

Summary
Intermittent PTH administration builds bone mass and prevents fractures, but its mechanism of action is unclear. We genetically deleted the PTH/PTHrP Receptor (PTH1R) in mesenchymal stem cells using Prx1Cre and found low bone formation, increased bone resorption and high bone marrow adipose tissue (BMAT). Bone marrow adipocytes traced to Prx1 and expressed classic adipogenic markers and high receptor activator of nuclear factor kappa-B ligand (Rankl) expression. RANKL levels were also elevated in bone marrow supernatant and serum, but undetectable in other adipose depots. By cell sorting, Pref1+RANKL+ marrow progenitors were twice as great in mutant versus control marrow. Intermittent PTH administration to control mice reduced BMAT significantly. A similar finding was noted in male osteoporotics. Thus, marrow adipocytes exhibit osteogenic and adipogenic characteristics, are uniquely responsive to PTH, and secrete RANKL. These studies reveal an important mechanism for PTH’s therapeutic action through its ability to direct mesenchymal cell fate.
Keywords: RANKL, PTH, Receptor, Lineage, Bone resorption
eTOC Blurb
XXX et al show that PTH regulates mesenchymal stem cell fate between bone and adipocyte in the marrow. Bone marrow adipocytes have distinct origins and properties from other adipocytes and are responsive to PTH, underlying the reduction in marrow adiposity in mouse models and idiopathic osteoporosis patients treated with PTH.
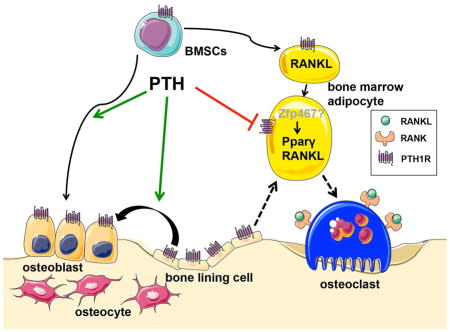
Introduction
Age-related osteoporosis is characterized by low bone mass, uncoupled bone remodeling, enhanced skeletal fragility and infiltration of the bone marrow with excessive numbers of adipocytes (Rosen and Bouxsein, 2006). The generation of excessive bone marrow adipose tissue (BMAT) is not well understood in part because there are both physiologic and pathologic changes in the marrow compartment with age in mice and humans. For example, during the growth of mammalian long bones, red marrow is gradually replaced by adipocytes. At the other extreme, with advanced age, BMAT replaces hematopoietic elements in the axial skeleton (Scheller and Rosen, 2014). Drugs (e.g. glucocorticoids and thiazolidinediones), calorie restriction or high fat diets, chemotherapy and/or radiation, and hormonal factors such as estrogen deprivation or leptin deficiency cause a significant increase in BMAT (Cawthorn et al., 2014; Georgiou et al., 2012; Naveiras et al., 2009; Prieur et al., 2013; Thomas et al., 2001). Several genetically engineered and inbred mouse strains exhibit high BMAT (Rosen et al., 2009). Notwithstanding, the mechanisms, whereby mesenchymal progenitors are directed towards the adipogenic rather than osteogenic lineage remain to be determined.
One possible mediator of mesenchymal stem cell fate is PTH or its related protein, PTHrP. PTH is well known to enhance differentiation of committed osteoblast precursors, to prevent apoptosis of osteoblasts and osteocytes, and to decrease sclerostin (Ishizuya et al., 1997; Jilka et al., 1999; Keller and Kneissel, 2005). Previously it was thought that intermittent PTH administration stimulated new bone formation by increasing the activity and effort of mature osteoblasts (Kim et al., 2012). However, recent studies suggest that PTH could recruit marrow stromal cells (MSCs) into the osteoblast lineage through PTH1R- and LRP6-dependent pathways and endocytosis of this PTH1R/LRP6 complex (Li et al., 2013; Yu et al., 2012). Consistent with this theory, targeted overexpression of PTH1R using the Col2.3 promoter led to increased osteoblast numbers, greater bone formation and suppressed marrow adiposity (Calvi et al., 2001; Tascau et al., 2016). By contrast, PTHrP haploinsufficient mice exhibited low bone mass and increased marrow adiposity (Amizuka et al., 1996). Moreover, PTHrP has been shown to inhibit adipocyte differentiation by suppressing PPARγ activity (Chan et al., 2001), or by interacting with bone morphogenetic protein 2 (BMP2) in pluripotent C3H10T1/2 mesenchymal cells (Chan et al., 2003). PTH1R is a G protein-coupled receptor, which can activate Gα subunits, such as Gs or Gi, which stimulate or inhibit cyclic AMP production, respectively (Weinstein et al., 2001). Loss of Gsα early in the osteoblast lineage favored adipogenic differentiation of MSCs and committed osteoblast precursors (Sinha et al., 2014). Importantly, the cell fate switch occurred relatively rapidly in another study upon PTH withdrawal implying the primary target cell could either trans-differentiate or is a unique progenitor (ASBMR 2015, #1021). Recently, lineage-tracing studies demonstrated that the bone-lining cell is a target for PTH’s anabolic action (Kim et al., 2012). Differentiation of these cells into functional osteoblasts is one means by which new bone formation occurs, but these cells may also be a reservoir for differentiation into other lineages (Matic et al., 2016).
Our study was designed to examine the mechanism of PTH action in bone marrow stromal cells. To address that question in vivo and in vitro, we generated a mouse model of PTH resistance that develops a form of age-related bone loss relatively early after birth. Conditional deletion of PTH1R using the Prx1Cre recombinase resulted in a substantial increase in BMAT by 3 weeks of age accompanied by high bone resorption despite the absence of functional PTH receptors in osteoprogenitors and osteoblasts. We isolated marrow adipocytes to gain further insights into the molecular signature of these cells as well as to establish a model for understanding marrow progenitor cell fate, its relationship to PTH1R and its role in bone resorption. Finally, we asked whether those findings in mice were translatable to male osteoporotic patients involved in a small clinical trial of intermittent PTH(1-34).
Results
Skeletal and marrow phenotype of Prx1Cre;PTH1Rfl/fl mice
We recently generated Prx1Cre;PTH1Rfl/fl mice (mutants), in which the PTH1R is ablated at the early limb bud stage (E9.5), thereby resulting in total lack of PTH1R expression in the adult limb and calvaria (Fan et al., 2016). To demonstrate the tissue specificity of the Prx1Cre transgene, Tomatofl/fl reporter mice (Madisen et al., 2010) were crossed with Prx1Cre mice (Logan et al., 2002) for further analyses. As shown in Figure S1 tomato staining was restricted to long bones and calvaria (Figure S1A) with no further expression found in any other tissues examined (Figure S1B). Mutants were born with the expected rate of Mendelian inheritance, were viable and showed severely shortened limbs at birth (Fan et al., 2016) and postnatally (Figure 1A). μCT analysis of the long bones showed a marked reduction in trabecular and cortical bone volume and thickness in mutants when compared to controls at 3 weeks of age (Fan et al., 2016). Hematoxilin/eosin staining of paraffin sections of the tibia confirmed the previously reported μCT data depicting the severe loss in trabecular and cortical bone in mutants (Figure 1B). To further investigate whether the cause for the reduction in bone volume was due to decreased osteoblast function we performed qRT-PCR analyses from RNA extracted from flushed tibia using Taqman primers. The majority of early osteoblast markers including Runt-related transcription factor 2 (Runx2), Osterix (Osx), alkaline phosphatase (Alp), Collagen Type I α1 (Col1α1), as well as osteocalcin (Ocn), an osteocyte marker, were significantly downregulated (Figure 1C). Most importantly however, bone marrow cells in mutants were replaced by mature adipocytes. Already by 3 weeks of age the marrow cavity of the distal tibia was occupied by large adipocytes (Figure 1D). Noteworthy, no adipocytes were detected in histological sections of the spine (L1) where PTH1R expression was normal, indicating that the appearance of marrow adipocytes coincided with the deletion of the PTH1R in long bones (Figure S1C). Using osmium tetroxide (OsO4) μCT analyses, we were able to visualize and quantitate marrow adipose tissue/total volume (control: 10.4 ± 1.1%, mutant: 20.3 ± 3.0%) in the intact tibia thereby supporting the histological findings (Figure 1E).
