

Abstract
Branchio-oto-renal (BOR) syndrome is an autosomal dominant disorder characterized by the coexistence of branchial cysts or fistulae, external ear malformation with pre-auricular pits or tags, hearing impairment and renal malformations. However, the presence of the main features varies in affected families. Here, we present a 16-year-old boy admitted to the Department of Nephrology at the Pediatric Clinic, University Clinical Center of Kosovo, Pristina, Republic of Kosovo because of severe renal insufficiency diagnosed 6 years ago, which progressed to end-stage renal failure. Clinical examination on readmission showed a pale, lethargic and edematous child, with auricular deformity, pre-auricular tags and pits as well as bilateral branchial fistulae. Laboratory tests revealed high blood urea nitrogen (BUN) 15.96 mmol/L and serum creatinine 633.0 µmol/L; low glomerular filtration rate (GFR) 12 mL/min./ 1.73 m2 and massive proteinuria 4+. Abdominal ultrasound showed bilateral kidney hypoplasia. A novel mutation of the EYA1 gene was confirmed. Daily hemodialysis is continuing until renal transplantation is done. This case is presented to increase awareness among general practitioners to consider BOR syndrome or other renal abnormalities in patients with branchial fistula and/ or external ear anomalies or similar findings in other family members.
Keywords: Branchial fistulae, Branchi-oto-renal (BOR) syndrome, Hearing impairment, Renal insufficiency
Introduction
Branchio-oto-renal (BOR) syndrome is an autosomal dominant disorder characterized by the coexistence of branchial cysts or fistulae, external ear malformation with pre-auricular pits or tags, hearing impairment and renal malformations. However, the presence of the main features varies in affected families [1]. Unilateral agenesis or unibilateral hypoplasia are the most common renal abnormalities that can lead to end-stage renal disease (ESRD). We here present a 16-year-old boy with ESRD from a Kosovar family with BOR syndrome.
Case Report
A 16-year-old boy was admitted to the Department of Nephrology at the Pediatric Clinic, University Clinical Center of Kosovo, Pristina, Republic of Kosovo because of severe renal insufficiency diagnosed 6 years ago. At that time, surgical correction of bilateral branchial cysts and fistulae at the Ear, Nose and Throat Department, University Clinical Center of Kosovo, Pristina, Republic of Kosovo, was planned. As laboratory evaluation revealed high serum creatinine levels, the patient was referred to our department and renal insufficiency was diagnosed. However, the parents refused hospitalization, thus we did not have any information on the patient until now. Clinical examination on readmission showed a pale, lethargic and edematous child, with auricular pinnae deformity, pre-auricular tags and pits as well as bilateral branchial fistulae as shown in Figures 1 and 2. His weight was 71 kg and height 163 cm.
Figure 1.
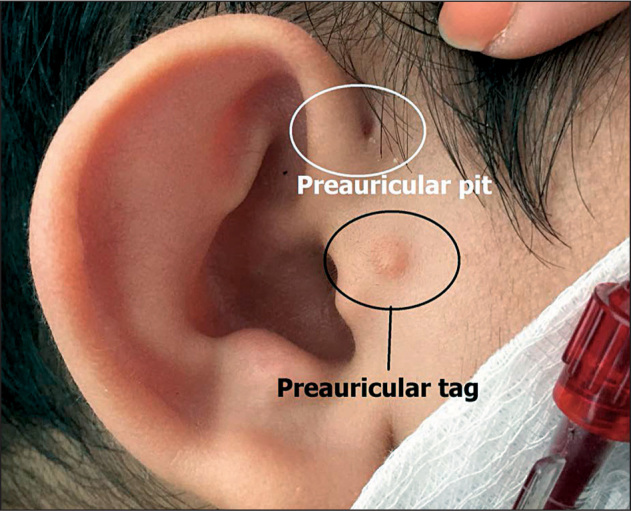
Auricular pinnae deformity with pre-auricular pit and tag (circled).
Figure 2.

Bilateral branchial fistulae.
Renal ultrasound showed bilateral kidney hypoplasia (Figure 3). In addition, bilateral fistulae were noticed in the neck of the patient (Figure 2), and the ultrasound of neck structures found communicating with an anechogen structure (possible puss) in the submandilar region.
Figure 3.
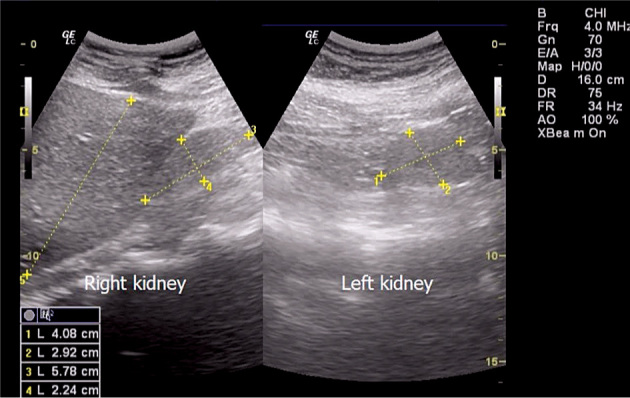
Bilateral kidney hypoplasia.
