

Summary
Various transcription factors are also known to enhance or suppress T helper type 17 (Th17) differentiation. We have shown previously that the development of collagen‐induced arthritis was suppressed in T‐bet transgenic (T‐bet Tg) mice, and T‐bet seemed to suppress Th17 differentiation through an interferon (IFN)‐γ‐independent pathway, although the precise mechanism remains to be clarified. The present study was designed to investigate further the mechanisms involved in the regulation of Th17 differentiation by T‐bet over‐expression, and we found the new relationship between T‐bet and aryl hydrocarbon receptor (AHR). Both T‐bet Tg mice and IFN‐γ–/–‐over‐expressing T‐bet (T‐bet Tg/IFN‐γ–/–) mice showed inhibition of retinoic acid‐related orphan receptor (ROR)γt expression and IL‐17 production by CD4+ T cells cultured under conditions that promote Th‐17 differentiation, and decreased IL‐6 receptor (IL‐6R) expression and signal transducer and activator of transcription‐3 (STAT‐3) phosphorylation in CD4+ T cells. The mRNA expression of ahr and rorc were suppressed in CD4+ T cells cultured under Th‐17 conditions from T‐bet Tg mice and T‐bet Tg/IFN‐γ–/– mice. CD4+ T cells of wild‐type (WT) and IFN‐γ–/– mice transduced with T‐bet‐expressing retrovirus also showed inhibition of IL‐17 production, whereas T‐bet transduction had no effect on IL‐6R expression and STAT‐3 phosphorylation. Interestingly, the mRNA expression of ahr and rorc were suppressed in CD4+ T cells with T‐bet transduction cultured under Th17 conditions. The enhancement of interleukin (IL)−17 production from CD4+ T cells by the addition of AHR ligand with Th17 conditions was cancelled by T‐bet over‐expression. Our findings suggest that T‐bet over‐expression‐induced suppression of Th17 differentiation is mediated through IFN‐γ‐independent AHR suppression.
Keywords: AHR, IL‐17, T‐bet over‐expression
Introduction
Helper T cell subsets have distinct function and cytokine profiles. For example, T helper type 1 (Th1) cells produce interferon (IFN)‐γ and play an important role in immunity against intracellular pathogens, whereas Th2 cells produce interleukin (IL)−4, IL‐5 and IL‐13, and are essential for defence against parasites and extracellular pathogens. Furthermore, Th17 cells produce IL‐17, IL‐21 and IL‐22, and are involved in immunity against bacterial and fungal infections 1 and the development of various autoimmune disorders 2.
Lineage commitment of each helper T cell subset from naive CD4+ T cells depends upon the expression of specific transcription factors induced by specific cytokine environment. For example, differentiation of Th‐1 depends upon the expression of transcription factor T‐bet, which is induced by T cell receptor (TCR) stimulation, IL‐12/signal transducer and activator of transcription‐4 (STAT‐4) signalling pathway and IFN‐γ/STAT‐1 signalling pathway. T‐bet activates the production of IFN‐γ directly 3, 4, 5. Similarly, Th17 cell differentiation in mice is dependent upon transcription factor retinoic acid‐related orphan receptor (ROR)γt induced by transforming growth factor (TGF)‐β and IL‐6 6. In this regard, down‐regulation of RORγt inhibits Th17 cell differentiation 6, 7, suggesting that RORγt is a master transcription factor in the differentiation of Th17 cells. Other transcription factors – STAT‐3, RORα, I kappa B zeta (IκBζ), interferon regulatory factor 4 (IRF4), runt‐related transcription factor 1 (Runx‐1) and aryl hydrocarbon receptor (AHR) – are also known to enhance Th17 differentiation 8, 9, 10, 11. For example, both IRF4 and Runx‐1 induce RORγt expression and also enhance IL‐17 expression directly, thus promoting Th17 differentiation 11, 12. AHR, a ligand‐dependent transcription factor, is another regulator of Th17 differentiation 13, 14, and enhances cytokine production such as IL‐17 and IL‐22 15. Previous studies reported that these transcription factors regulate the differentiation of other T cell subsets negatively by direct co‐interaction and/or indirect effect of cytokines produced by each T cell subset 16, 17. In an experimental model of autoimmune arthritis induced by Th17 cell response, T‐bet and IFN‐γ act as immunoregulatory factors 18, 19, 20. In this regard, we have suggested previously that the development of collagen‐induced arthritis (CIA) was suppressed in T‐bet transgenic (T‐bet Tg) mice under the CD2 gene promoter 21, and T‐bet seemed to suppress Th17 differentiation through an IFN‐γ‐independent pathway 22, although the precise mechanism remains to be clarified.
The present study was designed to investigate further the mechanisms involved in the regulation of Th17 differentiation by T‐bet over‐expression. For this purpose, we generated IFN‐γ–/– ‐over‐expressing T‐bet (T‐bet Tg/IFN‐γ–/–) mice by crossing T‐bet Tg mice with IFN‐γ‐deficient (IFN‐γ–/–) mice 22. Both T‐bet Tg mice and T‐bet Tg/IFN‐γ–/– mice showed inhibition of RORγt expression and IL‐17 production by CD4+ T cells cultured under conditions that promote Th17 differentiation. The mRNA expression of ahr and rorc were suppressed in T‐bet Tg mice and T‐bet Tg/IFN‐γ–/– mice. Interestingly, the results also showed inhibition of IL‐17 production and mRNA expression of ahr and rorc in CD4+ T cells of wild‐type C57BL/6 (WT) and IFN‐γ–/– mice transduced with T‐bet‐expressing retrovirus. Our study identified a new regulatory mechanism of Th17 cell differentiation involving IFN‐γ‐independent suppression of AHR mediated through T‐bet over‐expression.
Materials and methods
Mice
CD2–T‐bet Tg mice 21, which were prepared by back‐crossing mice with the C57BL/6 background, were provided by Professor S. Takahashi (University of Tsukuba, Ibaraki, Japan). C57BL/6J and IFN‐γ–/– mice were obtained from Jackson Laboratory Co. (Bar Harbor, ME, USA). T‐bet Tg/IFN‐γ–/– mice were generated by crossing T‐bet Tg mice with IFN‐γ–/– mice. All mice were maintained under specific pathogen‐free conditions in the Laboratory Animal Resource Center at the University of Tsukuba, and studied in 8–12‐week‐old male mice. All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals at the University of Tsukuba.
Cell isolation
Single‐cell suspensions from the spleen were prepared from each mouse, and then CD4+ T cells or CD4+ CD62L+ naive T cells were isolated by magnetic cell isolation and cell separation (MACS) using mouse CD4 microbeads or the CD4+CD62L+ T cell isolation kit II (Miltenyi Biotec, Bergisch Gladbach, Germany), according to the instructions provided by the manufacturer. The prepared cells were 92% pure CD4+ T cells or naive CD4+ T cells, as confirmed by fluorescence activated cell sorter (FACS) analyses.
