

Summary
The membrane ring that equatorially circumscribes the nuclear pore complex (NPC) in the perinuclear lumen of the nuclear envelope (NE) is comprised largely of Pom152 in yeast, and its ortholog Nup210 (or Gp210) in vertebrates. Here, we have used a combination of negative-stain EM, NMR, and SAXS methods to determine an integrative structure of the ~120 kDa luminal domain of Pom152. Our structural analysis reveals that the luminal domain is formed by a flexible string-of-pearls arrangement of nine repetitive cadherin-like Ig-like domains, indicating an evolutionary connection between NPCs and the cell adhesion machinery. The 16 Pom152 copies known to be present in the yeast NPC are long enough to form the observed membrane ring, suggesting how interactions between Pom152 molecules help establish and maintain the NPC architecture.
Introduction
The eukaryotic nucleus is delimited by the nuclear envelope (NE), comprised of two distinct membranes, the inner and the outer nuclear membranes, that enclose the perinuclear lumen. The outer and inner nuclear membranes join to form specialized circular apertures (nuclear pores), containing large proteinaceous assemblies termed nuclear pore complexes (NPCs) (Alber et al., 2007b). The yeast NPC is a large (~50 MDa) cylindrical assembly composed of multiple copies of ~30 different proteins, termed nucleoporins or Nups, arranged to form eight symmetrically arranged spokes linked by coaxial outer, inner, and membrane rings (Alber et al.; Rout et al., 2000). NPCs facilitate the active transport of macromolecules between the nucleoplasm and cytoplasm, and are involved in other multiple essential roles, including controlling genome organization and expression (Ibarra and Hetzer, 2015). As a consequence, disruptions of the NPC can lead to human disease (Simon and Rout, 2014).
It has been shown that the NPC has at its heart a cage-like core scaffold consisting of Nups composed entirely of either a β-propeller fold, an α-solenoid fold, or a distinctive arrangement of both folds, a combination unique to vesicle coating complexes (Devos et al., 2004). These similarities suggest a common evolutionary origin for NPCs and coated vesicles in an early membrane-curving module or “protocoatomer” that led to the formation of the internal membrane systems defining the features of modern eukaryotes (Devos et al., 2004; Sampathkumar et al., 2013). This coatomer-like core scaffold is anchored to the pore membrane through two different mechanisms. First, ALPS (Amphipathic Lipid Packing Sensor) motifs, membrane-binding α-helical “fingers”, are found on the membrane-facing surface of the NPC core scaffold (Drin et al., 2007; Kim et al., 2014; von Appen et al., 2015). Second, several Nups, termed pore membrane proteins or Poms, carry trans-membranous α-helices (Chial et al., 1998; Miao et al., 2006; Wozniak et al., 1989; Wozniak et al., 1994). Curiously, none of these transmembrane domains or ALPS motifs seem individually essential for NPC assembly or membrane anchoring, suggesting functional redundancy (Liu et al., 2009). One particular Pom stands out, by virtue of its size and its apparent homo-oligomerization (Tcheperegine et al., 1999; Yewdell et al.) to form the membrane ring that equatorially circumscribes the NPC in the perinuclear lumen of the NE (Alber et al., 2007b). In yeast, this protein is termed Pom152 (Wozniak et al., 1994), a type II integral membrane protein (Tcheperegine et al., 1999) that has an N-terminal NPC-associating region followed by a single transmembrane domain, whereas its presumed vertebrate homolog Nup210 (also known as Gp210) has its transmembrane domain near the C-terminus followed by the NPC-associating region (Greber et al., 1990; Wozniak et al., 1989). Both homologs have a large luminal domain that was previously suggested to be formed by repeated domains (Wozniak et al., 1994) of an Ig or cadherin-like fold (Devos et al., 2006). However, there is no available experimental evidence available defining the precise number and structure of these domains, neither for the protein as a whole. Though Pom152 is non-essential in yeast, its overexpression significantly inhibits cell growth (Wozniak et al., 1994), and it has been implicated in helping to form an early intermediate structure during NPC assembly (Marelli et al., 2001). In vertebrates, Nup210 is a key regulator of cell fate adaptation (D’Angelo et al., 2012; Gomez-Cavazos and Hetzer, 2015). Mutation and mis-regulation of Nup210 have been related to severe human diseases, including numerous cancers (Chapman et al., 2011; Rajkumar et al., 2011). Here, we have used a combination of negative-stain electron microscopy (EM), nuclear magnetic resonance (NMR), and small-angle X-ray scattering (SAXS) methods to determine an integrative structure of the ~120 kDa luminal domain of Pom152.
Results
Negative-stain electron microscopy analysis of Pom152
To determine the overall shape and dimensions of the native full-length Pom152 (Pom152FL; Figure 1A), we purified the endogenous protein as a monomer using affinity purification, native elution, and sucrose density gradients (Fernandez-Martinez et al., 2012) (Figures 1 and S1A). The samples were visualized by negative-stain EM. The individual particle images display varying degrees of curvature, though the dominant form was elongated with a pronounced curvature, convex on the side adjacent to the head (Figure 1). The particle images were analyzed using an iterative stable alignment and clustering (ISAC) method to generate class averages that were reproducible in multiple classification trials (Yang et al., 2012). All 35 resulting class averages showed a thin elongated shape for the isolated monomeric protein molecule (Figure 1B). The average end-to-end distance of Pom152FL class averages is 38.7 nm (±1.5 nm; 30 class averages) (Figure 1G).
Figure 1. Negative stain EM analysis shows that Pom152 has an extended, string-of-pearls shaped luminal domain.
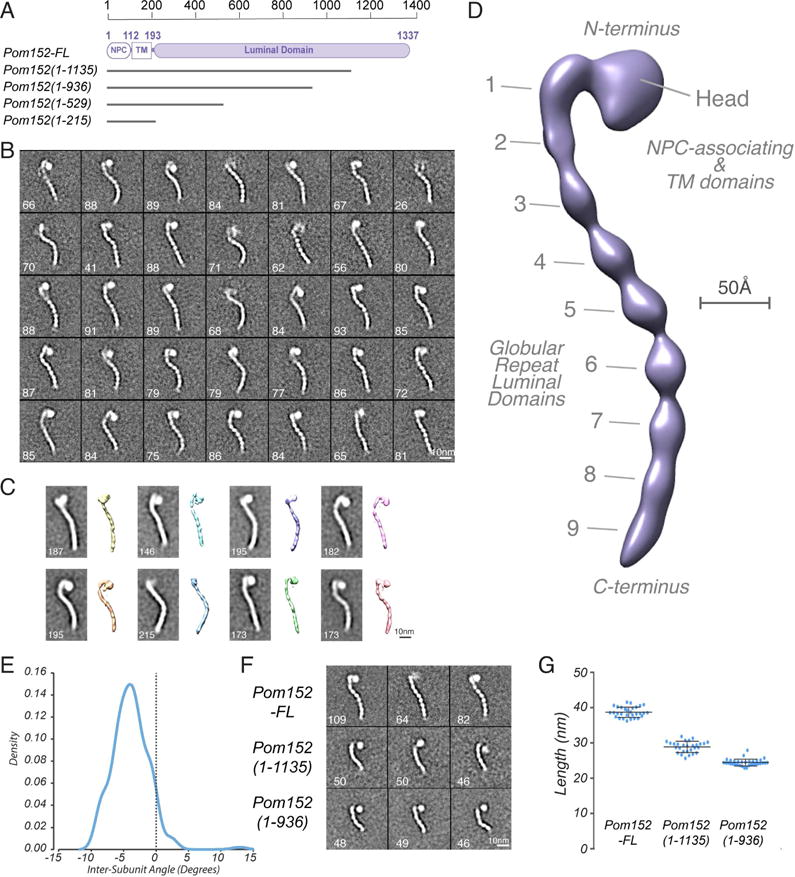
(A) Domain organization of Pom152FL and four truncations drawn to scale. Pom152FL exhibits a three-domain organization with the domain inside the NPC (NPC), the transmembrane segment (TM), and the domain inside the nuclear envelope lumen (luminal domain). The numbers indicate amino acid residue positions. Horizontal grey lines for the truncations represent the number of amino acid residues in each segment.
