche regulates breast tumour dormancy. Nature Cell Biology. 2013;15(7):807–817. doi: 10.1038/ncb2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiaur G, Yegnasubramanian S, Perkins B, Gucwa JL, Gerber JM, Jones RJ. Regulation of human hematopoietic stem cell self-renewal by the microenvironment’s control of retinoic acid signaling. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(40):16121–16126. doi: 10.1073/pnas.1305937110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuriato S, Ryeom S, Fan AC, Bachireddy P, Lynch RC, Rioth MJ, et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(44):16266–16271. doi: 10.1073/pnas.0608017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther EJ, Belka GK, Wertheim GB, Wang J, Hartman JL, Boxer RB, et al. A novel doxycycline-inducible system for the transgenic analysis of mammary gland biology. FASEB J. 2002;16(3):283–292. doi: 10.1096/fj.01-0551com. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Coussens LM. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- Hartkopf AD, Taran FA, Wallwiener M, Hahn M, Becker S, Solomayer EF, et al. Prognostic relevance of disseminated tumour cells from the bone marrow of early stage breast cancer patients—Results from a large single-centre analysis. European Journal of Cancer. 2014;50(15):2550–2559. doi: 10.1016/j.ejca.2014.06.025. [DOI] [PubMed] [Google Scholar]
- Hartkopf AD, Wallwiener M, Fehm TN, Hahn M, Walter CB, Gruber I, et al. Disseminated tumor cells from the bone marrow of patients with nonmetastatic primary breast cancer are predictive of locoregional relapse. Annals of Oncology. 2015;26(6):1155–1160. doi: 10.1093/annonc/mdv148. [DOI] [PubMed] [Google Scholar]
- Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13(1):58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nature Medicine. 2016;22(8):851–860. doi: 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. The Journal of Experimental Medicine. 2015;212(7):1043–1059. doi: 10.1084/jem.20141836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CA. Parallel progression of primary tumours and metastases. Nature Reviews Cancer. 2009;9(4):302–312. doi: 10.1038/nrc2627. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. The Journal of Experimental Medicine. 2011;208(13):2641–2655. doi: 10.1084/jem.20110840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450(7171):903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- Kopp A, Jonat W, Schmahl M, Knabbe C. Transforming growth factor beta 2 (TGF-beta 2) levels in plasma of patients with metastatic breast cancer treated with tamoxifen. Cancer Research. 1995;55(20):4512–4515. [PubMed] [Google Scholar]
- Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526(7571):131–135. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nature Communications. 2015;6:8983. doi: 10.1038/ncomms9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hively WP, Varmus HE. Use of MMTV-Wnt-1 transgenic mice for studying the genetic basis of breast cancer. Oncogene. 2000;19(8):1002–1009. doi: 10.1038/sj.onc.1203273. [DOI] [PubMed] [Google Scholar]
- Lilleby W, Stensvold A, Mills IG, Nesland JM. Disseminated tumor cells and their prognostic significance in nonmetastatic prostate cancer patients. International Journal of Cancer. 2013;133(1):149–155. doi: 10.1002/ijc.28002. [DOI] [PubMed] [Google Scholar]
- Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell. 2011;20(6):701–714. doi: 10.1016/j.ccr.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucia MS, Sporn MB, Roberts AB, Stewart LV, Danielpour D. The role of transforming growth factor-beta1, −beta2, and -beta3 in androgen-responsive growth of NRP-152 rat prostatic epithelial cells. Journal of Cellular Physiology. 1998;175(2):184–192. doi: 10.1002/(SICI)1097-4652(199805)175:2<184::AID-JCP8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- MacKie RM, Reid R, Junor B. Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. The New England Journal of Medicine. 2003;348(6):567–568. doi: 10.1056/NEJM200302063480620. [DOI] [PubMed] [Google Scholar]
- Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. 2016;165(1):45–60. doi: 10.1016/j.cell.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Marlow R, Dontu G. Modeling the breast cancer bone metastatic niche in complex three-dimensional cocultures. Methods in Molecular Biology. 2015;1293:213–220. doi: 10.1007/978-1-4939-2519-3_12. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Takeda Y, Narita M, Ozawa H. Survival of leukemic cells in a dormant state following cyclophosphamide-induced cure of strongly immunogenic mouse leukemia (DL811) International Journal of Cancer. 1991;49(2):303–309. doi: 10.1002/ijc.2910490227. [DOI] [PubMed] [Google Scholar]
