

Summary
Significant barriers to transplantation exist for individuals who are pre‐sensitized to donor antigen and have high titres of donor‐reactive antibody. We report the effect of autologous bone marrow transplantation (BMTx) after myeloablation in pre‐sensitized mice along with the use of monoclonal antibodies (mAbs) to tumour necrosis factor‐receptor super family 25 (TNFRSF25), expressed on regulatory T (Treg) cells. C57BL/6 mice, which had been sensitized earlier with BALB/c skin allografts, received secondary BALB/c grafts after the primary grafts had been rejected. Subsequently, recipient mice underwent myeloablation with cyclophosphamide and busulphan and were injected with T‐cell‐depleted bone marrow from CD45.1 congenic donors (BMTx). Recipient mice underwent immunosuppressive treatment with rapamycin. A subgroup of mice was also treated with mAbs to TNFRSF25. Control mice were pre‐sensitized mice that received cyclophosphamide and busulphan followed by rapamycin. BMTx‐treated mice had significantly prolonged skin graft survival versus control mice. These mice also showed attenuated donor‐specific mixed lymphocyte co‐culture responses relative to controls, increased splenic Treg cells and markedly diminished serum anti‐donor IgG. Infusion of anti‐TNFRSF25 mAbs further augmented graft survival and increased graft‐infiltrating Treg cells. These mAbs also expanded murine and human Treg cells in vitro with the capacity to attenuate mixed lymphocyte co‐cultures using fresh peripheral blood mononuclear cells. Overall, this study delineates the roles of autologous BMTx and anti‐TNFRSF25 mAbs in expanding Treg cells and attenuating alloimmune responses in pre‐sensitized mice.
Keywords: bone marrow transplantation, donor‐reactive antibodies, myeloablation, regulatory T cells, tumour necrosis factor‐receptor super family 25
Abbreviations
- Breg
regulatory B
- CTL
cytotoxic T lymphocyte
- IL‐2
interleukin‐2
- mAbs
monoclonal antibodies
- MLC
mixed lymphocyte co‐culture
- MLR
mixed lymphocyte reaction
- Treg
regulatory T
- TNFRSF25
tumour necrosis factor‐receptor super family 25
- Treg
regulatory T
Introduction
Transplantation is the preferred treatment for patients with end‐stage organ failure, although rejection, both acute and chronic, remains a significant pitfall to this process, along with immunosuppressive drug‐related toxicity, opportunistic infections and lymphoproliferative disorders.1 Using a skin graft model in naive mice we previously reported that by incorporating a chemotherapeutic procedure involving myelosuppression, followed by autologous bone marrow stem cell rescue, we could radically improve skin allograft survival (and indeed cardiac graft survival), with some 35% of recipients retaining their grafts, without immunosuppressive drugs, long‐term.2 Long‐term graft survival was associated with increased numbers of Foxp3+ regulatory T (Treg) cells in recipients, which could actively suppress anti‐graft reactivity, and expansion of Treg functional activity in vivo was reported in subgroups of mice receiving antibodies to the molecule tumour necrosis factor‐receptor super family 25 (TNFRSF25).2
TNFRSF25 (also known as DR3) is expressed primarily by CD4+ and CD8+ T and natural killer T cells.3, 4, 5, 6 The ligand for TNFRSF25, TL1A, is expressed by endothelial cell subsets and is induced on dendritic cells and macrophage/monocytes by triggering Toll‐like receptor 4 or FcγR.7, 8 In vitro TNFRSF25 signalling on CD4+, CD8+ or natural killer T cells has been reported to augment interleukin‐2 (IL‐2), IL‐4 and interferon‐γ production following T‐cell receptor activation.9 Despite these data, and reports that activation of TNFRSF25 by TL1A can exacerbate experimental asthma, inflammatory bowel disease, rheumatoid arthritis and experimental autoimmune encephalomyelitis,3, 7, 10, 11, 12 there is other evidence that the molecule is also expressed on Treg cells.10 As noted above, we ourselves reported that a heteroantibody to TNFRSF25 could expand Treg cells in vivo in mice receiving allogeneic skin transplants followed by autologous bone marrow transplantation in a tolerance‐inducing protocol,2 and Schreiber et al.13 reported on an anti‐TNFRSF25 antibody‐mediated expansion of Treg cells in an allergic lung inflammation model.