Plasmids and retroviral transduction
Murine T‐bet cDNA was transfected into pGCDNsam IRES‐EGFP (MSCV) retroviral vector (kindly provided by Dr Nishikii, University of Tsukuba, Ibaraki, Japan). The recombinant plasmid was transferred to the retroviral packaging cell, Plat‐E, by lipofectamine (Invitrogen, Carlsbad, CA, USA)‐mediated gene transfer. The retroviral transduction to naive CD4+ T cells was performed by the RetroNectin‐bound virus infection method (Takara Bio, Otsu, Japan), as described in detail previously 23. In brief, 48‐well plates were coated with 25 µg/ml of RetroNectin and 2 µg/ml anti‐CD3ɛ monoclonal antibody (mAb) (BioLegend, San Diego, CA, USA) overnight at 4°C. The retrovirus was added to the RetroNectin‐coated plate, and the plate was centrifuged for 2 h at 2000 g at 32°C and washed with phosphate‐buffered saline (PBS). Naive CD4+ T cells stimulated with plate‐bound 2 µg/ml anti‐CD3ε mAb, 1 µg/ml soluble anti‐CD28 mAb (BioLegend), 10 μg/ml of anti‐IFN‐γ antibody (BioLegend) and 10 μg/ml of anti‐IL‐4 antibody (BioLegend) for 24 h were added to the retrovirus‐bound RetroNectin and anti‐CD3 mAb‐coated plates and were cultured for 24 h at 37°C, and used in the experiments. After infection, green fluorescent protein (GFP)‐positive cells were sorted from transduced cells using the MoFlo cell sorter (DakoCytomation, Glostrup, Denmark), and cultured under neutral conditions or conditions favouring Th17 differentiation for another 4 days. The expression levels of T‐bet, RORγt, IFN‐γ, IL‐17 and AHR were analysed by intracellular staining.
In‐vitro T cell cultures
CD4+ cells (5 × 105 cells/well on the 48‐well plate) or naive CD4+ T cells transduced with the retrovirus (1 × 105 cells/well on the 96‐well plate) were cultured in RPMI‐1640 medium (Sigma‐Aldrich, St Louis, MO, USA) containing 10% fetal bovine serum, 100 units/ml of penicillin, 100 µg/ml of streptomycin and 50 µM 2‐mercaptoethanol with 2 µg/ml plate‐bound anti‐CD3ɛ mAb, 1 µg/ml soluble anti‐CD28 mAb, 10 μg/ml of anti‐IFN‐γ antibody and 10 μg/ml of anti‐IL‐4 antibody for neutral conditions.
For Th17 differentiation, the cells were cultured in a medium containing 2 µg/ml plate‐bound anti‐CD3ε mAb, 1 µg/ml soluble anti‐CD28 mAb, 2·5 ng/ml human TGF‐β (hTGF‐β; BioLegend), 50 ng/ml mouse IL‐6 (eBioscience, San Diego, CA, USA), 10 μg/ml of anti‐IFN‐γ antibody and 10 μg/ml of anti‐IL‐4 antibody. To analyse the effect of AHR on the differentiation of Th17 cells, 300 nM of 6‐formylindolo (3,2‐b) carbazole (FICZ) (Enzo Life Sciences, Exeter, UK and Abcam, Cambridge, MA, USA), the ligand of transcription factor AHR was added to the cultures used for Th17 differentiation.
Surface and intracellular staining and FACS analysis
For intracellular staining of cytokines, the cells were cultured with the cytokines cited above for 4 days, and then 50 ng/ml of phorbol myristate acetate (PMA) (Sigma‐Aldrich), 1 µg/ml of ionomycin (Sigma‐Aldrich) and GolgiStop (BD Biosciences, San Jose, CA, USA) were added during the last 6 h of each culture. First, dead cells were stained with fixable viability dye eFluor 780 (eBioscience). Next, cells were stained with anti‐CD4–peridinin chlorophyll/cyanin 5·5 (PerCP/Cy5·5) extracellularly, fixed and permeabilized with forkhead box protein 3 (FoxP3)/transcription factor, fixation/permeabilization concentrate and diluent (eBioscience), according to the protocol supplied by the manufacturer. GFP fluorescence was quenched by fixation. Intracellular cytokine staining was then performed using anti‐IFN‐γ–fluorescein isothiocyanate (FITC) (BioLegend), anti‐T‐bet–phycoerythrin (PE) (eBioscience), anti‐RORγt– allophycocyanin (APC) (eBioscience), anti‐IL‐17–PE/Cy7 (BioLegend) and anti‐AHR–eFluor 660 (eBioscience), according to the protocol supplied by the manufacturer. For analysis of IL‐6 receptor (IL‐6R) expression, dead cells were first stained with fixable viability dye, followed by cells stained with anti‐CD126–APC (BioLegend) and anti‐CD130–PE (eBioScience). For analysis of STAT‐3 phosphorylation, cells were stimulated with the cytokines cited above for 0, 15, 30 and 60 min, then fixed in lyse/fix buffer (BD Biosciences) at 37°C for 10 min and washed with FACS buffer. The staining experiments were performed according to the protocol supplied by the manufacturer. In brief, cells were permeabilized with cold Perm Buffer III (BD Biosciences) for 30 min on ice, then stained with anti‐STAT‐3–PE (pY705) (BD Biosciences) and anti‐STAT3–APC (BD Biosciences) for 60 min. Samples were analysed with a FACSVerse flow cytometer (Becton Dickinson; Mountain View, CA, USA), and data were analysed with FlowJo software (Tree Star; Ashland, OR, USA). The expression levels of T‐bet, RORγt, CD126, CD130 and AHR were expressed by the relative median fluorescence intensity (MFI) ratio [MFI (specific antibody)/MFI (isotype)].
Enzyme‐linked immunosorbent assay (ELISA)
The culture supernatants described above were collected and analysed for IFN‐γ, IL‐17 and IL‐22 production using the Duoset ELISA Development System (R&D Systems, Minneapolis, MN, USA).
Quantitative reverse transcription–polymerase chain reaction (qRT–PCR)
Total RNA was extracted from CD4+ T cells cultured as described above, using the Isogen (Nippon Gene, Tokyo, Japan) extraction method or the RNeasy Plus Micro kit (Qiagen, Hilden, Germany), according to the instructions provided by the manufacturer. cDNA was obtained by reverse transcription with a commercially available kit (PrimeScript RT Master Mix; Takara, Shiga, Japan). For qRT–PCR, we used a TaqMan Assay‐on‐Demand gene expression product (Applied Biosystems, Foster City, CA, USA). The expression levels of tbx21 (Mm00450960_m1), rorc (Mm01261022_m1), stat3 (Mm01219775_m1), stat1 (Mm00439531_m1), runx1 (Mm01213405_m1), irf4 (Mm00516431_m1), nfkbiz (Mm00600522_m1), ahr (Mm00478932_m1) and cyp1a1 (Mm00487218_m1) were normalized relative to the expression of GAPDH (Mm99999915_g1). All analyses were performed with an ABI Prism 7500 apparatus (Applied Biosystems) under the following conditions: inactivation of possible contaminating amplicons with AmpErase UNG for 2 min at 50°C, initial denaturation for 10 min at 95°C, followed by 45 thermal cycles of 15 s at 95°C and 60 s at 60°C.
Analysis of transcriptional factor gene expression of CII‐reactive CD4+ T cells
Native chicken CII (Sigma‐Aldrich) was dissolved in 0·01 M acetic acid and emulsified in Freund's complete adjuvant (CFA). CFA was prepared by mixing 5 mg of heat‐killed Mycobacterium tuberculosis H37Ra (Difco/BD Biosciences) and 1 ml of Freund's incomplete adjuvant (Sigma‐Aldrich). The 8–12‐week‐old mice were injected intradermally at the base of the tail with 200 μg of CII in CFA on day 0. Inguinal lymph nodes were harvested from each mouse and WT and T‐bet Tg on day 10 after immunization with CII. Single‐cell suspensions were prepared, and lymph node cells (1.0 × 105 cells/well on a 96‐well round‐bottomed plate) were cultured for 72 h in the presence of 100 μg/ml of denatured chicken CII. CD4+ T cells were isolated from cultured cells MACS, and total RNA were extracted.
