

Abstract
Drug innovation is characterized by painstaking molecular-level syntheses and modifications as the basic components of research and development. Similarly, natural products are chemically tailored and modified based upon their structural and biological properties. To some extent, the modification of natural products is quite different from de novo structure-based drug discovery. This review describes the general strategies and principles for the modification of natural products to drugs, as illustrated by several successful medicines that originated from natural products.
Key words: Natural products, Synthesis, Multi-dimensional optimization, Structure–acitivity reactivity, Artemisinin, Indirubin
Graphical abstract
This review describes the general strategies and principles for the modification of natural products to drugs, as illustrated by several successful medicines that originated from natural products.

1. Introduction
The species diversity of plants, animals, microorganisms, and marine organisms results in a multitude of secondary metabolites with diverse chemical structures, which have played, and will continue to play, a vital role in the drug discovery and development process. Indeed, the diversity of plant-based systems has provided an enormous number of lead compounds in healthcare. Similarly, the application of microorganismal metabolites as chemotherapeutics has created the field of antibiotics for anti-infectious and anti-cancer therapies. Many scaffolds of compounds obtained from marine microorganisms and phytoplankton, algae and sponges for example, similarly have provided more recent leads for drug discovery and development. And finally, endogenous neurotransmitters and active peptides from human beings and animals constitute an important source for drug research.
From the standpoint of drug innovation, it is necessary to modify natural product structures, because the aim in generation of secondary metabolites by organisms is to protect themselves from natural enemies as well as the environment. Natural product–based drug discovery is characterized with a starting-point of an active compound that necessitates “tailor-made” or individualized manipulation of the structure so as to reach a drug criterion. In this context, the molecular modification of natural products is quite different from the strategy of structure-based drug discovery. This article describes some aspects and principles of modifying natural compounds to lead to successful medicines.
2. The general features of natural products
Natural products, from simple salicylic acid to complex vancomycin, encompass compounds with various molecular sizes, chemotypes, and structural features that can be summarized as follows.
2.1. Structural diversity and complexity
Natural products are characterized by their structural diversity, much of which is even unexpected by chemists. For example in Fig. 1A, anti-malarial artemisinin (1) is composed of a fused trioxene system with peroxy, lactone, cyclic acetal and ketal moieties. The individual scaffold of artemisinin maintains both the oxidative potential as well as chemical stability. The well-known anti-cancer drug paclitaxel (2) possesses a 6,8,6,4-tetracycle-fused skeleton linking functional groups at special positions that ensures binding to tubulin. The non-peptide cholecystokinin (CCK) antagonist asperlicin (3), originated from extracts of Aspergillus alliaceus, is a chemically-complex molecule with a quinazolinone-fused benzodiazepine core.
Figure 1.

The general features of natural products.
Individual structures and multi-functional groups of natural products constitute the prerequisite for strong binding to targets, yet also increase the probability of additional interactions with biological molecules, as well as the difficulty in chemical synthesis. However, from the viewpoint of drug research, many structures of natural products exist as “redundant atoms”, which do not participate in the binding to target and convey disadvantageous effects on the physico-chemical, pharmacokinetic and biopharmaceutical properties. Therefore, those unnecessary moieties should be appropriately removed from lead compounds in structural modifications, raising the ligand efficiency (LE) of molecules1. LE is a measure of the binding energy per unit of mass for a given compound relative to its molecular target. High values of LE imply fewer unnecessary atoms relative to target-binding. This parameter is routinely used as one of the guiding factors in the early stages of lead optimization.
2.2. More sp3 carbon atoms and less nitrogen and halogen elements
Most natural products are composed of more sp3-hybrid carbons than sp2 carbons, making the tetrahedron carbons connect to each other to form flexible chain or cyclic structures. For example in Fig. 1B, both the immunological regulator tacrolimus (4) and the anti-cancer compound epothilone B (5) are macrolides containing many saturated carbon atoms. Another immune-regulator, ISP-1 (6), isolated from Cordyceps sinensis Sacc, is a linear and flexible molecule. An exception is the anti-cancer drug camptothecin (7), a fused and conjugated aromatic system with multiple sp2 carbons.
Most natural products consist of carbon, hydrogen, oxygen atoms but limited nitrogen incorporation, probably due to the weak nitrogen-fixing ability of microorganisms and plants (except Leguminoceae plants which produce alkaloids as secondary metabolites). These features provide various options for introducing nitrogen atoms during modification. Nitrogen is a versatile atom in many aspects, having a stronger nucleophilic property than carbon and oxygen, providing 3- or 5-valence bond, forming salts for alkaline amines or being a neutral amide, building heterocycles, aromatic and fused compounds, being able to be a terminal group or to serve as a linker to connect other groups. Therefore, introduction or modification of various nitrogen atoms is an important molecular operation in lead compound optimization. Natural products rarely contain halogen atoms, although marine products sometimes incorporate a bromine atom.
2.3. Existence of chiral centers and stereochemistry
Natural products are synthesized in organisms by enzymatic catalysis, which take place with stereochemical specificity, yielding stereospecific compounds with chiral centers, chiral axes, or cis/trans-configurations. These asymmetrical factors usually convey a critical contribution to pharmacological actions. However, those chiral centers not participating in the binding to targets are unnecessary. In manipulating or simplifying natural products the unnecessary chirality and stereochemistry have to be removed as much as possible, as compounds that lack chirality make the simplified analogs easy and economical to synthesize. For example in Fig. 1C, the old analgesic morphine (8) contains 21 heavy atoms (non-hydrogen atoms), 5 fused rings, and 5 chiral carbon atoms. The synthetic analgesic drug fentanyl (9) derived from morphine is a much simpler non-chiral molecule.
3. Modification strategies of natural products
3.1. Individualized manipulation based on the molecular size and complexity
For large and complicated natural products, simplified operations are normally carried out to eliminate structurally-unnecessary factors. Molecular dissection is a useful method. The linear molecules or peptides might be sheared along the chain consecutively. For compounds with fused rings, the scaffolds could be segmented into four quadrants to separately modify the structures or groups. Maintaining the structure-activity relationship (SAR) is absolutely necessary. Fig. 2A illustrates the SAR obtained from modifying the paclitaxel (2) molecule.
Figure 2.

Modification strategies for natural products.
The long-known alkaloid cocaine (10) was historically useful as a topical anesthetic in eye and nasal surgery, yet the major disadvantages of this use are cocaine׳s apostrophe intense vasoconstrictor activity and potential for cardiovascular toxicity. Cocaine has since been largely modified and simplified for synthetic local anesthetics, such as procaine (11), tetracaine (12) and lidocaine (13) (Fig. 2B).
Another natural product, physostigmine (14, Fig. 2C), is a parasympathomimetic alkaloid, specifically, a reversible cholinesterase inhibitor. Because of the chemical instability in vivo, physostigmine was modified to simple and non-chiral compounds, such as pyridostigmine bromide (15) and neostigmine bromide (16, Fig. 2C). Unlike physostigmine, which is a tertiary amine, these synthetic quaternary ammonium salts penetrate the central nervous system poorly and have a lower propensity to cause adverse effects such as orthostatic hypotension, while improving muscle tone in patients with myasthenia gravis.
Analog syntheses are usually performed for natural leads with appropriate molecular size. Using the principles of medicinal chemistry, such as bio-isosterism, chain-ring exchange, privileged structure and scaffold-hopping, new analogs are generated with superior properties and compound novelty.
Compounds with small size have structural space to add atoms, groups, or moieties, so that for example, introducing a hydrogen donor or acceptor may increase the affinity to receptors, or adding solubilizing groups raises the solubility or modulates the partition property to benefit or avoid crossing the blood brain barrier.
3.2. Analyzing SAR and designing novel structures
In the absence of information on target structures, classical medicinal chemistry methods are normally applied to the modification of natural products. SARs or quantitative SAR (QSAR) are explored to reveal and assign the pharmacophores, which guide the design of novel compounds with simplified or different scaffolds. Based upon the SAR of paclitaxel, two semi-synthetic analogs docetaxel (17) and cabazitaxel (18) have been launched (Fig. 2D), the modifications of which are restricted at “south-west” and “north-east” areas of the molecule.
3.3. Industrialized syntheses and protecting resources and environments
Achievement of total synthesis for natural products has multiple advantages: (a) to authenticate chemical structures; (b) to provide a series of intermediates for evaluating activities, which usually contain the identical pharmacophoric features as the original compounds and become simplified analogs; (c) to provide a basis for industrialized production in scale-up; (d) to protect the natural resources and environment.
3.4. Removal of unnecessary chiral centers
Chirality in drug molecules yields diploid characteristics. The positive side involves an increase of activity strength and selectivity because of the appropriate binding to sterically-complementary and asymmetrical targets. The negative side is the difficulty in synthesis, separation, and resolution of single eutomers. In fact, not all chiral centers in natural products are necessary for binding and activity. The redundant chiral factors should be removed in modifications as described later.
4. Key points in structural modulation of natural products
The final aim of modifying natural products is to develop active compounds into medicines. All aspects of pharmacological, toxicological, and druggable properties are included in the process of modification. Based on the adequacy of activity, safety, pharmacokinetics, or physico-chemical aspects, purposive modifications are performed as follows: (a) raising the activity strength and selectivity; (b) improving solubility and partition property; (c) increasing metabolic and chemical stability; (d) modulating pharmacokinetic parameters (ADME); (e) removing or alleviating toxicity and adverse reactions; (f) gaining novelty and intellectual property.
5. Examples of successful modifications
5.1. Simplifying structures
Natural products with a large size and complex structure are unfavorable for solubility, absorption and metabolism. One of the modification principles is to decrease the molecular size and to eliminate the unnecessary functional groups.
5.1.1. From halichondrin B to eribulin
Halichondrin B (19, Fig. 3), a marine natural product, was originally isolated from a Japanese rare marine sponge (Halichondria okadai). It is a large polyether macrolide with MW 1109, and reported to have strong anticancer activity against murine cancer cells both in vitro and in vivo. While antiproliferative patterns of halichondrin B were found to be similar to those of other antitubulin drugs, its biochemical mechanism of interaction with tubulin is distinct from all other known classes of antitubulin agents.
Figure 3.