- Mishra A, Wang J, Shiozawa Y, McGee S, Kim J, Jung Y, et al. Hypoxia stabilizes GAS6/Axl signaling in metastatic prostate cancer. Molecular Cancer Research. 2012;10(6):703–712. doi: 10.1158/1541-7786.MCR-11-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody SE, Sarkisian CJ, Hahn KT, Gunther EJ, Pickup S, Dugan KD, et al. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell. 2002;2(6):451–461. doi: 10.1016/s1535-6108(02)00212-x. [DOI] [PubMed] [Google Scholar]
- Morris VL, Tuck AB, Wilson SM, Percy D, Chambers AF. Tumor progression and metastasis in murine D2 hyperplastic alveolar nodule mammary tumor cell lines. Clinical & Experimental Metastasis. 1993;11(1):103–112. doi: 10.1007/BF00880071. [DOI] [PubMed] [Google Scholar]
- Morrissey C, Vessella RL, Lange PH, Lam HM. The biology and clinical implications of prostate cancer dormancy and metastasis. Journal of Molecular Medicine (Berlin, Germany) 2015;94:259–265. doi: 10.1007/s00109-015-1353-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller MM, Fusenig NE. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nature Reviews Cancer. 2004;4(11):839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- Muller-Hermelink N, Braumuller H, Pichler B, Wieder T, Mailhammer R, Schaak K, et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell. 2008;13(6):507–518. doi: 10.1016/j.ccr.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Najmi S, Korah R, Chandra R, Abdellatif M, Wieder R. Flavopiridol blocks integrin-mediated survival in dormant breast cancer cells. Clinical Cancer Research. 2005;11(5):2038–2046. doi: 10.1158/1078-0432.CCR-04-1083. [DOI] [PubMed] [Google Scholar]
- Ossowski L, Reich E. Experimental model for quantitative study of metastasis. Cancer Research. 1980;40(7):2300–2309. [PubMed] [Google Scholar]
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews. Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne KK, Keim RC, Graham L, Idowu MO, Wan W, Wang XY, et al. Tumor-reactive immune cells protect against metastatic tumor and induce immunoediting of indolent but not quiescent tumor cells. Journal of Leukocyte Biology. 2016:100. doi: 10.1189/jlb.5A1215-580R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierga JY, Bonneton C, Magdelenat H, Vincent-Salomon A, Nos C, Pouillart P, et al. Clinical significance of proliferative potential of occult metastatic cells in bone marrow of patients with breast cancer. British Journal of Cancer. 2003;89(3):539–545. doi: 10.1038/sj.bjc.6601121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisco AO, Brock A, Zhou J, Moor A, Mojtahedi M, Jackson D, et al. Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nature Communications. 2013;4:2467. doi: 10.1038/ncomms3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purton LE, Dworkin S, Olsen GH, Walkley CR, Fabb SA, Collins SJ, et al. RARgamma is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation. The Journal of Experimental Medicine. 2006;203(5):1283–1293. doi: 10.1084/jem.20052105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One. 2009;4(8):e6562. doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Zhang H, Li J, He T, Yeo EJ, Soong DY, et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. The Journal of Experimental Medicine. 2015;212(9):1433–1448. doi: 10.1084/jem.20141555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nature Medicine. 2013;19(11):1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell. 2010;18(5):485–498. doi: 10.1016/j.ccr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1–2):349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosol TJ, Tannehill-Gregg SH, Corn S, Schneider A, McCauley LK. Animal models of bone metastasis. Cancer Treatment and Research. 2004;118:47–81. doi: 10.1007/978-1-4419-9129-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross K. For organ transplant recipients, cancer threatens long-term survival. Journal of the National Cancer Institute. 2007;99(6):421–422. doi: 10.1093/jnci/djk141. [DOI] [PubMed] [Google Scholar]