Laboratory tests were as the follows: red blood cell (RBC) count: 3.57 × 1012/L; white blood cell (WBC) count: 6.0 × 109/L; platelets: 242 × 109/L; hemoglobin (Hb): 11.0 g/dL; hematocrit or packed cell volume (PCV): 0.30 L/L; blood urea nitrogen (BUN): 15.96 mmol/L; serum creatinine: 633.0 µmol/L; total proteins: 7.47 g/dL; albumin: 5.09 g/dL; glycemia: 5.19 mmol/L; erythrocyte sedimentation rate (ESR): 40 mm/hour; C-reactive protein (CRP): 24 mg/dL; procalcitonine: 1.07 ng/mL, urinalysis revealed massive proteinuria (4+) and 24 hour protein collection was 2.738 g/24 hour. Clotting screen was normal. Glomerular filtration rate (GFR) was 12 mL/min./1.73 m2 corresponding with grade 5 of chronic kidney disease. A central venous catheter was placed and hemodialysis was initiated.
Conductive hearing impairment was documented with 50-70 dB on audiogram, encountering another major phe-notypic feature. Ophthalmological examination was revealed to be normal except for small refractory errors. Furthermore, genetic analysis detected a premature stop codon (nonsense) mutation on p.Gln543* (c.1627C>T) of the EYA1 gene. It is a novel mutation that clearly causes severe damage on protein function.
Phenotypic features of his father and sister are consistent with BOR syndrome and are illustrated in Figure 4. Examination of other members of the family was attempted but we could not acquire their consent. Renal transplantation is planned. Until then, the patient is receiving hemodialysis on a daily basis.
Figure 4.
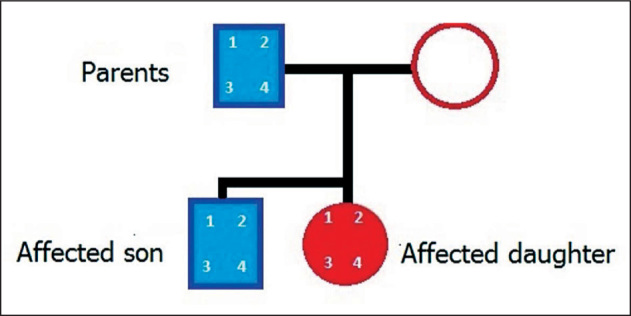
Family pedigree of the patient. Presence or absence of the four main features of BOR syndrome are marked by numbers. 1: Pre-auricular pits; 2: hearing loss; 3: branchial fistulae; 4: renal abnormalities.
Discussion
The incidence of BOR is estimated between 1:40,000 to 1:700,000. However, the incidence is higher among deaf children [2]. Fraser et al. [3] reported that 2.0% of deaf patients and 6.0% of ESRD patients have genetically proven BOR syndrome. Furthermore, more than 80 disorders presenting with sensorineural deafness and renal disturbances have been reported. Thus, phenotypic criteria should often be complemented with genetic analysis to avoid misdiagnosis.
Major phenotypic anomalies that occur in more than 20.0% of patients are: sensorineural or mixed hearing impairment (95.4%), followed by malformed auricles (86.8%), second branchial arch fistula/cyst (86.5%), pre-auricular sinus (87.0%), and renal anomalies ranging from mild hypoplasia to complete absence (58.3%) [4]. In addition, phenotypic anomalies occurring in less than 20.0% of patients are considered minor, i.e., pre-auricular tags, lacrimal duct aplasia, retrognathia, short palate, palatoschisis, facial paralysis, meatal atresia, etc. Likewise, at least three major criteria or two major and two minor criteria or one major criteria plus an affected first degree relative must be fulfilled to meet the clinical criteria for a BOR syndrome diagnosis [5]. Branchial fistulae, pre-auricular tags and conductive hearing impairment were present in all three members of the presented family. Also, renal abnormalities in the form of unilateral hypoplasia (father and sister) and bilateral hypoplasia (our patient) corresponding with BOR syndrome diagnosis were confirmed. However, variable phenotypic expressivity within families due to reduced penetrance has been observed [6]. Pierides et al. [1] followed 20 members of a three-generation Greek-Cypriot family for nearly three decades, and found only four members manifesting with all four BOR syndrome features of brachial fistulae, hearing loss, pre-auricular pits and renal abnormalities leading to renal failure.