Simplifying structures from halichondrin B to eribulin.
Halichondrin B contains 32 chiral carbon atoms distributed at polyether and macrolide moieties 18 and 12, respectively. Polyether and macrolide components are connected through C29 and C30 to form a fused macrocyclic compound. One of the junctions, C30, links a lactone group, which is easily hydrolyzed to release the macrolide and lose the activity, owing to the alteration in the conformation and spatial disposition of functional groups.
Kishi et al.2 of Havard University systematically investigated the total synthesis of halichondrin B. During the total synthesis they adopted a strategy of simplifying the structure. Comparing the repeated ethereal linkages of the polyether moiety to the multi-functional groups of the macrolide portion, the researchers concentrated on synthesizing the latter. Two critical points are modified in the synthesis of the macrolide: one is to replace the oxygen atom of the lactone linkage with a methylene group, not only to avoid the hydrolysis of the ester group linkage but also to keep the conformation unchanged; the other is to introduce a solubilizing group such as an amino group at the terminal of the side chain in order to increase solubility while forming a salt2.
From 180 synthesized target compounds, eribulin (20, Fig. 3) was found to be an optimal candidate and as a mesylate salt used in clinic trials3. Eribulin was approved by the US Food and Drug Administration (FDA) in 2010 to treat the patients with metastatic breast cancer.
Structurally, eribulin is a most complex drug by organic synthesis. There are nineteen chiral carbon atoms in the molecule. Through a 62-step reaction, eribulin is synthesized from simple organic starting materials. The synthetic scale-up of eribulin from microgram to multi-gram quantities is attributable to a ten-year technological study. The key coupling reactions include formation of the C30a to C1 carbon–carbon bond and macrocyclic ring closure through an intramolecular Nozaki–Hiyama–Kishi reaction, which is briefly shown in Scheme 14.
Scheme 1.

The final coupling reactions of eribulin.
5.1.2. From myriocin to fingolimod
The natural product myriocin (21, ISP-1, Fig. 4), a secondary metabolite isolated from the fungus Isaria sinclairii, possesses immuno-modulating activity ten times stronger than that of cephlosporin A in vitro and in vivo5. There exist 3 chiral carbon atoms and a trans double bond in compound 21. In addition, the existence of amino and carboxyl groups makes myriocin a zwitter ion at pH 7.4, which is disadvantageous to absorption in vivo. The aim of modifying compound 21 is to simplify the structure: alleviating the chiral centers, raising activity strength and selectivity, and improving the pharmacokinetic properties. Through a systematic examination of each functional groups and study of the structure-activity relationship, it was established that the C14 carbonyl group and the Δ6 double bond are unnecessary for binding to the receptor. Three chiral carbon atoms exhibit no stereo-specificity necessary for activity. The polar groups and the molecular size and shape are critical factors.
Figure 4.

Simplifying structures from myriocin to fingolimod.
Myriocin structurally resembles sphingosine (22, Fig. 4), and was later identified to be a modulator of the sphingosine-1-phosphate receptor6. Compound 23 (Fig. 4) reached the criterion of drug candidate, named fingolimod and entered the development phase7. Fingolimod is a linear symmetrical molecule lacking chiral and stereochemical factors. Insertion of a phenyl group to take the place of some saturated carbon atoms in the aliphatic chain partially reduces the molecular flexibility and makes chemical synthesis more facile. The position of the phenyl group in the aliphatic chain conveys a significant effect on the activity, indicating that the molecular shape and conformation play an important role. Fingolimod is the first oral disease-modifying drug approved by the US FDA in 2010 to reduce relapses and delay disability progression in patients with relapsing forms of multiple sclerosis. As a prodrug, one of the hydroxyl groups in fingolimod is phosphorylated by sphingosine kinases in the cell to yield the active form, fingolimod-1-phosphate (24, Fig. 4), which was developed as a new molecular entity.
5.1.3. From schizandrin C to diphenyl bicarboxylate (DDB) and bicyclol
It was reported that the Chinese traditional medicine Schizandra sinensis may improve liver function and reduce symptoms of patients infected with viral hepatitis B. Schizandrin C (25, Fig. 5), one of the lignins isolated from fruits of Schizandra, promotes hepatic anabolism and induces the activation of liver microsomal cytochromes P-450, as well as alters the metabolism of the carcinogen benzopyrene and its binding to DNA8. After completion of the total synthesis of schizandrin C and its region-isomer (26, Fig. 5) the target compounds and synthetic intermediates9 were pharmacologically evaluated and a serendipitous compound, DDB (27, Fig. 5), was found to be effective for protection against the hepatotoxicity induced by carbon tetrachloride, thioacetamide, and d-galactosamine in mice and rats.
Figure 5.

Simplifying structures from schizandrin C to DDB and bicyclol.
It is worth noting that DDB is one of the intermediates of the unnatural compound 26, and exerts stronger activity than those of 25. However, the corresponding intermediate of schizandrin C (28, Fig. 5) possesses much less activity than 27. It seems that the relative positions of methoxy and methylene dioxy groups on the biphenyl convey the effect of hepatoprotection. As expected, compound 29 with altered disposition of the groups shows intermediate activity (Fig. 5).
DDB (27) was selected as a candidate for clinical trials and approved by the China Food and Drug Administration (CFDA) in 1997, named bifendate, and used as a liver-protecting drug for the treatment of the patients with chronic hepatitis B.
To overcome the low solubility and poor biovailability of DDB a second generation of liver-protecting drugs were developed. After systematic structural modification and biological evaluation, bicyclol (30, Fig. 5) was found to be superior to bifendate both in pharmacological and pharmacokinetic properties. Different from the symmetrical structure of DDB, bicyclol breaks the molecular symmetry and improves physico-chemical properties and activity. Bicyclol was approved by CFDA in 2001.
5.2. Diminishing chiral centers
5.2.1. From lovastatin to “statin” drugs
Lovastatin (31, Fig. 6), a cholesterol-lowering natural product, is produced by certain higher fungi, such as Pleurotus ostreatus and the closely related Pleurotus spp. The mechanism of action consists of the reversible inhibition of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA reductase), an enzyme that catalyzes the conversion of HMG-CoA to mevalonate. Mevalonate is a required building block for cholesterol biosynthesis. Lovastatin binds to and inhibits HMG-CoA reductase. Although lovastatin was approved by US FDA in 1987 to be the first hypolipidemic agent, much research space remains for optimization10.
Figure 6.