Statistical analysis
Data are expressed as mean ± standard error of the mean (s.e.m.). Differences among groups were examined for statistical significance using Student's t‐test, Tukey's test and Kruskal–Wallis test. P‐values less than 0·05 were considered significant.
Results
Over‐expression of T‐bet suppresses Th17 differentiation through IFN‐γ‐independent pathway in T‐bet Tg mice
To clarify the effect of T‐bet on Th17 differentiation, CD4+ T cells were isolated from the spleen of WT and T‐bet Tg mice and then cultured for Th17 differentiation. FACS analysis demonstrated a smaller proportion of IL‐17‐producing CD4+ T cells in T‐bet Tg (7·6 ± 2·3%) than WT mice (14·6 ± 1·2%) (P < 0·05), but a larger proportion of IFN‐γ‐producing CD4+ T cells in T‐bet Tg mice (66·4 ± 5·9%) than WT mice (1·6 ± 0·1%) (P < 0·01) (Fig. 1a).
Figure 1.
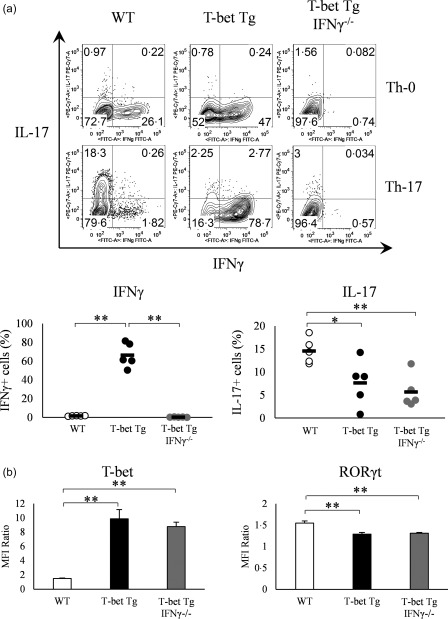
Over‐expression of T‐bet in transgenic (Tg) mice suppresses IL‐17 expression independently of interferon (IFN)‐γ. (a) IFN‐γ and interleukin (IL)−17 production by splenic CD4+ T cells of wild‐type (WT), T‐bet Tg and T‐bet Tg/IFN‐γ–/– mice CD4+ analysed by flow cytometry. Numbers in plots indicate percentage of cells that secrete the cytokines. Five mice per group were analysed under the T helper type 17 (Th17) condition and representative data are shown with neutral condition. (b) T‐bet and retinoic acid‐related orphan receptor (RORγt) expression on CD4+ cells analysed by flow cytometry. Five mice per group were analysed under the Th17 condition. Data are mean ± standard error of the mean (s.e.m.) of four mice. *P < 0·05; ** P < 0·01 (by Tukey's test).
To determine whether the suppression of Th17 differentiation was IFN‐γ‐dependent, we generated T‐bet Tg/IFN‐γ–/– mice, and CD4+ T cells isolated from these mice were cultured for Th17 differentiation. Similar to T‐bet Tg mice, the proportion of IL‐17‐producing CD4+ T cells was also smaller in T‐bet Tg/IFN‐γ–/– mice (5·7 ± 1·6%) (P < 0·01), although no IFN‐γ‐producing CD4+ T cells were detected in T‐bet Tg/IFN‐γ–/– mice (Fig. 1a). These results suggested that the suppression of Th17 differentiation by T‐bet was not due to over‐production of IFN‐γ.
We performed intracellular staining of transcription factors to examine whether T‐bet regulates RORγt expression, which was analysed by the relative MFI ratio. FACS analyses demonstrated higher expression of T‐bet on CD4+ T cells of T‐bet Tg mice and T‐bet Tg/IFN‐γ–/– mice than WT mice. In contrast, RORγt expression was lower on CD4+ T cells of T‐bet Tg mice and T‐bet Tg/IFN‐γ–/– mice than WT mice (Fig. 1b). These findings suggest that T‐bet suppressed RORγt expression as well as RORγt‐induced IL‐17 production, and this suppression was independent of IFN‐γ production.
T‐bet over‐expression suppresses IL‐6–STAT‐3 signalling and transcription factor expression that enhances Th17 differentiation
To clarify how over‐expression of T‐bet regulates the differentiation of Th17 cells, we first focused upon IL‐6 signalling, which is essential for the differentiation of Th17 cells 6. We examined the expression of IL‐6Rα (CD126) and IL‐6Rβ (CD130) in CD4+ T cells in WT, T‐bet Tg and T‐bet Tg/IFN‐γ–/– mice, and expressed the results by the relative MFI ratio. The expression levels of CD126 and CD130 were lower in T‐bet Tg and T‐bet Tg/IFN‐γ–/– mice than in WT mice (P < 0·05, P < 0·01) (Fig. 2a). In addition, we analysed the phosphorylation of STAT‐3 on IL‐6‐stimulated CD4+ T cells. FACS analyses showed suppression of STAT‐3 phosphorylation on CD4+ T cells in both T‐bet Tg mice (5.3 ± 2·0%) (P < 0·05) and T‐bet Tg/IFN‐γ–/– mice (7·4 ± 3·0%) (P < 0·05) compared with WT mice (29·6 ± 2·8%), although the total STAT‐3 in T‐bet Tg mice and T‐bet Tg/IFN‐γ–/– mice was equivalent to that in WT mice (Fig. 2b). We also examined the mRNA expression levels of molecules related to IL‐6–STAT‐3 signalling and certain transcription factors known to enhance the differentiation of Th17 cells independent of IL‐6–STAT‐3 signalling by qRT–PCR. Analysis of the expression of transcription factors in CD4+ T cells under culture conditions for Th17 differentiation confirmed tbx21 over‐expression (P < 0·01) and the repression of rorc in T‐bet Tg mice and T‐bet Tg/IFN‐γ–/– mice (P < 0·05) compared with WT mice. Over‐expression of T‐bet gene up‐regulated the expression of nfkbiz (P < 0·05) and stat1 (P < 0·05), while down‐regulating irf4 (P < 0·05) and ahr expression (P < 0·01). Moreover, the effect of T‐bet over‐expression on rorc, nfkbiz, stat1 and ahr expression was IFN‐γ‐independent (Fig. 2c). These results demonstrated two possible scenarios for inhibition of Th17 differentiation by T‐bet over‐expression: the suppression of STAT‐3 phosphorylation through inhibition of IL‐6R expression down‐regulation of RORγt and AHR.
Figure 2.