In 1992, the BOR gene was mapped to chromosome 8q12-22 [7]. Five years later, the BOR gene was identified as the human homologue of the Drosophila eyes absent gene (eya) and called it EYA1 [8]. However, mutations and deletions of the EYE1 gene have been identified in only about 40.0% of individuals with the BOR phenotype. Mutations of other genes such as SIX1 and SIX5 have also been related to BOR syndrome, but at a much lower incidence [9-11]. At least 80 mutations of the EYE1 gene resulting in BOR syndrome [12], suggests that the type of mutation has a dosage effect, explaining the variable ex-pressivity within families. Further studies are needed to better understand the phenotypical expressivity of BOR syndrome.
Branchial anomalies account for up to 45.0% of neck pathologies. It is usually the main complaint that families seek medical attention for [13]. Hence, BOR syndrome should always be considered in patients with branchial fistula and/or external ear anomalies or similar findings in other family members.
Declaration of Interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Pierides AM, Athanasiou Y, Demetriou K, Koptides M, Deltas CC. A family with the branchio-oto-renal syndrome: Clinical and genetic correlations. Nephrol Dial Transplant. 2002;17(6):1014–1018. doi: 10.1093/ndt/17.6.1014. [DOI] [PubMed] [Google Scholar]
- 2.Misra M, Nolph KD.. Renal failure and deafness: Branchio-oto-renal syndrome. Am J Kidney Dis. 1998;32(2):334–337. doi: 10.1053/ajkd.1998.v32.pm9708623. [DOI] [PubMed] [Google Scholar]
- 3.Fraser FC, Sproule JR, Halal F.. Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss. Am J Med Genet. 1980;7(3):341–349. doi: 10.1002/ajmg.1320070316. [DOI] [PubMed] [Google Scholar]
- 4.Stinckens C, Standaert L, Casselman JW, Huygen PL, Kumar S, Van de Wallen J. et al. The presence of a widened vestibular aqueduct and progressive sensorineural hearing loss in the branchio-oto-renal syndrome. A family study. Int J Pediatr Otorhinolaryngol. 2001;59(3):163–172. doi: 10.1016/s0165-5876(01)00473-6. [DOI] [PubMed] [Google Scholar]
- 5.Chang EH, Menezes M, Meyer NC, Cucci RA, Vervoort VS, Schwartz CE. et al. Branchio-oto-renal syndrome: The mutation spectrum in EYA1 and its phenotypic consequences. Hum Mutat. 2004;23(6):582–589. doi: 10.1002/humu.20048. [DOI] [PubMed] [Google Scholar]
- 6.Fraser FC, Ling D, Clogg D, Nogrady B.. Genetic aspects of the BOR syndrome – Branchial, fistulas ear, pits hearing loss, and renal anomalies. Am J Med Genet. 1978;2(3):241–252. doi: 10.1002/ajmg.1320020305. [DOI] [PubMed] [Google Scholar]
- 7.Kumar S, Kimberling WJ, Kenyon JB, Smith RJ, Marres HA, Cremers CW.. Autosomal dominant bran-chio-oto-renal syndrome – Localization of a disease gene to chromosome 8q by linkage in a Dutch family. Hum Mol Genet. 1992;1(7):491–495. doi: 10.1093/hmg/1.7.491. [DOI] [PubMed] [Google Scholar]
- 8.Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C. et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. 1997;15(2):157–164. doi: 10.1038/ng0297-157. [DOI] [PubMed] [Google Scholar]
- 9.Kochhar A, Orten DJ, Sorensen JL, Fischer SM, Cremers CW, Kimberling WJ. et al. SIX1 mutation screening in 247 branchio-oto-renal syndrome families: A recurrent missense mutation associated with BOR. Hum Mutat. 2008;29(4):565–570. doi: 10.1002/humu.20714. [DOI] [PubMed] [Google Scholar]
- 10.Hoskins BE, Cramer CH, Silvius D, Zou D, Raymond RM, Orten DJ. et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet. 2007;80(4):800–804. doi: 10.1086/513322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanggaard KM, Rendtorff ND, Kjaer KW, Eiberg H, Johnsen T, Gimsing S. et al. Branchio-oto-renal syndrome: Detection of EYA1 and SIX1 mutations in five out of six Danish families by combining linkage, MLPA and sequencing analyses. Eur J Hum Genet. 2007;15(11):1121–1131. doi: 10.1038/sj.ejhg.5201900. [DOI] [PubMed] [Google Scholar]
- 12.Orten DJ, Fischer SM, Sorensen JL, Radhakrishna U, Cremers CW, Marres HA. et al. Branchio-oto-renal syndrome (BOR): Novel mutations in the EYA1 gene, and a review of the mutational genetics of BOR. Hum Mutat. 2008;29(4):537–544. doi: 10.1002/humu.20691. [DOI] [PubMed] [Google Scholar]
- 13.Lindau TA, Cardoso AC, Rossi NF, Giacheti CM.. Anatomical changes and audiological profile in bran-chio-oto-renal syndrome: A literature review. Int Arch Otorhinolaryngol. 2014;18(1):68–76. doi: 10.1055/s-0033-1358659. [DOI] [PMC free article] [PubMed] [Google Scholar]