Simplifying structures from lovastatin to “statin” drugs.
Lovastatin contains eight asymmetrical carbon atoms, two located at the lactone moiety of the upper part and the others distributed to the lower hexahydronaphthalene moiety as indicated with red dots in structure 31. The lactone ring opens in cells by enzymatic catalysis to form a (R,R)-β,δ-dihydroxypentanoic acid moiety, which structurally mimics HMG and constitutes the pharmacophoric feature. It is evident that the configuration of the two carbon atoms is crucial for binding to HMG-CoA reductase. The lower part of the molecule binds to a hydrophobic pocket and plays the inhibitory role. In fact, the chirality of this part is unnecessary for binding. Based on the lovastatin structure, several synthetic HMG-CoA reductase inhibitors have been developed, such as simvastatin (32), fluvastatin (33), atorvastatin (34) and rosuvastatin (35), all of which contain the same fragment (R,R)-β,δ-dihydroxypentanoic acid as in lovastatin (Fig. 6). The hydrophobic moieties, however, are various and none of them has asymmetric factors except simvastatin.
Crystallographic studies showed that the interactions between the dihydroxypentanoic acid moieties of the statins and the enzyme are mostly ionic or polar. The binding modes are similar for all statin drugs. The rigid hydrophobic groups of the statins are located in a shallow groove between helices Lα1 and Lα10 in the same manner, though the scaffolds are different. It is worthwhile to emphasize that atorvastatin and rosuvastatin contain carbamide and sulfonamide moieties, respectively, which form hydrogen bonds with amino acid residues11. Only these two statins form hydrogen bonds between Ser565 and a carbonyl oxygen atom or a sulfone oxygen atom. Because of the hydrogen bond the thermodynamic parameters of 34 and 35 are different from those of others. The binding energy between rosuvastatin and the enzyme are mainly contributed by enthalpy gain (ΔH: 76%, —TΔS: 24%), for atorvastatin (ΔH: 39%, —TΔS: 61%), but for the other statins the enthalpy contributes less than 20%. The hydrogen bonding increases the specific binding12.
5.2.2. From trichostatin A to vorinostat
Trichostatin A (36, TSA, Fig. 7), isolated from Streptomyces hygroscopicus in the 1970s, was initially investigated as an antifungal antibiotic. This linear polyketide was later found to be a potent reversible inhibitor of histone deacetylases (HDAC). HDACs׳ function is to remove acetyl groups from acetylated histones. Another enzyme histone acetyltransferase (HAT) catalyzes acetylation of histones to neutralize positive charges on their tail regions, reducing their ability to bind DNA and thus loosening the structure of chromatin. Whereas, HDACs remove acetyl groups and HATs add them, the balance of these activities modulates the transcriptional process. By inhibiting HDAC action, trichostatin A mimics HAT activity, leading to hyperacetylation of chromatin. HDAC is a target for anti-cancer and anti-inflammatory therapy.
Figure 7.

Simplifying structures from trichostatin A to vorinostat.
The first US FDA-approved HDAC inhibitor was vorinostat (37, SAHA, Fig. 7) in 2006 for the treatment of cutaneous T cell lymphoma (CTCL) when the disease persists, gets worse, or comes back during or after treatment with other medicines. At first glance, vorinostat seems to be a simplified version of TSA. Actually, vorinostat originated not from TSA, but from the simple organic molecule dimethyl sulfoxide (DMSO). Starting from DMSO׳s induction of cell differentiation, Breslow and Marks et al.13 40 years ago designed and evaluated aliphatic bifunctional compounds using a phenotype screening model. By means of medicinal chemistry and SAR analysis, hundreds of compounds were investigated, from which vorinostat (37) was selected to be developed as an anti-cancer agent. Comparing the structural features of vorinostat with TSA, they have similar molecular shape and length, and pharmacophoric distribution: the hydroxamic acid group at one end, and one hydrophobic phenyl ring at the other end. Both compounds exhibit the same inhibitory activity for HDACs. Crystallographic studies of the HDAC-vorinostat and HDAC-TSA complexes indicate that the binding features of both compounds are almost identical13.
As the proverb says “All roads lead to Rome”, the success of vorinostat is independent of the natural product TSA, yet the target and the mechanism of action of TSA provided the guide for elucidating the molecular mechanism of vorinostat. On the other hand, the simplified structure of vorinostat suggests the trans–trans conjugated double bond and the chiral center of TSA are unlikely to be necessary for the inhibition.
Entinostat (38, Fig. 7), a derivative of 2-aminophenyl benzamides, is a potent and orally-available inhibitor of HDACs. The 2-aminophenyl benzamide moiety serves as a bio-isoster of hydroxamic acid group and binds to the zinc binding area. Entinostat has been widely investigated for the treatment of cancer in phase III clinical trials14, 15.
5.3. Increasing activity or selectivity
5.3.1. From vancomycin to telavancin and dalbavancin
Vancomycin (39, Fig. 8) is a natural antibiotic used to treat a number of bacterial infections. It is recommended intravenously as a first-line treatment for complicated skin and bloodstream infections.
Figure 8.

Simplifying structures from vancomycin to telavancin and dalbavancin.
Being a large hydrophilic molecule, vancomycin is able to form 5 hydrogen bonds with the terminal d-alanyl-d-alanine moieties of the N-acetylmuramic acid/N-acetylglucosamine (NAM/NAG)-peptides under normal circumstances. The interactions prevent cell wall synthesis of the long polymers of NAM and NAG that form the backbone strands of the bacterial cell wall, and prevent the backbone polymers that do manage to form through cross-linking with each other. However, in resistant bacteria, the terminal d-ala residue has been replaced by a d-lactate, so vancomycin cannot bind, and cross-links are successfully formed.
To overcome resistance to vancomycin, second generation drugs were developed. Telavancin (40, launched in 2009, Fig. 8), a semisynthetic multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. It binds to the D-alanyl-D-alanyl residues on the growing peptidoglycan chains and prevents transpeptidation from occurring, preventing peptidoglycan elongation and cell wall formation. In the telavancin molecule a long aliphatic chain is introduced into the aminosugar moiety, which anchors in the lipophilic bacterial membrane, thereby increasing its stability in the target environment and its affinity for peptidoglycan. In addition, a polar fragment amino-phosphonic acid is linked to the biphenyl moiety to balance the hydrophilic–lipophilic properties. Telavancin was approved by the US FDA in 200916.
Another semisynthetic analog, dalbavancin (41, Fig. 8), was launched in 2014. A similar lipophilic chain is also attached to the aminosugar and functions in the hydrophobic interactions with the cell membrane as described above. The modification of other parts in the molecule is to improve the affinity and physico-chemical properties17.
5.3.2. From enkephalin to eluxadoline
Compounds that modulate opioid receptors (ORs) have long been explored to search for analgesics. Morphine (8, Fig. 1C), as a mimic of the endogenous pentapeptide enkephalin (42, Fig. 9), agonizes ORs and has been a strong analgesic through history. Constipation, one of morphine׳s adverse effects, is due to the activation of ORs in the intestinal tract. Recently this “negative effect” has been become appreciated as therapeutics for gastrointestinal motility modulation.
Figure 9.

Simplifying structures from enkephalin to eluxadoline.
The ORs are categorized into three major subclasses, μ, δ, and κ, with their classifications based on well-defined pharmacological profiles. Starting from enkephalin as the lead compound, eluxadoline (43, Fig. 9) was developed as a dual modulator (μ receptor agonist and δ antagonist) to treat diarrhea in irritable bowel syndrome. Eluxadoline was approved by US FDA and launched in 201518.
The main points in the modification of enkephaline into eluxadoline are outlined as follows: (a) to change the peptide scaffold into a small organic molecule; (b) to increase chemical and metabolic stability; (c) to avoid penetrating blood brain barrier; (d) to remove the analgesic effect and addiction; (e) to restrict locally to the gastrointestinal tract.
Fig. 10 concisely illustrates the optimization course from enkephaline to eluxadoline. The N-terminal amido linkage of 42 is replaced by a tetrahydroisoquinoline moiety to limit the flexible conformation (42a). To avoid hydrolysis of the amide by intramolecular nucleophilic attack of free amino group of 42a the amido fragment is bioisosterically substituted by an imidazole ring. Simultaneously, the phenylalanine amide is removed to yield the dipeptidomimetic compound 42b. Optimizing the phenyl ring of 42b affords 42c, which is separately cut at the two single bonds of the tetrahydroisoquinoline to yield 42d. Alteration of R1 and R2 of 42d gives rise to the optimal scaffold 42e, in which R on phenyl group is verified by various substituents to tune the selectivity for μ and δ receptors. Finally eluxadoline (43) is validated as the final candidate. It is worthwhile to note that the presence of free carboxyl and amino groups in 43 yield a zwitterion that prevents the drug from being absorbed into the blood stream19.
Figure 10.