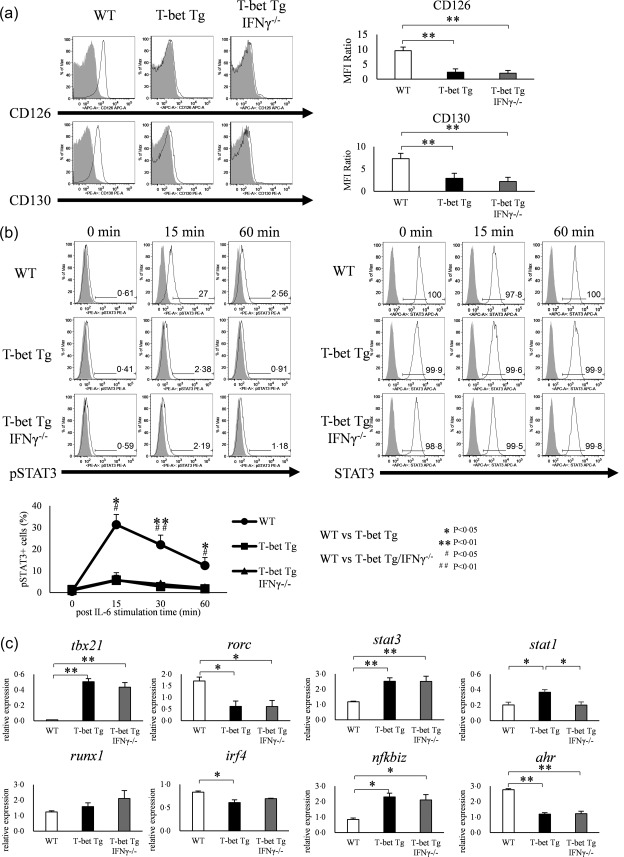
Over‐expression of T‐bet in transgenic (Tg) mice modulates the interleukin (IL)−6/signal transducer and activation of transcription‐3 (STAT‐3) pathway and regulates the expression and function of other transcription factors. (a) Expression of CD126 (IL‐6Rα) and CD130 (IL‐6Rβ) on CD4+ T cells of wild‐type (WT), T‐bet Tg and T‐bet Tg/interferon (IFN)‐γ–/– mice analysed by flow cytometry (open histogram; CD126 or CD130, shaded histogram; isotype control). Five mice per group were analysed and representative data are shown. *P < 0·05; **P < 0·01 (by Tukey's test). (b) STAT‐3 phosphorylation analyzed by flow cytometry in splenic CD4+ T cells of WT, T‐bet Tg and T‐bet Tg/IFN‐γ–/– mice stimulated with IL‐6 for 0, 15, 30 and 60 min (open histogram; phosphorylated STAT‐3 or STAT‐3, shaded histogram; isotype control or blank). Five mice per group were analysed and representative data are shown. *P < 0·05; **P < 0·01 (by Tukey's test and Kruskal–Wallis test). (c) Expression of tbx21, rorc, stat3, stat1, runx1, irf4, nfkbiz and ahr in CD4+ T cells of WT, T‐bet Tg and T‐bet Tg/IFN‐γ–/– mice analysed by quantitative reverse transcription–polymerase chain reaction (qRT–PCR). Four mice per group were analysed. Data are mean ± standard error of the mean (s.e.m.) of four mice. *P< 0·05; **P < 0·01 (by Tukey's test).
Transduction of T‐bet gene regulates Th17 differentiation
To investigate whether or not the suppression of Th17 differentiation by T‐bet was specific for T‐bet Tg mice, we performed retroviral transduction of T‐bet gene into naive CD4+ T cells isolated from WT and IFN‐γ–/– mice. FACS analysis demonstrated T‐bet over‐expressing cells infected with retrovirus expressing T‐bet (T‐bet RV), but not with empty vector (empty RV). Infection with T‐bet RV, but not empty RV, tended to be suppressed RORγt expression, even when retroviral‐transduced CD4+ T cells were cultured under conditions favouring Th17 differentiation (Fig. 3a). Furthermore, the proportion of IL‐17‐positive cells was reduced significantly among CD4+ T cells in both WT mice and IFN‐γ–/– mice transduced with T‐bet RV (1·7 ± 0·4%, 1·5 ± 0·3%) (P < 0·01) compared with empty RV (6·7 ± 0·9%, 6·5 ± 0·7%) (Fig. 3b). These FACS data were confirmed by ELISA. The level of IL‐17 level in the culture supernatant was decreased significantly in the culture supernatant of CD4+ T cells transfected with T‐bet RV (65.3 ± 25.0, 67.1 ± 18.9 pg/ml) (P < 0.05) compared with empty RV (2512 ± 274, 1436 ± 215 pg/ml) (Fig. 3c). These findings suggest that inhibition of Th17 differentiation by retroviral transduction of T‐bet gene could involve the IFN‐γ‐independent pathway.
Figure 3.
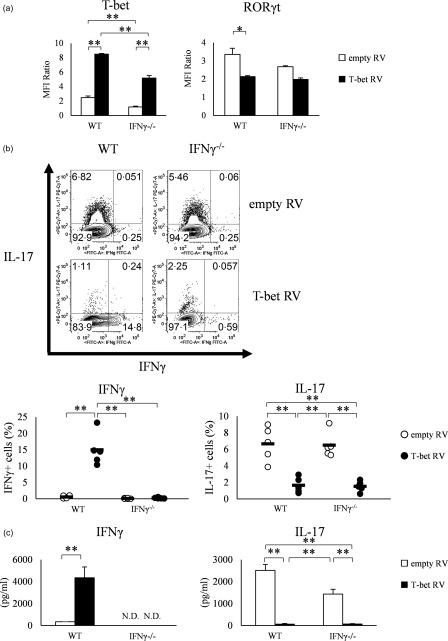
Over‐expression of T‐bet by retroviral transduction into naive CD4+ T cells of wild‐type (WT) and interferon (IFN)‐γ–/– mice suppresses retinoic acid‐related orphan receptor (RORγt) expression and interleukin (IL)−17 production. (a) T‐bet and RORγt expression in retroviral transfected splenic CD4+ T cells of WT and IFN‐γ–/– mice, analysed by flow cytometry. Data of five experiments were analysed. (b) IFN‐γ and IL‐17 production analysed by flow cytometry. Data of five experiments were analysed and representative data are shown. Numbers in plots indicate percentage of cells that secrete the indicated cytokines. (c) IFN‐γ and IL‐17 production in the culture supernatant, as determined by enzyme‐linked immunosorbent assay (ELISA). Data are mean ± standard error of the mean (s.e.m.) of four experiments; n.d. = not detected. *P < 0·05; **P < 0·01 (by Tukey's test).
Retroviral transduction of T‐bet gene had no effect on IL‐6–STAT‐3 signalling but suppresses AHR gene expression
To assess whether the IL‐6–STAT‐3 signaling was inhibited by the forced expression of T‐bet by retroviral vector, we first analysed the expression of IL‐6R, CD126 and CD130 of empty RV‐ and T‐bet RV‐transduced cells, but no difference was observed in both CD126 and CD130 between empty RV and T‐bet RV‐transduced cells (Fig. 4a). FACS analysis also showed no difference in STAT‐3 phosphorylation, irrespective of T‐bet over‐expression with retroviral vector (Fig. 4b). These results suggest that T‐bet gene transduction did not result in inhibition of IL‐6–STAT‐3 signalling.
Figure 4.