(B) 35 representative negative-stain EM class averages of Pom152FL. The number of particles in each class is shown. Bar, 10 nm.
(C) Representative random conical tilt 3D maps (right) are aligned to each of the corresponding class averages (left). The number of particles in each class is shown. Bars, 10 nm.
(D) Negative-stain EM density map of Pom152FL. The nine globular domains in C-terminal lumen (1 to 9) and the N-terminal head region containing the NPC-associating and TM domains are indicated. Bar, 50 Å.
(E) The average inter-domain angle for the last 7 repetitive regions was estimated in 37 representative Pom152FL class averages using the ImageJ angle tool. One angle was measured between domains 3 and 9, its difference from 180° was determined and the resulting value divided between the seven globular domains involved in the estimation. Distribution of the resulting inter-domain angles is shown in a Kernel density plot with a peak of −4.1° indicating a small negative curvature for the particles.
(F) Representative negative-stain EM class averages of three assigned views of Pom152FL, Pom1521–1135, and Pom1521–936. The number of particles in each class is shown. Bar, 10 nm.
(G) End-to-end distances of Pom152FL, Pom1521–1135, and Pom1521–936 in samples of 30, 28, and 33 class averages, respectively. The lines on the data points indicate the mean and SD: 38.7 ±1.5 nm, 28.9 ±1.6 nm and 24.4 ±1.0 nm for the samples, respectively.
To further assess the structural features of Pom152FL, we relied on the random conical tilt method (Radermacher, 1988) to compute an initial 3D-dimensional map (Figure 1C) and then used the Relion program to compute the final 3D map at 25 Å resolution (Scheres, 2012) (Fig 1D). The resolution is limited by incomplete angular coverage of particle views and conformational heterogeneity (Figure S1D and E). Nevertheless, the map faithfully recapitulates the major features seen in the 2D class averages. Pom152FL has a prominent head that is attached to a long tail resembling a string of pearls (Figure 1B). This tail is apparently formed by 9 consecutive globular domains (Figure 1D), and exhibits heterogeneity in the observed conformational states, probably due to changes in the relative orientations of the globular domains with respect to each other (Figure 1B–C). The arrangement of the first three domains (the neck) is relatively linear, while domains 3 to 9 form a curved shape in all observed classes. The estimated inter-domain angle between the latter globular domains (3 to 9) in most particle images ranges from −10° to +5°, with an average of −4.1° (Figure 1E, 1B, and S1C). Some curvature is retained even as the domains are sequentially removed, supporting the idea that the curvature is an intrinsic property of the tail (Figure 1F; below).
To define which of the morphological features correspond to the N- and C-terminal domains of Pom152, we used negative-stain EM to analyze two C-terminally truncated versions of the protein. All resulting class averages for the C-terminal truncations Pom1521–1135 and Pom1521–936 showed an intact head, but were missing a number of globular domains proportional to the size of the deletion, as reflected by their average end-to-end distance of 28.9 nm (±1.6 nm; 28 class averages) for Pom1521–1135 and 24.4 nm (±1.0 nm; 33 class averages) for Pom1521–936 (Figure 1A, F, and G). This finding suggested that the tail corresponds to the C-terminal luminal domain of Pom152 (Pom152LD) and that the head contains the N-terminal NPC-associating region and the transmembrane domain. The truncated Pom152 particles showed a degree of heterogeneity similar to that of Pom152FL. The difference between the end-to-end distances of Pom152FL and the truncated forms (Pom1521–1135 and Pom1521–936) suggested an average size of 4.0 nm (±2.4 nm; 20 class averages) for each globular domain. The average width of the domains is 2.9 nm (±1.6 nm; 37 class averages).
The luminal domain of Pom152 stabilizes the NPC’s association with the pore membrane
Complete deletions of Pom152 have not been observed to change the fitness phenotype (Wozniak et al., 1994), but have been shown to cause synthetic defects when combined with mutations affecting inner ring nucleoporins (Tcheperegine et al., 1999) or other integral membrane proteins involved in NPC biogenesis, such as Heh1 (Yewdell et al., 2011) or Apq12 (Scarcelli et al., 2007). Pom152 is a part of the NPC membrane ring and has been suggested to be involved in shaping and stabilizing the NPC’s pore membrane (Alber et al., 2007b; Onischenko et al., 2009; Yewdell et al., 2011). Indeed, Nups with ALPS-motif associated with the pore membrane also appear to help stabilize the membrane (Drin et al., 2007; Marelli et al., 2001; Meszaros et al., 2015), as is exemplified by sensitivity to growth in the membrane-destabilizing reagent benzyl-alcohol (Fernandez-Martinez et al., 2012). We thus decided to test the phenotype of Pom152LD truncation mutants in the presence of benzyl-alcohol.
Addition of benzyl-alcohol caused especially clear growth defects in the luminal domain mutants (Figure 2A). A complete deletion of Pom152 does not show such obvious defects, suggesting that the observed phenotype is mainly related to the functional role of the luminal domain. When the same mutants were tested for thermosensitivity, no growth defect was observed (Figure 2A), showing that the benzyl-alcohol phenotype is a specific and not general response to stress; also, addition of the chemical chaperone tauroursodeoxycholic acid (TUDCA) did not compensate for the benzyl-alcohol growth defect (not shown), indicating that the phenotype is not associated with the ER stress response pathway, as shown for hNup210 (Gomez-Cavazos and Hetzer, 2015). To control for the possibility of subcellular mislocalization causing the Pom152 truncation mutant phenotypes, we used fluorescently tagged reporters to observe the localization of the Pom152 truncations in relation to other NPC markers. As shown in Figure 2B, the truncated form of Pom152 shows the nuclear rim staining characteristic of a nucleoporin; the localization and distribution of NPCs, revealed by co-localization of the inner ring component Nup188, appears normal. However, upon treatment with benzyl-alcohol, both Nup188 and the truncated Pom152 start to co-accumulate at cytoplasmic foci. Our results thus show that the luminal domain of Pom152 is functionally relevant, and suggest that it plays key roles in shaping and stabilizing the NPC structure.
Figure 2. Functional analysis of truncations affecting the Pom152 luminal domain.
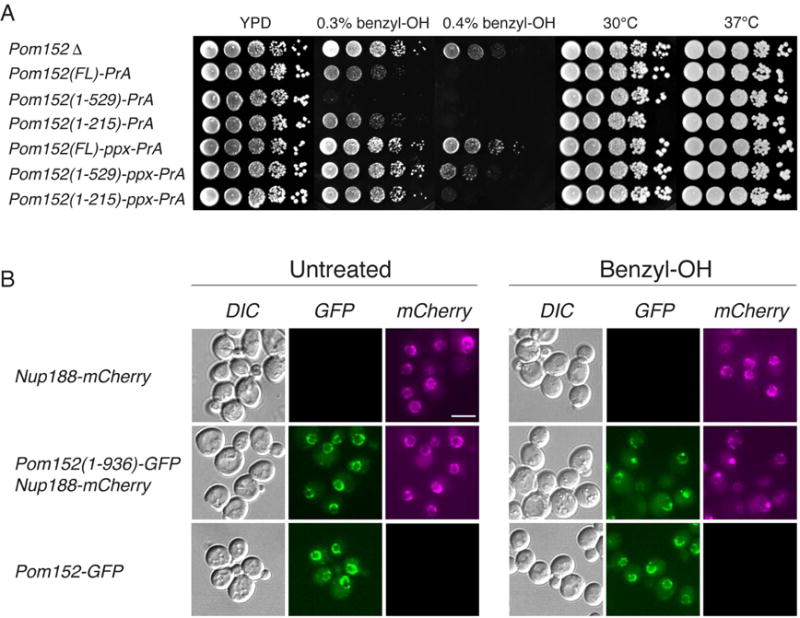
(A) Growth phenotypes of Pom152FL tagged with Protein A or prescission protease cleavage site (PPX)-Protein A and related truncation mutants (see Figure 1A). Serial 10-fold dilutions of cells were spotted on YPD plates in the absence or presence of 0.3% and 0.4% benzyl-OH at 30°C or on YPD plates an d grown at the indicated temperatures for 1–3 days.