The optimization from enkephalin to eluxadoline.
5.3.3. From himbacine to vorapaxar—profit from chemical syntheses
Himbacine (44, Fig. 11) is a piperidine alkaloid with a tricyclic lactone scaffold isolated from Galbulimima baccata of Magnoliaceae in Australia. Since the discovery of himbacine in the 1960s, half century has passed until vorapaxar (45, Fig. 11) was marketed as an antithrombotic agent in 2014. The success of varapaxar is attributable to the skilled methods of total synthesis and chemistry-driven research and development, despite switching the project target during the course of development.
Figure 11.
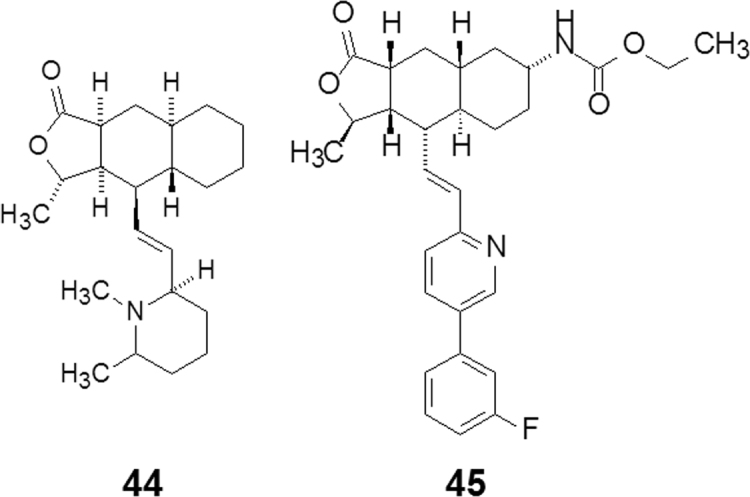
Simplifying structures from himbacine to vorapaxar.
Himbacine was originally investigated as an anti-Alzheimer agent owing to the potent inhibitory activity on the M2 receptor (Ki=4.5 nmol/L) with a 10-fold selectivity over the M1 receptor. In order to identify a therapeutically useful target, both the potency and selectivity of himbacine needed to be optimized through a rigorous structure–activity relationship study. A total synthesis of himbacine was carried out as briefly listed in Scheme 2.
Scheme 2.

The concise scheme of total synthesis of himacidine.
The achievement of synthesis resulted in the design and preparation of analogs with various scaffolds. Compounds 46 and 47 (Fig. 12) were synthesized for evaluating the inhibition of the M2 receptor, but none of them showed activity, indicating that the tricyclic lactone might be a pharmacophoric feature.
Figure 12.
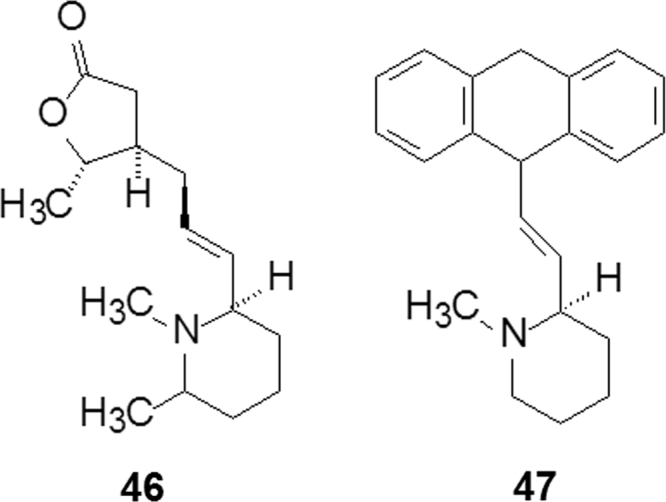
Structures of compounds 46 and 47.
Keeping the tricyclic moiety constant, the piperidine part was modified by various heterocyclic groups, and a new synthetic route was designed as shown in Scheme 3 for preparing compound 48.
Scheme 3.

Synthetic scheme of compound 48.
Compound 48, a gem-dimethyl pyrrolidine derivative of hibmacine, possesses the same inhibition against M2 receptor as himbacine. The activities of the N-ethyl compound or the diastereomer of 48 notably decreased20.
Simultaneously, the compounds were evaluated for inhibition of the thrombin receptor (protease activated receptor 1, PAR-1), because himbacine exhibits strong antagonism on PAR-1. The PAR-1 receptor is widely distributed on human platelets, endothelial cells, and smooth muscle cells. Thrombin is the most potent activator of platelets, and plays a critical role in the pathophysio–logy of thrombosis. Activated platelets bind to fibrinogen, causing platelets to aggregate at the site of cardiovascular injury to form a thrombus that is further stabilized by a thrombin-generated fibrin network. Since PAR-1 antagonists are expected to be specific for the cellular activation of thrombin and do not inhibit fibrin generation, such an agent is likely to have less bleeding liability than the currently-existing antithrombotic drugs.
Compound 49 (Fig. 13), a 6-methylpyridine-substituted analog, was identified as a PAR-1 lead in a high-throughput assay (IC50=300 nmol/L). A new synthetic route was carried out to prepare the congeners as illustrated in Scheme 4.
Figure 13.
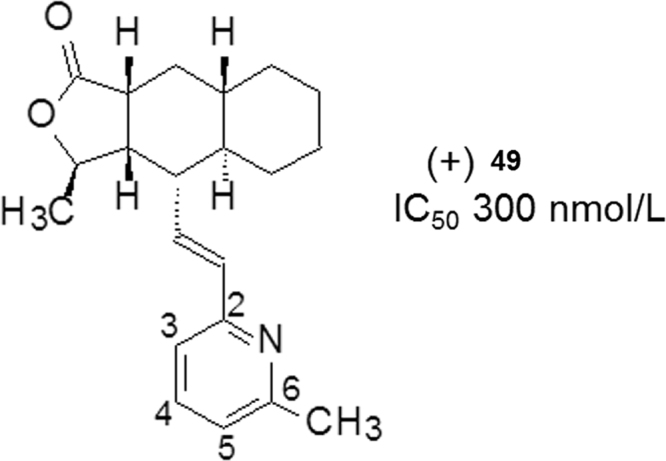
Structures of compounds 49.
Scheme 4.

Synthetic route of pyridine-containing analogs.
Structure–activity relationship studies led to the identification of the 5-phenyl-substituted analog 50 with higher activity. Compound 50 (Fig. 14) was optically resolved and it was found that the enantiomer with absolute chirality opposite to that of himbacine in the tricyclic system was more active. The (+)-50 compound with an IC50 of 27 nmol/L is 10 times more active than the racemate. To increase the metabolic stability of 43, several analogs with substituents on the phenyl group were synthesized21.
Figure 14.

Pyridine-containing analogs.
A metabolic study on compound 50 by LC–MS/MS analysis revealed that three pairs of hydroxylated diastereoisomers were produced. One of the metabolites, the 7R-OH compound (51, Fig. 14) was synthesized and showed 3 times more activity (IC50 11 nmol/L) than that of 50. Compound 51 exhibits reasonable bioavailability (F = 30% and 50% for rats and monkey, respectively) and stability. In the ex vivo platelet aggregation assay in the cynomolgus monkey, 51 showed complete and sustained inhibition of platelet aggregation at 3 mg/kg after oral administration. Compound 51 is the most potent PAR-1 antagonist reported to date. However, it still showed a less than optimal clearance profile, owing to the generation of a considerable amount of 7,8-dihydroxy metabolites. Therefore, 51 as a milestone compound was modified by extensively incorporating heteroatoms into the C-ring of the tricyclic motif and the terminal phenyl ring. In this series, a benchmark compound, 52 (Fig. 14), showed a Ki of 4.5 nmol/L against PAR-1. In cynomolgus monkeys, 52 exhibited an oral bioavailability of 62% at a dose of 3 mg/kg and the half-life was 6.2 h following intravenous administration. Compound 52 was the first candidate entered into preclinical trials22.
Simultaneously various derivatives of the C-7 amine were explored with derivatization of the amine to the corresponding amide, urea, sulfonamide, and carbamate. Among them, the carbamate compound 45 showed a very promising profile. Its unique properties are underscored by the excellent oral bioavailability in multiple animal species, high potency in a series of in vitro functional assays, and potent oral activity in an ex vivo cynomolgus monkey model of platelet aggregation. Compound 45 was 30 times more potent than the initial development candidate in the series, showing complete obliteration of agonist-induced platelet activation at 0.1 mg/kg with >24 h duration of activity (versus comparable efficacy at 3 mg/kg for the initial candidate 45). After a successful outcome in phase III clinical studies, 45 was approved by the FDA in 2014 as a new chemical entity (NCE) for the treatment of for acute coronary syndrome and secondary prevention of cardiovascular events in high-risk patients23.
5.4. Increasing metabolic stability
5.4.1. From phlorizin to canagliflozin and other “gliflozins”
More than 99% of the plasma glucose that is filtered in the kidney glomerulus is reabsorbed. This reabsorption process is mediated by two sodium-dependent glucose cotransporters (SGLTs): SGLT1, a low-capacity, high-affinity transporter expressed in the gut, heart and kidney, and SGLT2, a high-capacity, low-affinity transporter that is expressed mainly in the kidney. Selective inhibition of SGLT2 has been proposed to aid in the normalization of plasma glucose levels in patients with diabetes by preventing the renal glucose reabsorption process and promoting glucose excretion in urine. Selective SGLT2 inhibitors would be desirable, since gastrointestinal side effects associated with SGLT1 inhibition would be minimized.
The natural product O-glucoside phlorizin (53, Fig. 15) is a well-documented, potent glucosuric agent that was subsequently shown to be a nonselective SGLT inhibitor24. Because of its inhibition of SGLT1 and also its poor metabolic stability due to its susceptibility to β-glucosidase-mediated cleavage, 53, as a lead compound, was modified in several respects: (a) altering its molecular structure to increase the selectivity for SGLT2; (b) removing phenolic groups to reduce phase 2 metabolism; (c) altering the O-glycoside link into a C-glycoside to increase resistance to β-glucosidase.
Figure 15.

