

SUMMARY
Type 1 diabetes (T1D) is a chronic autoimmune disease that involves immune-mediated destruction of β cells. How β cells respond to immune attack is unknown. We identified a population of β cells during the progression of T1D in non-obese diabetic (NOD) mice that survives immune attack. This population develops from normal β cells confronted with islet infiltrates. Pathways involving cell movement, growth and proliferation, immune responses, and cell death and survival are activated in these cells. There is reduced expression of β cell identity genes and diabetes antigens and increased immune inhibitory markers and stemness genes. This new subpopulation is resistant to killing when diabetes is precipitated with cyclophosphamide. Human β cells show similar changes when cultured with immune cells. These changes may account for the chronicity of the disease and the long-term survival of β cells in some patients.
In Brief
Type 1 diabetes (T1D) is due to the immune-mediated destruction of β cells. Rui et al. identify a population of β cells that survives immune attack during T1D progression in non-obese diabetic mice, which may account for the long-term survival of some β cells in patients.
INTRODUCTION
Type 1 diabetes (T1D) is a chronic autoimmune disease involving the destruction of insulin-producing β cells by diabetes-antigen-specific T cells. Despite widespread acceptance of this paradigm, several key questions about the disease pathogenesis remain. For example, it is not clear why β cell killing takes such an extended period of time. Some have proposed that there is waxing and waning of the immune response, possibly due to environmental factors, and our previous studies support the periodic nature of β cell killing during the period prior to dysglycemia in individuals at risk (Herold et al., 2015; von Herrath et al., 2007).
In addition, β cell destruction is not always complete, and some β cells survive immune attack. Recent observational studies have shown that there are detectable levels of insulin production in individuals who have had diabetes for many years (Keenan et al., 2010; Liu et al., 2009). Consistent with this is the finding that the immunologic destruction is not uniform throughout the islets, and regions that are heavily infiltrated with immune cells may be located next to regions that are free of inflammation (Atkinson et al., 2011). These observations point to heterogeneity of β cells, but the nature and distribution of subpopulations, their ontogeny, and immunologic properties have not been identified.
Tissues can adapt to chronic inflammation. Adaptation may involve expression of inflammatory mediators, such as cytokines, or factors that may improve the viability of the cells, such as VEGF (Akirav et al., 2011; Eizirik et al., 2009). Little is known about how immune responses change β cells and how β cells may respond to immune mediators. The question is of clinical importance because despite the success of some immune mediators in reducing decline in β cell function short term, permanent remission of T1D has not been achieved. It is possible that intrinsic β cell factors lead to destruction.
Our lab and others have shown increased rates of β cell proliferation during the prediabetes period in non-obese diabetic (NOD) mice (Sherry et al., 2006; Sreenan et al., 1999). At diabetes onset, there is a population of degranulated β cells that may recover function. These β cells were assumed to have released all of their insulin granules, but the characteristics and durability of these cells are not known. In other settings of metabolic stressors, Talchai et al. described adaptation of β cells to metabolic stressors involving dedifferentiation with expression of transcripts associated with β cell precursors such as Neurog3, Oct4, and Nanog (Talchai et al., 2012).
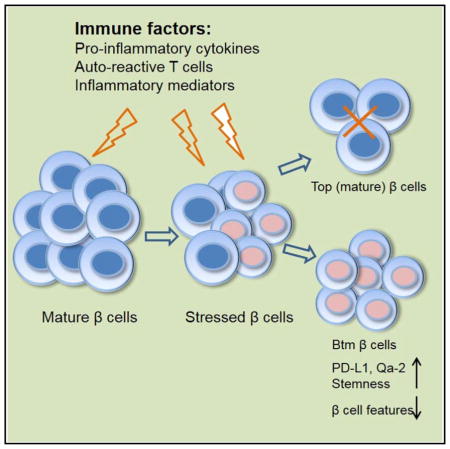
Studies of β cells in humans, particularly those at risk of T1D, are difficult because of the inaccessibility of tissue for study. Herein, we describe how β cells adapt to chronic auto-immunity in NOD mice by development of a novel subpopulation that can resist immune attack. This subpopulation has reduced expression of genes associated with their parent cells and acquires stem-like features, including increased rates of proliferation. These cells avoid immune attack by reducing the expression of diabetogenic antigens and increasing expression of immunomodulatory molecules. We postulate that adaptation in these cells may account for the chronicity of the disease and persistence of β cells in patients with T1D.
RESULTS
Subpopulations of β Cells Are Found during Progression of Diabetes in NOD Mice
We analyzed the cellular composition of islets from NOD and B6 mice of different ages by flow cytometry. Dissociated islet cells were stained with FluoZin and tetramethylrhodamine ethyl ester perchlorate (TMRE) to identify viable β cells (Jayaraman, 2011; Rui et al., 2016) and with anti-CD45 monoclonal antibody (mAb) to identify islet infiltrates. Representative β cell profiles showing side scatter (SSC) and forward scatter (FSC) in mice of different ages are shown (Figure 1A). At the beginning of infiltration with CD45+ cells at 4 weeks of age, a population of β cells with lower granularity (lower SSC) was seen. The proportion of lower-granularity cells among the total number of insulin-positive cells (Pearson r2 = 0.9537, p = 0.0043), and total CD45 islet cells (i.e., all CD45− islet cells; Pearson r2 = 0.984, p = 0.0009) significantly increased over time. The frequency of the lower-granularity population positively correlated with the appearance of CD45+ intra-islet immunocytes (Pearson r2 = 0.9741, p = 0.0018) (Figure 1C). By 12 weeks of age, when the mice were still normoglycemic, the lower-granularity population accounted for ~50% of all Zn+ β cells. We did not find similar β cell subsets in B6 mice (even up to 18 months of age) or in NOD severe combined immunodeficiency (SCID) mice (data not shown). In order to distinguish the different β cell populations, we refer to the parental β cells as Top β cells and the β cells with lower granularity as Btm (bottom) β cells (Figure 1A, arrows).
Figure 1. Changes in β Cell Composition during Diabetes Progression in NOD Mice<.

br>(A) Islets from non-obese diabetic (NOD) and B6 mice of different age were hand-picked, dispersed into single cells, and stained with FluoZin and tetramethylrhodamine ethyl ester perchlorate (TMRE) to identify viable β cells. Top and bottom (Btm) β cells were detected by differences in side scatter (SSC) and forward scatter (FSC) (numbers refer to the percentage of total Zn+TMRE+ cells). Shown are representative FACS profiles from one of three independent experiments, each with three to five mice at each time point.
(B) FACS side/forward scatter (SSC/FSC) profiles of GFP+ β cells from either mouse insulin-promoter (MIP) NOD-GFP or age-matched B6-GFP transgenic mice. Numbers refer to the percentage of either Top or Btm β cells among all GFP+ cells. Data are from one individual experiment representative of three individual experiments with three or four mice for each time point.
