

Abstract
Human genetic disease needs differential diagnosis to optimize clinical management, enable prenatal detection, and genetic counselling. The current methods of robust DNA sequencing also require next generation phenotyping to match with for better interpretation of genotypic and phenotypic heterogeneity commonly observed. We report use of human ontology based phenotypic characterization with Phenomizer that gives statistical score for possible diagnoses based on which, the gene mutation was studied.
A case of craniosynostosis which refers to a group of syndromes characterized by a premature fusion of skull was studied. The phenotypic features viz, dental crowding and dental malocclusion, bulbous nose, downslanted palpebral fissures, radial deviation of thumb, syndactyly of fingers, macrocephaly, and oxycephaly were entered to query the web-based tool Phenomizer which indicated high probability of mutation in FGFR2 gene. The proband, a 13-year-old male born to non-consanguineous parents showed mutation on FGFR2 gene at c.755C>G indicative of Apert syndrome. Apert syndrome is one of the most severe craniosynostosis syndromes with two possible mutations in the exon IIIa of FGFR2 gene reported in majority of the cases. This case study shows the importance of Phenomizer and molecular genetic analysis in differential diagnosis of genetic diseases.
Abbreviations: FGFR, fibroblast growth factor receptor; BKT, Binet Kamat intelligence test; ADHD, attention deficit hyperactivity disorder
Keywords: Craniosynostosis, Apert syndrome, FGFR2, Phenomizer, Genetic diagnosis
1. Introduction
The human genetic diseases require team of clinician, geneticist, and laboratory personnel in addition to proband and family. The important part in clinical examination of genetic diseases is description of phenotype which can be subjective and superfluous to specific and harmonized based on the expertise of the person involved. The current whole genome sequencing approaches are highly informative due to increased sensitivity and thus give more scope of finding causative genetic changes, provided phenotyping is also next generation to match with. The Phenomizer serves to carry out phenotypic analysis using standard and globally harmonized terminology for each trait, offering computation advantage for similarity searches.1 Phenomizer is freely available web based tool which employs semantic similarity in an ontology to rank candidate diseases (the differential diagnosis) based on the query terms and p value that indicates whether the similarity scores of best-matching candidate diseases are significant than would be expected by chance.1 The Phenomizer itself does not make diagnoses instead provides ranked list of possibilities that can be used as a part of the diagnostic workup and thus narrow down the diagnosis process.
Craniosynostosis is a heterogeneous group of syndromes characterized by a premature sutural fusion as a sole or with other anomalies.2 It exhibits considerable phenotypic and genetic heterogeneity with incidence of 1 in 2500 live births.3 There are more than 180 syndromes included in the category of craniosynostosis; out of which around eight types are linked with mutations in fibroblast growth factor receptor (FGFR2) gene i.e. isolated coronal synostosis, Pfeiffer syndrome, Crouzon syndrome, Apert syndrome, Beare–Stevenson syndrome, Jackson–Weiss syndrome, Crouzon syndrome with acanthosis nigricans, and Muenke syndrome.4
Inspite of recent advances in laboratory medicine, in the majority of cases, diagnosis remains a challenge unless that requires use of strategies developed and validated over the last 30 years.5 The molecular genetics rearrangement is vital to get accurate diagnosis and thus customized treatment, disease management, and genetic counselling. The molecular diagnosis helps clinician to persuade the family for acceptance of the disability, and connect with other parents and support groups.
1.1. Case report
We describe here a 13-year-old male with intellectual disability and major dysmorphisms. The proband is second child born to non-consanguineous, healthy parents when the mother was 24-year-old with a normal male child of 4 years (Fig. 1). At age of 2 months plain CT scan of brain showed prominent cisterna magna, fused coronal sutures, hypoplastic appearance of frontal and sphenoid bones and sagittal and lambdoid sutures appeared open. The X-ray report of chest at age of 5 months showed a prominent broncho-vascular markings at both hilar and parahilar zones. The global developmental delay was noticed from early infancy, he started sitting at 1 year, unclear speech at 1.5 years and walking at 2 years. Clinical history revealed that at the age of 3 the proband had low haemoglobin, low plasma electrolytes namely sodium, chlorides, bicarbonates with anionic gap. The C-reactive protein test was positive (24 mg/L). At the age of 12 years the proband was tested for thyroid function test including T3, T4 and TSH (thyroid stimulating hormone) which was normal.