Simplifying structures from phlorizin to canagliflozin and other “gliflozins”.
Through a series of explorations, the structure activity relationships of 53 were disclosed, and resulted in a general scaffold of selective and potent SGLT2 inhibitors, such as formula 54 (Fig. 15), which was characterized with C-aryl glucosides and meta-substituted diarylmethanes. A number of compounds with preferred C-4′ and C-4 substitutions were synthesized and evaluated. Aryl groups A and B were also replaced by heteroaromatic rings. Compound 55 (Fig. 15) showed strong inhibition and selectivity against SGLT2 with an IC50 of 2.2 nmol/L over SGLT1 with IC50=910 nmol/L (more than 400-fold). Compound 48 exerted oral bioavailability in rats of F=78%, a half-life in plasma of 5 h, and pronounced anti-hyperglycemic effects in high-fat diet-fed KK mice. Compound 55, named canagliflozin, entered into clinical trials and was approved by the FDA in 2013 for the treatment of type 2 diabetes mellitus25.
Almost at the same time, successive SGLT2 inhibitors were launched (Fig. 15), including dapagliflozin (56)26, empagliflozin (57)27, and ipragliflozin (58)28 in 2014, and tofogliflozin (59)29 is in the phase III clinical trials.
The activity, selectivity, and bioavailability of these “gliflozins” are listed in Table 1, indicating that all of these SGLT2 inhibitors possess high selectivity and good absorption characteristics. It is worth noting that compounds 48—52 were independently developed in different companies, and based on the common lead compound O-glucoside phlorizin (53); the target molecules have the same pharmacophore features and similar scaffolds.
Table 1.
Activity, selectivity and bioavailability of SGLT2 inhibitors.
| No. | Name | SGLT2 IC50 (nmol/L) | SGLT1 IC50 (nmol/L) | Selectivity (fold) | Bioavailability (%) | Condition |
|---|---|---|---|---|---|---|
| 55 | Canagliflozin | 2.2 | 910 | 414 | 78 | Launched in 2013 |
| 56 | Dapagliflozin | 1.1 | 1390 | 1263 | 78 | Launched in 2014 |
| 57 | Empagliflozin | 3.1 | > 930 | >300 | >90 | Launched in 2014 |
| 58 | Ipragliflozin | 7.4 | 1880 | 254 | 71.7 | Launched in 2014 |
| 59 | Tofogliflozin | 2.9 | 8444 | 2912 | 85 | Phase III |
5.4.2. From retinoic acid to tamibarotene
All-trans-retinoic acid (60, ATRA, Fig. 16) is a metabolite of vitamin A (retinol) that mediates and induces growth and differentiation of epithelial cells. ATRA acts by binding to and activating the retinoic acid receptor (RAR); clinically ATRA is indicated for the treatment of acute promyelocytic leukemia and acne. However, serious adverse effects and its chemical instability restrict its application. The structure of the conjugated polyene is fast and easy to generate at room temperature. Structure–activity relationships reveal that the pharmacophore of ATRA features a polar group (carboxyl) at one end, a steric and hydrophobic moiety at the other end, with a linker connecting them. Tamibarotene (61, Fig. 16), an RAR agonist, is structurally quite different from that of ATRA, yet it resembles the pharmacophore of ATRA: the carboxyl group and the tetramethyltetraline moiety correspond to trimethylcyclohexene part, the two phenyl rings include several double bonds, and the amide linkage makes the whole molecule conjugated by p—π conjugation. Compound 61 is more active and stable than 6030. Tamibarotene was approved by the FDA in 2005 for the treatment of acute promyelocytic leukemia31.
Figure 16.

Simplifying structures from retinoic acid to tamibarotene.
9-cis-Retinoic acid (62, Fig. 16), an ATRA stereoisomer, is an agonist of retinoid X receptor (RXR). Compound 55 was used to treat Capocci sarcoma and dermatitis. The same poor stability prompted modification of the structure. Based on the same strategy of ATRA modification, bexarotene (63, Fig. 16) was designed and developed. It is a chemically-stable agonist of the RXR and was approved in 2000 to treat psoriasis and dermatitis32.
5.4.3. From patupilone to ixabepilone
Patupilone (64, epothilone B, Fig. 17) was originally identified as one of the 16-membered polyketides produced by Soragium cellulosum. Like paclitaxel, patupilone prevents cancer cells from dividing by interfering with tubulin, because they share the same binding site. Patupilone binds to the αβ-tubulin heterodimer subunit. Once bound, the rate of αβ-tubulin dissociation decreases, thus stabilizing the microtubules. Furthermore, it also causes cell cycle arrest at the G2-M transition phase, thus leading to cytotoxicity and eventually cell apoptosis33, 34.
Figure 17.

Simplifying structures from patupilone to ixabepilone.
While patupilone showed potent antineoplastic activity in vitro, this effect was not seen in preclinical in vivo models due to its poor metabolic stability and unfavorable pharmacokinetics. The reason is that it is subject to inactivation via esterase cleavage of the lactone moiety. In medicinal chemistry amides, as bioisosters of esters, generally possess significant stability over ester groups. A semisynthetic approach was taken to prepare analogs without this liability. Ixabepilone (65, Fig. 17), the 16-aza-patupilone, showed increased metabolic stability (t1/2=52 h), potent tubulin polymerization activity, and retained activity against paclitaxel-resistant lines. Based on its shown efficacy in clinical trials, ixabepilone was approved by the US FDA in 2007 for the treatment of aggressive metastasis of locally advanced breast cancer35.
Initially ixabepilone was prepared by a semi-synthetic method starting from patupilone, which involved by Pd-catalysed opening of the lactone bond, and through azide anion nucleophilic attack on C15. The reduction of the azide yielded an amino compound, which cyclized to ixabepilone, as briefly shown in Scheme 5 36. The first total synthesis of epothilone was achieved by the Danishefsky’s group37. The same group also realized the total synthesis of ixabepilone38. One of the synthetic route is listed in Scheme 6.
Scheme 5.

The semisynthetic route of ixabepilone.
Scheme 6.

The total synthesis of ixbepilone.
5.5. Improving physico-chemical properties
5.5.1. From artemisinin to dihydtoartemisinin, artemether, artesunate, and artether
In search of new anti-malarial drugs against chloroquine-resistant Plasmodium falciparum, a new natural product, artemisinin (1) was discovered and isolated from Artemisia plant by Tu and her co-workers in 1970 s. Artemisinin is a sesquiterpene containing 5 interweaving oxygen atoms to form a cyclic ether and a peroxy ether, cyclic acetal and ketal, as well as a lactone. The special scaffold and the presence of the peroxy moiety resulted in a new mechanism of action and less cross-resistance for the inhibition of plasmodia. Structure–activity relationships revealed that the peroxy group is a critical pharmacophoric feature. Reduction of the peroxy moiety of 1 into an ether leads to loss of activity. However, reducing the lactone moiety with NaBH4 under low temperature to the hemiacetal affords dihydroatemisinin (66, Fig. 18), which exerted stronger activities than did the lead compound 1, suggesting that alterations in the lactone moiety retain or increase activity.
Figure 18.