Figure 1. Skeletal and marrow phenotype of Prx1Cre;PTH1Rfl/fl.

(A) Alizarin red/alcian blue staining of hindlimb of 3-wk-old PTH1Rfl/fl and Prx1Cre;PTH1Rfl/fl. (B) HE stained paraffin sections of control and mutant tibia at 3 wks showed significant reduced trabecular (upper panel, scale bar: 500μm) and cortical bone (lower panel, scale bar: 50μm) in mutant tibia. n=6. Arrowheads indicate area of trabecular bone. (C) Gene expression pattern of osteoblast markers from cortical bone RNA of PTH1Rfl/fl and Prx1Cre;PTH1Rfl/fl at 3 wks. n=6, *p<0.05, ***p<0.001 versus control. Graph shows mean ± SEM. (D) HE staining showed bone marrow space in mutant distal tibia is filled with adipocytes compared to controls. n=6. Scale bar: 500μm. Boxed areas show high-magnification, scale bar: 200μm. (E) OsO4 μCT image depicting the increase in bone marrow adiposity in Prx1Cre;PTH1Rfl/fl, particularly in the distal tibia, but also extending more proximal, when compared to PTH1Rfl/fl at 3 wks. n=6. Scale bar: 1mm. See also Figure S1.
BMAT exhibits classic markers of adipogenesis
To further characterize the cell population occupying the bone marrow space we purified adipocytes from the spun bone marrow using a specific centrifugation protocol (see experimental procedures) and performed gene expression analyses. First, we were able to confirm the efficient deletion of Pth1r in mutants when compared to controls (Figure 2A). Furthermore, the cells were not only strongly positive for the adipocytic transcription factors CCAAT/enhancer binding proteins (Cebpα, β, Δ), nuclear receptor peroxisome proliferator-activated receptor-γ (Pparγ) and Zinc finger protein 467 (Zfp467), but also expressed differentiated adipose cell markers including fatty acid binding protein 4 (Fabp4), Adiponectin (Adipo), and Perilipin (Figure 2B), genes prominent in peripheral fat depots including epididymal (eWAT), inguinal (iWAT) and interscapular (iBAT) adipose depots (Figure S2A). Next, we cultured bone marrow stromal cells (BMSCs) of 3-wk-old control and mutant mice under adipogenic condition, followed by oil red O (ORO) staining and quantification by spectrophotometry at OD500. The results demonstrated a marked enrichment in adipocyte differentiation in the mutants (Figure 2C), strongly indicating that loss of PTH1R in mesenchymal precursors affects cell fate decision early on. Enhanced adipogenic differentiation was confirmed by testing the gene expression pattern of the cultured cells (Figure 2D).
Figure 2. Characterization of BMAT.

(A, B) Schematic representation of bone marrow adipocytes isolated for qRT-PCR analyses. mRNA expression levels of Pth1r (A) and adipocyte specific genes (B) in isolated BMAT of PTH1Rfl/fl and Prx1Cre;PTH1Rfl/fl at 3 wks. n=4/control, 5/mutant. (C) Schematic representation of BMSCs cultured for qRT-PCR analyses. Oil red O staining and quantification of BMSCs of PTH1Rfl/fl and Prx1Cre;PTH1Rfl/fl mice cultured under adipogenic conditions. Scale bar: 200μm. (D) qRT-PCR analyses of BMSCs under adipogenic condition for adipocyte markers. n=6–9. *p<0.05, **p<0.01, ***p<0.001,****p<0.0001 versus control. All graphs show mean ± SEM. White bar represent PTH1Rfl/fl and black bar represent Prx1Cre;PTH1Rfl/fl in all panels. See also Figure S2.
In vitro deletion of PTH1R from BMSCs and calvarial osteoblasts results in adipogenesis
We also examined whether deletion of PTH1R from postnatal BMSCs (3 wks) and calvarial osteoblasts (P4) of PTH1Rfl/fl mice using Adenovirus-mediated Cre (Ad-CRE) results in enhanced adipogenesis. First, the efficient deletion of the Pth1r in both cell types (Figures S2B and C) was confirmed. Furthermore, qRT-PCR analyses demonstrated significantly increased expression of adipogenic markers in Pth1r-deficient BMSCs and also in osteoblasts when compared to Ad-GFP treated control cells (Figures S2B and C).
PTH regulates BMAT in vivo and in vitro
To test whether PTH could influence the formation of BMAT in vivo, we daily injected control and mutant mice with 25 nmol/kg PTH(1-34) for 2 weeks (P8-P21). 3-wk-old tibiae were used to perform OsO4 μCT analyses and showed greater BMAT volume in mutants versus controls. Interestingly, PTH administration to control mice resulted in a marked reduction in volume of adipose tissue/total volume in 2 regions of interest (ROIs); proximal epiphyses and the growth plate to the tibial fibular junction. Most importantly however, mutants exhibited higher basal BMAT levels, which were not significantly affected by PTH administration (Figure 3A). To further observe the effect of PTH(1-34) in control mice and to validate the lack of response to PTH(1-34) in mutants, we cut histological sections of their tibiae (Figure 3B) and counted bone marrow adipocyte ghosts. No statistically significant decrease in adipocytes could be detected between vehicle injected (77.4 ± 12.0 cells/hpf) and PTH(1-34) injected (53.6 ± 12.8 cells/hpf) mutant mice. These data are the first to show that PTH administration can reduce bone marrow adipogenesis in vivo and support the tenet that PTH controls a regulatory mechanism in adipocyte differentiation.
Figure 3. Control of BMAT formation by PTH in vivo and in vitro.