(B) Subcellular localization of benzyl-OH treated Pom152-GFP constructs. Panels show the localization of the indicated genomically tagged Pom152-GFP or Nup188-mCherry (used as NPC localization control) constructs as determined by fluorescence microscopy. Cells were grown on liquid yeast minimal media supplemented with 2% glucose at 30° C (untreated) or with an additional 0.1% benzyl-OH for 3 h at 30° C (benzyl-OH). Differential interference contrast (DIC). Bar, 5 microns.
Structure determination of Pom152718–820, the luminal Ig-like domain, using NMR spectroscopy
Negative-stain EM revealed that Pom152LD is formed by 9 globular domains whose arrangement displays a significant but limited degree of heterogeneity and/or flexibility (Figure 1), making this domain challenging for structure determination. However, their small size made individual domains suitable for structure determination by solution NMR spectroscopy. Two segments (Pom152603–820 corresponding to domains 3–4 and Pom152718–820 corresponding to domain 4) were evaluated by recording their 2D 1H-15N HSQC (heteronuclear single-quantum coherence) spectra (Figure S2). The line shapes and intensities of the peaks seen for Pom152603–820 were non-uniform, making this segment intractable by NMR spectroscopy, whereas Pom152718–820 HSQC spectra showed well-dispersed peaks with uniform line shapes and intensities. Pom152718–820 was thus chosen for full structure characterization due to its apparently higher conformational homogeneity.
The backbone and side-chain resonances as well as distance restraints were determined for a [U-13C, 15N] Pom152718–820 sample. The 20 best-scoring structures in the ensemble superimpose well on each other, with the root-mean-square deviation (RMSD) of 0.60 ± 0.11 Å over N, Cα, and C backbone atoms; they also have good stereochemistry (Figure 3A and Table 1). The solution structure of Pom152718–820 revealed an Ig-like fold (Figure 3C–D) containing 9 β-strands that form two β-sheets with a typical β-sandwich topology (Figure 3B). The two β-sheets are made of the ABE and C′CFGG′A′ β-strands (using the standard Ig-like domain annotation (Halaby et al., 1999)) (Figure 3B). The fold does not contain any inter-sheet disulfide-bonds, resulting in a less compact β-sandwich with a distance between the two sheets of 11.4 Å (between B3 (Ser739) and F3 (Ile792) Cα atoms) that is larger than that in related Ig-like fold structures, such as cadherins (e.g., 9.4 Å between I33 and V79 in the neural cell adhesion molecule (NCAM; PDB code 1EPF)) (Kasper et al., 2000). Accordingly, we consider the Ig-like domain of Pom152718–820 to be a variant of the C3-subtype Ig-like fold family (Table 2 in (Halaby et al., 1999)). Structural mapping of sequence conservation of Pom152718–820 among 37 fungal species reveals a conserved surface region lined by residues from the F, G, and G′ β-strands and the FG loop, as well as highly conserved cysteine residues at the termini of the Ig domains (Figures 3E–F and S6). The former region has a net negative electrostatic potential in its center, partly bounded by patches with a net positive potential (Figure 3G–H). We tested the possibility that this region is a binding site for calcium ions, as is the case for many cadherin-like proteins. However, addition of EDTA or calcium chloride did not significantly alter chemical shifts of Pom152718–820, nor the negative-stain EM images (not shown), suggesting that Pom152 does not bind calcium ions, unlike many other cadherin-like proteins.
Figure 3. NMR structure determination of Pom152718–820 reveals a conserved Ig-like fold domain.
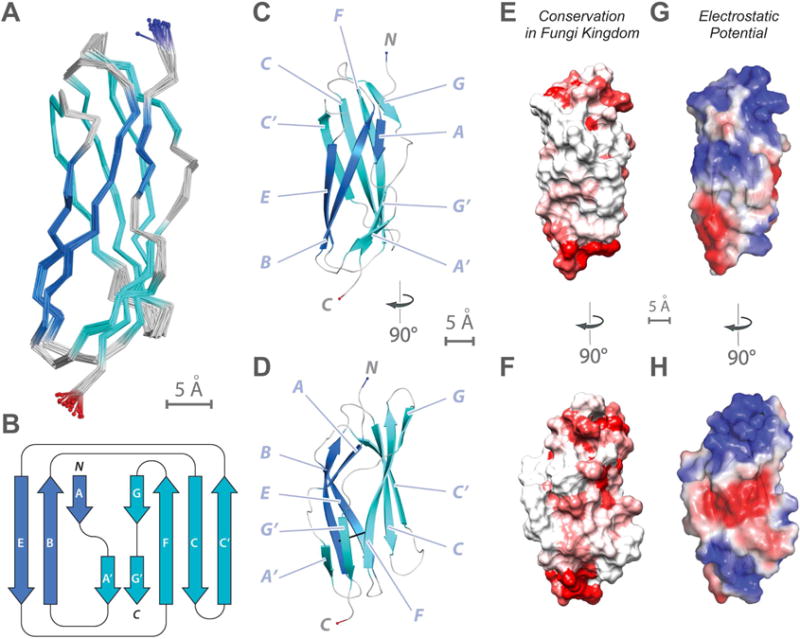
(A) Superimposition of 20 lowest-energy structures of Pom152718–820 that satisfy the NMR restraints best. The two β-sheets are made of the ABE (blue) and C′CFGG′A′ β-strands (cyan).
(B) Topology of the Pom152718–820 fold. β-strands are represented as thick arrows and the linkers connecting them as lines. Each strand is named following the standard Ig-like domain annotation (Halaby et al., 1999). Scale bar, 5 Å.
(C and D) Two views of the best-scoring structure, showing the arrangement of β-strands and features labeled as in panel B.
(E and F) Sequence conservation of Pom152718–820 among 37 fungal homologs (Figure S6), plotted on the surface of the Pom152718–820 structure; low conservation in white, high conservation in red.
(G and H) Electrostatic potential on the surface of the Pom152718–820 structure, calculated using the PyMOL tool APBS; negative (−2 kT/e) and positive (+2 kT/e) potentials are shown in red and blue, respectively.
Table 1.
NMR restraints and structural statistics for the 20 lowest-energy structures of Pom152718–820. See also Figures 3 and S2.
| Restraints and statistics | WT |
|---|---|
|
| |
| Total number of restraints | 2590 |
| NOE restraints | 2346 |
| Unambiguous | 2086 |
| Intraresidue | 863 |
| Sequential | 478 |
| Short-range | 115 |
| Medium-range | 27 |
| Long-range | 603 |
| Ambiguous | 260 |
| Inter-Molecular | |
| Dihedral angle restraints | 176 |
| Hydrogen bond restraintsa | 68 |
| Structure Statisticsb | |
| NOE violations > 0.5 Å | 0% |
| Dihedral violations > 5 ° | 0% |
| RMSD from averagec, d | |
| Backbone (N, Cα, C) (Å) | 0.60 ± 0.11 |
| Heavy atoms (Å) | 1.20 ± 0.10 |
| Ramachandran Statisticse | |
| Most favored region (%) | 78.9 (89.7) |
| Additionally allowed (%) | 18.4 (8.9) |
| Generously allowed (%) | 2.6 (1.4) |
| Disallowed (%) | 0.0 (0.0) |
Hydrogen bond restraints were HN-O distance of 1.8–2.3 Å and an N-O distance of 2.8–3.3 Å.
Structural characteristics for the final ensemble of 20 water-refined structures.
RMSD of the mean structure from individual structures in the ensemble.
RMSD for residues 718–820 shown.
Ramachandran plot data shown for residues 718–820 (in bracket structured region: 722–725; 731–733; 737–746; 751–759; 766–773; 778–785; 789–800; 804–807; 813–817).
Pom152 luminal domain is formed by an array of Ig-like domains
We predicted that the remaining 8 domains in the luminal domain of Pom152 are also Ig-like domains, as was already revealed by NMR spectroscopy for Pom152718–820. This prediction was based on the following four considerations. First, sequence-based predictions of secondary structure, disordered regions, and domain boundaries are consistent with an Ig-like fold (Figure S3A–C). Second, the 8 domains share statistically significant sequence alignments to Pom152718–820 (E-values ranging from 3.3×10−49 to 6.8×10−39) (Figure 4B–C and S3D). Third, comparative models of these domains, constructed with MODELLER (Sali and Blundell, 1993) using the NMR structure of Pom152718–820 as a template (Figure 4B–C), have reasonable stereochemistry, indicating all 9 domains might assume the same fold. Finally, the cysteine residues in Pom152718–820 that we identified as conserved across different species (above) are also conserved in the remaining 8 domains (Figures 3E–F and S6). As an aside, it is conceivable that putative disulfide bridges involving these cysteine residues help stabilize interfaces between the Ig-like domains of Pom152, intra- and/or inter-molecularly.