Simplifying structures from artemisinin to dihydtoartemisinin, artemether, artesunate, and artether.
Given the absence of a solubilizing moiety in its structure, artemisinin is insoluble in water, and poorly soluble in lipids. The poor biopharmaceutic properties and low bioavailability of artemisinin restricts its clinical application. Li and co-workers39 synthesized hundreds derivatives of dihydroartemisinin with two types of functional groups: ethers and esters. The methyl ether of dihydroartemisinin (67, artemether, Fig. 18) showed strong inhibitory activities in several plasmodia-infected animal models and as a candidate entered into clinical trials in 1978. Artemether was approved by the CFDA in 1987 as a new molecule entity and launched in several formulations39.
To develop water-soluble artemisinin-related drugs for injectable formulations, dihydroartemisinin C10-monoesters of diacids were designed and synthesized by Liu and co-workers40. One of the compounds, succinic acid monoester (68, artesunate, Fig. 18) possesses excellent antimalarial activity and physico-chemical properties. Its sodium salt is soluble in water and suitable for injection dosage. Clinical trials indicated that artesunate exerts a significant therapeutic effect for the treatment of patients with tertiary falciparum and cerebral malaria. Artesunate was approved by CFDA in 1987 as a new molecule entity and launched in several formulations40.
Brossi et al.41 investigated dihydroartemisinin-C10-β-ethylether (69, Fig. 18), which was developed by Brocacef Co. in The Netherlands and launched in 2000. As a homolog of artemether and a follow-up medicine, arteether does not show advantage over artemether42.
Artemether, arteether, and artesunate are all prodrugs. These molecules are rapidly hydrolyzed in plasma to dihydroartemisinin, which is the active form and is rapidly conjugated to glucuronic acid and excreted in the bile, resulting in short durations of action. Moreover, dihydroartemisinin exerts some neurotoxicity.
To prolong the period of action and avoid the production of dihydroartemisinin, a series of nitrogen-containing compounds were synthesized. Compound 70 (Fig. 18), which has excellent physico–chemical and pharmacological properties, was validated to be a safe and effective antimalarial in aotus monkeys; the compound, named artemisone, is now in a phase II clinical trial43.
5.5.2. From indirubin to meisoindigo
The old Chinese traditional medicine Angelica-Aloe-Pill (Danggui Luhui Wan) has been used for the treatment of chronic granulocyte leukemia. Through systematic investigation of the six-ingredient pill Qingdai, processed Indigofera tictoria was singled out as the effective medicine of the pill. Indirubin (71, Fig. 19), in turn, was found to have antileukemic active principles in this traditional medicine44. Animal tests showed that indirubin exhibits inhibitory activity against the growth of transplantable tumors. Clinically indirubin is indicated for the treatment of chronic granulocytic leukemia by oral administration. However, the adverse effects of indirubin on gastro-intestinal systems and poor bioavailability prevent it from being widely applied45.
Figure 19.

Simplifying structures from indirubin to meisoindigo.
Indirubin is a 2,3′-bis(dihydroindolone) compound. Its high melting point (348–353 °C) and low solubility is attributed to the planar structure and hydrogen bond with high lattice energy. To break the intermolecular hydrogen, bond a series of N-alkyl substituted indrubin were synthesized to optimize the balance between the anti-cancer activity, solubility, and in vivo effects46. Simultaneously, substituted isoindigo [3,3′-bis(2,3-dihydro-2-oxoindolylidene] compounds were also explored47. Among them compound 72 (Fig. 19) with optimal activity and physico-chemical properties was selected to be a candidate for clinical– trials. Compound 72 is 3,3′-bis(1-methyldihydro-2,2′-dioxoindolydene), named meisoindigo, and proved to be an effective drug for chronic myeloid leukemia and was approved by the CFDA in 1984.
Meisoinigo was also reported to have efficacy in acute myeloid leukemia (AML). In the AML cell lines, HL-60, NB4 and U937, meisoindigo induces apoptosis in both caspase-dependent and -independent pathways. The effect induced by meisoindigo is likely to be mediated through the intrinsic mitochondrial pathway. Meisoindigo also induces cell cycle arrest with more cells in sub-G1 and G0/G1 phases and fewer cells in the S phase48.
Following the indirubin scaffold, Chan and co-workers49 optimized the solubility and the ability to penetrate into cells. Compound 73 (Fig. 19), indirubin-3׳-(2,3-dihydroxypropyl)-oximether, E804) exhibited potent angiosuppressive effects. An in vitro study showed that E804 could significantly inhibit human umbilical vein endothelial cell proliferation, migration, and tube formation in a concentration-dependent manner (0.4–40 μmol/L). E804 would be a new potential candidate in the treatment of angiogenesis-dependent diseases49.
5.6. Synthesis-driven drug innovation: the platform for tetracycline synthesis and the new antibiotic eravacycline
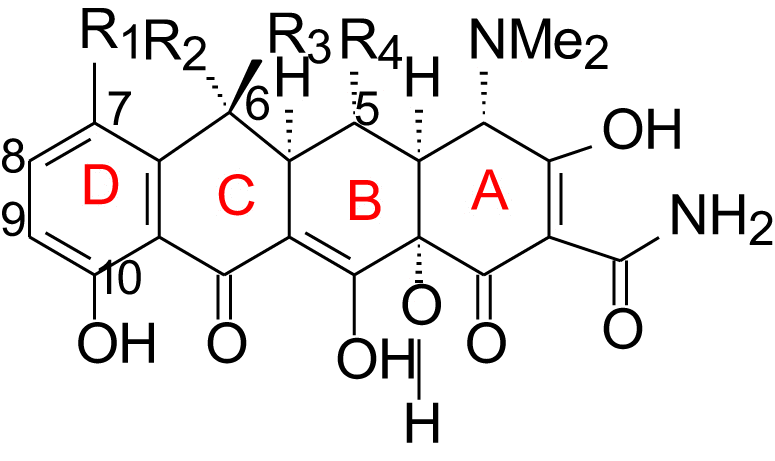
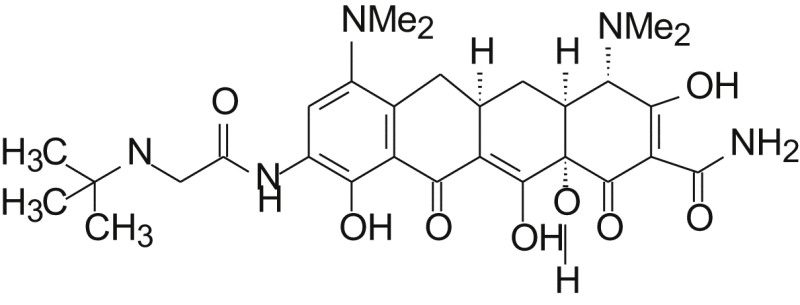
Since the first tetracycline antibiotic aureomycine (74, Table 2) was discovered in 1948, all tetracyclines approved for human or veterinary use today are fermentation products or are derived from fermentation products by semi-synthesis. The market of both natural and semi-synthetic tetracyclines (74—81) is listed in Table 2.
Table 2.
The structure of natural and semi-synthetic tetracyclines.
 |
 |
|||||
|---|---|---|---|---|---|---|
|
74—80 |
81 |
|||||
| No. | Name | R1 | R2 | R3 | R4 | Origin |
| 74 | Aureomycine | Cl | CH3 | OH | H | Natural |
| 75 | Terramycine | H | CH3 | OH | OH | Natural |
| 76 | Tetracycline | H | CH3 | OH | H | Natural |
| 77 | 6-Demethyltetracycline | H | H | OH | H | Natural |
| 78 | 6-Deoxytetracycline | H | CH3 | H | H | Semi-synthetic |
| 79 | Doxycycline | H | CH3 | H | OH | Semi-synthetic |
| 80 | Minocycline | N(CH3)2 | H | H | H | Semi-synthetic |
| 81 | Tigecycline | – | Semi-synthetic | |||
The early synthesis of sancycline (6-deoxy-6-demethyltetracycline) was realized by Woodward and coworkers50 in 1962, Strategically, the route developed by Woodward for the synthesis employed a “left-to-right” or D→A mode of construction. With the benefit of more than 50 years of structure–activity relationship data, as well as X-ray crystal structures of tetracycline bound to the bacterial ribosome, the left-to-right mode of construction used in these pioneering synthetic efforts can be seen to present a practical disadvantage to the discovery of new tetracycline antibiotics, because the D ring has emerged as one of the most promising sites for structural variation. This was a primary consideration guiding Myers and co-workers to assemble tetracyclines by a convergent coupling of D- and AB-ring precursors. The AB plus D strategy for tetracycline synthesis by C-ring construction is shown to be robust across a range of different carbocyclic and heterocyclic D-ring precursors, proceeding reliably and with a high degree of stereochemical control. A series of tetracyclines with various substitutions on the phenyl ring or the heteroaromatic rings (D ring) were synthesized to explore potent and anti-MRSA antibiotics. Among them, eravacycline (75, Table 2) and 7-fluorotetracycline exhibited broad spectrum antibacterial activity and are now in phase III clinical trials.
The synthetic process utilizes two key intermediates in a convergent approach as concisely shown in Scheme 7. The key transformation is a Micheal–Dieckmann reaction between a suitable substituted aromatic moiety (82a, D-ring) and a key cyclocyclohexanone derivative (82b, AB-ring) to give rise to 82c with a desired configuration. Subsequent deprotection (82d) and hydrogenation to open the isooxazole ring (82e), and then acylation yields the target molecule eravacycline. As a platform for tetracycline synthesis, this technology gives access to a broad range of molecules that would be inaccessible by semi-synthetic methods and provides a powerful engine for the discovery and development of new tetracycline antibiotics51.
Scheme 7.