(C) The frequency of intra-islet lymphocytes and Btm β cells as a proportion of β cells or total islet endocrine cells increased over time. The lines show Btm β cells as a proportion of insulin-positive islet cells (○) (Pearson r2 = 0.9537, p = 0.0043) and as a proportion of total islet cells (Δ) (i.e., CD45−, Pearson r2 = 0.984, p = 0.0009). The frequency of islets infiltrates (i.e., CD45+) increased over time (⋄) (Pearson r2 = 0.9083, p = 0.0121). Data represent mean ± SEM (n = 6 mice each time point). There was a close relationship between the frequency of intra-islet lymphocytes and bottom (Btm) cells (Pearson r2 = 0.987, p = 0.002).
(D) The results of IPGTT and FACS profiles of β cells from two 9-week-old NOD mice are shown.
(E) The ratio of the mean fluorescence intensity (MFI) of FACS-sorted Top and Btm β cell islets of NOD mice of different ages, stained with anti-insulin-APC (Allophycocyanin) antibody, is shown. Column statistics refer to a one-sample t test compared to a mean of 1. Data represent mean ± SEM from individual mice (**p < 0.01, ***p < 0.001, and ****p < 0.0001).
(F) Top and Btm β cells were sorted from islets from six mice, and insulin was measured in cultures with 1 g/L or 4.5 g/L glucose for 8 hr. Top cells made more insulin (p < 0.0001) than Btm cells and responded to high glucose. Data represent individual wells (50,000 cells in each well) from pools of three to four mice (p < 0.0001, two-way ANOVA, multiple comparisons).
To confirm that the cells with lower granularity were β cells and not α or other cells that may have Zn+ granules, we gated on the GFP+ β cells in NOD-GFPTg and B6- GFPTg mice in which GFP expression is driven by the mouse Ins1 promoter (Figure 1B). Diabetes occurs earlier (between 6 and 9 weeks after birth) in NOD GFPTg mice than in NOD mice, and we therefore analyzed these mice beginning at 3 weeks of age. As in wild-type NOD mice, we identified two populations, based on SSC, among GFP+ β cells in transgenic NOD, but not B6, mice.
Glucose itself may affect β cell mass and granularity (Jonas et al., 1999; Laybutt et al., 2003). To determine whether increasing glucose was associated with the changes in β cells, we performed intraperitoneal glucose tolerance tests (IPGTTs) on NOD mice aged 6–13 weeks and B6 mice aged 7–10 weeks and compared the glucose area under the curve (AUC) and peak glucose during the 120-min test. The AUC and peak glucose levels did not increase significantly in NOD mice over this age range and were significantly lower in NOD mice than in B6 mice at each time point (Figure S1, available online). In addition, we compared IPGTT results in NOD and NOD/ SCIDγc−/− mice, which do not have an immune system, and found that neither the AUC nor peak glucose levels were significantly different in these strains (Figure S1). Figure 1D shows the normal IPGTT and the fluorescence-activated cell sorting (FACS) plots of β cells from two NOD mice aged 9 weeks.
The reduced granularity was associated with reduced insulin content per β cell. We compared the mean fluorescence intensity (MFI) of insulin in Btm cells and Top cells in NOD mice of different ages and found a decrease in insulin protein in Btm versus Top cells beginning after 4 weeks of age, with a precipitous decline at 12 weeks (Figure 1E). Insulin release from FACS-sorted Top cells was higher, and Top, but not Btm, cells responded to high glucose (Figure 1F).
Previous reports have highlighted the role of endoplasmic reticulum (ER) stress in β cell function and survival (Tersey et al., 2012). To determine whether Btm cells have increased ER stress and cell death, we compared the expression of ER stress markers in FACS-sorted subpopulations from islets of 8- to 10-week-old NOD mice. We did not identify significant differences in expression of Chop, Xbp1, Bip, Aft4, or Wfs1 (Figure S2A). In addition, the expression of the autophagy genes Atg5, LC3b, Becn1, Bnip3, and Gabarap as well as the cell apoptosis gene Casp3 was similar between the two subpopulations (Figure S2B).
To test the role of immune cells in the development of β cell subpopulations, we treated 9-week-old NOD mice with F(ab′)2 fragments of α-CD3 mAb, which arrests immune-mediated killing of β cells by T cells (Chatenoud et al., 1997). Nine days later, the frequency of Btm cells was lower in anti-CD3 mAb-treated mice (Figure 2B). To determine whether arresting the attack would lead to recovery of the Top population, we followed mice after α-CD3 mAb treatment at 7 weeks of age for an additional 3 weeks (Figure 2C). Treatment with anti-CD3 mAb maintained the frequency of subpopulations at levels similar to the pre-treatment group, but the frequency of the Btm subpopulation did not increase beyond the level measured at the start of immune treatment (Figure 2C).
Figure 2. Immune Therapy with Anti-CD3 mAb Arrests Changes in β Cells.

F(ab′)2 fragments of anti-CD3 mAb 145-2C11 or control Ig were given to 9-week-old NOD mice (n = 6–9 mice each experiment). The mice were sacrificed after 9 days, and β cell subpopulations were analyzed by flow cytometry.
(A) Representative data showing SSC/FCS of β cells are shown from a single experiment representative of three independent experiments. The numbers indicate the percentage of Top and Btm β cells.
(B) The percentage of Top β cells was compared between anti-CD3 Tx mice and those in the immunoglobulin control and untreated groups (Kruskal-Wallis test, **p < 0.01).
(C) Human immunoglobulin (hIg) or anti-CD3 mAb was given to 7-week-old NOD mice, and these mice were followed for 3 weeks after treatment (Kruskal-Wallis, pre-treatment [Pre-Tx] or hIg versus anti-CD3, ***p < 0.001). Ig, immunoglobulin; IgG, immunoglobulin G; Tx, treatment. Data from individual mice are shown. Bars are mean ± SEM.
Thus, these studies indicate that a new subpopulation of viable β cells with reduced insulin granularity appears with immune infiltrates. Immune therapy blocks the development of this new subpopulation.
Changes in the Transcriptome of β Cell Subpopulations
We then compared gene expression in the Top and Btm subpopulations by RNA sequencing (RNA-seq). The cells were sorted from dissociated islets from 8- to 10-week-old NOD mice (n = 4–6 mice per sorting, four sortings). We identified 457 genes that showed significant differences (false discovery rate [FDR] < 0.05 and >2-fold change) between the two subpopulations (Figure S3). The most differentially expressed 50 genes with the smallest FDR and a volcano plot of the 457 genes are shown in Figures 3A and 3B. The affected transcripts were from genes associated with β cell function (e.g., Glut2), metabolism (Acot7, ApoE, Arg1, and Pcx), and cell mobility (CCL19, CD24, and CXCL12). Ingenuity Pathway Analysis (IPA) was used to identify affected pathways (Table 1). Among those most highly affected were pathways associated with immune cell trafficking and signaling of inflammatory mediators. The apoptosis pathway was decreased in Btm cells (p = 4.19 × 10−7), and pathways associated with islet cell development were affected (p = 1.89 × 10−3). Genes associated with cell proliferation were increased in Btm cells (p = 2.06 × 10−4).
Figure 3. RNA-Seq Analysis of the Subpopulations of β Cells from NOD Mice.