Figure 4. Comparative models of 8 Ig-like domains and comparison to an Ig-like domain in human Nup210.
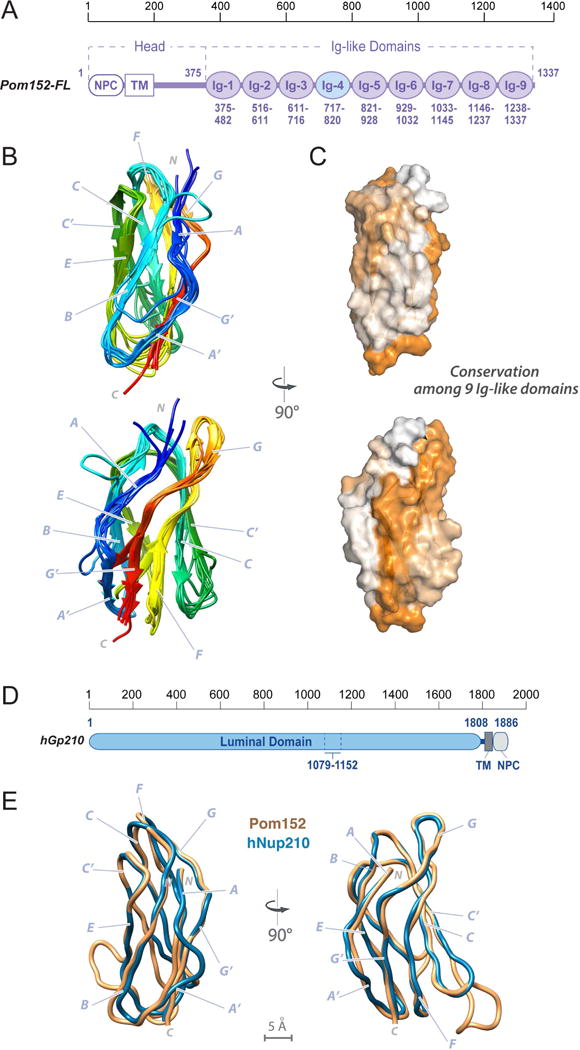
(A) Domain organization of Pom152FL and its domains drawn to scale. The head region contains the domain that faces the NPC inner ring (NPC) and the transmembrane segment (TM), followed by the lumenal domain composed of nine Ig-like repeats. The amino acid boundaries for each Ig-like repeat are indicated below them. The repeat analyzed by NMR (Ig-4, Pom152718–820) is highlighted in light blue.
(B) Comparative models of the remaining 8 Ig-like domains were built using the Pom152718–820 NMR structure as the template, followed by superposing them on the Pom152718–820 structure. The 8 domains share statistically significant sequence alignments to Pom152718–820 (E-values ranging from 3.3×10−49 to 6.8×10−39).
(C) Sequence conservation among the 9 Ig-like domains was mapped on the structure of Pom152718–820 using ConSurf (Ashkenazy et al., 2016). Low conservation, white; high conservation, orange.
(D) Domain organization of human Nup210. See legend of Figure 1A.
(E) Superposition of comparative models for yeast Pom152718–820 (orange) and human Nup2101079–1152 (blue).
See also Figure S3.
A structural model of a luminal Ig-like domain in human Nup210
Nup210 is the human ortholog of Pom152. Its luminal domain is a key regulator of cell differentiation (D’Angelo et al., 2012; Gomez-Cavazos and Hetzer, 2015). The two orthologs share domain composition, although the order of the domains is swapped, with the luminal domain located at the N-terminus of Nup210, while the transmembrane and NPC-associating domains are at its C-terminus (Figure 4D). The low sequence identity between the yeast and human luminal domains (~20%) prevented us from unambiguously defining the number and boundaries of the Nup210 Ig-like domains. Nevertheless, we were able to construct a comparative model of Nup2101079–1152 by relying on our structure of Pom152718–820 as a template (Figure 4E and Supplemental Experimental Procedures). The NCBI Conserved Domain (Marchler-Bauer et al., 2015) server predicts that Nup2101079–1152 is an Ig-like fold domain, based on an alignment (E-value 1.02×10−16) to the Pfam family PF02368 (a bacterial Ig-like domain family), suggesting that the luminal domain of Nup210 is organized similarly to that of Pom152.
Integrative structure determination of Pom152FL
We determined the structure of Pom152FL through an integrative modeling approach (Figure 5) that has proven useful for structural analysis of flexible and thus conformationally heterogeneous proteins, such as Pom152 (Fernandez-Martinez et al., 2016; Kim et al., 2014). We represented Pom152FL in a coarse-grained fashion to reflect the relatively low resolution of structural information about it, as follows. Each of the 9 Ig-like domains was represented as a rigid body at the resolution of 1 residue per bead, computed from the atomic NMR structure (Figure 3C) or a comparative model (Figure 4B–C). In addition to these 9 domains connected by flexible linkers (from 4 to 10 residues per bead), a Pom152FL model also included a flexible string of beads (from 20 to 100 residues per bead) corresponding to the N-terminal NPC-associating domain (residues 1–100), the transmembrane domain (residues 101–200), and the linker domain (residues 201–374). Next, 100,000 Pom152FL models were computed by flexibly fitting random initial models into the negative-stain EM density map while avoiding steric clashes and retaining sequence connectivity.
Figure 5. 4-stage scheme for integrative structure determination of Pom152FL.
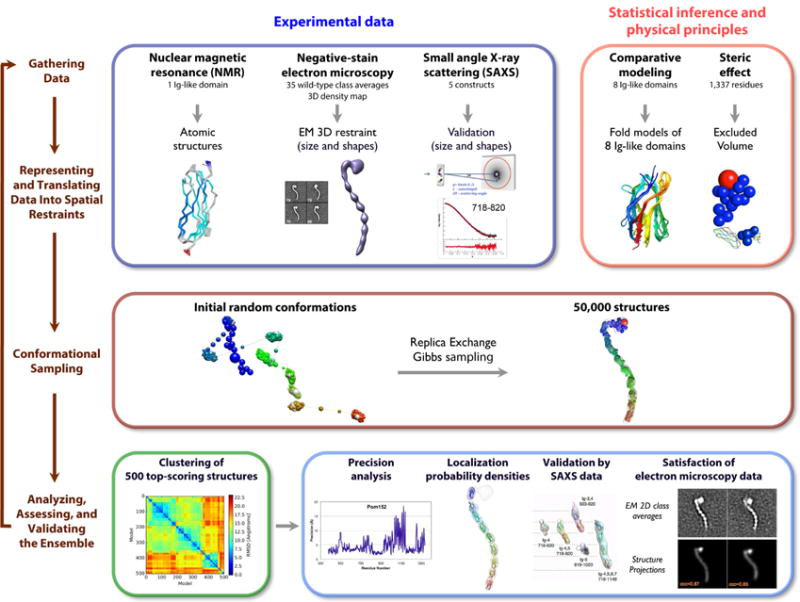
The integrative structure determination of Pom152FL proceeds through 4 stages: (1) gathering data, (2) representing and translating data into spatial restraints, (3) conformational sampling to produce an ensemble of structures that satisfies the restraints, and (4) analyzing, assessing, and validating the ensemble structures. The modeling protocol (i.e., stages 2, 3, and 4) was scripted using the Python Modeling Interface (PMI), version 4d97507, a library for modeling macromolecular complexes based on our open-source Integrative Modeling Platform (IMP) package, version 2.6 (http://integrativemodeling.org) (Russel et al., 2012).
The 500 best-scoring models (i.e., the ensemble) fit the EM map as well as satisfy the excluded volume and sequence connectivity restraints used to compute the models. The structures also fit the EM class averages, with cross-correlation coefficients ranging from 0.84 to 0.87 for representative class averages (Figure S4C).