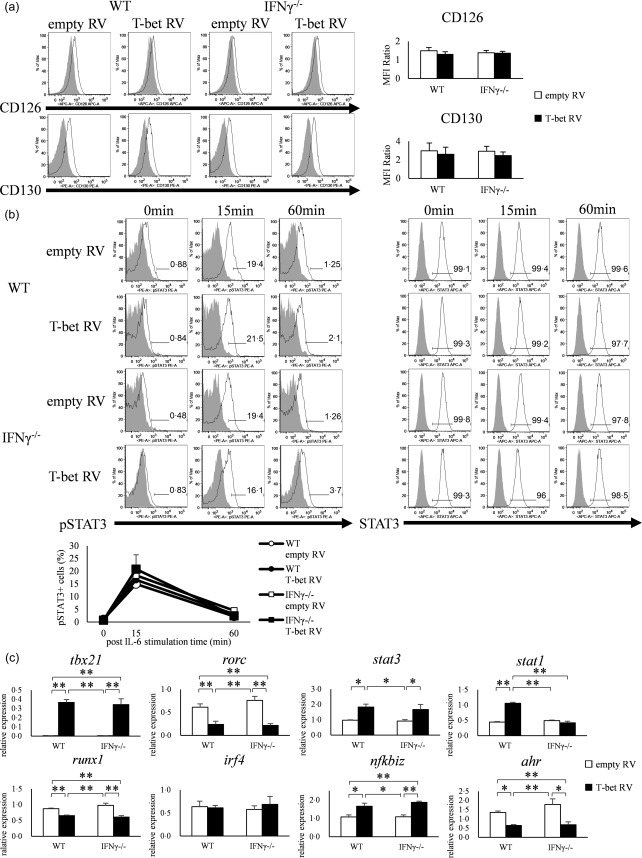
Over‐expression of T‐bet in CD4+ T cells neither suppresses the expression of interleukin (IL)−6R nor phosphorylation of signal transducer and activation of transcription‐3 (STAT‐3). (a) Expression of CD126 (IL‐6Rα) and CD130 (IL‐6Rβ) on CD4+ T cells of wild‐type (WT) and interferon (IFN)‐γ–/– mice transfected with retroviral vector (open histogram; CD126 or CD130, shaded histogram; isotype control). Data are mean ± standard error of the mean (s.e.m.) of four experiments. (b) signal transducer and activation of transcription‐3 (STAT‐3) phosphorylation in CD4+ T cells of WT and IFN‐γ–/– mice transfected with retroviral vector and stimulated with IL‐6 for 0, 15 and 60 min, as analysed by flow cytometry (open histogram; phosphorylated STAT‐3 or STAT‐3, shaded histogram; isotype control or blank). Three experiments were analysed and representative data are shown. Data are mean ± s.e.m. of three experiments. *P < 0·05; **P < 0·01 (by Tukey's test). (c) CD4+ T cells of WT and IFN‐γ–/– mice transfected with retroviral vector and analysed for the expression of tbx21, rorc, stat3, stat1, runx1, irf4, nfkbiz and ahr by quantitative reverse transcription–polymerase chain reaction (qRT–PCR). Data are mean ± s.e.m. of five experiments. *P < 0·05; **P < 0·01 (by Tukey's test).
To clarify whether transduction of T‐bet with retroviral vectors affects the expression of transcription factors, we analysed the expression of eight transcription factors (tbx21, rorc, stat3, stat1, runx1, irf4, nfkbiz and ahr) by qRT–PCR. The expression of tbx21 was increased significantly in T‐bet RV‐transfected cells of both WT and IFN‐γ–/– mice compared with empty RV (P < 0·01), while the expression of rorc was decreased significantly in T‐bet RV‐transfected cells compared with empty RV (P < 0·05). Furthermore, the expression levels of runx1 and ahr were also lower in T‐bet RV‐transduced cells compared with empty RV (P < 0·01, P < 0·05). The expression levels of nfkbiz (WT; P < 0·05, IFN‐γ–/–; P < 0·01) and stat1 were higher in T‐bet RV‐transduced cells of WT mice (P < 0·05), and the expression of stat3 was higher in both WT and IFN‐γ–/– mice (P < 0·05). T‐bet transduction had no effect on the expression of irf4 in our examination (Fig. 4c). These findings suggest that inhibition of Th17 differentiation by forced expression of T‐bet is probably related to the IFN‐γ‐independent under‐expression of RORγt, Runx‐1 and AHR.
T‐bet over‐expression suppresses AHR expression
Intracellular staining of AHR was performed to confirm the under‐expression of AHR in both T‐bet Tg mice and retroviral transduction of the T‐bet gene. FACS analysis confirmed a significant decrease in AHR expression in both T‐bet Tg and T‐bet Tg/IFN‐γ–/– mice (P < 0·05) as well as in T‐bet RV‐transduced cells of both WT and IFN‐γ–/– mice, compared with empty RV‐transduced cells (P < 0·05) (Fig. 5a,b). As FICZ, which is the ligand of AHR, can activate AHR and promote Th17 differentiation, we investigated whether the facilitation of Th17 differentiation by the addition of FICZ was cancelled in T‐bet over‐expression conditions. Although the addition of FICZ promoted the differentiation of Th17 cells in WT mice (P < 0·01), enhancement of Th17 differentiation by FICZ was cancelled in T‐bet Tg mice and T‐bet Tg/IFN‐γ–/– mice (Fig. 5c). Similarly, Th17 differentiation was facilitated by FICZ in empty RV‐transfected cells (P < 0·01), whereas no difference in Th17 differentiation was found with and without FICZ in T‐bet RV‐transduced cells (Fig. 5d). Furthermore, the stimulation of FICZ did not promote IL‐22 production in T‐bet over‐expressed conditions (P < 0·01) (Fig. 5e,f). Moreover, FICZ did not induce the expression of AHR target gene cyp1a1, which encodes a cytochrome p450 protein, under T‐bet over‐expression (P < 0·01) (Fig. 5g,h). These results support the hypothesis that T‐bet over‐expression suppressed the induction of Th17 differentiation through inhibition of AHR expression.
Figure 5.
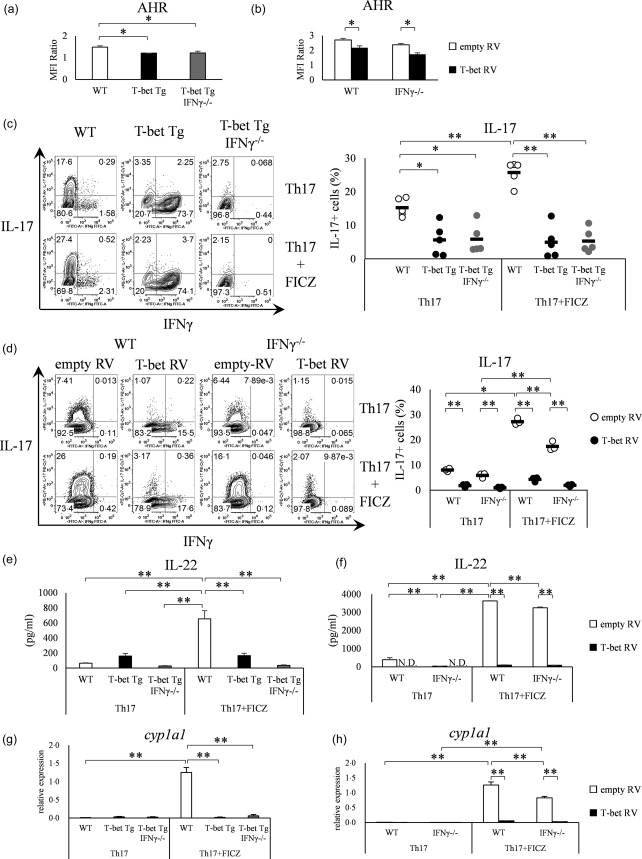
T‐bet over‐expression suppresses aryl hydrocarbon receptor (AHR) expression independent of interferon (IFN)‐γ. (a) CD4+ T cells of wild‐type (WT), T‐bet transgenic (Tg) and T‐bet Tg/IFN‐γ–/– mice analysed for the AHR expression by flow cytometry. Four mice per group were analysed. (b) CD4+ T cells of WT and IFN‐γ–/– mice transfected with retroviral vector and analysed for AHR expression by flow cytometry. Four experiments per group were analysed. (c) CD4+ T cells of WT, T‐bet Tg and T‐bet Tg/IFN‐γ–/– mice cultured with or without 6‐formylindolo (3,2‐b) carbazole (FICZ) and analysed for IFN‐γ and interleukin (IL)−17 production by flow cytometry. Five mice per group were analysed and representative data are shown. (d) CD4+ T cells of WT and IFN‐γ–/– mice transfected with retroviral vector cultured with or without FICZ, and analysed for IFN‐γ and IL‐17 production by flow cytometry. Three experiments per group were analysed and representative data are shown. (e) The production of IL‐22 analysed by enzyme‐linked immunosorbent assay (ELISA) in the experiment described in (c). (f) The production of IL‐22 analysed by ELISA in the experiment described in (d). (g) Expression levels of cyp1a1 analysed by quantitative reverse transcription–polymerase chain reaction (qRT–PCR) in the experiment described in (c). (h) Expression level of cyp1a1 analysed by qRT–PCR in the experiment described in (d). *P < 0·05; **P < 0·01 (by Tukey's test).