Fig. 1.
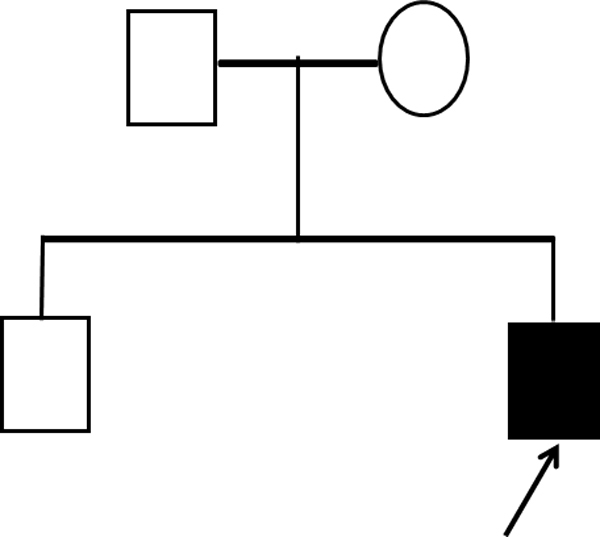
Pedigree of proband.
Current medical history includes mild mental retardation with IQ (intelligence quotient) 60% based on BKT (Binet Kamat intelligence test) and ADHD (attention deficit hyperactivity disorder). The phenotypic features of proband are depicted in Fig. 2. The X-ray reports of skull and mandibular region showed deformities (Fig. 3). The variations in size of body parts with respect to standards were recorded.6 Minor changes and dysmorphism are depicted in Table 1, Table 2, Table 3.
Fig. 2.

Figure depicts major phenotypic anomalies of the patient viz. (A and B) Flat facial features, (C) craniosynostosis, (D) dental crowding and dental-malocclusion, (E) bulbous nose, (F) downslanted palpebral fissures, (G and H) radial deviation of thumb, 1–2 finger syndactyly, 2–3 finger syndactyly and 3–4 finger syndactyly and (I) macrocephaly and oxycephaly (corrective surgeries performed).
Fig. 3.

X-ray reports of skull and mandibular region showing deformity.
Table 1.
Upper part measurement (in cm).
| Proband | Standard range | Remarks | |
|---|---|---|---|
| Philtrum | 0.8 | 1–3.5 | Normal |
| Ear length | 6.5 | 5.4–6.7 | Normal |
| Head circumference | 56.5 | 52–58 | Normal |
| Chest circumference | 89.5 | 61–98 | Normal |
| Shoulder width | 38.5 | 34–42 | Normal |
Table 2.
Eye measurements (in cm).
| Proband | Standard range | Remarks | |
|---|---|---|---|
| Outer canthal | 11.5 | 7.5–9.8 | Variation |
| Inner canthal | 4.5 | 2.75–3.75 | Variation |
| Inter pupillary | 9 | 5.1–6.6 | Variation |
| Palpebral fissures | 3.5 | 2.8–3.3 | Normal |
Table 3.
Limbs measurements (in cm).
| Proband | Standard Range | Remarks | |
|---|---|---|---|
| Upper limb | 69.5 | 60–80 | Normal |
| Full hand length | 14.5 | 16–22 | Normal |
| Palm length | 8.5 | 9.5–12 | Variation |
| Middle finger length | 6.0 | 7–9 | Variation |
| Lower limb length | 94.5 | 92–115 | Normal |
| Foot lengths | 24 | 23.5–28 | Normal |
2. Materials and methods
2.1. Karyotyping
The GTG banded karyotyping was done following standard protocol.7 Peripheral blood was collected in heparinised vial aseptically after obtaining written informed consent from parents. Short term culture of whole blood with PHA as mitogen was carried out. Air dried slides were used for GTG banding and karyotype was done as per the International System for Human Cytogenetic Nomenclature 20138 using digital image analysis system and IKAROS karyotyping software (Metasystems, Germany).