In addition to satisfying information used to compute them, the models are also similar to each other. The clustering of the best-scoring models identified a single dominant cluster of 364 similar structures, with a precision (Supplemental Experimental Procedures) of 7.0 Å for the luminal domain (Figure S4A).
In general, an ensemble of good-scoring models can be visualized as a localization probability density map. The map gives the probability of any volume element being occupied by a certain bead in superposed good-scoring models. Figure 6A shows the localization density for each of the 9 Ig-like domains, as sampled by the 364 good-scoring models in the dominant cluster. The 7.0 Å precision of this cluster is sufficiently high to pinpoint the locations, but not the orientations, of the constituent Ig-like domains (Figure S4B).
Figure 6. Integrative structure of Pom152FL based on NMR spectroscopy, negative-stain EM, and SAXS.
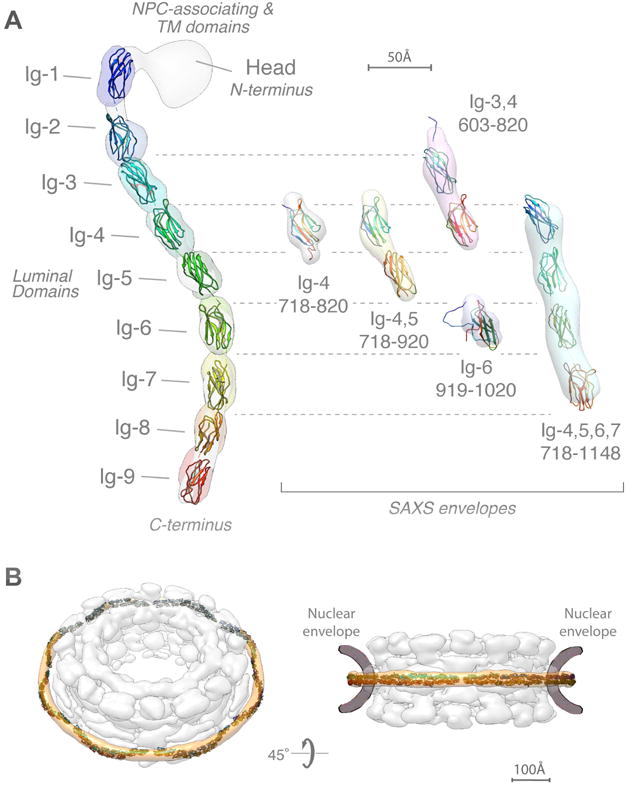
(A) (left) The localization probability density map computed from 364 superposed structures that satisfy the input spatial restraints shows the location of each of the 9 Ig-like domains (ranging from blue to red). The negative-stain EM density map is superposed in grey. A representative molecular model of Pom152LD (ribbon plot) was obtained by adjusting the relative orientations of adjacent Ig-like domains to resemble those in known cadherin structures (PDB codes 1L3W, 1EPF, 1NCI, 4ZI9, and 5K8R) (Boggon et al., 2002; Kasper et al., 2000; Nicoludis et al., 2015; Nicoludis et al., 2016; Shapiro et al., 1995).
(right) Validation of the integrative structure of Pom152LD by SAXS data for five Pom152 segments, spanning residues 718–820 (Ig-4), 718–920 (Ig-4,5), 603–820 (Ig-3,4), 919–1020 (Ig-6), and 718–1148 (Ig-4,5,6,7). The shapes of these segments in our integrative structure match the envelopes (ab initio shapes) computed from the corresponding SAXS profiles.
(B) Fit of 16 copies of Pom152LD into the yeast NPC map (Alber et al., 2007b). A good fit positions two copies of the extended Pom152LD molecule in an anti-parallel fashion on top of each other, forming a homodimer; we only show the potential arrangement of the antiparallel homodimer, which is implied by the C2-symmetry of the NPC (Alber et al., 2007b; Kosinski et al., 2016; Lin et al., 2016).
The integrative structure further supports the similarity between the molecular architectures of Pom152LD and cadherins (Figure 6A) (Devos et al., 2006).
Validation of the Pom152LD structure using SAXS data
We validated our integrative structure of Pom152LD by SAXS data. Specifically, the computed SAXS profiles match measured SAXS profiles for all five experimentally characterized segments, spanning residues 718–820 (Ig-4), 718–920 (Ig-4,5), 603–820 (Ig-3,4), 919–1020 (Ig-6), and 718–1148 (Ig-4,5,6,7) (Figure S5 and Table S1). In addition, the shapes of these segments in our model also match the envelopes (ab initio shapes) computed from the corresponding SAXS profiles (Figures 6 and S5). The linearity of the Guinier plots confirms a high degree of homogeneity for each of the five Pom152 SAXS samples. Each radius of gyration (Rg) and maximum particle size (Dmax) are consistent with those of the corresponding Pom152 segments (Table S1). Notably, all five SAXS profiles show well-defined “bell-shaped” curves in Kratky plots (Figure S5, middle), indicating relatively rigid conformations of the individual Ig-like domains. In addition, plateaus at the high q region (0.2 – 0.3 Å−1) in the Kratky plots indicate some flexibility between Ig-like domains in solution, consistent with the heterogeneity inferred from the negative-stain EM class averages (Figure 1B).
Position of Pom152LD within the NPC
Pom152 is the main component of the membrane ring of the NPC (Alber et al., 2007b; Tcheperegine et al., 1999; Wozniak et al., 1994). To determine whether or not the shape and length of our integrative structure of Pom152 account for the formation of the membrane ring, we fitted the luminal domains of 16 copies of Pom152 into our previously published yeast NPC map (Alber et al., 2007b) (Figures 6B and S7). A good fit positions two copies of the extended Pom152LD molecule in an anti-parallel fashion on top of each other, forming a homodimer; it is ambiguous whether the tail-to-head direction is clockwise or counterclockwise. The length of the elongated Pom152LD (~38.7 nm) is sufficient to span the spoke width. The arrangement suggests that the membrane ring is made possible by tight interactions between the Ig-like domains in the two antiparallel luminal domains, in agreement with the observed homo-dimerization of Pom152 in vivo (Alber et al., 2007b; Tcheperegine et al., 1999).
Discussion
Since the first high-resolution structure of a nucleoporin domain was solved in 2002 (Hodel et al., 2002), the combined efforts of many groups have provided a detailed structural picture for most of the major components of the NPC (Schwartz, 2016). However, conspicuously absent was any such information for the NPC membrane ring. Our structure of Pom152FL is thus the first detailed view of the molecular architecture of the NPC’s transmembrane components. The structure confirmed our previous suggestion (Devos et al., 2006) that the domain arrangement in Pom152, with repeated Ig-like β-sandwich folds forming an extended luminal module, most strongly resembles the organization of another conserved eukaryotic family of proteins, classical (Type I) cadherins (Ishiyama and Ikura, 2012). The similarities between these two types of proteins extend beyond each repetitive unit, including: i) they share an overall arch-shape (Figure 1 and (Shapiro and Weis, 2009)); ii) they are both able to dimerize (Shapiro and Weis, 2009; Tcheperegine et al., 1999; Yewdell et al.); and iii) they share a similar domain organization, with a segment of repetitive Ig-like domains connected through a single-pass transmembrane region to a short protein-protein interaction domain (though in cadherins the orientation of the domains is equivalent to that in Nup210, see Figures 1, 4 and (Shapiro and Weis, 2009)). In the case of cadherins, their cytoplasmic short region is extended and largely unstructured (Huber et al., 2001) and has been shown to interact with the arm-repeat α-solenoid proteins p120 catenin (Ishiyama et al., 2010) and β-catenin (Huber et al., 2001). The NPC-associating domains of Pom152 and Nup210 are of similar size (~150 amino acid residues) and both are predicted to be largely disordered, though the orientation of this domain in Nup210 is at the C-terminus while that of Pom152 is at the N-terminus (Wozniak et al., 1989; Wozniak et al., 1994). Variability in the position and type of the membrane domain between potential Nup homologs has previously been seen in the Trypanosome NPC (Obado et al., 2016); interestingly, Pom152 localizes to the NPC when expressed in mammalian cells (Wozniak et al.), suggesting conservation in assembly mechanism and even function between vertebrates and fungi.