(A) OsO4 μCT analyses of 3-wk- old tibiae of PTH1Rfl/fl and Prx1Cre;PTH1Rfl/fl under vehicle or PTH(1-34) administration. Control mice showed marked reduction in volume of adipose tissue/total volume, whereas Prx1Cre;PTH1Rfl/fl mice exhibited higher basal BMAT levels, which were not significantly affected by PTH administration. ***p<0.001 versus PTH1Rfl/fl under vehicle treatment. Two-way ANOVA and Bonferroni posttests were used to evaluate individual samples responses related to PTH(1-34) treatment. Genotype effect: ##p=0.0014, PTH(1-34) effect: $p=0.0264. (B) Toluidine blue stained paraffin sections of tibiae of PTH1Rfl/fl and Prx1Cre;PTH1Rfl/fl under vehicle or PTH(1-34) administration n=6. Scale bar: 500μm. (C) Oil red O staining and quantification of PTH1Rfl/fl and Prx1Cre;PTH1Rfl/fl BMSC cultures under adipogenic condition, treated with vehicle or 100 nM PTH(1-34). PTH treatment reduced oil red O staining in PTH1Rfl/fl BMSCs. In contrast, Prx1Cre;PTH1Rfl/fl cells showed higher numbers of adipocytes at basal stage and did not respond to PTH(1-34). n=3. Scale bar: 200μm. (D) qRT-PCR analyses of adipocyte specific genes in BMSCs under adipogenic condition, with vehicle or PTH(1-34) treatment. PTH1Rfl/fl BMSCs showed significantly reduced adipogenic markers (Fabp4, Adiponectin, Perilipin, Cebpα and Pparγ) after PTH administration whereas Prx1Cre;PTH1Rfl/fl BMSCs did not respond to PTH(1-34). n=3. *p<0.05, **p<0.01, ***p<0.001 versus PTH1Rfl/fl under vehicle treatment. All graphs show mean ± SEM. See also Figure S3.
To complement our in vivo findings with in vitro data we cultured BMSCs of controls or mutants with medium alone or containing 100 nM PTH(1-34) under adipogenic condition. Control BMSCs efficiently responded to PTH by markedly decreasing their number of adipocytes as shown by reduced ORO staining (OD500nm). In contrast, no changes could be detected in mutant cells, which showed significantly increased basal numbers of adipocytes and stronger ORO staining (Figure 3C). To further support reduced adipogenic differentiation of cells upon PTH(1-34) treatment on a molecular level, we performed qRT-PCR analyses and found significant decrease in some adipogenic markers in PTH1Rfl/fl derived cells, whereas no reduction could be observed in Prx1Cre;PTH1Rfl/fl cells (Figure 3D). Similarly exposure to 100 nM PTH(1-34) (Con-PTH) from day 1 reduced ORO staining when primary BMSCs from B6 wild-type mice were cultured in adipogenic media (Figure S3A). In contrast, treatment of cells with PTH at a later stage during adipogenic differentiation (day 6, Post-PTH) had no effect and ORO staining continued to be high (Figure S3B). Moreover, a single treatment of 3T3-L1 cells with 100 nM PTH(1-34) prior to adipogenic differentiation resulted in less adipogenesis and was similar to continuous PTH treatment, as determined by decreased ORO (Figure S3C).
Intermittent PTH(1-34) reduces BMAT in osteoporotic men
There are very few studies that detail the effect of PTH on bone marrow adiposity. But Yang et al. reported reduced bone marrow adiposity by MRI spectroscopy in women treated with PTH(1-34) (Yang et al., 2016). To test the hypothesis that PTH treatment reduces marrow adipocyte number in humans, we analyzed paired bone biopsies (baseline and 18 months) from 7 males with idiopathic osteoporosis treated with PTH(1-34) (Dempster et al., 2001). Marrow adipocyte number (126.4 ± 19.0) was reduced by 27% after 18 months of PTH (92.1 ± 11.3, p=0.055) whereas adipocyte size was not lowered by PTH treatment (p=0.99) (Figure 4).
Figure 4. Control of BMAT by PTH(1-34) treatment in men.

Representative paired iliac crest biopsy from a male subject treated with PTH(1-34), at baseline and after 18 months of treatment (Dempster et al., 2001). 3 sections were analyzed per region of tissue. The cells were counted and marrow adipocyte volume and marrow volume were measured.
Bone marrow adipocytes are the source of RANKL in the absence of PTH1R signaling
Our data indicated that the low bone mass in mutants could be due to a decrease in osteoblast function and activity (Figure 1C), however we also detected an increased number of tartrate resistant alkaline phosphatase (TRAP) positive osteoclasts (Figure 5A). This is in concert with our previous report where we have shown a marked upregulation of serum carboxy-terminal collagen crosslink levels (CTX) (Fan et al., 2016), indicating enhanced bone resorption. This increase was accompanied by a significant elevation of Rankl and receptor activator of nuclear factor kappa-B (Rank) expression but no changes in osteoprotegerin (Opg) in the cortical bone of mutants. In contrast, no changes in expression of any of these genes could be observed in the spine of the same animals, indicating that the increased Rankl/Opg ratio is a consequence of loss of PTH1R signaling selectively in long bones due to Prx1Cre (Figure 5B).
Figure 5. Bone marrow adipocytes are the source of RANKL in the absence of PTH1R signaling.

(A) Paraffin sections of Prx1Cre;PTH1Rfl/fl femurs show more TRAP positive cells than controls. n=3. Scale bar: 500μm in low magnification, 200μm in high magnification. (B) Expression of Opg, Rankl and Rank mRNA and Rankl/Opg ratio in long bones and spine of PTH1Rfl/fl and Prx1Cre;PTH1Rfl/fl mice at 3 wks. n=12. (C) Histological section of proximal tibia of a Prx1Cre; PTH1Rfl/fl mouse at 3 weeks. TRAP-positive osteoclasts lining the bone surface are found in close vicinity to the many large adipocytes located in the bone marrow space. Scale bar: 200μm. (D) qRT-PCR analysis for Rankl mRNA expression in flushed whole bone marrow (whole BM) and isolated bone marrow adipose tissue (BMAT). n=6/control, 7/mutant. (E) Serum and BM supernatant RANKL protein levels were significantly elevated in Prx1Cre;PTH1Rfl/fl when compared to PTH1Rfl/fl. n=17/control, 16/mutant. *p<0.05, **p<0.01 versus control. All graphs show mean ± SEM. See also Figure S4 and S5.
The increase in bone resorption in the mutants was accompanied by a 4-fold increase in urinary calcium excretion (Fan et al., 2016) confirming enhanced bone resorption despite the absence of functional PTH1R. These data were rather surprising since loss of PTH1R in long bones excludes PTH as a stimulator of Rankl (Figure S4A). We then tested serum levels of other RANKL regulators such as 1,25(OH)2D3 (Fan et al., 2016), tumor necrosis factor α(TNFα), interleukin 6 (IL-6) and leptin, but only the latter one was markedly decreased in mutants (Figure S4B). Changes in any of these circulating factors could have also affected Rankl expression in the spine, however no such changes were noted between controls and mutants (Figure 5B). To further address this question, we performed more detailed histological analyses which revealed the provocative finding that many of the larger adipocytes located in the mutant marrow space were situated in very close proximity to the bone surface, which was covered by TRAP-positive osteoclasts suggesting potential cross-communication (Figure 5C).