Top and Btm β cells, sorted from 8- to 10-week-old NOD mice, were used for RNA-seq.
(A) Heatmap generated from 50 genes with the smallest false discovery rate (FDR) is shown.
(B) Volcano plot of the 457 differentially affected genes categorized by pathway (n = 4 separate sortings with islet cells from five or six mice in each). Column headings refer to individual sortings.
Table 1.
Top Diseases and Biofunctions Identified by Ingenuity Pathway Analysis of RNA-Seq Data
| p Value | Number of Molecules | |
|---|---|---|
| Diseases and Disorders | ||
| Name | ||
| Inflammatory response | 1.05E–03–4.57E–13 | 222 |
| Organismal injury and abnormalities | 1.05E–03–1.53E–11 | 290 |
| Inflammatory disease | 1.40E–04–4.61E–11 | 65 |
| Molecular and Cellular Functions | ||
| Name | ||
| Cellular movement | 1.05E–03–5.07E–20 | 201 |
| Cellular growth and proliferation | 9.78E–04–2.70E–13 | 331 |
| Cell death and survival | 1.11E–03–6.86E–13 | 264 |
| Lipid metabolism | 1.05E–03–3.18E–12 | 154 |
| Molecular transport | 1.05E–03–3.18E–12 | 226 |
| Physiological System Development and Function | ||
| Name | ||
| Organismal development | 1.10E–03–2.11E–20 | 446 |
| Immune cell trafficking | 1.05E–03–1.76E–15 | 146 |
| Tissue morphology | 1.02E–03–2.18E–12 | 339 |
Top diseases and biofunctions identified by Ingenuity Pathway Analysis of RNA-seq data from Top and Btm subpopulations of β cells sorted from 8- to 10-week-old NOD mice (n = 5–6 mice over four separate sortings). Results were summarized into different diseases and biofunctions. The name of the specific pathway, as well as the p value and number of molecules involved in each pathway, is shown.
To confirm and extend our findings, we analyzed specific genes in the two sub-populations by real-time qPCR. There was reduced expression of Ins1, Ins2, Glut2, Mafa, Foxo1, Nkx6.1, Pdx1, and Chga in Btm cells (Figure 4A; n = 3–11 per group, t test with FDR < 0.05). Neurog3, which marks endocrine progenitors, was increased ~8-fold in Btm cells (p < 0.0001). Compared to Top cells, there was increased expression of both Gcg and Sst in Btm β cells. Similar changes were seen in β cells from NOD MIP-GFPTg mice, but expression of FoxO1, Mafa, Chga, and Neurog3 was not significantly different (Figure S4). We did not find any insulin-glucagon dual-positive cells by flow cytometry, but we did identify an increase in islet cells that were negative for both insulin and glucagon in NOD islets when compared to age-matched B6 islets (Figure 4B).
Figure 4. Transcription Profiles of the Two β Cell Subpopulations by Real-Time qPCR.

(A) Top and Btm β cells were sorted from 8- to 10-week-old NOD mice. RNA was recovered, and the transcription levels of the β cell genes shown were measured by real-time qPCR. The Ct values were normalized to Actb mRNA levels (ΔCt = Ct of Actb-Ct of target gene + 20). Data represent mean ± SEM of three experiments, each with pools of four to six mice. Student’s t tests were performed with an FDR of 5% (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
(B) FACS analysis of insulin and glucagon expression in islet cells pooled from 10-week-old NOD and B6 mice (numbers refer to the percent of the cells gated on CD45− cells). Data are from a single experiment representative of three experiments.
(C) Eight-week-old NOD mice were given bromodeoxyuridine (BrdU) in drinking water for 9 days, followed by normal water for another week before sacrifice. The incorporation of BrdU was studied in unsorted or sorted Top and Btm β cells. The number refers to the percentage of BrdU+ cells of total β cells. Data are from pooled cells from two or three mice per experiment and are representative of three independent experiments.
(D) Data points represent pools of two or three mice from three experiments. Values represent mean ± SEM (***p = 0.0002, Mann-Whitney test).
(E) The transcription levels of Sox9 and the stem cell factors Sox2, Oct4, and L-Myc were studied by real-time qPCR in Top and Btm β cells from 8- to 10-week-old NOD mice. RNA input was normalized to Actb. Data represent mean ± SEM of three experiments, with β cells sorted from four to six mice. Btm and Top cells from the same isolation were compared using a single-sample t test to a mean of 1 (n = 4; *p < 0.05, **p < 0.01, and ***p < 0.001). Comparisons were also made between the stem cell line SCRC-1002 and Top β cells (p < 0.0001, two-way ANOVA).
(F) Top and Btm β cells were sorted from 8- to 10-week-old NOD mice, and ALDH activity was measured by FACS and compared between Top and Btm β cells. One paired sample from three independent experiments is shown (n = 6–8 mice per experiment). ALDH, aldehyde dehydrogenase. DEAB is an inhibitor of ALDH.
Our pathway studies showed increased expression of genes associated with cellular proliferation in the Btm subpopulation. To measure this, we added BrdU to the drinking water of 8-week-old mice for 9 days and analyzed the frequency of BrdU+ subpopulations of β cells. The incorporation of BrdU into the cells in the Btm subpopulation was increased 2-fold compared to the Top population (Figures 4C and 4D).
The loss of the β cell markers and the increase in Neurog3 implied dedifferentiation of the Btm β cells. We measured genes associated with “stemness” and found increased expression of Sox9, Sox2, Oct4, and L-Myc by real-time qPCR in Btm cells versus Top cells (Figure 4E). However, the expression level of these transcription factors was ~10-fold lower than that found in the mouse stem cell line SCRC-1002 (Figure 4E). Consistent with enhanced progenitor signatures, we also detected higher aldehyde dehydrogenase (ALDH) activity in Btm β cells than in Top cells in islets from 8- to 10-week-old NOD mice (Figure 4F).
β Cell Survival during Immune Attack
The increased frequency of the Btm β cells over time and reduced expression of genes associated with apoptosis and cell death in our IPA analysis suggested that Btm cells might be resistant to immune killing. According to the results of RNA-seq, relevant antigens, Gad1, Chga, and Ins1/Ins2 had reduced gene expression. We analyzed expression of other antigens and other immune-regulatory molecules suggested in the transcriptional analysis. Real-time qPCR determined that expression of IGRP, ZnT8, IA-2, and Gad1 was lower in Btm populations than in Top populations (Figure 5A). In addition, real-time qPCR detected increased expression of PD-L1, the ligand for PD-1, a negative costimulatory molecule for T cells (Figure 5B), and the minor class I major histocompatibility complex (MHC) antigen Qa-2 (Figure 5B), which is known to modulate cellular immune responses (Riteau et al., 2001), in Btm cells. We confirmed increased expression of PD-L1 in Btm β cells by flow cytometry and also found decreased expression of CD40 and insulin (Figures 5C and 5D). Interestingly, there was a strong correlation between PD-L1-expressing β cells and CD45+ islet infiltrates (Figure 5E).
Figure 5. Immune Features of Btm β Cells.