The N-terminal Pom152 NPC-associating domain is positioned close to the NPC inner ring (Alber et al., 2007b). Indeed genetic interactions with inner ring components (Nic96, Nup59, Nup170 and Nup188) that require the presence of the Pom152 N-terminus have been described (Tcheperegine et al., 1999); moreover, Pom152 appears to physically interact with several inner ring proteins (Alber et al., 2007b). It is thus reasonable to suggest that, similar to the cadherin-catenin interactions, the disordered N-terminus of Pom152 connects to the α-solenoids of the NPC inner ring components (Alber et al., 2007b; Kosinski et al., 2016; Lin et al., 2016), just as cadherins use a largely unstructured linker to interact with the α-solenoid catenin proteins.
Despite their common organization, some clear differences are also observed between Pom152 and cadherins. First, Pom152 lacks the conserved tryptophans that mediate the “strand-swap” mechanism of dimerization in cadherins (Shapiro and Weis, 2009). Not a single Trp residue is present at the C-terminus of Pom152 (Figure S6) and our fitting into the whole NPC suggests that Pom152 molecules likely dimerize through more extensive contacts (Figures 6B and S7). The distribution of electrostatic potential and sequence conservation on the structure of Pom152718–820 (Figures 3E–H and S6) indicates a mechanism where facing Ig-like domains from opposite strands generate complementary interaction surfaces. Second, unlike cadherins, Pom152 does not seem to be able to bind calcium. Cadherin Ig-like domains coordinate calcium through highly conserved residues, stiffening the connections between successive domains and imparting a strong curvature to the full-length ectodomain (Boggon et al., 2002; Nagar et al., 1996). Removal of calcium leads to a disordering of interdomain orientations that can be observed by negative-stain EM (Pokutta et al., 1994). However, we were not able to detect calcium binding, nor any obvious calcium-dependent Pom152 shape changes (data not shown). Perhaps this observation is unsurprising in proteins that diverged more than a billion years ago.
Pom152 and Nup210 homologs exist beyond the opisthokonts (Fungi and Metazoa). Clear homologs are found in Amoebozoa and in plants, though all these are more similar to Nup210, the closer homologs of Pom152 being restricted to the Fungi. Though clearly structurally and functionally similar, the precise nature of the evolutionary relationship between the Pom152-like and Nup210-like homologs therefore remains somewhat unclear. Even so, cadherin-like proteins predate the eukaryota, being found in bacteria where they may mediate cell-cell contact (Fraiberg et al., 2010), consistent with an ancient evolutionary origin for Pom152 and Nup210, and the entire NPC (Alber et al., 2007b; Devos et al., 2004; Devos et al., 2006; Field and Dacks, 2009).
Experimental Procedures
Affinity purification of endogenous Pom152 and truncation mutants
Native Pom152, and the truncation mutants Pom1521–1135 and Pom1521–936 were isolated by affinity purified, natively eluted and further purified in 5–20% sucrose gradients as previously described (Sampathkumar et al., 2013).
Yeast strains
Yeast strains (Table S2) were constructed in a W303 (Mata/alpha ade2-1 ura3-1 his3-11, 15 trp 1-1 leu2- 3,112 can1-100) background using standard techniques (Fernandez-Martinez et al., 2012). Otherwise stated, strains were grown at 30°C in YPD media (1% yeast extract, 2% bactopeptone, and 2% glucose).
Electron microscopy analyses and 3D reconstruction of Pom152
Purified Pom152FL and the truncated versions Pom1521–1135 and Pom1521–936 were applied to glow-discharged carbon-coated copper grids and stained with 1% uranyl formate. Images were collected on a JEOL JEM-2100F transmission electron microscope (JEOL USA Inc., Peabody MA) or a Philips CM200 transmission electron microscope (FEI; Eindhoven, The Netherlands) and analyzed using the Iterative Stable Alignment and Clustering (ISAC) (Yang et al., 2012). The 3D density map of Pom152FL was generated through the random conical tilt reconstruction method and Relion (Scheres, 2012). Angles were measured with ImageJ angle tool. Further details are provided in the Supplemental Experimental Procedures.
Phenotypic assays
To analyze the growth phenotype, 10-fold dilutions of yeast cultures were spotted on YEPD plates in the presence or absence of 0.4% benzyl-alcohol and incubated at the indicated temperatures (Fernandez-Martinez et al., 2012). Indicated fluorescently tagged proteins were visualized, in the presence of absence of 0.1% benzyl-alcohol, using a 63× 1.4 NA Plan-Apochromat objective using a microscope (Axioplan 2) equipped with a cooled charge-coupled device camera (ORCA-ER) (Fernandez-Martinez et al., 2012).
Expression and Purification of Pom152 constructs
Pom152 constructs used for SAXS studies were expressed as SeMET labeled proteins and purified following a standard procedure (Kim et al., 2014; Sampathkumar et al., 2012). Pom152 constructs used for the NMR studies were expressed in minimal media following a standard protocol (Weber et al., 1992) with minor modifications. See Supplemental Experimental Procedures for details.
NMR resonances assignments and structure calculation of Pom152718–820
The [U-13C, 15N] Pom152718–820 samples were used for backbone and side chain resonances assignments using multidimensional NMR experiments (Sattler et al., 1999). All NMR data was acquired at 25°C using either Varian 600 MHz or Bruker 600 and 900 MHz spectrometers equipped with cryogenic probes capable of applying pulse-field gradients along the z-axis. Structure calculations were carried out using distance, dihedral, and hydrogen-bond restraints using the ARIA/CNS program (Linge et al., 2003). A total of 2,095 restraints were used to solve the structure of Pom152718–820 (Figure 3A). 20 best-scoring structures with no distance restraint violations larger than 0.5 Å and no dihedral restraint violations larger than 5° were chosen to represent the structural ensemble consistent with the NMR data. Further details are provided in the Supplemental Experimental Procedures.
Small angle X-ray scattering (SAXS) experiments
SAXS measurements for five Pom152 segments were carried out at the SSRL Beamline 4-2 in the SLAC National Accelerator Laboratory (Menlo Park, CA) (Figures 6A and S5 and Table S1). Further details of SAXS analysis are provided in Supplemental Experimental Procedures and our previous publications (Kim et al., 2014; Sampathkumar et al., 2012).
Comparative modeling of luminal Ig-like domains in Pom152 and human Nup210
Comparative models of 8 luminal Ig-like domains of Pom152 (Figure 4B) and a single luminal Ig-like domain of human Nup2101079–1152 (Figure 4E) were computed with MODELLER (Sali and Blundell, 1993), using the NMR structure of Pom152718–820 as the template. Further details are provided in Supplemental Experimental Procedures.
Integrative structure determination of Pom152FL
The integrative structure determination of Pom152FL proceeded through four stages (Figure 5) (Alber et al., 2007a; Alber et al., 2007b; Fernandez-Martinez et al., 2012; Lasker et al., 2012; Lasker et al., 2010): (1) gathering data, (2) representing and translating data into spatial restraints, (3) conformational sampling to produce an ensemble of structures that satisfies the restraints, and (4) analyzing, assessing, and validating the ensemble structures. The modeling protocol (i.e., stages 2, 3, and 4) was scripted using the Python Modeling Interface (PMI), version 4d97507, a library for modeling macromolecular complexes based on our open-source Integrative Modeling Platform (IMP) package, version 2.6 (http://integrativemodeling.org) (Russel et al., 2012). Further details are provided in Supplemental Experimental Procedures and our previous publications (Fernandez-Martinez et al., 2016; Kim et al., 2014; Shi et al., 2014).