2.2. Phenomizer query
The phenotypic features of the proband were added to the Phenomizer viz, 3–4 finger syndactyly, dental crowding, craniosynostosis, macrocephaly, downslanted palpebral fissures, 2–3 finger syndactyly, oxycephaly, brachycephaly, radial deviation of thumb terminal phalanx, 1–2 finger syndactyly and dental malocclusion.
The dysmorphic features entered in the form of query in Phenomizer indicated possibility of APERT SYNDROME; ACROCEPHALOSYNDACTYLY, TYPE I; ACS1; ACS-I APERT-CROUZON DISEASE, INCLUDED; ACROCEPHALOSYNDACTYLY, TYPE II, INCLUDED; ACS II, INCLUDED; VOGT CEPHALODACTYLY, INCLUDED (OMIM:101200), PFEIFFER SYNDROME (OMIM:101600) and FAMILIAL SCAPHOCEPHALY SYNDROME, MCGILLIVRAY TYPE (OMIM:609579) with 3.8920, 3.7762 and 2.8926 scores respectively having 0.0016 p values. The Phenomizer query results showed the FGFR2 as a common gene that may be mutated out of which first two showed higher score (Table 4).
Table 4.
Phenomizer result based on query using combination of traits.
| p-Value | Score | Disease entry | Known genes |
|---|---|---|---|
| 0.0016 | 3.8920 | #101200 APERT SYNDROME;ACROCEPHALOSYNDACTYLY, TYPE I; ACS1;ACS IAPERT-CROUZON DISEASE, INCLUDED;ACROCEPHALOSYNDACTYLY, TYPE II, INCLUDED;ACS II, INCLUDED; VOGT CEPHALODACTYLY, INCLUDED (OMIM:101200) | FGFR2 |
| 0.0016 | 3.7762 | PFEIFFER SYNDROME (OMIM:101600) | FGFR1, FGFR2 |
| 0.0016 | 3.5832 | ACROCEPHALOPOLYSYNDACTYLY TYPE III (OMIM:101120) | |
| 0.0016 | 3.1731 | #614976 CARPENTER SYNDROME 2; CRPT2 (OMIM:614976) | MEGF8 |
| 0.0016 | 3.0998 | #269500 SCLEROSTEOSIS 1; SOST1;SOST;CORTICAL HYPEROSTOSIS WITH SYNDACTYLY (OMIM:269500) | SOST |
| 0.0016 | 3.0677 | %227330 FACIODIGITOGENITAL SYNDROME, AUTOSOMAL RECESSIVE;AARSKOG-LIKE SYNDROME;KUWAIT TYPE FACIODIGITOGENITAL SYNDROME (OMIM:227330) | |
| 0.0016 | 3.0505 | SUMMITT SYNDROME (OMIM:272350) | |
| 0.0016 | 2.9670 | POTOCKI-SHAFFER SYNDROME (OMIM:601224) | |
| 0.0016 | 2.9342 | ACROFRONTOFACIONASAL DYSOSTOSIS 2 (OMIM:239710) | |
| 0.0016 | 2.8926 | FAMILIAL SCAPHOCEPHALY SYNDROME, MCGILLIVRAY TYPE (OMIM:609579) | FGFR2 |
2.3. Genomic DNA Isolation and FGFR-2 gene analysis
Based on the possible clinical diagnosis and result of Phenomizer the FGFR2 gene analysis was carried out by Sanger method for targeted sequencing using the genetic analyzer 3500. A 300-bp fragment containing the p.Ser252Phe and p.Pro253Arg was amplified and amplicons were sequenced using BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Sequencing products were purified by 125 mm of EDTA prior to loading on the ABI 3500 DNA analyzer. ABI files were analyzed with the v5.2 sequence analysis software or Codon code Aligner Software.