(A and B) Top and Btm β cells were sorted from 8- to 10-week-old NOD mice, and the transcription level of the autoantigens IGRP, ZnT8, GAD1, and IA-2 (A) and the immunomodulatory ligands PD-L1 and Qa-2 (B) were measured by RT-PCR. Ct values were normalized to Actb (ΔCt = Ct of Actb-Ct of target gene + 20). Data points are from pooled samples analyzed from four to six mice analyzed in four independent experiments. Values represent mean ± SEM (Student’s t test in A with an FDR of 5%; *p < 0.05 and **p < 0.01).
(C and D) Top and Btm β cells from NOD mice at Q1 8–10 weeks of age were sorted and stained for insulin, PD-L1, and CD40 and analyzed by FACS. Histogram overlay of each protein in Btm versus Top β cells is shown. A representative result from a single experiment of two and four experiments is shown. The MFIs of insulin, PD-L1, and CD40 in Btm versus Top β cells in each paired sample are shown. The ratios were compared in a one-sample t test to a value of 1 (*p < 0.05 and ****p < 0.0001). Each point represents data from four, three, and two independent experiments, each with a pool of three to six mice.
(E) The correlation between PD-L1-expression on total β cells and the CD45+ islets infiltrates. Data points represent individual mice from six experiments (total n = 38; Pearson r2 = 0.822, p < 0.0001).
(F–I) Increased killing of the Top subpopulation of β cells: 9-week-old NOD mice were given a single dose of cyclophosphamide (250 mg/kg, i.p.).
(F) Twelve days after treatment, β cell composition was compared with saline controls by flow cytometry. Representative plots are from one of three experiments. The numbers indicate the percentage of cells in each gate after gating on TMRE+CD45− cells.
(G) The frequency of Top β cells is shown (mean ± SEM). Data from individual mice from four separate experiments are shown (Mann Whitney test, ***p < 0.0008).
(H) Histogram of FACS Live/Dead staining of Top and Btm β cells that were sorted and cultured with CD45+ islet-infiltrating cells at a 1:5 ratio or with IL-1β, IFN-γ, and IL-6 for 24 hr. The numbers show the percentage live cells from a single experiment representative of five experiments.
(I) Percentage of live cells after 24 hr in culture (n = 5–8 per group, two-way ANOVA, multiple comparison; *p < 0.05, ***p < 0.001, and ****p < 0.0001).
To directly test the susceptibility of the β cell subsets to immune attack, we synchronized the disease mechanisms by treating 9-week-old NOD mice with cyclophosphamide, which is thought to induce diabetes by reducing regulatory T cells (Brode et al., 2006). Two weeks later, the frequency of the two β cell subsets was compared to that of mice treated with saline. Over the course of the experiment, 1 out of 13 mice in the saline-treated group and 4 out of 11 mice in the cyclophosphamide-treated group developed overt hyperglycemia. (These mice are included in the analysis shown in Figures 5F and 5G.) The proportion of Btm cells was significantly higher in cyclophosphamide-treated mice than in saline-treated mice, indicating that there was preferential loss of the Top subset. When diabetic mice were excluded from the analysis, there was still a significantly lower proportion of Top cells in cyclophosphamide-treated mice (p = 0.003, Mann-Whitney U test). Moreover, we also analyzed the proportions of cells in NOD mice that had developed diabetes spontaneously or following treatment with cyclophosphamide (Figure S5). Btm cells comprised the majority of total β cells, indicating that these cells persist even after the onset of hyperglycemia.
To further test the susceptibility of the two populations to killing, we cultured FACS-sorted Top and Btm β cells with islet immune infiltrates (CD45+ T cells) or proinflammatory cytokines (interferon γ [IFN-γ] + interleukin-1β [IL-1β] + IL-6) and compared cell death by flow cytometry (Figures 5H and 5I). Compared to culture with media alone, there was increased cell killing when Top, but not Btm, β cells were cultured with infiltrating lymphoid cells or cytokines, respectively, and the proportion of live Top cells was lower at the end of the cultures (Figures 5H and 5I).
Changes in Human β Cells in Culture with Allogeneic Lymphoid Cells
To test whether similar changes occur in human β cells upon encountering immune stressors, we cultured human islets from non-diabetic donors with allogeneic peripheral blood mononuclear cells (PBMCs) from patients with T1D or healthy control subjects (HC) for 4 days and then analyzed β cell composition by flow cytometry. When the islets were cultured with both PBMC donors, we found a subpopulation of human β cells similar to those found in NOD mice with insulitis (Figures 6A and 6B).
Figure 6. Changes in Human β Cells in Culture with Allogeneic Lymphoid Cells from Patients with T1D.
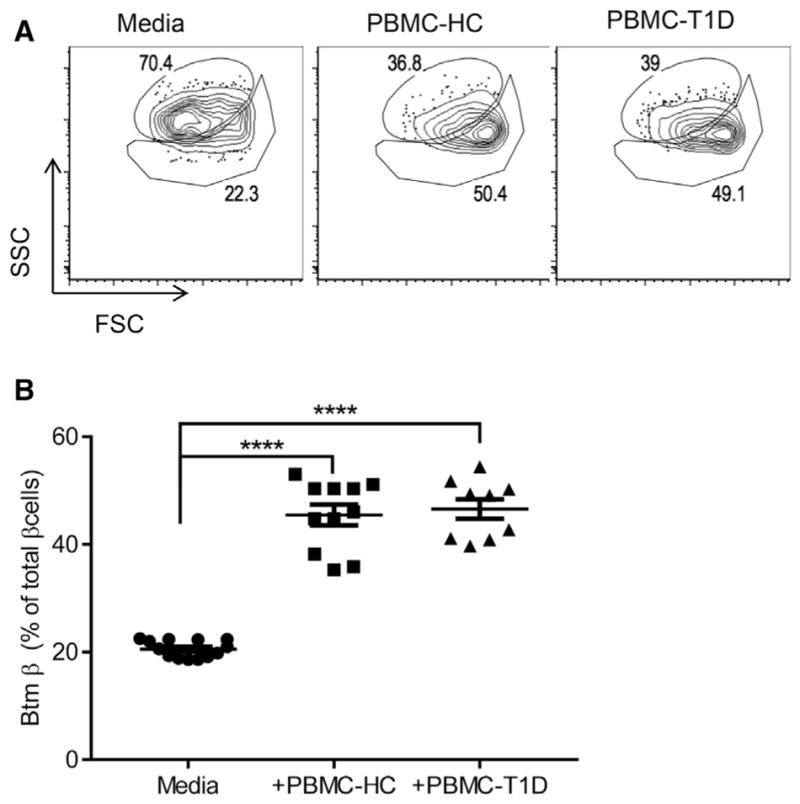
PBMCs from patients with T1D or healthy control subjects (HC) were cultured with human islets for 4 days. Single β cell composition was analyzed by FACS after staining with FluoZin and TMRE. Representative data showing SSC/FSC from one of four experiments are shown. The numbers indicate the percentage of Top and Btm β cells (A). (B) The percentage of Btm β cells from each well from four independent experiments was calculated and compared. Each data point represents a pool of islet cells from four separate experiments (mean ± SEM; ****p = 0.0001, Dunnett’s multiple comparison test).