An additional problem in clinical transplantation is associated with the dilemma of understanding how best to treat patients for whom a first graft has been irrevocably rejected, and/or for whom pre‐transplant analysis reveals significant titres of donor‐specific antibody and immune reactivity.14 In the renal transplant field it has been estimated that up to 30% of individuals on the transplant waiting list express significant titres of such antibodies (through previous transfusion, failed grafts or pregnancy), and additional data indicate that such titres are generally associated with an increased risk of antibody‐mediated rejection and poor graft survival.15 It has long been recognized that it is important to develop techniques that can obviate the clear disadvantages to graft survival associated with such pre‐sensitization.16 Both alloantibodies per se, and anti‐donor reactive B cells, capable of acting as antigen‐presenting cells17 or producing pathological cytokines, are thought to be causally implicated in enhanced rejection in sensitized individuals, though it should be noted that there is also some thought that regulatory B (Breg) cells may in turn counter the adverse effect of sensitized B cells.18
Current protocols have investigated mechanisms for desensitization of immune hosts, with the aim of facilitating engraftment to such recipients, using plasmapheresis, B‐cell‐depleting (anti‐CD20) antibodies and intravenous immunoglobulin.19 More recently, additional agents have come under investigation, including proteasome inhibitors (Bortezomib),20 anti‐cytokine agents (Belimumab : anti‐BAFF)21 and reagents interfering with IL‐6 : IL‐6 receptor interactions.22 However, none of these approaches has shown a systematic and reproducible efficacy in the transplant scenario with pre‐sensitized recipients. Given the success achieved in naive allograft recipients in terms of prolongation of graft acceptance using autologous bone marrow infusion, we have asked whether a similar approach can be used to promote graft survival in pre‐sensitized recipients. The data shown below, using a mouse skin allograft model, argue convincingly that autologous marrow transplants can indeed improve graft survival in such recipients, with augmented numbers/function of Treg cells and attenuation of donor‐specific antibody levels. We show that monoclonal antibodies (mAbs) to TNFRSF25 further improved graft survival in pre‐sensitized mice and when used in culture these mAbs expanded populations of both murine and human Treg cells capable of attenuating mixed lymphocyte co‐culture (MLC) reactivity and development of cytotoxic T lymphocytes (CTL) in vitro.2, 13
Materials and methods
Mice
Stock wild‐type male C3H/HeJ, BALB/c, C57BL/6.CD45.2 (BL/6) and BL/6.CD45.1 congenic mice were purchased from the Jackson Laboratories (Bar Harbour, ME) and housed as described elsewhere.2 All animals were handled according to the recommendations of the Canadian Council for Animal Care and all animal protocols were approved by the Animal Resource Center, University Health Network.
Media and cell lines
All in vitro assays were performed using complete α‐modified Eagle's medium (αF10) supplemented with 10% fetal calf serum and 2‐mercaptoethanol. Media were supplied by the Tissue Culture Media Facility at the Princess Margaret Hospital, Toronto, ON. Cells were counted using a haemocytometer and viability was assessed using trypan blue. For MLC and FACS assays, single‐cell spleen suspensions were prepared by pressing spleens through a 40‐μm filter and performing red blood cell lysis using ACK lysis buffer. Tumour cell lines (EL4 and P815) used for CTL targets, were cultured throughout in αF10.
Monoclonal antibodies
The following mAbs were used: from BD Pharmingen (San Diego, CA): FITC anti‐mouse CD4 (clone‐RM4‐4); from BioLegend (San Diego, CA): FITC anti‐human CD4 (OKT4), phycoerythrin (PE)/Cy7 anti‐human CD25 (BC96), PE anti‐human FOXP3 (206D), allophycocyanin (APC) anti‐mouse CD25 (3C7), APC anti‐mouse CD3ε (145‐2C11), PE anti‐mouse FOXP3 (150D), CD45.1 (A20), CD45.2 (104); from Cedarlane Laboratories, (Hornby, ON, Canada), anti‐Thy 1.2 (5a‐8); and from Bio‐rad (Hercules, CA), FITC‐anti‐mouse CD3 (MCA500F). FITC anti‐rat IgM (MRM‐47) was used for secondary staining of anti‐DR3 mAbs.