The presence of mutation was determined by PCR with specific primers of FGFR2 (Fig. 4). PCR was performed in a reaction mix containing 13.3 μl of DH2O, 2 μl of Taq buffer A 15 mM (Invitrogen®), 0.5 μl of 2.5 mM dNTP (Gen Nei®), 0.2 μl of 3 U/μl of Hot Start Taq polymerase (Gen Nei®), 2 μl of DNA, and 1 μl of 10 pmol of each primer. The cycling PCR condition for amplifying a region of 300 bp by PCR system Viriti DX PCR machine was 95 °C for 5 min for initial denaturation, followed by 45 cycles of denaturation at 95 °C for 20 s, annealing at 59 °C for 20 s, and extension at 72 °C for 30 s, with the final extension at 72 °C for 5 min and hold at 4 °C. The reaction product was determined by ethidium bromide staining (10 μg/ml) with 2.5% of agarose gel and examined by UV. The PCR products were purified by ExoSAP-IT reagent (USB®) for downstream processing. Purification was programmed at 37 °C for 15 min and 80 °C for 15 min.
Fig. 4.

Primers and conditions for genomic amplification of FGFR2.
Downstream processing i.e., cycle sequencing was done by specific sequencing primers. Cycle sequencing was performed in a reaction containing 1.80 μl of 5× sequencing buffer (Invitrogen®), 0.5 μl of RR-100 (Invitrogen®), 1.0 μl of 10 pmol sequencing with forward and reverse primers, 1 μl of EXO-SAP product and 5.7 μl of DH2O. Cycling condition for cycle sequencing was initial denaturation at 96 °C for 1 min, followed by 25 cycles of melt at 96 °C for 10 s, annealing at 50 °C for 5 s, extension 60 °C for 4 min and hold at 4 °C.
3. Results and discussion
The karyotype was normal. The genomic DNA sequencing of proband detected mutation on FGFR2 gene at c.755C>G [p.Ser252Trp] indicative of Apert syndrome (Fig. 5).
Fig. 5.

Genomic DNA sequencing data.
Eugene Apert, a French physician described Apert syndrome in 1906.9 The Apert syndrome follows an autosomal dominant pattern of inheritance with mutations in fibroblast growth factor receptor 2 (FGFR2) gene located on chromosome 10q25.3-26.10 Apert syndrome is one of the most severe craniosynostosis syndromes. The prevalence of Apert syndrome is estimated to be 1 in 65,000 newborns and accounts for about 4.5% of all the cases of craniosynostosis.2 It is present since in utero with premature fusion of the cranial sutures and severe syndactyly of the hands and feet. Cerebral, cardiac, tracheal and genitourinary malformations may also be present. Additional skeletal manifestations, acne lesions and intellectual disability are also common. A major study done by Cohen et al. on around 135 Apert patients shows that some craniofacial features are very common in people with Apert syndrome including divergent upgaze, eso-tropic downgaze, hyperacrobrachycephaly, steep wide forehead, craniofacial asymmetry, flat occiput, marked depression of the nasal bridge, ocular hepertelorism and proptosis, downslanting palpebral fissures, short and wide nose having bulbous tip and reduced anterior facial height.11
It was reported in 1995 that two mutations, p.Ser252Trp (c.755C>G) and p.Pro253Arg (c.758C>G), within exon IIIa of FGFR2 were the main causes of Apert syndrome,12 detected in approximately 85% and 15% of cases respectively.13 Apart from the common mutation of FGFR2 at 252 and 253 position in exon IIIa; a case with 1372 bp deletion between exons IIIb and IIIc originating from recombination between 13 bp of identical DNA sequence present in both exons has also been reported.14
There are no significant differences in the phenotypic features of Apert syndrome patients having different mutations; any alteration in FGFR2 is associated with similar biological and phenotypic consequences.15 The phenotypic heterogeneity in the Apert syndrome makes molecular genetic analysis an important aspect in diagnosis and clinical management due to genetic heterogeneity described above. The rarity of the Apert syndrome and similarity of features with other craniosynostosis syndromes like Crouzon, Pfieffer also makes it a diagnostic dilemma.2 To the best our knowledge, this is third case of Apert syndrome in addition to a previous report of two cases of FGFR2 mutation from India.16 The case report emphasize on Phenomizer which uses the semantic structure of the Human Phenotype Ontology17 to find clinical features on the basis of specificity and also to identify those clinical features that best distinguish among the top candidate differential diagnoses.1
Conflicts of interest
The authors have none to declare.