Supplementary Material
Acknowledgments
We thank NYULMC OCS Microscopy Core for assistance with negative-stain EM, NYSGRC for providing samples for SAXS and NMR analyses; and T. Matsui and T.M. Weiss at SSRL, SLAC National Accelerator Laboratory, for assistance with collecting SAXS data. The instrumentation in the Einstein NMR Resource is supported by the Albert Einstein College of Medicine and the Bruker 600 NMR instrument was purchased using funds from NIH award 1S10OD016305. Some of the NMR experiments were performed at the New York Structural Biology Center, which is a STAR center supported by the New York Office of Science, Technology and Academic Research. The New York Structural Biology Center is supported by grant number 349247 from the Simons Foundation. Support was provided by NSF GRF (#1650113) (I.E.C.); and the NIH grants U54 RR022220 to B.C., A.S., and M.R.; R01 GM112108 to M.R.; U54GM094662 to S.C.A; P41 GM109824 to M.R., A.S. and B.C.; U01GM098256 to M.P.R.; P41 GM103314 to B.C.; and R01 GM083960 to A.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession codes
The coordinates and spatial restraints for NMR structure determination were deposited in the Protein Data Bank (accession code 5TVZ). NMR resonance assignments were deposited to BMRB (accession numbers 30201). 3D negative stain EM reconstruction of Pom152 was deposited in the Electron Microscopy Data Bank (accession number EMD-8543). SAXS profiles for five Pom152 segments were deposited in SASBDB (preliminary accession codes SASDB239 to SASDB243). Files containing the input data, scripts, and output structures are available online (https://salilab.org/pom152; https://github.com/salilab/pom152).
Author Contributions
Conceptualization, P.U., S.J.K., P.S., A.S., J.F-M. and M.P.R.; Investigation, P.U., S.J.K., P.S., K.D., I.E.C., S.M.C., R.W. and J.F-M.; Formal Analysis, P.U., S.J.K., P.S., K.D., I.E.C., S.M.C., J.B.B., W.J.R., D.L.S., D.C., A.S., M.P.R. and J.F.M.; Writing, J.F-M., S.J.K., I.E.C., A.S., P.S., K.D., D.C. and M.P.R.; Funding Acquisition, D.L.S., D.C., S.C.A., A.S., and M.P.R.; Supervision, J.F.M., D.L.S., D.C., S.C.A., A.S. and M.P.R.
Competing Financial Interests
The authors declare no competing financial interest.
References
- Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, et al. Determining the architectures of macromolecular assemblies. Nature. 2007a;450:683–694. doi: 10.1038/nature06404. [DOI] [PubMed] [Google Scholar]
- Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, et al. The molecular architecture of the nuclear pore complex. Nature. 2007b;450:695–701. doi: 10.1038/nature06405. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic acids research. 2016;44:W344–350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chial HJ, Rout MP, Giddings TH, Winey M. Saccharomyces cerevisiae Ndc1p is a shared component of nuclear pore complexes and spindle pole bodies. J Cell Biol. 1998;143:1789–1800. doi: 10.1083/jcb.143.7.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo MA, Gomez-Cavazos JS, Mei A, Lackner DH, Hetzer MW. A change in nuclear pore complex composition regulates cell differentiation. Developmental cell. 2012;22:446–458. doi: 10.1016/j.devcel.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos D, Dokudovskaya S, Alber F, Williams R, Chait BT, Sali A, Rout MP. Components of coated vesicles and nuclear pore complexes share a common molecular architecture. PLoS biology. 2004;2:e380. doi: 10.1371/journal.pbio.0020380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos D, Dokudovskaya S, Williams R, Alber F, Eswar N, Chait BT, Rout MP, Sali A. Simple fold composition and modular architecture of the nuclear pore complex. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2172–2177. doi: 10.1073/pnas.0506345103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drin G, Casella JF, Gautier R, Boehmer T, Schwartz TU, Antonny B. A general amphipathic alpha-helical motif for sensing membrane curvature. Nature structural & molecular biology. 2007;14:138–146. doi: 10.1038/nsmb1194. [DOI] [PubMed] [Google Scholar]
- Fernandez-Martinez J, Kim SJ, Shi Y, Upla P, Pellarin R, Gagnon M, Chemmama IE, Wang J, Nudelman I, Zhang W, et al. Structure and Function of the Nuclear Pore Complex Cytoplasmic mRNA Export Platform. Cell. 2016;167:1215–1228 e1225. doi: 10.1016/j.cell.2016.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Martinez J, Phillips J, Sekedat MD, Diaz-Avalos R, Velazquez-Muriel J, Franke JD, Williams R, Stokes DL, Chait BT, Sali A, et al. Structure-function mapping of a heptameric module in the nuclear pore complex. J Cell Biol. 2012;196:419–434. doi: 10.1083/jcb.201109008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MC, Dacks JB. First and last ancestors: reconstructing evolution of the endomembrane system with ESCRTs, vesicle coat proteins, and nuclear pore complexes. Current opinion in cell biology. 2009;21:4–13. doi: 10.1016/j.ceb.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Fraiberg M, Borovok I, Weiner RM, Lamed R. Discovery and characterization of cadherin domains in Saccharophagus degradans 2–40. Journal of bacteriology. 2010;192:1066–1074. doi: 10.1128/JB.01236-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Cavazos JS, Hetzer MW. The nucleoporin gp210/Nup210 controls muscle differentiation by regulating nuclear envelope/ER homeostasis. J Cell Biol. 2015;208:671–681. doi: 10.1083/jcb.201410047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber UF, Senior A, Gerace L. A major glycoprotein of the nuclear pore complex is a membrane-spanning polypeptide with a large lumenal domain and a small cytoplasmic tail. The EMBO journal. 1990;9:1495–1502. doi: 10.1002/j.1460-2075.1990.tb08267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaby DM, Poupon A, Mornon J. The immunoglobulin fold family: sequence analysis and 3D structure comparisons. Protein engineering. 1999;12:563–571. doi: 10.1093/protein/12.7.563. [DOI] [PubMed] [Google Scholar]
- Hodel AE, Hodel MR, Griffis ER, Hennig KA, Ratner GA, Xu S, Powers MA. The three-dimensional structure of the autoproteolytic, nuclear pore-targeting domain of the human nucleoporin Nup98. Molecular cell. 2002;10:347–358. doi: 10.1016/s1097-2765(02)00589-0. [DOI] [PubMed] [Google Scholar]
- Huber AH, Stewart DB, Laurents DV, Nelson WJ, Weis WI. The cadherin cytoplasmic domain is unstructured in the absence of beta-catenin. A possible mechanism for regulating cadherin turnover. The Journal of biological chemistry. 2001;276:12301–12309. doi: 10.1074/jbc.M010377200. [DOI] [PubMed] [Google Scholar]
- Ibarra A, Hetzer MW. Nuclear pore proteins and the control of genome functions. Genes & development. 2015;29:337–349. doi: 10.1101/gad.256495.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama N, Ikura M. The three-dimensional structure of the cadherin-catenin complex. Sub-cellular biochemistry. 2012;60:39–62. doi: 10.1007/978-94-007-4186-7_3. [DOI] [PubMed] [Google Scholar]
- Ishiyama N, Lee SH, Liu S, Li GY, Smith MJ, Reichardt LF, Ikura M. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell. 2010;141:117–128. doi: 10.1016/j.cell.2010.01.017. [DOI] [PubMed] [Google Scholar]
- Kasper C, Rasmussen H, Kastrup JS, Ikemizu S, Jones EY, Berezin V, Bock E, Larsen IK. Structural basis of cell-cell adhesion by NCAM. Nature structural biology. 2000;7:389–393. doi: 10.1038/75165. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Fernandez-Martinez J, Sampathkumar P, Martel A, Matsui T, Tsuruta H, Weiss TM, Shi Y, Markina-Inarrairaegui A, Bonanno JB, et al. Integrative structure-function mapping of the nucleoporin Nup133 suggests a conserved mechanism for membrane anchoring of the nuclear pore complex. Molecular & cellular proteomics: MCP. 2014;13:2911–2926. doi: 10.1074/mcp.M114.040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinski J, Mosalaganti S, von Appen A, Teimer R, DiGuilio AL, Wan W, Bui KH, Hagen WJ, Briggs JA, Glavy JS, et al. Molecular architecture of the inner ring scaffold of the human nuclear pore complex. Science. 2016;352:363–365. doi: 10.1126/science.aaf0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker K, Forster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker K, Phillips JL, Russel D, Velazquez-Muriel J, Schneidman-Duhovny D, Tjioe E, Webb B, Schlessinger A, Sali A. Integrative structure modeling of macromolecular assemblies from proteomics data. Molecular & cellular proteomics: MCP. 2010;9:1689–1702. doi: 10.1074/mcp.R110.000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DH, Stuwe T, Schilbach S, Rundlet EJ, Perriches T, Mobbs G, Fan Y, Thierbach K, Huber FM, Collins LN, et al. Architecture of the symmetric core of the nuclear pore. Science. 2016;352:aaf1015. doi: 10.1126/science.aaf1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linge JP, Habeck M, Rieping W, Nilges M. ARIA: automated NOE assignment and NMR structure calculation. Bioinformatics (Oxford, England) 2003;19:315–316. doi: 10.1093/bioinformatics/19.2.315. [DOI] [PubMed] [Google Scholar]