Next, to determine the source of the increased Rankl levels in mutants we isolated RNA from whole bone marrow and isolated BMAT from controls and mutants to perform qRT-PCR analyses. Rankl expression was significantly upregulated in mutant whole BM and BMAT (Figure 5D). Moreover, we measured RANKL protein levels by ELISA in the serum and BM supernatant of control and mutant mice. Interestingly, PTH1R deficient samples showed a marked elevation in RANKL protein indicating again that early loss of PTH1R signaling results in uncontrolled differentiation of mesenchymal cells to bone marrow adipocytes that can produce high levels of RANKL to induce bone resorption (Figure 5E).
RANKL production is limited to BMAT
We then examined the capability of other adipose depots to secrete RANKL. To explore this possibility and to clarify whether RANKL production was limited to bone marrow adipocytes of mutants, we analyzed Rankl gene expression in three peripheral adipose depots: eWAT, iWAT and iBAT. We found virtually undetectable Rankl expression in either genotype when compared to BMAT (Figures S4C and D). Thus the marrow adipocytes in Prx1Cre;PTH1Rfl/fl are unique in their expression signature, possibly reflecting their site of origin.
In addition, we tested whether genetic absence of the PTH1R was temporally specific such that loss of the receptor later in adipocyte differentiation resulted in greater peripheral RANKL expression. PTH1R was ablated using AdiponectinCre, which showed significantly reduced Pth1r expression in peripheral adipose depots (eWAT, iWAT and iBAT). Virtually undetectable Rankl expression was found in peripheral fat of either PTH1Rfl/fl or AdiponectinCre;PTH1Rfl/fl mice (Figure S4E). Moreover, primary stromal vascular fraction pre-adipocytes from iWAT did not respond to PTH treatment in vitro (Figure S4F).
To be absolutely certain that Rankl was not expressed in mature adipocytes outside the marrow, we examined RANKL ribosomal RNA expression in peripheral fat depots from AdiponectinCre;eGFP-L10a mice using the translating ribosome affinity purification (TRAP) technology (Liu et al., 2014). No detectable RANKL was noted in the inguinal depot of those mice (Figure S5A), whereas all other peripheral markers of adipogenesis were enriched in AdiponectinCre;eGFP-L10a mice (data not shown). Similarly, Zfp467 mRNA, a marker of adipocytes that is suppressed by PTH, was 60% enriched in the eGFP+ cells from the AdiponectinCre;eGFP-L10a mice versus Cre-negative controls (Figure S5A). In addition, to exclude any potential contribution of RANKL by macrophages present in isolated BMAT preparation we performed flow cytometry using CD11b as marker and found less than 2% contamination excluding macrophages as main source of RANKL in BMAT (Figure S5B). Moreover, we evaluated RANKL expression in other tissues and found no differences in RANKL expression between control and mutant mice in thymus or spleen (Figure S5C and D), and Rankl was undetectable in liver (data not shown).
Increased bone marrow Rankl is independent of culture conditions
In order to determine whether the increased Rankl expression found in the bone marrow of Prx1Cre;PTH1Rfl/fl mice could be altered by various culture conditions, we grew control and mutant cells in either osteogenic (OM), adipogenic (AM), or normal (no treatment) medium. The results showed that independent of which culture condition was used, mutant BMSCs had consistently and significantly higher Rankl and markedly reduced Opg expression levels leading to an increased Rankl/Opg ratio under every circumstance (Figure S6A). We also asked whether lack of PTH1R in MSCs would change the number of colony forming units and performed colony-forming unit fibroblastic (CFU-F), CFU-alkaline phosphatase (CFU-ALP), and CFU-Adipocyte (CFU-Adip) assays and found that the frequency of CFU-F mesenchymal progenitors, CFU-ALP osteoblast precursors as well as CFU-adipo adipocyte precursors is significantly increased in mutants with a higher increase in CFU-Adipo adipocyte precursors (Figure S6B).
Loss of PTH1R in MSCs increases the number of RANKL expressing pre-adipocytes
To further characterize RANKL positive cells in the bone marrow we performed extensive flow cytometry using the RANKL antibody in combination with Pref-1 as the marker of pre-adipocytes (Takeshita et al., 2014) and B220 as a B cell lineage marker (Figure 6) (Smas and Sul, 1993). FACS analysis revealed that in B220 negative cells, the percentage of Pref-1 positive cells was increased in Prx1Cre;PTH1Rfl/fl,Tmfl/+ mice compared to control heterozygous littermates (Figure 6A left, Q1+Q2). Moreover, mutants showed more RANKL positive cells in bone marrow mesenchymal cells (Figure 6A right, Q2+Q3). Importantly, we observed higher Pref-1 and RANKL double positive cells in the mutant mice in B220− cells (Figure 6A, Q2). All measurements between control and mutant littermates were significantly different (genotype effect p<0.01). When mutant bone marrow cells were gated using Pref-1 and B220, the Pref-1+/B220− population had a significantly higher mean fluorescence signal for RANKL expression (p<0.05) compared to the ones in Pref-1−/B220− (Figure 6B, R1:1738 vs. R2:947). Prx1Cre;PTH1Rfl/+,Tmfl/+ heterozygous mice were used as littermate controls.
Figure 6. Loss of PTH1R in MSCs increases the number of RANKL expressing pre-adipocytes.

Bone marrow cells of 3-wk-old controls (Prx1Cre;PTH1Rfl/+,Tmfl/+, PTH1Rfl/fl) and mutants (Prx1Cre;PTH1Rfl/fl,Tmfl/+, Prx1Cre;PTH1Rfl/fl) were stained with FITC-anti-B220 antibody, PE-anti-Pref-1 and APC/Cy7-anti-RANKL antibody and analyzed by flow cytometry. (A) Cells were gated on B220− cells and characterized with Pref-1 and RANKL (separated into Q1–Q4). Q1+Q2 represent Pref1+B220− population, Q2+Q3 represent RANKL+B220− population and Q2 represents Pref-1+RANKL+B220− population. Table shows the percentage of each population. (B) Mutant cells were gated on B220 and Pref-1 and separated into R1 and R2, and each fraction was analyzed with RANKL expression. R1 represents Pref-1+RANKL+B220− and R2 represents Pref-1-RANKL+B220− population. Table shows mean fluorescence intensity of RANKL expression. Results shown are representative of three independent experiments displaying similar results.
Next, to determine whether the large number of bone marrow adipocytes in mutants derives from Prx1 positive mesenchymal progenitors, we cultured BMSCs from 3wk old Prx1Cre;Tmfl/+ and Prx1Cre;PTH1Rfl/fl,Tmfl/+ mice and double stained them with antibodies for i) Tomato to affirm the Prx1 lineage and ii) Adiponectin to reassert the adipocyte phenotype. The merged image shown in Figure 7A illustrated many double positive cells (yellow), and their number was statistically increased in mutant BMSCs (Figure 7B), providing firm evidence that mesenchymal progenitor cells void of PTH1R preferentially differentiate into adipocytes.
Figure 7. Cells ablated for PTH1R preferentially developed into adipocytes in vitro.

(A) Immunofluorescence staining of BMSCs cultured under adipogenic conditions derived from 3wk old Prx1Cre;Tmfl/+ and Prx1Cre;PTH1Rfl/fl,Tmfl/+ mice. Red: Prx1-Tomato positive cells, green: adipocytes expressing adiponectin, blue: DAPI stain for cell nuclei and yellow: double positive (red/green) cells rendering BMSCs undergoing adipogenic differentiation. Scale bar: 100μm. (B) The percentage of double positive cells in Prx1Cre;PTH1Rfl/fl,Tmfl/+ mice was significantly higher than in controls. n=3. **p<0.01 versus control. All graphs show mean ± SEM.