DISCUSSION
It takes years for T1D to develop in humans. The extended course of the disease has been attributed to a waxing and waning of the immune response and the focal nature of β cell killing in the pancreas, but previous studies have not addressed how β cells respond to immune assault, which may also account for the kinetics and the finding that residual β cells escape destruction in patients with long-duration T1D. In these studies, we have addressed how β cells respond to immune attack. During progression of insulitis in the NOD model of T1D, a population of β cells appears that, compared to normal cells, show reduced insulin granularity and reduced expression of markers that define mature β cells. This population develops in response to the activity of immune cells, because its formation correlates with infiltration of immune cells in the islets, is not found in mice without insulitis, and is prevented with anti-CD3 mAb. A comparison of gene transcripts between the original (Top) and new (Btm) populations identified pathways involved in cell movement, growth and proliferation, immune responses, and cell death and survival. Our analysis by real-time qPCR showed changes in the expression of genes associated with β cell ontogeny and genes that activate the diabetogenic T cell response: antigens and costimulatory molecules. With human islets, a similar population of β cells appears when islets are cultured with PBMCs from patients with T1D or healthy control subjects. The immune responses in these experiments were driven by the allogeneic difference. These studies identify a novel mechanism of cell protection from immunologic destruction in which β cells lose features of their normal identity, increase expression of immune inhibitory molecules and proliferation, and resist killing. This mechanism may lead to an ongoing cycle of neogenesis of β cells that are killed by an immune response or even long-term survival of a subpopulation of β cells, as has been described in individuals with longstanding T1D (Keenan et al., 2010; Liu et al., 2009).
A trivial explanation for our findings is that the lower-granularity population of cells represents non-β islet cells that can harbor Zn+ granules (Bosco et al., 2010). However, the same two populations of β cells were identified in GFP+ β cells from NOD mice, in which GFP was driven by Ins1 promoter. It was also possible that the lower-granularity population represented dying or stressed β cells; activation of ER stress pathways in β cells has been seen prior to onset of T1D in NOD mice (Tersey et al., 2012). However, we did not identify differences in the expression of individual genes associated with ER stress and apoptosis by real-time qPCR between Top and Btm cells, and our IPA analysis of the RNA-seq data showed reduced expression of apoptosis pathway genes in Btm cells (181 genes, Z score −2.142, p = 4.19E–07).
Degranulated β cells have been described by our group and others at the time of diagnosis in NOD mice (Sherry et al., 2006). It was thought that β cells released all of their insulin-containing granules to maintain glycemic control. However, our previous observations and these new findings suggest that Btm β cells are the consequence of islet infiltrates rather than metabolic stressors, since glucose levels were normal in NOD mice. Moreover, if cell infiltration is treated with anti-CD3 mAb, the development of Btm cells is prevented, although the frequency of Top, fully granulated β cells does not recover beyond what was found when immune therapy commenced. There may be additional “recovery” of Top cells with more extended follow-up.
Btm cells differ from Top cells in a number of ways in addition to reduced insulin granules. We found increased transcription of heat shock proteins (both increased by RNA-seq [HspB1, 6.46-fold, p = 0.005] and decreased [HspB6e, 180-fold, p = 0.0003]), which has been seen in islet stress responses (Eizirik and Cnop, 2010; Tsang et al., 2010). It has been proposed that the deficit in β cell mass in T1D may be in part due to β cell degranulation to chromogranin-positive hormone-negative (CPHN) cells (Akirav et al., 2008). A recent study with tissue from human T1D subjects suggests that CPHN cells are more frequent in T1D and distributed in a pattern comparable to neonatal pancreas, implying possible attempted regeneration (Md Moin et al., 2016).
Our analysis also shows a dynamic interplay between immune and β cells. In Btm cells, there was increased expression of PD-L1, the ligand for PD-1 that is expressed in diabetogenic CD8+ T cells as a result of antigen encounter but whose ligation inhibits T cell activity (Karwacz et al., 2011). Expression of transcripts for diabetes-associated antigens such as IGRP, Chga, Gad1, and Ins1/Ins2 gene expression was reduced (more than 80-fold for Gad1 by RNA-seq, p = 6.86 × 10−12). Immune inhibitory ligands such as Qa-2 (also known as H2-Q9), the murine homolog of human HLA-G, which plays a major role in maternal-fetal and self-tolerance (Rouas-Freiss et al., 1997), protects cells from the lytic activity of both innate (natural killer [NK] cells) and effectors when transfected in M8 melanoma cells, (Lee et al., 2010; Riteau et al., 2001) were expressed. In addition, there was increased expression of chemokines (e.g., CCL5 [p = 1 × 10−6], CCL19 [p = 1.1 × 10−11], and others) involved in immune cell movement. Altogether, our data show resistance of Btm cells to immune killing through five complementary approaches: (1) An anti-immune agent, anti-CD3 mAb, which inhibits T cells, prevents further predominance of the bottom subpopulation. (2) There are higher rates of cell death in the Top subpopulation when the two subpopulations are cultured with the same islet-infiltrating immune cells. (3) There are higher rates of cell death in the Top subpopulation when the two subpopulations are cultured with proinflammatory cytokines. (4) Cyclophosphamide treatment, which precipitates diabetes, presumably by inhibiting immune regulatory mechanisms, leads to preferential loss of the top subpopulation. (5) After hyperglycemia develops, the Btm cells are present and represent the majority of cells.
We also found an increase in several genes associated with the deposition of matrix and cell migration, such as collagen and fibronectin, but many of these molecules may also play an important role in recruiting immune cells by serving as ligands for integrin ligands such as α5 (Tzima et al., 2005). For example, TNFRSF9 (CD137, 41BB), which is found in this pathway, can serve as a costimulatory ligand for T cells and was reduced 19.7-fold (p = 0.028). We cannot exclude that there was migration of Btm β cells from the sites of immunopathology, possibly from the adjacent exocrine pancreas, but our studies did not address this possibility.
Btm β cells have many features of a dedifferentiated state. Talchai et al. described differentiation of β cells as a mechanism that can lead to β cell loss in mice with metabolic stress but may also create a pool of potential precursor cells that can ultimately develop into insulin producing cells (Talchai et al., 2012). Several transcription factors associated with mature β cells showed significant differences when tested by real-time qPCR, such as Chga, Mafa, Foxo1, Pdx1, and Nkx6-1. Dnmt3a, which is required for β cell function maturation (Dhawan et al., 2015), was decreased by 2.11-fold by RNA-seq (p = ns), as was Dnmt3b, which was decreased 20-fold (p = 0.0065) by RNA-seq and has been shown to be induced by oxidative stress (Wei and Loeken, 2014). The relative expression of Gcg as well as Sst is increased in Btm cells compared to normal β cells, but we did not find dual-hormone-positive cells by flow cytometry or electron microscopy (EM) (data not shown), indicating that the cells did not acquire features of true endocrine precursor cells. Likewise, the lower-granularity subpopulation showed increased expression of genes associated with endocrine progenitors and cell “stemness” and increased ALDH activity, which is widely used as a hallmark of stem/progenitor cells (Pearce et al., 2005). However, the expression level of these genes was relatively low. These findings suggest that the Btm population has features of dedifferentiated β cells rather than true stem cells. In addition, Btm β cells showed higher rates of replication compared to Top cells. Although the uptake of bromodeoxyuridine (BrdU) was increased, it is difficult to determine whether the net result of the proliferation was an increase in β cell mass, since while cell replication is occurring, cell killing is also taking place.