Anti‐Thy‐1.2 and anti‐CD45.1 antibody treatment
Bone marrow was flushed from femurs and red blood cell lysis was performed using ACK lysis buffer. Cells used to reconstitute BL/6 mice were treated at a concentration of 5 × 106 cells/ml with anti‐Thy‐1.2 antibody (Cedarlane Laboratories) and rabbit complement for 60 min at 37°. T‐cell depletion (≥ 99%) was confirmed by FACS staining with commercial FITC rat anti‐mouse CD3 mAb (Serotec). In some experiments, cells harvested from mice were treated with anti‐CD45.1 antibody (BioLegend) and rabbit complement before use in assays, as described in the text. Both anti‐CD45.1 and anti‐CD45.2 antibodies (BioLegend) were also used in FACS analysis with cells from mice following bone marrow transplantation (see below).
Skin grafts
Skin grafts were performed as described in a previous manuscript2. To produce pre‐sensitized recipients naive BL/6 or C3H mice received C3H or BL/6 skin grafts, respectively, with no additional treatment thereafter.23 Grafts were inspected visually from day 7 post transplant. All grafts were rejected by 16 days post transplantation (median survival across all recipients 13·5 ± 2·4 days). Mice were rested for 14 days following rejection in all animals, and entered into studies as ‘pre‐sensitized’, along with age‐matched naive mice as controls. All experimental naive/pre‐sensitized transplanted mice subsequently received rapamycin (Wyeth 1 mg/kg at 36‐hr intervals) post transplantation. Skin grafts were transplanted to the flank of anaesthetized recipients, with grafts examined at daily intervals by an investigator blinded to the groups.
Where grafted mice received myeloablation before receiving autologous bone marrow transplants, rapamycin was stopped and individual mice received busulphan (20 mg/kg/day × 4 days) intraperitoneally in 100 μl PBS followed by cyclophosphamide (100 mg/kg/day × 2 days) intraperitoneally in 100 μl PBS, before resting for 2 days.2 Animals then received 5 × 106 T‐cell‐depleted autologous marrow intravenously in 0·5 ml PBS. Five days after bone marrow infusion, subgroups of mice were restarted on rapamycin for a further 21 days until day 35 post transplant.
Chimerism in BL/6 recipients (with BALB/c or C3H grafts) was assessed by infusion of CD45.1 or CD45.2 bone marrow, and FACS staining with anti‐CD45.1 or anti‐CD45.2 antibodies of spleen and lymph nodes of mice at > 40 days post bone marrow transplantation. For FACS analysis, CD45.1‐positive versus CD45.2‐positive events were gated on singlet cells. The degree of chimerism in individual mice, both naive and pre‐sensitized, ranged from 25 to 65% of CD45.1+ cells present, with no significant difference in levels of chimerism between naive or immune recipients.
Quantification of donor‐specific IgG and MLC cytotoxicity assays
Donor‐specific antibody levels were determined by a flow cross‐match assay as described previously.24 Blood was obtained from control (naive) mice and graft recipients by cardiac puncture under anaesthesia. Serum was obtained by centrifugation (10,621 × 20 min at 4°) and stored at −20° until use. Splenocytes (0·5 × 106) from donor BL/6 were incubated with 2 μl of serum for 30 min. Cells were washed and subsequently stained with secondary FITC‐conjugated goat anti‐mouse IgG polyclonal antibody (Immunology Consultants Laboratory, Portland, OR) and finally with APC‐conjugated anti‐mouse CD3ε (BioLegend). Donor‐specific antibody levels were assessed with flow cytometric analysis using a BD LSRII (BD Pharmingen). Data analysis was performed using flowjo VX (Treestar, Ashland, OR) and titres were reported as median fluorescence intensity. FACS analysis was performed by gating for CD3+ events on singlet cells and assessing the median fluorescence intensity values for FITC in the CD3+ fraction.