Discussion
In this study, we demonstrated that genetic loss of PTH receptor signaling in mesenchymal progenitors results in a rapid increase in BMAT accompanied by very low bone mass due in part to a high rate of bone resorption. We also showed that marrow adipocytes, unlike peripheral adipocytes secrete RANKL, their progenitors are responsive to PTH, and their origin differs from other adipocytes. Moreover, we found the impact on mesenchymal cell fate from deletion of the PTH1R was temporally specific and that loss of the PTH1R later in osteoprogenitor differentiation had no effect on cell fate. Importantly, we provided the first evidence from paired bone biopsies that patients treated with PTH(1-34) show a significant reduction in marrow adipocyte number reinforcing our tenet that activation of the PTH1R regulates mesenchymal cell fate in both mice and humans.
BMAT comprises a significant proportion of total body fat stores in humans, yet its origin and function have not been clearly defined (Sulston and Cawthorn, 2016). Marrow adipocytes are dynamic; i.e. their size and number can change in response to environmental (e.g. cold temperature via sympathetic activation), nutritional (e.g. calorie restriction or high fat diet), and hormonal (e.g. estrogen deficiency, leptin) cues. Previous work suggested that PTH could influence MSCs allocation in the bone marrow niche (Kim et al., 2012) and that constitutive activation of the PTH1R experimentally (Col1caPPR) caused high bone mass, less hematopoiesis and delayed development of bone marrow constituents, particularly adipocytes (Calvi et al., 2001). Cain et al. demonstrated that targeting osteoblastic Rs1, an activator of Gs signaling, using the Col2.3 promoter, enhanced bone mass and increased insulin sensitivity, but reduced both bone marrow and peripheral adiposity (Cain et al., 2016). In contrast, conditional deletion of Gsα in early osteoprogenitors by OsxCre resulted in impaired bone formation and accelerated marrow adipogenesis (Sinha et al., 2014). Recently it was reported that marrow adipocytes trace with Osterix, a marker of the osteogenic lineage (Chen et al., 2014; Liu et al., 2013). However, using OsxCre for deletion of PTH1R in slightly more mature pre-osteoblasts, we were unable to detect an increase in BMAT (Figure S7). This lends further support to the tenet that PTH1R activation impacts cell fate in the marrow in a context specific manner. However, the precise temporal sequence of lineage allocation in mesenchymal progenitors has not been determined.
In our studies, we isolated both marrow adipocytes and their progenitors, and found those cells express all the markers of classic adipocytes. However, we were not able to define the earliest transcriptional event in this process. One possible candidate is Zfp467. Previously, the Martin group noted from a gene array study of a stromal cell line that Zfp467 was downregulated by PTH (Quach et al., 2011). Moreover, silencing of Zfp467 was found to induce osteogenesis (You et al., 2012). Those authors also reported that Zfp467 could not only upregulate PPARγ but also induce RANKL and that it was a critical co-factor in adipogenic differentiation and in the development of marrow adipocytes in vivo. You et al. also reported that Zfp467 was an important lineage determinant, and that it regulated Sost independent of its effects on PPARγ, thereby targeting the Wnt signaling pathway (You et al., 2015). In our studies we found that PTH administration suppressed Zfp467 expression in cortical bone from control but not mutant mice (data not shown), and that Zfp467 was up-regulated 8-fold in RANKL producing marrow adipocytes from Prx1Cre;PTH1Rfl/fl mice when compared to controls (Figure 2B). In support of that observation, primary BMSCs differentiated in adipogenic media had a four-fold increase in expression relative to baseline, gene expression for other markers of adipogenesis (Pparγ, Fabp4, Adipo, Perilipin) were also 5–10 fold increased in the marrow adipocytes of mutants versus controls (data not shown). Thus the marrow adipocyte in our model exhibited characteristic features of classic white adipocytes but also had the capacity to secrete RANKL and impact bone resorption.
The origin of marrow adipocytes is not known, but lineage tracing has provided insights into the nature of osteo-progenitors. For example, Yue et al. demonstrated that sustained increases in leptin, acting via the leptin receptor promoted adipocyte differentiation by skeletal stem cells during high fat feeding (Yue et al., 2016). Moreover, genetic deletion in the limbs of the LepR using the Prx1Cre prevented high fat diet induced marrow adipogenesis as well as bone loss from accelerated resorption. In our studies, we also found that deletion of the PTH1R with Prx1Cre, had a major impact on cell fate, with a significant reduction in osteoprogenitors and a marked increased in marrow adipocytes. But in contrast to the Yue work, the Prx1Cre;PTH1Rfl/fl mice had low leptin levels and lower body weight, suggesting that other determinants were operative in driving adipogenic differentiation within the niche.
BMAT is often accompanied by bone loss due to increased bone resorption. In Prx1Cre;PTH1Rfl/fl mice high resorption rates contributed to low bone mass in both trabecular and cortical compartments. But this seemed counter-intuitive. First, bone marrow osteoprogenitors are an important source of RANKL, and are activated by PTH (O'Brien, 2010). In the absence of functional PTH activity it would seem unlikely that bone resorption could be high. Second, a previous report had noted that overexpression of the PTH1R, rather than deletion, led to greater bone resorption (Calvi et al., 2001). To address this apparent paradox we asked whether another cell could drive the high resorption phenotype. Osteocytic osteolysis due to enhanced expression of RANKL could have contributed to the high rate of bone resorption but we found no differences in osteocyte number/area or lacunar area in mutant versus control mice (Fan et al., 2016).
But, one clue came from histology in which we noted that many of the marrow adipocytes were located adjacent to the endosteal bone surface and closely associated with TRAP+ osteoclasts (Figure 5C). Moreover, a similar feature was noted in human biopsies. Thus, we hypothesized that an early progenitor harboring adipogenic properties in the bone marrow could secrete RANKL and was responsible for high bone resorption in mutants. Several lines of experimental evidence supported that tenet. For example, we found high Rankl expression in isolated bone marrow adipocytes from the mutant mice (Figure 5D) and high RANKL protein in the bone marrow supernatant and in the serum of Prx1Cre;PTH1Rfl/fl mice (Figure 5E). Also in gating experiments we found that the proportion of Pref1+/RANKL+/B220− cells was two-fold higher in the marrow of mutants than controls (Figure 6). It is of note that Sinha et al. also reported that GsαOsxKO bones showed an 8-fold increase in Pref-1 expression, a marker of pre-adipocytes, compared to controls (Sinha et al., 2014). And previously, Takeshita et al. noted that in aging mouse marrow, with significant adiposity, the proportion of RANKL+/Pref1+ cells was markedly increased (Takeshita et al., 2014). In addition, we noted that BMSCs derived from the mutants when grown in osteogenic, adipogenic or no differentiation media, expressed consistently higher levels of Rankl and greater Rankl/Opg ratios than controls (Figure S6A). Finally, we were able to exclude any significant contribution of Rankl production by additional tissues outside of BMAT. The TRAP technology (Liu et al., 2014) was useful to exclude other adipose depots such as iWAT as contributor. FACS and qRT-PCR analyses eliminated spleen, thymus and liver as main sources of Rankl in Prx1Cre;PTH1Rfl/fl mice, respectively. Moreover, we validated our BMAT preparation and could show that our cell population is almost void of any macrophages. Thus, it is conceivable that early mesenchymal marrow progenitor cells destined to become adipocytes have the capacity to secrete RANKL.