Dorrell et al. recently identified four antigenically distinct subtypes of human β cells, distinguished by expression of ST8SlA1 and CD9, that were altered in distribution in the islets of patients with T2D (Dorrell et al., 2016). Interestingly, the Btm β cells that we have studied showed significantly higher expression of CD9 (increase 7.8-fold, p = 0.0005) but ST8SlA1 was decreased (2.64-fold, p = 0.227). Bader found proliferative and mature β cells in the islets that could be distinguished by Fltp that differentiated mature β cells from those with proliferative capacity (Bader et al., 2016). However, our studies pertain to cells that have already fully differentiated, and therefore, the factors that affect β cell lineage and development may not be altered.
Even subtle elevations of glucose may affect β cells (Jonas et al., 1999; Laybutt et al., 2003). In addition, placement of islet from a hyperglycemic environment into a normoglycemic environment can restore insulin secretion (Krogvold et al., 2015). However, the changes that we found in β cells occurred in the absence of any detectable impairment in glucose tolerance (Figure S1). We do not know which immune factors lead to changes in β cells. Blodgett et al. identified inflammatory gene products in fetal human β cells, raising the possibility that immune cell products can directly affect β cell differentiation (Blodgett et al., 2015). The relatively rapid appearance of the subpopulation most likely reflects epigenetic changes in these cells, but further studies will be needed to address this mechanism. Our previous studies showed that cytokines may cause changes in DNA methyltransferases and Ins1/2 DNA methylation (Rui et al., 2016), and we found increased expression of several inflammatory pathways in Btm cells by RNA-seq, including HMGB1(p = 2.11E00), Cxcl15(p = 1.91E00), and IL-6 (p = 2.18E00), among others. Epigenetic changes in other β cell genes are likely to occur, but further study will be needed to identify these mechanisms.
Due to the practical difficulties in studying β cells from individuals at risk of T1D, we carried out our studies in NOD mice, a widely recognized model of human disease. Studies using this rodent system clearly have limitations. First, some evidence suggests that β cell proliferation may be more vigorous in mice than in humans, where clear evidence for cell replication and increases in β cell mass after early childhood is lacking. Previous studies of NOD diabetes have described proliferation of β cells in the prediabetes period, which may be accounted for the sub-population we have described (Sherry et al., 2006; Sreenan et al., 1999). Whether the same changes occur in human subjects in whom T1D develops has not been studied. Metabolic studies do suggest that there is dysfunction of β cells, reflected by a delayed secretory response to glucose during progression and after onset of T1D that may reflect the changes in β cells that we have studied (Ferrannini et al., 2010). Interestingly, in an analysis of β cell decline after the onset of T1D, we previously showed that individuals with a delayed response to an oral meal lose the insulin secretory response at a slower rate than those who show a normal rapid response (Steele et al., 2004). Finally, it is important to note that the studies in NOD mice reflect occurrences in an inbred strain, where the events leading to disease are more or less synchronized. In humans, there may be other environmental factors that affect progression of β cell and immunologic responses. Nonetheless, our studies suggest that the features we observed are not restricted to mice, since similar changes occur in human β cells.
In summary, our findings describe modifications of β cells that occur during autoimmune attack that lead to T1D. Eventually, in NOD mice as in humans, the majority of (if not all) β cells are destroyed by immune effectors and products. However, the process is protracted. We have identified mechanisms that β cells use to survive. Future studies that can recover mature β cells from the pool of modified cells may identify ways of restoring normal metabolic function together with immune therapy.
EXPERIMENTAL PROCEDURES
Mice
Female NOD, B6, and NOD/SCID mice were obtained from The Jackson Laboratory and maintained under pathogen-free conditions. Female MIP-GFPTg mice were a gift from M. Hara (University of Chicago), and B6-GFPTg mice were purchased from The Jackson Laboratory and later bred in our own animal specific-pathogen-free (SPF) facility. All protocols were approved by the Yale Institutional Animal Care and Use Committee.
Human Islets and Cell Lines
Human islet samples were obtained from adult, non-diabetic organ donors from the Integrated Islet Distribution Program (Dhawan et al., 2015). Mouse stem cell line SCRC-1002, derived from B6 mice, was provided by Dr. Shangqin Guo. The use of human tissues was approved by the Yale Institutional Review Board.
Mouse Islet Isolation and β Cell Staining and Purification
Mouse islets were handpicked with a stereomicroscope after collagenase digestion of pancreases (Rui et al., 2016). Single-cell suspensions were prepared after a second collagenase digestion. Cells were stained with FluoZin-3 (Invitrogen) and TMRE (Life Technologies) and anti-CD45 mAb (eBioscience). In some experiments, single islet cells were stained with anti-insulin (R&D Systems) and anti-glucagon (Abcam) antibodies labeled with fluorescein isothiocyanate (FITC), PD-L1 (BioLegend), or CD40 (BioLegend) and analyzed with LSRFortessa (BD). For purification, β cells stained with TMRE and FluoZin-3 were sorted using an FACSAria II (BD).
In other studies, β cell subpopulations were sorted from 8- to 10-week-old NOD mice (n = 6–8 per experiment), and aldehyde dehydrogenase (ALDH) activity was detected by flow cytometry after processing the cells using an ALDEFLUOR kit (STEMCELL Technologies) according to the manufacturer’s instructions.
Low-Input RNA-Seq and Data Analysis
Subpopulations of β cells were sorted from 8- to 10-week-old NOD mice. Cell viability and concentration were assessed using the Countess Automated Cell Counter (Thermo Fisher Scientific). Reverse transcription and cDNA amplification were performed from ~1,000 cells using the SMARTer Ultra Low RNA kit (Clontech Laboratories). Sequencing libraries were prepared using Nextera XT DNA Sample Preparation kit (Illumina). Libraries were pooled and sequenced on an Illumina HiSeq2000 using single-end 50-bp reads. Mapping of reads to mouse reference transcriptome mm9 and quantification of mRNA expression levels were done using RSEM (Li and Dewey, 2011). Identification of differentially expressed genes was performed using the Bioconductor package EdgeR (Robinson et al., 2010), employing a generalized linear model taking into account both the technical batch and the cell type. The resulting p values were adjusted using Benjamini and Hochberg’s procedure for controlling the FDR (Benjamini and Hochberg, 1995). A heatmap was generated with the row Z score of the log2 (counts per minute [CPM] + 1) using R heatmap.2 function. Data were also analyzed with QIAGEN IPA with the following analysis settings: fold change > 1.5, p value < 0.05, Ingenuity Knowledge Base reference set, direct and indirect relationships, for tissue and cell lines, and all mutations. This exploratory analysis was not corrected for multiple comparisons.