The brief synthetic route of eravacycline.
5.7. Protecting resource and environment: development of houttuynium
Developing medicines cannot be at the sacrifice of natural resources and the environment. The research and development of the antibacterial agent houttuynium is an example. In folklore medicine, Houttuynia Cordala is topically used for the treatment of colpitis of women. The plant is not available in the winter seasons. In 1970, the scientists at Institute of Materia Medica, Chinese Academy of Medical Sciences (CAMS), extracted the volatile oil from Houttuynia Cordala and identified compound 83 (Fig. 20) as the main component of the oily mixture by GC–MS.
Starting from simple organic materials, the preparation of 83 was easily realized as shown in Scheme 8. To increase the stability 83 (a liquid) was transformed to the sodium bisulfite adduct (84, Fig. 20) as white crystals. After preclinical and clinical trials, 84, named houttuynium, was approved by the CFDA as an anti-infectious drug52.
Scheme 8.

The synthetic route of houttuynium.
6. Brief remarks
The secondary metabolites of living organisms do not exist for the healthcare of human beings. Although the natural products are normally valuable lead compounds, seldom can they be directly used in clinical applications. Structural modifications are necessary and involve several aspects. The strategy for structural modification is to increase potency and selectivity, to improve physico-chemical, biochemical, and pharmacokinetic properties, to eliminate or reduce adverse effects, to simplify the structural complexity, including removal of redundant atoms and chirality while retaining activities, and to generate patentable compounds. To accomplish these multi-dimensional operations, sophisticated syntheses and skillful preparation of complicated molecules are essential. Mutual cooperation and interaction between organic and medicinal chemists play an essential role in modifying natural products for commercial use through drug research and development.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Hopkins A.L., Groom C.R., Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 2.Aicher T.D., Buszek K.R., Fang F.G., Forsyth C.J., Jung S.H., Kishi Y. Total synthesis of halichondrin B and norhalichondrin B. J Am Chem Soc. 1992;114:3162–3164. [Google Scholar]
- 3.Towle M.J., Salvato K.A., Budrow J., Wels B.F., Kuznetsov G., Aalfs K.K. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001;61:1013–1021. [PubMed] [Google Scholar]
- 4.Yu M.J., Zheng W.J., Seletsky M.B. From micrograms to grams: scale-up synthesis of eribulin mesylate. Nat Prod Rep. 2013;30:1158–1164. doi: 10.1039/c3np70051h. [DOI] [PubMed] [Google Scholar]
- 5.Fujita T., Inoue K., Yamamoto S., Ikumoto T., Sasaki S., Toyama R. Fungal metabolites. Part 11. A potent immunosuppressive activity found in Isaria sinclairii metabolite. J Antibiot. 1994;47:208–215. doi: 10.7164/antibiotics.47.208. [DOI] [PubMed] [Google Scholar]
- 6.Fujita T., Hirose R., Yoneta M., Sasaki S., Inoue K., Kiuchi M. Fingolimod (FTY720): potent immunosuppressants, 2-alkyl-2-aminopropane-1,3-diols. J Med Chem. 1996;39:4451–4459. doi: 10.1021/jm960391l. [DOI] [PubMed] [Google Scholar]
- 7.Fujita T., Yoneta M., Hiros R., Sasaki S., Inoue K., Kiuchi M. Simple compounds, 2-alkyl-2-amino-1,3-propanediols have potent immunosuppressive activity. Bioorg Med Chem Lett. 1995;5:847–852. [Google Scholar]
- 8.Liu G.T. From the study of Schizandra to the discovery of DDB. Acta Pharm Sin. 1983;18:714–720. [PubMed] [Google Scholar]
- 9.Xiw J.X., Zhou J., Zhang C.Z., Yang J.H., Chen J.X., Jin H.Q. Synthesis of schizandrin C analogs. Acta Pharm Sin. 1981;16:306–309. [PubMed] [Google Scholar]
- 10.Alberts A.W., Chen J., Kuron G., Hunt V., Huff J., Hoffman C. Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci U S A. 1980;77:3957–3961. doi: 10.1073/pnas.77.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Istvan E.S., Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2012;292:1160–1164. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- 12.Sarver R.W., Bills E., Bolton G., Bratton L.D., Caspers N.L., Dunbar J.B. Thermodynamic and structure guided design of statin based inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J Med Chem. 2008;51:3804–3813. doi: 10.1021/jm7015057. [DOI] [PubMed] [Google Scholar]
- 13.Finnin M.S., Donigian J.R., Cohen A., Richon V.M., Rifkind R.A., Marks P.A. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki T., Ando T., Tsuchiya K., Fukazawa N., Saito A., Mariko Y. Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J Med Chem. 1999;42:3001–3003. doi: 10.1021/jm980565u. [DOI] [PubMed] [Google Scholar]
- 15.Heltweg B., Dequiedt F., Marshall B.L., Brauch C., Yoshida M., Nishino N. Subtype selective substrates for histone deacetylases. J Med Chem. 2004;47:5235–5243. doi: 10.1021/jm0497592. [DOI] [PubMed] [Google Scholar]
- 16.Higgins D.L., Chang R., Debabov D.V., Leung J., Wu T., Krause K.M. Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2005;49:1127–1134. doi: 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malabarba A., Ciabatti R., Scotti R., Goldstein B.P., Ferrari P., Kurz M. New semisynthetic glycopeptides MDL 63,246 and MDL 63,042, and other amide derivatives of antibiotic A-40,926 active against highly glycopeptide-resistant VanA enterococci. J Antibiot. 1995;48:869–993. doi: 10.7164/antibiotics.48.869. [DOI] [PubMed] [Google Scholar]
- 18.Dolle R.E., Machaut M., Martinez-Teipel B., Belanger S., Cassel J.A., Stabley G.J. 4-Carboxamido)phenylalanine is a surrogate for tyrosine in opioid receptor peptide ligands. Bioorg Med Chem Lett. 2004;14:3545–3548. doi: 10.1016/j.bmcl.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 19.Breslin H.J., Miskowski T.A., Rafferty B.M., Coutinho S.V., Palmer J.M., Wallace N.H. Rationale, design, and synthesis of novel phenyl imidazoles as opioid receptor agonists for gastrointestinal disorders. J Med Chem. 2004;47:5009–5020. doi: 10.1021/jm030548r. [DOI] [PubMed] [Google Scholar]
- 20.Chackalamannil S., Doller D., McQuade R., Ruperto V. Himbacine analogs as muscarinic receptor antagonists––effects of tether and heterocyclic variations. Bioorg Med Chem Lett. 2004;14:3967–3970. doi: 10.1016/j.bmcl.2004.05.047. [DOI] [PubMed] [Google Scholar]
- 21.Chackalamannil S., Xia Y., Greenlee W.J., Clasby M., Doller D., Tsai H. Discovery of potent orally active thrombin receptor (protease activated receptor 1) antagonists as novel antithrombotic agents. J Med Chem. 2005;48:5884–5887. doi: 10.1021/jm0502236. [DOI] [PubMed] [Google Scholar]
- 22.Clasby M.C., Chackalamannil S., Czarniecki M., Doller D., Eagen K., Greenlee W. Metabolism-based identification of a potent thrombin receptor antagonist. J Med Chem. 2007;50:129–138. doi: 10.1021/jm061043e. [DOI] [PubMed] [Google Scholar]