An important translational finding from our work was the demonstration that in vivo PTH treatment reduces BMAT in both mice and humans. Intermittent administration of teriparatide (PTH 1-34) is widely used for the treatment of postmenopausal osteoporosis because it reduces fractures and increases bone mineral density (Neer et al., 2001). Changes in bone remodeling with intermittent PTH administration are generally anabolic and reflect an early but marked increase in bone formation. BMAT changes have not been reported in randomized trials of teriparatide or PTH(1-84). But in a small pilot study, Cohen et al. reported that adipocyte volume per marrow volume, perimeter and area but not number were significantly reduced in a small group (n=21) of women with idiopathic osteoporosis (Cohen et al., 2013). More recently, Yang et al. demonstrated with MRI spectroscopy of the femur, that PTH(1-34) treatment in postmenopausal women reduced BMAT (Yang et al., 2016).
Recent studies have shown that PTH can act directly on mature adipocytes to induce lipolysis and these cells can also respond to PTHrP by changing their adipogenic program towards a more thermogenic pattern (Kir et al., 2016). Importantly in the Yang et al. study where MRI demonstrated loss of BMAT in response to PTH(1-34), they failed to find any changes in peripheral fat depots (subcutaneous or visceral), nor have there been any reports of changes in peripheral fat mass in patients treated with PTH(1-34) (Yang et al., 2016). In our analysis of paired bone biopsies from 7 males in a small randomized trial of PTH for male idiopathic osteoporosis (Dempster et al., 2001), we found a 27% reduction in adipocyte number but not size at 18 months in men treated with PTH. Although the trial was not powered for change in marrow fat cell volume or number, these data support the need for larger studies from clinical trials.
There are several limitations to our current work. First, we cannot exclude the possibility that other systemic factors contributed to enhanced marrow adiposity in the mutant mice. We measured several circulating factors, including IL-6 and TNFα that could cause increased numbers of marrow adipocytes, but these hormones were not elevated. On the other hand, leptin levels were low in mutants. In some models such as the Ob/Ob mouse, or in women with anorexia, low circulating leptin concentrations have been associated with higher marrow adiposity in the limbs of mice and vertebrae of humans (Bredella et al., 2009; Hamrick et al., 2004). Second, although calorie restriction can induce marrow adiposity, it is unlikely that nutritional factors played a role in the induction of adipogenesis since the mice were just weaned at the time of sacrifice. Moreover, in mutants the greatest enhancement in marrow adiposity was noted in the distal tibia, a site of constitutive marrow adipose tissue, and one in which few or no changes have been noted with calorie restriction (Scheller et al., 2015). Third, although we identified Zfp467 as a transcriptional factor down regulated by PTH during osteogenesis that may mediate RANKL production, we have not established that it is the principle driver of marrow adipogenesis in the absence of PTH1R. Further genetic studies are needed to delineate the precise role of Zfp467 in mediating marrow adipogenesis. Fourth, to determine the temporal importance of PTH1R deletion from progenitor cells we used OsxCre tg mice to target more mature pre-osteoblasts. We compared the BMAT phenotype of OsxCre;PTH1Rfl/fl and PTH1Rfl/fl mice and found no differences. But since OsterixCre mice are slightly runted, a non-floxed OsxCre tg mouse would have served as a better control.
In summary, we showed that deletion of the PTH1R in bone marrow mesenchymal progenitors results in the phenotypic presentation of low bone mass, high bone marrow adiposity and increased bone resorption. We established that the bone marrow adipocyte is unique in supporting osteoclastogenesis. We also showed for the first time, that the anabolic skeletal effect of PTH can be attributed, at least in part, to its ability to change cell fate.
Experimental Procedures
Mouse strains
Prx1Cre;PTH1Rfl/fl mice were generated by crossing PTH1Rfl/fl mice (Kobayashi et al., 2002) to Prx1Cre transgenic mice (Logan et al., 2002) as described previously (Fan et al., 2016). B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J mice (Tmfl/fl) and AdiponectinCre mice were purchased from Jackson Laboratory. Prx1Cre;Tmfl/+ mice were generated by mating Prx1Cre to Tmfl/fl mice. PTH1Rfl/fl mice (± AdiponectinCre) (Kir et al., 2016) and AdiponectinCre;eGFP-L10a mice (Liu et al., 2014) were described previously. Mice lacking PTH1R in osteoprogenitors were generated by crossing PTH1Rfl/fl with OsterixCre mice (Panaroni et al., 2015). All studies performed were approved by the Institutional Animal Care and Use Committee at the Harvard Medical School.
Isolation of bone marrow adipocytes and supernatant
Both femurs and tibiae were collected and cleaned in sterile PBS. Both ends of femurs and tibias were snipped. The bone marrow adipocytes were isolated as previously described (Liu et al., 2011; Scheller et al., 2015). Bones were placed in a 0.6 mL microcentrifuge tube that was cut open at the bottom and placed into a 1.5 mL microcentrifuge tube. Fresh bone marrow was spun out by quick centrifuge (from 0 to 10,000 rpm, 9 seconds, room temperature (RT)). Red blood cells were lysed using RBC lysing buffer (Sigma). After centrifugation (3000 rpm, 5 minutes, RT), floating adipocytes were collected from the top layer and washed with PBS for 3 times. The remaining middle layer of the bone marrow was collected as bone marrow supernatant for RANKL measurement using an ELISA.
Analysis of paired bone biopsies from males with idiopathic osteoporosis
We analyzed bone biopsy samples from a previous trial that included eight men with paired (before and after) iliac crest bone biopsies treated with PTH(1-34) (Dempster et al., 2001). Analysis was performed using a reader blinded to treatment assignment using the semi-automated Bio-Quant methodology for histomorphometry. The primary outcomes were adipocyte number/marrow volume and adipocyte volume/marrow volume. Cell ghosts and ghost volume were measured in three regions of the iliac crest biopsy section from each subject before and after treatment and the mean was calculated for adipocyte volume/marrow volume, and adipocyte number/marrow volume. Seven of the 8 biopsies pairs were readable in respect to measurements of discrete adipocytes within the marrow cavity. This study was approved by the Institutional Review Boards of Helen Hayes Hospital and Columbia-Presbyterian Medical Center. All subjects gave informed consent.
Statistics
For statistical analysis, GraphPad Prism 5.0 (Graphpad Software Inc.) was used. Unpaired two-tailed Student’s t test was used between two groups in mice and one-way ANOVA followed by Tukey’s test was used for multiple comparisons. Variables between vehicle and PTH treatment groups were evaluated by two-way ANOVA. Two-way ANOVA with multiple comparisons was used for flow cytometry to calculate the difference between control and mutant mice. For the translational study in men with paired biopsies, we sought to test the hypothesis from the mouse data that PTH would reduce adipocyte number, so we employed a one-tailed t-test with paired samples to examine the treatment effect (Dempster et al., 2001). All values are expressed as mean ± SEM. P values <0.05 were considered significant for all analyses. See Supplemental Experimental Procedures for detailed methods.