Probes, Primers, and Real-Time qPCR
The TaqMan probes (Thermo Fisher Scientific) used to perform real-time qPCR using a TaqMan Gene Expression Master Mix are listed in Table S1. In some experiments, primer pairs were used together with a QuantiFast SYBR green PCR kit (QIAGEN). The sequences of primer pairs used in this study are listed in Table S2. The Actb housekeeping gene was used to normalize the input RNA in all real-time qPCR assays. Gene transcription was presented as ΔCt = (Ct of Actb −Ct of target gene) + 20 or in other cases as relative fold to the comparator subpopulation.
BrdU Administration and Incorporation Analysis
BrdU (0.8 mg/mL, Sigma) was added to the drinking water of 8-week-old NOD mice (n = 4–6 per experiment) with 1% glucose for 9 days. One week later, the mice were sacrificed and the incorporation of BrdU in β cells was studied by flow cytometry. One aliquot of the single islet cells was set aside to enumerate the total number of β cells, and the incorporation of BrdU into subpopulations of β cells was identified with a BrdU staining kit (BioLegend).
Induction of Diabetes with Cyclophosphamide
Nine-week-old NOD mice were given a single dose of cyclophosphamide (250 mg/kg intraperitoneally [i.p.], Sigma), and glucose was measured for up to 12 days. Islets were then harvested, and β cell composition was compared with saline-treated controls by flow cytometry.
In Vitro β Cell Killing Assay
Top and Btm β cells and islet infiltrates (CD45+ T cells) were sorted from 8- to 10-week-old NOD mice and placed in RPMI 1640 media for 4 hr before adding cytokine cocktails (10 U/mL IL-1β, 100 U/mL IFN-γ + 100 U/mL IL-6), or CD45+ islet cell infiltrates at a ratio of 5:1, to either Top or Btm β cells for 24 hr. The cells were then harvested, and cell death was analyzed by flow using fixable viability dyes (Thermo Fisher Scientific).
Intraperitoneal Glucose Tolerance Test
NOD, NSG, or B6 mice were fasted for 16 hr. Blood glucose levels were measured 0, 15, 30, 60, and 120 min after glucose injection (2 g/kg body weight).
Measurement of Insulin Response to Glucose
Top and Btm β cells were sorted from islets from six mice between 8 and 10 weeks of age. Sorted cells were placed in CMRL1066 media for 4 hr, rinsed, and placed in low- (1 g/L) or high-glucose (4.5 g/L) CMRL for 8 hr. Insulin was measured in supernatants using an Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem).
Therapy with Anti-CD3 mAb
F(ab′)2 fragments of anti-CD3 mAb 145-2C11 or control immunoglobulin (10 μg/day i.p., five times) was given to 7- or 9-week-old NOD mice. The treated mice were then sacrificed at the indicated times, and islets were analyzed by flow cytometry.
Human Islet Cultures
Human islets were placed in culture in CMRL 1066 medium with 2.5 × 106 PBMCs from patients with T1D for 4 days. The islets were then collected, dispersed into single cells, and stained with FluoZin and TMRE to determine β cell composition by flow cytometry.
Statistical Analysis
Unless otherwise indicated, data are expressed as means ± SEM. Differences between experimental groups and time points were compared using non-parametric tests (Mann-Whitney U test), one-way or two-way ANOVA, or t tests with an FDR of 5%. Statistical analyses were done with GraphPad Prism7. Differences with a p value < 0.05 were considered statistically significant.
Supplementary Material
Highlights.
Novel β cells with lower granularity develop during progression of T1D in NOD mice
The novel β cells show decreased expression of markers of mature β cells
The novel β cells are protected from immune killing
The novel β cells are less differentiated and show stem-like features
Acknowledgments
We thank Domenico Accili for advice regarding the ALDH assay. This work was supported by NIH grants R01 DK057846, DP3 DK101122, and UC4DK104205; by JDRF grant 17-2012-546; and by a gift from the Howalt family.
Footnotes
ACCESSION NUMBERS
The accession number for the XXX data reported in this paper is XXX: ■■■.
Supplemental Information includes five figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2017.01.005.
AUTHOR CONTRIBUTIONS
J.R. and K.C.H. conceived and designed the experiments. J.R., S.D., and A.L.P. conducted experiments. A.A. and Z.L. analyzed the RNA-seq data. J.R. and K.C.H. wrote the paper with input from all the authors.
References
- Akirav E, Kushner JA, Herold KC. Beta-cell mass and type 1 diabetes: going, going, gone? Diabetes. 2008;57:2883–2888. doi: 10.2337/db07-1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akirav EM, Baquero MT, Opare-Addo LW, Akirav M, Galvan E, Kushner JA, Rimm DL, Herold KC. Glucose and inflammation control islet vascular density and beta-cell function in NOD mice: control of islet vasculature and vascular endothelial growth factor by glucose. Diabetes. 2011;60:876–883. doi: 10.2337/db10-0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson MA, Bluestone JA, Eisenbarth GS, Hebrok M, Herold KC, Accili D, Pietropaolo M, Arvan PR, Von Herrath M, Markel DS, Rhodes CJ. How does type 1 diabetes develop?: the notion of homicide or β-cell suicide revisited. Diabetes. 2011;60:1370–1379. doi: 10.2337/db10-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader E, Migliorini A, Gegg M, Moruzzi N, Gerdes J, Roscioni SS, Bakhti M, Brandl E, Irmler M, Beckers J, et al. Identification of proliferative and mature β-cells in the islets of Langerhans. Nature. 2016;535:430–434. doi: 10.1038/nature18624. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- Blodgett DM, Nowosielska A, Afik S, Pechhold S, Cura AJ, Kennedy NJ, Kim S, Kucukural A, Davis RJ, Kent SC, et al. Novel observations from next-generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes. 2015;64:3172–3181. doi: 10.2337/db15-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco MD, Mohanasundaram DM, Drogemuller CJ, Lang CJ, Zalewski PD, Coates PT. Zinc and zinc transporter regulation in pancreatic islets and the potential role of zinc in islet transplantation. Rev Diabet Stud. 2010;7:263–274. doi: 10.1900/RDS.2010.7.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brode S, Raine T, Zaccone P, Cooke A. Cyclophosphamide-induced type-1 diabetes in the NOD mouse is associated with a reduction of CD4+CD25+Foxp3+ regulatory T cells. J Immunol. 2006;177:6603–6612. doi: 10.4049/jimmunol.177.10.6603. [DOI] [PubMed] [Google Scholar]
- Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol. 1997;158:2947–2954. [PubMed] [Google Scholar]