AHR expression under T‐bet over‐expression in CIA model
We have reported previously that the over‐expression of T‐bet suppressed CIA through the inhibition of antigen‐specific response of Th17 cells 22; however, the involvement of AHR in this model was unknown. To evaluate whether our observations in vitro hold true in in‐vivo inflammatory conditions, we examined the AHR expression of T‐bet Tg mice in the CIA model. mRNA expression of rorc and ahr of the CD4+ T cells of inguinal lymph nodes from T‐bet Tg mice after CII immunization was depressed compared with WT mice (P < 0·01) (Fig. 6).
Figure 6.
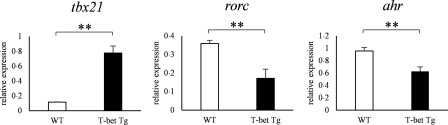
The mRNA expression of ahr was suppressed in collagen‐induced arthritis (model) of T‐bet transgenic (Tg) mice. Ten days after the collagen type II (CII) immunization, lymph node cells from wild‐type (WT) and T‐bet Tg mice were cultured for 72 h in the presence of 100 μg/ml of denatured CII. After culture of lymph node cells with CII, CD4+ cells were purified by magnetic cell isolation and cell separation (MACS), and cDNA was obtained. The levels of tbx21, rorc and ahr expression were analysed by reverse transcription–polymerase chain reaction (RT–PCR). Data are mean ± standard error of the mean (s.e.m.) of three mice. **P < 0·01 (by Student's t‐test).
Figure 7 summarizes schematically the IFN‐γ‐dependent and independent pathways involved in the suppression of IL‐17 production by T‐bet over‐expression.
Figure 7.
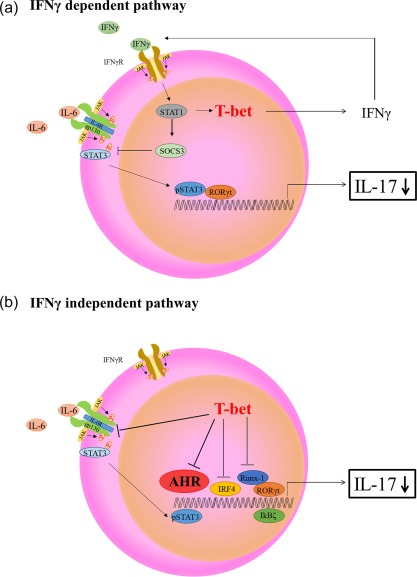
Schematic diagram of potential mechanisms involved in T helper type 17 (Th17) differentiation under T‐bet over‐expression conditions. (a) Interferon (IFN)‐γ‐dependent T‐bet suppression of Th17 differentiation. IFN‐γ signal transducer and activation of transcription‐1 (STAT‐1) signalling induces suppressor of cytokine signaling 3 (SOCS3) expression, inhibition of IL‐6/STAT‐3 and retinoic acid‐related orphan receptor (RORγt) expression, resulting in suppression of interleukin (IL)−17 production. (b) IFN‐γ‐independent T‐bet suppression of Th‐17 differentiation. T‐bet over‐expression regulates various transcription factors [RORγt, runt‐related transcription factor 1 (Runx‐1), I kappa B zeta (IκBζ), IFN regulatory factor 4 (IRF4) and aryl hydrocarbon receptor (AHR)], resulting in suppression of IL‐17 production. Our findings particularly support the possibility that the regulatory mechanism of Th17 differentiation might be due to the suppression of AHR.
Discussion
We reported previously that over‐expression of T‐bet in T cells suppressed CIA through inhibition of Th17 differentiation 21. The present study was designed to clarify the molecular mechanism of suppression of Th17 differentiation by T‐bet over‐expression. For this purpose, we analysed the expression of several genes in WT, T‐bet Tg and T‐bet Tg/IFN‐γ–/– mice. Our results showed that T‐bet inhibited the differentiation of Th17 independently of IFN‐γ, and that the suppression of IL‐6R could result in the inhibition of STAT‐3 phosphorylation. Moreover, the results also showed up‐regulation of nfkbiz, which encodes I kappa B zeta (IκBζ), and down‐regulation of rorc, irf4 and ahr under T‐bet over‐expression. The reduced expression of rorc and irf4 was in agreement with the results of Lazarevic et al. 24 and Gökmen et al. 25, who showed inhibition of RORγt transcription by T‐bet through direct interaction with Runx‐1 and IRF4. Whereas IκBζ can induce Th17 differentiation through co‐operation with RORγt 9, the high expression of nfkbiz might be not sufficient to enhance IL‐17 in T‐bet Tg and T‐bet Tg/IFN‐γ–/– mice. The levels of stat1 were up‐regulated IFN‐γ dependently, and the level of stat3 was also up‐regulated IFN‐γ independently. The increase of stat1 expression was accountable by the IFN‐γ–STAT‐1 pathway 26. As STAT‐3 was regulated negatively by suppressor of cytokine signalling (SOCS3) 27, and SOCS3 expression was depressed by T‐bet over‐expression 28, we speculated that T‐bet over‐expression increased STAT‐3 expression by SOCS3 depression. The relation of T‐bet and Iκbζ has not been reported. The mRNA expression of irf4 was not suppressed by T‐bet over‐expression, unlike in the Gökmen et al. study 25. The reason for this disparity might be related to the different mice backgrounds, BALB/c or C57BL/6, and discordance in the Th17 induction between the study by Gökmen et al. and our assessment, in which IL‐1β was not used in the induction of Th17 cells in our study 29, 30.