- 23.Chackalamannil S., Wang Y.G., Greenlee W.J., Hu Z.Y., Xia Y., Ahn H.S. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J Med Chem. 2008;51:3061–3064. doi: 10.1021/jm800180e. [DOI] [PubMed] [Google Scholar]
- 24.Ehrenkranz J.R.L., Lewis N.G., Kahn C.R., Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21:31–38. doi: 10.1002/dmrr.532. [DOI] [PubMed] [Google Scholar]
- 25.Nomura S., Sakamaki S., Hongu M., Kawanishi E., Koga Y., Sakamoto T. Discovery of canagliflozin, a novel C-glucoside with thiophene ring, as sodium-dependent glucose cotransporter 2 inhibitor for the treatment of type 2 diabetes mellitus. J Med Chem. 2010;53:6355–6360. doi: 10.1021/jm100332n. [DOI] [PubMed] [Google Scholar]
- 26.Meng W., Ellsworth B.A., Nirschl A.A., McCann P.J., Patel M., Girotra R.N. Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem. 2008;51:1145–1149. doi: 10.1021/jm701272q. [DOI] [PubMed] [Google Scholar]
- 27.Grempler R., Thomas L., Eckhardt M., Himmelsbach F., Sauer A., Sharp D.E. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012;14:83–90. doi: 10.1111/j.1463-1326.2011.01517.x. [DOI] [PubMed] [Google Scholar]
- 28.Imamura M., Nakanishi K., Suzuki T., Ikegai K., Shiraki R., Ogiyama T. Discovery of ipragliflozin (ASP1941): a novel C-glucoside with benzothiophene structure as a potent and selective sodium glucose co-transporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes mellitus. Bioorg Med Chem. 2012;20:3263–3279. doi: 10.1016/j.bmc.2012.03.051. [DOI] [PubMed] [Google Scholar]
- 29.Ohtake Y., Sato T., Kobayashi T., Nishimoto M., Taka N., Takano K. Discovery of tofogliflozin, a novel C-arylglucoside with an O-spiroketal ring system, as a highly selective sodium glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem. 2012;55:7828–7840. doi: 10.1021/jm300884k. [DOI] [PubMed] [Google Scholar]
- 30.Kagechika H., Kawachi E., Hashimoto Y., Shudo K., Himi T. Retinobenzoic acids. 1. Structure-activity relationships of aromatic amides with retinoidal activity. J Med Chem. 1988;31:2182–2192. doi: 10.1021/jm00119a021. [DOI] [PubMed] [Google Scholar]
- 31.Sanda T., Kuwano T., Nakao S., Iida S., Ishida T., Komatsu H. Antimyeloma effects of a novel synthetic retinoid Am80 (tamibarotene) through inhibition of angiogenesis. Leukemia. 2005;19:901–909. doi: 10.1038/sj.leu.2403754. [DOI] [PubMed] [Google Scholar]
- 32.Boehm M.F., Zhang L., Badea B.A., White S.K., Mais D.E., Berger E. Synthesis and structure-activity relationships of novel retinoid X receptor-selective retinoids. J Med Chem. 1994;37:2930–2941. doi: 10.1021/jm00044a014. [DOI] [PubMed] [Google Scholar]
- 33.Gerth K., Bedorf N., Höfle G., Irschik H., Reichenbach H. Epothilons A and B: antifungal and cytotoxic compounds from Sorangium cellulosum (myxobacteria) production, physico-chemical and biological properties. J Antibiot. 1996;49:560–563. doi: 10.7164/antibiotics.49.560. [DOI] [PubMed] [Google Scholar]
- 34.Winkler J.D., Axelsen P.H. A model for the taxol (paclitaxel)/epothilone pharmacophore. Bioorg Med Chem Lett. 1996;6:2963–2966. [Google Scholar]
- 35.Lee F.Y., Borzilleri R., Fairchild C.R., Kamath A., Smykla R., Kramer R. Preclinical discovery of ixabepilone, a highly active antineoplastic agent. Cancer Chemother Pharmacol. 2008;63:157–166. doi: 10.1007/s00280-008-0724-8. [DOI] [PubMed] [Google Scholar]
- 36.Hunt J.T. Discovery of ixabepilone. Mol Cancer Ther. 2009;8:275–281. doi: 10.1158/1535-7163.MCT-08-0999. [DOI] [PubMed] [Google Scholar]
- 37.Balog A, Meng D, Kamenecka T, Bertinato P, Su DS, Sorensen EJ, et al. A total synthesis of (–)-Epothilone A. Angew Chem Int Ed. Engl 1996;35:2801–03.
- 38.Stachel SJ, Lee CB, Chappell MSD, Bornmann WG, Danishefsky SJ, Chou TC, et al. On the interactivity of complex synthesis and tumor pharmacology in the drug discovery process: total synthesis and comparative in vivo evaluations of the 15-aza epothilones. J Org Chem 2001;66:4369–78. [DOI] [PubMed]
- 39.Li Y, Yu PL, Chen YX, Li LQ, Gai YZ, Wang DS, etal. Studies of analogs of artemisinine. I. The synthesis of ethers, carboxylic esters and carbonates of dihydroartemisinine. Acta Pharm Sin 1981;16:429–39 [PubMed]
- 40.Liu X. Study on artemisinin derivatives. Acta Pharm Sin. 1980;15:39. [Google Scholar]
- 41.Brossi A., Venugopalan B., Dominguez Gerpe L., Yeh H.J., Flippen-Anderson J.L., Buchs P. Arteether, a new antimalarial drug: synthesis and antimalarial properties. J Med Chem. 1988;31:645–650. doi: 10.1021/jm00398a026. [DOI] [PubMed] [Google Scholar]
- 42.Pu JL, Chen RG, Cai JL. The structur edetermination of artemisinin derivatives. Proceedings of 2nd Annual Meeting. Guangxi Pharma-ceutical Society; 1979, p. 82.
- 43.Haynes R.K., Fugmann B., Stetter J., Rieckmann K., Heilmann H.D., Chan H.W. Artemisone—a highly active antimalarial drug of the artemisinin class. Angew Chem Int Ed. 2006;45:2082–2088. doi: 10.1002/anie.200503071. [DOI] [PubMed] [Google Scholar]
- 44.Istitute of Hematology, CAMS Clinical and experimental atudies in the treatment of chronic granulocytic leukemia with indirubin. Chin J Intern Med. 1979;18:83–91. [PubMed] [Google Scholar]
- 45.Li C., Ji X.J. Comparative study on the physiological disposition of 3H-indirubin and its derivatives, 3H-79002 and 3H-79005 in animals. Acta Pharm Sin. 1983;18:332–338. [PubMed] [Google Scholar]
- 46.Wu K.M., Zhang M.Y., Fang Z., Huang L. Synthesis of N1-substituted derivatives of indirubin, an antileukemic compound. Acta Pharm Sin. 1984;19:513–518. [Google Scholar]
- 47.Wu K.M., Zhang M.Y., Fang Z., Huang L. Potential antileukemic agents, synthesis of derivatives of indirubin, indigo, and isoindigotin. Acta Pharm Sin. 1985;20:821–826. [PubMed] [Google Scholar]
- 48.Lee C.C., Lin C.P., Lee Y.L., Wang G.C., Cheng Y.C., Liu H.E. Meisoindigo is a promising agent with in vitro and in vivo activity against human acute myeloid leukemia. Leuk Lymphoma. 2010;51:897–905. doi: 10.3109/10428191003672115. [DOI] [PubMed] [Google Scholar]
- 49.Chan Y.K., Kwok H.H., Chan L.S., Leung K.S.Y., Shi J., Mak N.K. An indirubin derivative, E804, exhibits potent angiosuppressive activity. Biochem Pharmacol. 2012;83:598–607. doi: 10.1016/j.bcp.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 50.Conover L.H., Butler K., Johnston J.D., Korst J.J., Woodward R.B. The total synthesis of 6-demethyl-6-deoxytetracycline. J Am Chem Soc. 1962;84:3222–3224. [Google Scholar]
- 51.Sun C.X., Wang Q., Brubaker J.D., Wright P.M., Lerner C.D., Noson K. A robust platform for the synthesis of new tetracycline antibiotics. J Am Chem Soc. 2008;130:17913–17927. doi: 10.1021/ja806629e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pharmaceutical Factory Jiangxi “5,7 Cadre School”. Preparation and clinical effect of synthetic houttuynium. Pharm Ind. 1972;5:8. [Google Scholar]