- Ruppender N, Larson S, Lakely B, Kollath L, Brown L, Coleman I, et al. Cellular adhesion promotes prostate cancer cells escape from dormancy. PLoS One. 2015;10(6):e0130565. doi: 10.1371/journal.pone.0130565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger N, Effenberger KE, Riethdorf S, Van Haasteren V, Gauwerky J, Wiegratz I, et al. Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. International Journal of Cancer. 2011;129(10):2522–2526. doi: 10.1002/ijc.25895. [DOI] [PubMed] [Google Scholar]
- Sansone P, Ceccarelli C, Berishaj M, Chang Q, Rajasekhar VK, Perna F, et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nature Communications. 2016;7:10442. doi: 10.1038/ncomms10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudemont A, Quesnel B. In a model of tumor dormancy, long-term persistent leukemic cells have increased B7-H1 and B7.1 expression and resist CTL-mediated lysis. Blood. 2004;104(7):2124–2133. doi: 10.1182/blood-2004-01-0064. [DOI] [PubMed] [Google Scholar]
- Schardt JA, Meyer M, Hartmann CH, Schubert F, Schmidt-Kittler O, Fuhrmann C, et al. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell. 2005;8(3):227–239. doi: 10.1016/j.ccr.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(30):10519–10524. doi: 10.1073/pnas.0800939105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Scognamiglio R, Cabezas-Wallscheid N, Thier MC, Altamura S, Reyes A, Prendergast AM, et al. Myc depletion induces a pluripotent dormant state mimicking diapause. Cell. 2016;164(4):668–680. doi: 10.1016/j.cell.2015.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry MM, Greco FA, Johnson DH, Hainsworth JD. Metastatic breast cancer confined to the skeletal system. An indolent disease. The American Journal of Medicine. 1986;81(3):381–386. doi: 10.1016/0002-9343(86)90286-x. [DOI] [PubMed] [Google Scholar]
- Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. The Journal of Clinical Investigation. 2011;121(4):1298–1312. doi: 10.1172/JCI43414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozawa Y, Pedersen EA, Patel LR, Ziegler AM, Havens AM, Jung Y, et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia. 2010;12(2):116–127. doi: 10.1593/neo.91384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MS, Avivar-Valderas A, Bragado P, Wen HC, Aguirre-Ghiso JA. ERK1/2 and p38alpha/beta signaling in tumor cell quiescence: Opportunities to control dormant residual disease. Clinical Cancer Research. 2011;17(18):5850–5857. doi: 10.1158/1078-0432.CCR-10-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nature Reviews. Cancer. 2014;14(9):611–622. doi: 10.1038/nrc3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, et al. NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nature Communications. 2015;6:6170. doi: 10.1038/ncomms7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taichman RS, Patel LR, Bedenis R, Wang J, Weidner S, Schumann T, et al. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS One. 2013;8(4):e61873. doi: 10.1371/journal.pone.0061873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorban S, Rosenberg R, Busch R, Roder RJ. Epithelial cells in bone marrow of oesophageal cancer patients: A significant prognostic factor in multivariate analysis. British Journal of Cancer. 2000;83(1):35–39. doi: 10.1054/bjoc.2000.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352(6282):169–175. doi: 10.1126/science.aaf2784. [DOI] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnoi M, Peddibhotla S, Yin W, Scamardo AT, George GC, Hong DS, et al. The isolation and characterization of CTC subsets related to breast cancer dormancy. Scientific Reports. 2015;5:17533. doi: 10.1038/srep17533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell. 2015;27(2):193–210. doi: 10.1016/j.ccell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhold KJ, Miller DA, Wheelock EF. The tumor dormant state. Comparison of L5178Y cells used to establish dormancy with those that emerge after its termination. The Journal of Experimental Medicine. 1979;149(3):745–757. doi: 10.1084/jem.149.3.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollenberg B, Walz A, Kolbow K, Pauli C, Chaubal S, Andratschke M. Clinical relevance of circulating tumour cells in the bone marrow of patients with SCCHN. Onkologie. 2004;27(4):358–362. doi: 10.1159/000079088. [DOI] [PubMed] [Google Scholar]
- Zou L, Barnett B, Safah H, Larussa VF, Evdemon-Hogan M, Mottram P, et al. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Research. 2004;64(22):8451–8455. doi: 10.1158/0008-5472.CAN-04-1987. [DOI] [PubMed] [Google Scholar]