- Liu HL, De Souza CP, Osmani AH, Osmani SA. The three fungal transmembrane nuclear pore complex proteins of Aspergillus nidulans are dispensable in the presence of an intact An-Nup84–120 complex. Molecular biology of the cell. 2009;20:616–630. doi: 10.1091/mbc.E08-06-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, et al. CDD: NCBI’s conserved domain database. Nucleic acids research. 2015;43:D222–226. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marelli M, Lusk CP, Chan H, Aitchison JD, Wozniak RW. A link between the synthesis of nucleoporins and the biogenesis of the nuclear envelope. J Cell Biol. 2001;153:709–724. doi: 10.1083/jcb.153.4.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meszaros N, Cibulka J, Mendiburo MJ, Romanauska A, Schneider M, Kohler A. Nuclear pore basket proteins are tethered to the nuclear envelope and can regulate membrane curvature. Developmental cell. 2015;33:285–298. doi: 10.1016/j.devcel.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao M, Ryan KJ, Wente SR. The integral membrane protein Pom34p functionally links nucleoporin subcomplexes. Genetics. 2006;172:1441–1457. doi: 10.1534/genetics.105.052068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar B, Overduin M, Ikura M, Rini JM. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature. 1996;380:360–364. doi: 10.1038/380360a0. [DOI] [PubMed] [Google Scholar]
- Nicoludis JM, Lau SY, Scharfe CP, Marks DS, Weihofen WA, Gaudet R. Structure and Sequence Analyses of Clustered Protocadherins Reveal Antiparallel Interactions that Mediate Homophilic Specificity. Structure. 2015;23:2087–2098. doi: 10.1016/j.str.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoludis JM, Vogt BE, Green AG, Scharfe CP, Marks DS, Gaudet R. Antiparallel protocadherin homodimers use distinct affinity- and specificity-mediating regions in cadherin repeats 1–4. eLife. 2016:5. doi: 10.7554/eLife.18449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obado SO, Brillantes M, Uryu K, Zhang WZ, Ketaren NE, Chait BT, Field MC, Rout MP. Interactome Mapping Reveals the Evolutionary History of the Nuclear Pore Complex. PLoS biology. 2016;14 doi: 10.1371/journal.pbio.1002365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onischenko E, Stanton LH, Madrid AS, Kieselbach T, Weis K. Role of the Ndc1 interaction network in yeast nuclear pore complex assembly and maintenance. J Cell Biol. 2009;185:475–491. doi: 10.1083/jcb.200810030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokutta S, Herrenknecht K, Kemler R, Engel J. Conformational changes of the recombinant extracellular domain of E-cadherin upon calcium binding. European journal of biochemistry. 1994;223:1019–1026. doi: 10.1111/j.1432-1033.1994.tb19080.x. [DOI] [PubMed] [Google Scholar]
- Radermacher M. Three-dimensional reconstruction of single particles from random and nonrandom tilt series. Journal of electron microscopy technique. 1988;9:359–394. doi: 10.1002/jemt.1060090405. [DOI] [PubMed] [Google Scholar]
- Rajkumar T, Sabitha K, Vijayalakshmi N, Shirley S, Bose MV, Gopal G, Selvaluxmy G. Identification and validation of genes involved in cervical tumourigenesis. BMC cancer. 2011;11:80. doi: 10.1186/1471-2407-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout, Aitchison, Suprapto A, Hjertaas K, Zhao Y, Chait BT. The yeast nuclear pore complex: composition, architecture, and transport mechanism. In J Cell Biol. 2000:635–651. doi: 10.1083/jcb.148.4.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russel D, Lasker K, Webb B, Velazquez-Muriel J, Tjioe E, Schneidman-Duhovny D, Peterson B, Sali A. Putting the pieces together: integrative modeling platform software for structure determination of macromolecular assemblies. PLoS biology. 2012;10:e1001244. doi: 10.1371/journal.pbio.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. Journal of molecular biology. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Sampathkumar P, Kim SJ, Manglicmot D, Bain KT, Gilmore J, Gheyi T, Phillips J, Pieper U, Fernandez-Martinez J, Franke JD, et al. Atomic structure of the nuclear pore complex targeting domain of a Nup116 homologue from the yeast, Candida glabrata. Proteins. 2012;80:2110–2116. doi: 10.1002/prot.24102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampathkumar P, Kim SJ, Upla P, Rice WJ, Phillips J, Timney BL, Pieper U, Bonanno JB, Fernandez-Martinez J, Hakhverdyan Z, et al. Structure, dynamics, evolution, and function of a major scaffold component in the nuclear pore complex. Structure. 2013;21:560–571. doi: 10.1016/j.str.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Progress in Nuclear Magnetic Resonance Spectroscopy. 1999;34:93–158. [Google Scholar]
- Scarcelli JJ, Hodge CA, Cole CN. The yeast integral membrane protein Apq12 potentially links membrane dynamics to assembly of nuclear pore complexes. J Cell Biol. 2007;178:799–812. doi: 10.1083/jcb.200702120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz TU. The Structure Inventory of the Nuclear Pore Complex. Journal of molecular biology. 2016;428:1986–2000. doi: 10.1016/j.jmb.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grubel G, Legrand JF, Als-Nielsen J, Colman DR, Hendrickson WA. Structural basis of cell-cell adhesion by cadherins. Nature. 1995;374:327–337. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Weis WI. Structure and biochemistry of cadherins and catenins. Cold Spring Harbor perspectives in biology. 2009;1:a003053. doi: 10.1101/cshperspect.a003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Fernandez-Martinez J, Tjioe E, Pellarin R, Kim SJ, Williams R, Schneidman-Duhovny D, Sali A, Rout MP, Chait BT. Structural characterization by cross-linking reveals the detailed architecture of a coatomer-related heptameric module from the nuclear pore complex. Molecular & cellular proteomics: MCP. 2014;13:2927–2943. doi: 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DN, Rout MP. Cancer and the nuclear pore complex. Advances in experimental medicine and biology. 2014;773:285–307. doi: 10.1007/978-1-4899-8032-8_13. [DOI] [PubMed] [Google Scholar]
- Tcheperegine SE, Marelli M, Wozniak RW. Topology and functional domains of the yeast pore membrane protein Pom152p. The Journal of biological chemistry. 1999;274:5252–5258. doi: 10.1074/jbc.274.8.5252. [DOI] [PubMed] [Google Scholar]
- von Appen A, Kosinski J, Sparks L, Ori A, DiGuilio AL, Vollmer B, Mackmull MT, Banterle N, Parca L, Kastritis P, et al. In situ structural analysis of the human nuclear pore complex. Nature. 2015;526:140–143. doi: 10.1038/nature15381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber DJ, Gittis AG, Mullen GP, Abeygunawardana C, Lattman EE, Mildvan AS. NMR docking of a substrate into the X-ray structure of staphylococcal nuclease. Proteins. 1992;13:275–287. doi: 10.1002/prot.340130402. [DOI] [PubMed] [Google Scholar]
- Wozniak RW, Bartnik E, Blobel G. Primary structure analysis of an integral membrane glycoprotein of the nuclear pore. J Cell Biol. 1989;108:2083–2092. doi: 10.1083/jcb.108.6.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak RW, Blobel G, Rout MP. POM152 is an integral protein of the pore membrane domain of the yeast nuclear envelope. J Cell Biol. 1994;125:31–42. doi: 10.1083/jcb.125.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure. 2012;20:237–247. doi: 10.1016/j.str.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell WT, Colombi P, Makhnevych T, Lusk CP. Lumenal interactions in nuclear pore complex assembly and stability. Molecular biology of the cell. 2011;22:1375–1388. doi: 10.1091/mbc.E10-06-0554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.