Supplementary Material
Highlights.
PTH1R regulates lineage allocation in the marrow
Bone marrow adipocytes compose a unique adipose depot and produce RANKL
PTH reduced marrow adipogenesis in mice and man
Acknowledgments
We thank Drs. Noriko Ide, William N. Addison, and Tadatoshi Sato for help with experimental protocols and Mr. Hao Wang for assistance with animal care. We would also like to thank Dr. TJ Martin for discussing the data and providing valuable input. This work was supported by grants from NIH-DK097105 (BL), DK100584 (MM), DK092759-06 (CR), Dean’s Scholarship from Harvard School of Dental Medicine (YF), and State Key Laboratory of Oral Diseases Open Funding (SKLOD2015OF01). Baron R. is on the advisory board of Eli Lilly and Radius Health, and Dempster D. obtains research support, consulting fees and honoraria for speaking at Eli Lilly.
Footnotes
Author Contributions
Y.F. designed and performed most of the experiments. J.H., P.L., R.B., V.D., D.M., and C.F. performed experiments. R.T.B. performed mice necropsy. S.K., M.H. and J.W. provided mouse samples and J.B., D.D. provided human biopsies. J.H., X.Z., R.B., M.M., C.R. and B.L. conceived, designed and supervised the project. Y.F., C.R. and B.L. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amizuka N, Karaplis AC, Henderson JE, Warshawsky H, Lipman ML, Matsuki Y, Ejiri S, Tanaka M, Izumi N, Ozawa H, et al. Haploinsufficiency of parathyroid hormone-related peptide (PTHrP) results in abnormal postnatal bone development. Dev Biol. 1996;175:166–176. doi: 10.1006/dbio.1996.0104. [DOI] [PubMed] [Google Scholar]
- Bredella MA, Fazeli PK, Miller KK, Misra M, Torriani M, Thomas BJ, Ghomi RH, Rosen CJ, Klibanski A. Increased bone marrow fat in anorexia nervosa. J Clin Endocr Metab. 2009;94:2129–2136. doi: 10.1210/jc.2008-2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain CJ, Valencia JT, Ho S, Jordan K, Mattingly A, Morales BM, Hsiao EC. Increased Gs Signaling in Osteoblasts Reduces Bone Marrow and Whole-Body Adiposity in Male Mice. Endocrinology. 2016;157:1481–1494. doi: 10.1210/en.2015-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, Kronenberg HM, Baron R, Schipani E. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest. 2001;107:277–286. doi: 10.1172/JCI11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthorn WP, Scheller EL, Learman BS, Parlee SD, Simon BR, Mori H, Ning X, Bree AJ, Schell B, Broome DT, et al. Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab. 2014;20:368–375. doi: 10.1016/j.cmet.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan GK, Deckelbaum RA, Bolivar I, Goltzman D, Karaplis AC. PTHrP inhibits adipocyte differentiation by down-regulating PPAR gamma activity via a MAPK-dependent pathway. Endocrinology. 2001;142:4900–4909. doi: 10.1210/endo.142.11.8515. [DOI] [PubMed] [Google Scholar]
- Chan GK, Miao D, Deckelbaum R, Bolivar I, Karaplis A, Goltzman D. Parathyroid hormone-related peptide interacts with bone morphogenetic protein 2 to increase osteoblastogenesis and decrease adipogenesis in pluripotent C3H10T 1/2 mesenchymal cells. Endocrinology. 2003;144:5511–5520. doi: 10.1210/en.2003-0273. [DOI] [PubMed] [Google Scholar]
- Chen J, Shi Y, Regan J, Karuppaiah K, Ornitz DM, Long F. Osx-Cre targets multiple cell types besides osteoblast lineage in postnatal mice. PloS One. 2014;9:e85161. doi: 10.1371/journal.pone.0085161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A, Stein EM, Recker RR, Lappe JM, Dempster DW, Zhou H, Cremers S, McMahon DJ, Nickolas TL, Muller R, et al. Teriparatide for idiopathic osteoporosis in premenopausal women: a pilot study. J Clin Endocr Metab. 2013;98:1971–1981. doi: 10.1210/jc.2013-1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster DW, Cosman F, Kurland ES, Zhou H, Nieves J, Woelfert L, Shane E, Plavetić K, Müller R, Bilezikian J, et al. Effects of Daily Treatment with Parathyroid Hormone on Bone Microarchitecture and Turnover in Patients with Osteoporosis: A Paired Biopsy Study. J Bone Miner Res. 2001;16:1846–1853. doi: 10.1359/jbmr.2001.16.10.1846. [DOI] [PubMed] [Google Scholar]
- Fan Y, Bi R, Densmore MJ, Sato T, Kobayashi T, Yuan Q, Zhou X, Erben RG, Lanske B. Parathyroid hormone 1 receptor is essential to induce FGF23 production and maintain systemic mineral ion homeostasis. FASEB J. 2016;30:428–440. doi: 10.1096/fj.15-278184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou KR, Hui SK, Xian CJ. Regulatory pathways associated with bone loss and bone marrow adiposity caused by aging, chemotherapy, glucocorticoid therapy and radiotherapy. Am J Stem Cells. 2012;1:205–224. [PMC free article] [PubMed] [Google Scholar]
- Hamrick MW, Pennington C, Newton D, Xie D, Isales C. Leptin deficiency produces contrasting phenotypes in bones of the limb and spine. Bone. 2004;34:376–383. doi: 10.1016/j.bone.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Ishizuya T, Yokose S, Hori M, Noda T, Suda T, Yoshiki S, Yamaguchi A. Parathyroid hormone exerts disparate effects on osteoblast differentiation depending on exposure time in rat osteoblastic cells. J Clin Invest. 1997;99:2961–2970. doi: 10.1172/JCI119491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. 1999;104:439–446. doi: 10.1172/JCI6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37:148–158. doi: 10.1016/j.bone.2005.03.018. [DOI] [PubMed] [Google Scholar]
- Kim SW, Pajevic PD, Selig M, Barry KJ, Yang JY, Shin CS, Baek WY, Kim JE, Kronenberg HM. Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J Bone Miner Res. 2012;27:2075–2084. doi: 10.1002/jbmr.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kir S, Komaba H, Garcia AP, Economopoulos KP, Liu W, Lanske B, Hodin RA, Spiegelman BM. PTH/PTHrP Receptor Mediates Cachexia in Models of Kidney Failure and Cancer. Cell Metab. 2016;23:315–323. doi: 10.1016/j.cmet.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Chung UI, Schipani E, Starbuck M, Karsenty G, Katagiri T, Goad DL, Lanske B, Kronenberg HM. PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development. 2002;129:2977–2986. doi: 10.1242/dev.129.12.2977. [DOI] [PubMed] [Google Scholar]