Acknowledgements
The authors acknowledge funding support from GSBTM (Gujarat State Biotechnology Mission) (4QD5ZD), NERF (Nirma Education and Research Foundation) and BM Institute of Mental Health, Ahmedabad (Gujarat). Technical help from Dr. Krati Shah (Clinical geneticist), Dr. Sanjeev Mehta (Paediatric neurologist), Supratech Micropath Laboratory Research Institute, Ahmedabad; proband, and his parents for their kind support and consent for providing samples. The research was prospectively reviewed and approved by a duly constituted Institutional Human ethical committee.
References
- 1.Kohler S., Schulz M.H., Krawitz P. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet. 2009;85(4):457–464. doi: 10.1016/j.ajhg.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatia P.V., Patel P.S., Jani Y.V., Soni N.C. Apert's syndrome: report of a rare case. J Oral Maxillofac Pathol. 2013;17(2):294–297. doi: 10.4103/0973-029X.119782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panigrahi I. Craniosynostosis genetics: the mystery unfolds. Indian J Hum Genet. 2011;17(2):48–53. doi: 10.4103/0971-6866.86171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robin N.H., Falk M.J., Haldeman-Englert C.R. FGFR-related craniosynostosis syndromes. GeneReviews. 1998 [Google Scholar]
- 5.Newell-Price J., Trainer P., Besser M., Grossman A. The diagnosis and differential diagnosis of Cushing's syndrome and pseudo-Cushing's states. Endocr Rev. 1998;19(5):647–672. doi: 10.1210/edrv.19.5.0346. [DOI] [PubMed] [Google Scholar]
- 6.Hall J.G., Allanson J.E., Gripp K.W., Slavotinek A.M. 2nd ed. Oxford University Press; 2007. Handbook of Physical Measurements. [Google Scholar]
- 7.Moorhead P.S., Nowell P.C., Mellman W.J., Battips D.M., Hungerford D.A. Chromosome preparations of leukocytes cultured from human peripheral blood. Exp Cell Res. 1960;20:613–616. doi: 10.1016/0014-4827(60)90138-5. [DOI] [PubMed] [Google Scholar]
- 8.Shaffer L.G., McGowan-Jorda J., Schmid M. Karger Publication; 2013. An International System for Human Cytogenetic Nomenclature. [Google Scholar]
- 9.Mukhopadhyay A.K., Mukherjee D. Apert's syndrome. Indian J Dermatol Venereol Leprol. 2004;70(2):105–107. [PubMed] [Google Scholar]
- 10.Kannan V.P., Madhu P. Apert syndrome. J Indian Soc Pedod Prev Dent. 2010;28:322–325. doi: 10.4103/0970-4388.76169. [DOI] [PubMed] [Google Scholar]
- 11.Cohen M.M., Jr., Kreiborg S. A clinical study of the craniofacial features in Apert syndrome. Int J Oral Maxillofac Surg. 1996;25(1):45–53. doi: 10.1016/s0901-5027(96)80011-7. [DOI] [PubMed] [Google Scholar]
- 12.Wilkie A.O., Slaney S.F., Oldridge M. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9(2):165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- 13.Mundhofir F.E., Sistermans E.A., Faradz S.M., Hamel B.C. p.Ser252Trp and p.Pro253Arg mutations in FGFR2 gene causing Apert syndrome: the first clinical and molecular report of Indonesian patients. Singap Med J. 2013;54(3):e72–e75. doi: 10.11622/smedj.2013055. [DOI] [PubMed] [Google Scholar]
- 14.Fenwick A.L., Bowdin S.C., Klatt R.E., Wilkie A.O. A deletion of FGFR2 creating a chimeric IIIb/IIIc exon in a child with Apert syndrome. BMC Med Genet. 2011;12:122. doi: 10.1186/1471-2350-12-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park W.J., Theda C., Maestri N.E. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet. 1995;57(2):321–328. [PMC free article] [PubMed] [Google Scholar]
- 16.Girisha K.M., Phadke S.R., Khan F., Agrawal S. S252W mutation in Indian patients of Apert syndrome. Indian Pediatr. 2006;43(8):733–735. [PubMed] [Google Scholar]
- 17.Robinson P.N., Kohler S., Bauer S., Seelow D., Horn D., Mundlos S. The human phenotype ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;83(5):610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]