To determine the exact mechanism involved in T‐bet over‐expression‐induced inhibition of Th17 differentiation, we performed retroviral transduction of T‐bet gene into naive CD4+ T cells isolated from WT and IFN‐γ–/– mice, and examined the effect of T‐bet over‐expression. Retroviral induction of T‐bet suppressed Th17 differentiation independently of IFN‐γ, similar to findings of the experiment of T‐bet Tg and T‐bet Tg/IFN‐γ–/– mice. However, the forced expression of T‐bet with retroviral vector did not inhibit the suppression of IL‐6R expression and STAT‐3 phosphorylation, suggesting that the suppression of Th17 differentiation by retroviral induction of the T‐bet gene did not involve the IL‐6–STAT‐3 pathway. These findings were different to those involving T‐bet Tg and T‐bet Tg/IFN‐γ–/– mice. Although the reason for discrepancy in IL‐6–STAT‐3 signalling between T‐bet Tg mice and retroviral transduction of the T‐bet gene was not elucidated, we speculate that the effect of the T‐bet gene on Th17 cell differentiation is different between short‐term expression induced by retroviral vector and long‐term expression induced by genetically enforced expression in T‐bet Tg mice. Because steady expression of T‐bet in Th‐1 cells is established in the late stage of differentiation 31, the CD4+ T cells from T‐bet Tg mice might not be affecting IL‐6–STAT‐3 signalling. T‐bet transduction in the early stage of differentiation might respond to IL‐6 stimulation. In addition, it is known that repetitive polarizing stimulations and sustained expression of lineage‐specific transcription factors stabilize the lineage commitment of T helper cell subsets with the epigenetic modification of the genes related to the lineage commitment 32. Brown et al. showed that epigenetic repression of IL‐6Rα gene expression is related to the stability of Th‐1 cell lineage commitment 33. We speculated that the epigenetic remodelling of some factors associated with IL‐6R might be induced by sustained expression of T‐bet with genetic over‐expression (T‐bet Tg mice), but not by temporary expression of T‐bet with retroviral transduction.
It is noteworthy that rorc and ahr mRNA expressions were suppressed by T‐bet gene transduction as well as in T‐bet Tg and T‐bet Tg/IFN‐γ–/– mice. Previous studies reported that these transcription factors regulate the differentiation of other T cell subsets negatively by direct co‐interaction and/or indirect effect of cytokines produced by each T cell subset 16, 17. In an experimental model of autoimmune arthritis induced by Th17 cell response, T‐bet and IFN‐γ act as immunoregulatory factors 18, 19, 20. AHR plays a key role for the differentiation and activation of Th17 cells and production of IL‐17 and IL‐22 15. As the IL‐17, IL‐22 and AHR‐deficient mice had reduced disease severity in the CIA model 34, 35, 36, 37, we formed a hypothesis that CIA was suppressed in T‐bet Tg mice not only by RORγt suppression but also by AHR suppression. Although AHR suppression reduced endogenous transcription of IL‐6 38, the reduction had very little to do with the regulation of Th17 differentiation in this study.
Our findings indicate that T‐bet over‐expression could suppress AHR expression independently of IFN‐γ. Moreover, over‐expression of T‐bet cancelled FICZ‐induced facilitation of Th17 differentiation, suggesting the involvement of AHR down‐regulation in T‐bet over‐expression‐induced inhibition of Th17 differentiation. To investigate the function of AHR, the cytokine associated with AHR, IL‐22, was examined. Although the addition of FICZ induces the significant augmentation of IL‐22 production in both WT CD4+ T cells and empty RV‐transfected CD4+ T cells in Th17, FICZ‐inducing IL‐22 production was cancelled by T‐bet over‐expression, indicating that the over‐expression of T‐bet inhibits IL‐22 production induced by AHR. While AHR was the master transcription factor of IL‐22, some reports showed that IL‐22 is produced by IFN‐γ‐secreting cells 39, 40. In the results of T‐bet transduction, IL‐22 was higher in WT than IFN‐γ−/− mice, indicating the possibility that IL‐22 production was dependent partially upon IFN‐γ. Although the relationship between T‐bet and AHR has not been reported previously, AHR could be a key molecule in the regulation of Th17 differentiation by T‐bet.
To analyse further the regulation of AHR gene expression by T‐bet over‐expression and its role in Th17 cell differentiation, we focused upon the potential of direct or indirect interaction of T‐bet with the AHR expression. First, we considered two possibilities of direct suppression of AHR gene expression. One was that the T‐bet gene binds to the promoter region of the AHR gene, similar to the previously described interaction between T‐bet and IRF4 11. The other was T‐bet binding to another transcription factor that could inhibit the transcriptional activity against AHR gene similar to the T‐bet and RORγt relation on Runx‐1 12. Concerning the indirect mechanism of AHR suppression, it is possible that T‐bet suppressed AHR gene expression by inhibiting signal transduction inducing AHR. It is known that AHR is induced by both IL‐6 and TGF‐β, which subsequently induces Th17 cell differentiation 14, 41. As retroviral transduction of the T‐bet gene neither affected IL‐6R expression nor STAT‐3 phosphorylation through IL‐6 stimulation, we speculate that T‐bet could block the TGF‐β signalling pathway, resulting in AHR suppression. However, we determined the mechanism of AHR depression by T‐bet over‐expression incompletely. A limitation of this study was that, regrettably, we did not perform the Chip sequence and promotor assay. Further experiments are needed to determine the mechanisms involved in the regulation of Th17 differentiation.
To clarify whether AHR was suppressed in the CIA model of T‐bet Tg mice, we examined the mRNA expression of ahr in CD4+ T cells isolated from draining lymph node cells after the stimulation of CII in vitro in CII immunized mice. According to the results in vitro, ahr expression was suppressed as well as rorc in T‐bet Tg mice. AHR‐deficient mice were resistant to CIA through the suppression of Th17 differentiation 37, similar to T‐bet Tg mice 22. Therefore, these results showed that the mechanism of the attenuation of CIA in T‐bet Tg mice involved AHR suppression. As IL‐22 was not detected as a CII‐reactive cytokine, the relation of IL‐22 to CIA in T‐bet Tg mice was unclear.
Figure 7 shows schematically two T‐bet‐based pathways involved in Th17 regulation. The first is the IFN‐γ‐dependent pathway (Fig. 7a), while the other is the IFN‐γ‐independent pathway (Fig. 7b) 42, 43. In the IFN‐γ‐dependent pathway, IFN‐γ–STAT‐1 signalling up‐regulates SOCS3 and inhibits IL‐6–STAT‐3 and RORγt, followed by suppression of IL‐17 production 42. In the IFN‐γ‐independent pathway, T‐bet suppresses several molecules such as IRF4, Runx‐1, IL‐6R and pSTAT‐3. In the present study, T‐bet‐induced under‐expression of AHR regulated IL‐17 production.
In conclusion, our findings suggest that T‐bet over‐expression in T cells regulates Th17 differentiation and that this action is mediated by AHR in addition to the RORγt pathway.
Disclosure
None.
Acknowledgements
We thank Dr Nisikii for the providing of retroviral vector. We also thank Dr F.G. Issa for the critical reading of the manuscript. This study was supported in part by the Japanese Ministry of Science and Culture, and the Japanese Ministry of Health, Labor and Welfare.