- Dhawan S, Tschen SI, Zeng C, Guo T, Hebrok M, Matveyenko A, Bhushan A. DNA methylation directs functional maturation of pancreatic β cells. J Clin Invest. 2015;125:2851–2860. doi: 10.1172/JCI79956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell C, Schug J, Canaday PS, Russ HA, Tarlow BD, Grompe MT, Horton T, Hebrok M, Streeter PR, Kaestner KH, Grompe M. Human islets contain four distinct subtypes of β cells. Nat Commun. 2016;7:11756. doi: 10.1038/ncomms11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik DL, Cnop M. ER stress in pancreatic beta cells: the thin red line between adaptation and failure. Sci Signal. 2010;3:pe7. doi: 10.1126/scisignal.3110pe7. [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–226. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- Ferrannini E, Mari A, Nofrate V, Sosenko JM, Skyler JS DPT-1 Study Group. Progression to diabetes in relatives of type 1 diabetic patients: mechanisms and mode of onset. Diabetes. 2010;59:679–685. doi: 10.2337/db09-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold KC, Usmani-Brown S, Ghazi T, Lebastchi J, Beam CA, Bellin MD, Ledizet M, Sosenko JM, Krischer JP, Palmer JP Type 1 Diabetes TrialNet Study Group. β cell death and dysfunction during type 1 diabetes development in at-risk individuals. J Clin Invest. 2015;125:1163–1173. doi: 10.1172/JCI78142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman S. Assessment of beta cell viability. Curr Protoc Cytom. 2011;Chapter 6(Unit 6.27) doi: 10.1002/0471142956.cy0627s55. [DOI] [PubMed] [Google Scholar]
- Jonas JC, Sharma A, Hasenkamp W, Ilkova H, Patanè G, Laybutt R, Bonner-Weir S, Weir GC. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J Biol Chem. 1999;274:14112–14121. doi: 10.1074/jbc.274.20.14112. [DOI] [PubMed] [Google Scholar]
- Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, Escors D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med. 2011;3:581–592. doi: 10.1002/emmm.201100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan HA, Sun JK, Levine J, Doria A, Aiello LP, Eisenbarth G, Bonner-Weir S, King GL. Residual insulin production and pancreatic β-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes. 2010;59:2846–2853. doi: 10.2337/db10-0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogvold L, Skog O, Sundström G, Edwin B, Buanes T, Hanssen KF, Ludvigsson J, Grabherr M, Korsgren O, Dahl-Jørgensen K. Function of isolated pancreatic islets from patients at onset of type 1 diabetes: insulin secretion can be restored after some days in a nondiabetogenic environment in vitro: results from the DiViD study. Diabetes. 2015;64:2506–2512. doi: 10.2337/db14-1911. [DOI] [PubMed] [Google Scholar]
- Laybutt DR, Glandt M, Xu G, Ahn YB, Trivedi N, Bonner-Weir S, Weir GC. Critical reduction in beta-cell mass results in two distinct outcomes over time. Adaptation with impaired glucose tolerance or decompensated diabetes. J Biol Chem. 2003;278:2997–3005. doi: 10.1074/jbc.M210581200. [DOI] [PubMed] [Google Scholar]
- Lee M, Choi B, Kwon HJ, Shim JA, Park KS, Lee ES, Sohn S. The role of Qa-2, the functional homolog of HLA-G, in a Behcet’s disease-like mouse model induced by the herpes virus simplex. J Inflamm (Lond) 2010;7:31. doi: 10.1186/1476-9255-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu EH, Digon BJ, 3rd, Hirshberg B, Chang R, Wood BJ, Neeman Z, Kam A, Wesley RA, Polly SM, Hofmann RM, et al. Pancreatic beta cell function persists in many patients with chronic type 1 diabetes, but is not dramatically improved by prolonged immunosuppression and euglycaemia from a beta cell allograft. Diabetologia. 2009;52:1369–1380. doi: 10.1007/s00125-009-1342-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Md Moin AS, Dhawan S, Shieh C, Butler PC, Cory M, Butler AE. Increased hormone-negative endocrine cells in the pancreas in type 1 diabetes. J Clin Endocrinol Metab. 2016;101:3487–3496. doi: 10.1210/jc.2016-1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce DJ, Taussig D, Simpson C, Allen K, Rohatiner AZ, Lister TA, Bonnet D. Characterization of cells with a high aldehyde dehydrogenase activity from cord blood and acute myeloid leukemia samples. Stem Cells. 2005;23:752–760. doi: 10.1634/stemcells.2004-0292. [DOI] [PubMed] [Google Scholar]
- Riteau B, Rouas-Freiss N, Menier C, Paul P, Dausset J, Carosella ED. HLA-G2, -G3, and -G4 isoforms expressed as nonmature cell surface glycoproteins inhibit NK and antigen-specific CTL cytolysis. J Immunol. 2001;166:5018–5026. doi: 10.4049/jimmunol.166.8.5018. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouas-Freiss N, Gonçalves RM, Menier C, Dausset J, Carosella ED. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc Natl Acad Sci USA. 1997;94:11520–11525. doi: 10.1073/pnas.94.21.11520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui J, Deng S, Lebastchi J, Clark PL, Usmani-Brown S, Herold KC. Methylation of insulin DNA in response to proinflammatory cytokines during the progression of autoimmune diabetes in NOD mice. Diabetologia. 2016;59:1021–1029. doi: 10.1007/s00125-016-3897-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry NA, Kushner JA, Glandt M, Kitamura T, Brillantes AM, Herold KC. Effects of autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes. Diabetes. 2006;55:3238–3245. doi: 10.2337/db05-1034. [DOI] [PubMed] [Google Scholar]
- Sreenan S, Pick AJ, Levisetti M, Baldwin AC, Pugh W, Polonsky KS. Increased beta-cell proliferation and reduced mass before diabetes onset in the nonobese diabetic mouse. Diabetes. 1999;48:989–996. doi: 10.2337/diabetes.48.5.989. [DOI] [PubMed] [Google Scholar]
- Steele C, Hagopian WA, Gitelman S, Masharani U, Cavaghan M, Rother KI, Donaldson D, Harlan DM, Bluestone J, Herold KC. Insulin secretion in type 1 diabetes. Diabetes. 2004;53:426–433. doi: 10.2337/diabetes.53.2.426. [DOI] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012;150:1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, Evans-Molina C, Rickus JL, Maier B, Mirmira RG. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the non-obese diabetic mouse model. Diabetes. 2012;61:818–827. doi: 10.2337/db11-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang KY, Chan D, Bateman JF, Cheah KS. In vivo cellular adaptation to ER stress: survival strategies with double-edged consequences. J Cell Sci. 2010;123:2145–2154. doi: 10.1242/jcs.068833. [DOI] [PubMed] [Google Scholar]
- Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- von Herrath M, Sanda S, Herold K. Type 1 diabetes as a relapsing-remitting disease? Nat Rev Immunol. 2007;7:988–994. doi: 10.1038/nri2192. [DOI] [PubMed] [Google Scholar]
- Wei D, Loeken MR. Increased DNA methyltransferase 3b (Dnmt3b)-mediated CpG island methylation stimulated by oxidative stress inhibits expression of a gene required for neural tube and neural crest development in diabetic pregnancy. Diabetes. 2014;63:3512–3522. doi: 10.2337/db14-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.