- Li C, Xing Q, Yu B, Xie H, Wang W, Shi C, Crane JL, Cao X, Wan M. Disruption of LRP6 in osteoblasts blunts the bone anabolic activity of PTH. J Bone Miner Res. 2013;28:2094–2108. doi: 10.1002/jbmr.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Krautzberger AM, Sui SH, Hofmann OM, Chen Y, Baetscher M, Grgic I, Kumar S, Humphreys BD, Hide WA, et al. Cell-specific translational profiling in acute kidney injury. J Clin Invest. 2014;124:1242–1254. doi: 10.1172/JCI72126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LF, Shen WJ, Ueno M, Patel S, Kraemer FB. Characterization of age-related gene expression profiling in bone marrow and epididymal adipocytes. BMC Genomics. 2011;12:212. doi: 10.1186/1471-2164-12-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Strecker S, Wang L, Kronenberg MS, Wang W, Rowe DW, Maye P. Osterix-cre labeled progenitor cells contribute to the formation and maintenance of the bone marrow stroma. PloS One. 2013;8:e71318. doi: 10.1371/journal.pone.0071318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matic I, Matthews BG, Wang X, Dyment NA, Worthley DL, Rowe DW, Grcevic D, Kalajzic I. Quiescent Bone Lining Cells Are a Major Source of Osteoblasts During Adulthood. Stem Cells. 2016;34:2930–2942. doi: 10.1002/stem.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009;460:259–263. doi: 10.1038/nature08099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–1441. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- O'Brien CA. Control of RANKL gene expression. Bone. 2010;46:911–919. doi: 10.1016/j.bone.2009.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaroni C, Fulzele K, Saini V, Chubb R, Pajevic PD, Wu JY. PTH Signaling in Osteoprogenitors Is Essential for B-Lymphocyte Differentiation and Mobilization. J Bone Miner Res. 2015;30:2273–2286. doi: 10.1002/jbmr.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieur X, Dollet L, Takahashi M, Nemani M, Pillot B, Le May C, Mounier C, Takigawa-Imamura H, Zelenika D, Matsuda F, et al. Thiazolidinediones partially reverse the metabolic disturbances observed in Bscl2/seipin-deficient mice. Diabetologia. 2013;56:1813–1825. doi: 10.1007/s00125-013-2926-9. [DOI] [PubMed] [Google Scholar]
- Quach JM, Walker EC, Allan E, Solano M, Yokoyama A, Kato S, Sims NA, Gillespie MT, Martin TJ. Zinc finger protein 467 is a novel regulator of osteoblast and adipocyte commitment. J Biol Chem. 2011;286:4186–4198. doi: 10.1074/jbc.M110.178251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen CJ, Ackert-Bicknell C, Rodriguez JP, Pino AM. Marrow fat and the bone microenvironment: developmental, functional, and pathological implications. Crit Rev Eukaryot Gene Expr. 2009;19:109–124. doi: 10.1615/critreveukargeneexpr.v19.i2.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen CJ, Bouxsein ML. Mechanisms of disease: is osteoporosis the obesity of bone? Nature clinical practice Rheumatol. 2006;2:35–43. doi: 10.1038/ncprheum0070. [DOI] [PubMed] [Google Scholar]
- Scheller EL, Doucette CR, Learman BS, Cawthorn WP, Khandaker S, Schell B, Wu B, Ding SY, Bredella MA, Fazeli PK, et al. Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues. Nature Commun. 2015;6:7808. doi: 10.1038/ncomms8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller EL, Rosen CJ. What's the matter with MAT? Marrow adipose tissue, metabolism, and skeletal health. Annals New York Acad Sciences. 2014;1311:14–30. doi: 10.1111/nyas.12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha P, Aarnisalo P, Chubb R, Ono N, Fulzele K, Selig M, Saeed H, Chen M, Weinstein LS, Pajevic PD, et al. Loss of Gsalpha early in the osteoblast lineage favors adipogenic differentiation of mesenchymal progenitors and committed osteoblast precursors. J Bone Miner Res. 2014;29:2414–2426. doi: 10.1002/jbmr.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smas CM, Sul HS. Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell. 1993;73:725–734. doi: 10.1016/0092-8674(93)90252-l. [DOI] [PubMed] [Google Scholar]
- Sulston RJ, Cawthorn WP. Bone marrow adipose tissue as an endocrine organ: close to the bone? Horm Mol Biol Clin Investig. 2016;28:21–38. doi: 10.1515/hmbci-2016-0012. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Fumoto T, Naoe Y, Ikeda K. Age-related marrow adipogenesis is linked to increased expression of RANKL. J Biol Chem. 2014;289:16699–16710. doi: 10.1074/jbc.M114.547919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tascau L, Gardner T, Anan H, Yongpravat C, Cardozo CP, Bauman WA, Lee FY, Oh DS, Tawfeek HA. Activation of Protein Kinase A in Mature Osteoblasts Promotes a Major Bone Anabolic Response. Endocrinology. 2016;157:112–126. doi: 10.1210/en.2015-1614. [DOI] [PubMed] [Google Scholar]
- Thomas T, Burguera B, Melton LJ, 3rd, Atkinson EJ, O'Fallon WM, Riggs BL, Khosla S. Role of serum leptin, insulin, and estrogen levels as potential mediators of the relationship between fat mass and bone mineral density in men versus women. Bone. 2001;29:114–120. doi: 10.1016/s8756-3282(01)00487-2. [DOI] [PubMed] [Google Scholar]
- Weinstein LS, Yu S, Warner DR, Liu J. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev. 2001;22:675–705. doi: 10.1210/edrv.22.5.0439. [DOI] [PubMed] [Google Scholar]
- Yang Y, Luo X, Xie X, Yan F, Chen G, Zhao W, Jiang Z, Fang C, Shen J. Influences of teriparatide administration on marrow fat content in postmenopausal osteopenic women using MR spectroscopy. Climacteric. 2016;19:285–291. doi: 10.3109/13697137.2015.1126576. [DOI] [PubMed] [Google Scholar]
- You L, Chen L, Pan L, Gu WS, Chen JY. Zinc finger protein 467 regulates Wnt signaling by modulating the expression of sclerostin in adipose derived stem cells. Biochem Biophys Res Commun. 2015;456:598–604. doi: 10.1016/j.bbrc.2014.11.120. [DOI] [PubMed] [Google Scholar]
- You L, Pan L, Chen L, Chen JY, Zhang X, Lv Z, Fu D. Suppression of zinc finger protein 467 alleviates osteoporosis through promoting differentiation of adipose derived stem cells to osteoblasts. J Transl Med. 2012;10:11. doi: 10.1186/1479-5876-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Zhao X, Yang C, Crane J, Xian L, Lu W, Wan M, Cao X. Parathyroid hormone induces differentiation of mesenchymal stromal/stem cells by enhancing bone morphogenetic protein signaling. J Bone Miner Res. 2012;27:2001–2014. doi: 10.1002/jbmr.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue R, Zhou BO, Shimada IS, Zhao Z, Morrison SJ. Leptin Receptor Promotes Adipogenesis and Reduces Osteogenesis by Regulating Mesenchymal Stromal Cells in Adult Bone Marrow. Cell Stem Cell. 2016;18:782–796. doi: 10.1016/j.stem.2016.02.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.