References
- 1. Jetten AM. Retinoid‐related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal 2009; 7:e003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev 2008; 223:87–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T‐bet, directs Th1 lineage commitment. Cell 2000; 100:655–69. [DOI] [PubMed] [Google Scholar]
- 4. Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T‐bet in TH1 lineage commitment and IFN‐γ production in CD4 and CD8 T cells. Science 2002; 295:338–42. [DOI] [PubMed] [Google Scholar]
- 5. Afkarian M, Sedy JR, Yang J et al T‐bet is a STAT1‐induced regulator of IL‐12R expression in naïve CD4+ T cells. Nat Immunol 2002; 3:549–57. [DOI] [PubMed] [Google Scholar]
- 6. Ivanov II, McKenzie BS, Zhou L et al The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL‐17+ T helper cells. Cell 2006; 126:1121–33. [DOI] [PubMed] [Google Scholar]
- 7. Zhou L. AHR function in lymphocytes: emerging concepts. Trends Immunol 2016; 37:17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Manel N, Unutmaz D, Littman DR. The differentiation of human TH−17 cells requires transforming growth factor‐β and induction of the nuclear receptor RORγt. Nat Immunol 2008; 9:641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Okamoto K, Iwai Y, Oh‐Hora M et al IκBζ regulates TH17 development by cooperating with ROR nuclear receptors. Nature 2010; 464:1381–5. [DOI] [PubMed] [Google Scholar]
- 10. Ciofani M, Madar A, Galan C et al A validated regulatory network for Th17 cell specification. Cell 2012; 151:289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brüstle A, Heink S, Huber M et al The development of inflammatory TH−17 cells requires interferon‐regulatory factor 4. Nat Immunol 2007; 8:958–66. [DOI] [PubMed] [Google Scholar]
- 12. Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORγt and Foxp3 regulate the differentiation of interleukin 17‐producing T cells. Nat Immunol 2008; 9:1297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Veldhoen M, Hirota K, Westendorf AM et al The aryl hydrocarbon receptor links TH17‐cell‐mediated autoimmunity to environmental toxins. Nature 2008; 453:106–9. [DOI] [PubMed] [Google Scholar]
- 14. Quintana FJ, Basso AS, Iglesias AH et al Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008; 453:65–71. [DOI] [PubMed] [Google Scholar]
- 15. Monteleone I, Rizzo A, Sarra M et al Aryl hydrocarbon receptor‐induced signals up‐regulate IL‐22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 2011; 141:237–48. [DOI] [PubMed] [Google Scholar]
- 16. Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. T helper cell fate specified by kinase‐mediated interaction of T‐bet with GATA‐3. Science 2005; 307:430–3. [DOI] [PubMed] [Google Scholar]
- 17. Zhou L, Lopes JE, Chong MM et al TGF‐β‐induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature 2008; 453:236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hultgren OH, Verdrengh M, Tarkowski A. T‐box transcription‐factor‐deficient mice display increased joint pathology and failure of infection control during staphylococcal arthritis. Microbes Infect 2004; 6:529–35. [DOI] [PubMed] [Google Scholar]
- 19. Doodes PD, Cao Y, Hamel KM et al IFNγ regulates the requirement for IL‐17 in proteoglycan‐induced arthritis. J Immunol 2010; 184:1552–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manoury‐Schwartz B, Chiocchia G, Bessis N et al High susceptibility to collagen‐induced arthritis in mice lacking IFNγ receptors. J Immunol 1997; 158:5501–6. [PubMed] [Google Scholar]
- 21. Ishizaki K, Yamada A, Yoh K et al Th1 and type 1 cytotoxic T cells dominate responses in T‐bet overexpression transgenic mice that develop contact dermatitis. J Immunol 2007; 178:605–12. [DOI] [PubMed] [Google Scholar]
- 22. Kondo Y, Iizuka M, Wakamatsu E et al Overexpression of T‐bet gene regulates murine autoimmune arthritis. Arthritis Rheum 2012; 64:162–72. [DOI] [PubMed] [Google Scholar]
- 23. Suto A, Kashiwakuma D, Kagami S et al Development and characterization of IL‐21‐producing CD4+ T cells. J Exp Med 2008; 205:1369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lazarevic V, Chen X, Shim JH et al T‐bet represses TH17 differentiation by preventing Runx1‐mediated activation of the gene encoding RORγt. Nat Immunol 2011; 12:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gökmen MR, Dong R, Kanhere A et al Genome‐wide regulatory analysis reveals that T‐bet controls Th17 lineage differentiation through direct suppression of IRF4. J Immunol 2013; 191:5925–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1‐dependent and –independent pathways in IFN‐gamma‐dependent signaling. Trends Immunol 2002; 23:96–101. [DOI] [PubMed] [Google Scholar]
- 27. Zhu BM, Ishida Y, Robinson GW et al SOCS3 negatively regulates the gp130‐STAT3 pathway in mouse skin wound healing. J Invest Dermatol 2008; 128:1821–9. [DOI] [PubMed] [Google Scholar]
- 28. Oestreich KJ, Huang AC, Weinmann AS. The lineage‐defining factors T‐bet and Bcl‐6 collaborate to regulate Th1 gene expression patterns. J Exp Med 2011; 208:1001–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chung Y, Chang SH, Martinez GJ et al Critical regulation of early Th17 cell differentiation by interleukin‐1 signaling. Immunity 2009; 30:576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Végran F, Berger H, Boidot R et al The transcription factor IRF1 dictates the IL‐21‐dependent anticancer functions of Th9 cells. Nat Immunol 2014; 15:758–66. [DOI] [PubMed] [Google Scholar]
- 31. Nakayamada S, Kanno Y, Takahashi H et al Early Th1 cell differentiation is marked by a Tfh cell‐like transition. Immunity 2011; 35:919–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wilson CB, Rowell E, Sekimata M. Epigenetic control of T‐helper‐cell differentiation. Nat Rev Immunol 2009; 9:91–105. [DOI] [PubMed] [Google Scholar]
- 33. Brown CC, Esterhazy D, Sarde A et al Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 2015; 42:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen‐induced arthritis in IL‐17 deficient mice. J Immunol 2003; 171:6173–7. [DOI] [PubMed] [Google Scholar]
- 35. Geboes L, Dumoutier L, Kelchtermans H et al Proinflammatory role of the Th17 cytokine interleukin‐22 in collagen‐induced arthritis in C57BL/6 mice. Arthritis Rheum 2009; 60:390–5. [DOI] [PubMed] [Google Scholar]
- 36. Corneth OB, Reijmers RM, Mus AM et al Loss of IL‐22 inhibits autoantibody formation in collagen‐induced arthritis in mice. Eur J Immunol 2016; 46:1404–14. [DOI] [PubMed] [Google Scholar]
- 37. Nakahama T, Kimura A, Nguyen NT et al Aryl hydrocarbon receptor deficiency in T cells suppresses the development of collagen‐induced arthritis. Proc Natl Acad Sci USA 2011; 108:14222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hollingshead BD, Beischlag TV, Dinatale BC, Ramadoss P, Perdew GH. Inflammatory signaling and aryl hydrocarbon receptor mediate synergistic induction of interleukin 6 in MCF‐7 cells. Cancer Res 2008; 68:3609–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gurney AL. IL‐22, a Th1 cytokine that targets the pancreas and select other peripheral tissues. Immunopharmacology 2004; 4:669–77. [DOI] [PubMed] [Google Scholar]
- 40. Behrends J, Renauld JC, Ehlers S, Hölscher C. IL‐22 is mainly produced by IFNγ‐secreting cells but is dispensable for host protection against Mycobacterium tuberculosis infection. PLOS ONE 2013; 8:e57379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kimura A, Naka T, Nohara K, Fujii‐Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci USA 2008; 105:9721–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tanaka K, Ichiyama K, Hashimoto M et al Loss of suppressor of cytokine signaling 1 in helper T cells leads to defective Th17 differentiation by enhancing antagonistic effects of IFN‐γ on STAT3 and Smads. J Immunol 2008; 180:3746–56. [DOI] [PubMed] [Google Scholar]
- 43. Mori T, Miyamoto T, Yoshida H et al IL‐1β and TNFα‐initiated IL‐6–STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int Immunol 2011; 23:701–12. [DOI] [PubMed] [Google Scholar]