In MLC, 1·0 × 106 responder cells from the spleens of graft recipients were stimulated in triplicate in 250 μl αF10 in V‐bottomed tissue‐culture plates with equal numbers of irradiated (20 Gray) donor or third‐party stimulator splenocytes with 50 ng/ml recombinant human IL‐2 (BioLegend). Cells from replicate wells were pooled at 5 days and titrated at different effector : target ratios for killing (4 hr at 37°) of 1 × 103 51Cr‐labelled EL4 or P815 tumour target cells, or 72 hr Concanavalin‐A‐activated splenocyte blasts (for C3H targets). Data in all figures show % lysis for 20 : 1 effector : target ratios at 4 hr. In separate assays [3H]TdR incorporation into similar cultures was assayed 72 hr after initiation, by pulsing for 14 hr with 1 μCi/well of [3H]TdR followed by harvesting and quantification in a β‐cell counter.
Where splenocytes were assessed for their ability to attenuate responses in MLC from a pool of fresh naive responder cells, 1 × 106 splenocytes (or CD4+‐enriched cells from MACS columns) were added to 3 × 106 naive splenocytes stimulated with 2 × 106 irradiated target spleen cells for 5 days, with CTL enumerated again in 51Cr‐release assays.
Preparation of rat mAbs to mouse TNFRSF25
Rats were immunized against mouse TNFRSF25 as previously described.2, 13 A C‐terminal region peptide of murine TNFRSF25 was synthesized and four Lewis rats were immunized three times at 21‐day intervals with 500 μg emulsified incomplete Freund's adjuvant (Cedarlane Laboratories). Pre‐immunization bleeding (yield ~2 ml) served as normal rat serum control. Fourteen days after the third immunization, splenocytes were harvested from the rat with the highest titre (ELISA with plates coated with peptide) and used for fusion to produce mAbs using a rat parent myeloma cell line. Culture supernatants were initially screened in ELISAs with peptide coupled to plates. Following selection of positive clones, supernatant samples were further tested in FACS (1 : 5) for staining of Treg cells harvested from 5‐day cultures of CD4+ splenocytes (MACS column purification: Miltenyi Biotec, Bergisch Gladbach, Germany) stimulated on anti‐CD3‐coated dishes with 2 μg/ml anti‐CD28 and 10 ng/ml transforming growth factor‐β (TGF‐β).13 Τreg cells cultured in vitro were stained using FITC anti‐CD4, APC anti‐CD25 and PE anti‐FOXP3 antibodies (BioLegend). For determination of in vivo Treg cell frequencies, data were analysed by gating for CD4 and Foxp3 double‐positive cells to delineate Treg cells. The mAbs were also assayed in ELISA (again 1 : 5 final concentration) with the same Treg cells adherent to poly‐l‐lysine (100 ng/well) coated wells and fixed with glutaraldehyde. Immunoglobulin samples purified from four selected hybridoma supernatants were used to expand Treg cells in vitro (of human or mouse origin) by addition (2 μg/ml final concentration) at 72 hr to similar cultures (on anti‐CD3‐coated plates with anti‐CD28 + TGF‐β) before harvest of cells (5 days) and addition in vitro (7·5 × 105 Treg cells) to fresh MLC (2 × 106 responder mononuclear cells with 1 × 106 irradiated simulator cells). CTL were assayed in the latter at 5·5 days. Control MLC contained no Treg cells, or Treg cells from cultures without the addition of mAbs at 72 hr. The two most active mAbs were used in vivo as described.
Statistics
Most studies compared groups using analysis of variance followed by paired t‐tests. For skin graft survival, groups were compared using non‐parametric tests (Mann–Whitney U‐test). All data are expressed as mean ± SD unless stated otherwise.
Results
Increased graft survival in pre‐sensitized mice after autologous bone marrow transplantation
Previous studies showed that skin allograft survival was markedly improved in naive mice after myeloablation using a short course of busulphan/cyclophosphamide, followed by autologous marrow transplantation and ongoing rapamycin treatment.2 Rapamycin with busulphan/cyclophosphamide, in the absence of marrow transplantation, produced no such effect. To assess whether a similar protocol would extend survival in pre‐sensitized recipients we performed a similar study, using 10 mice per group, in which the recipients were re‐transplanted 14 days following rejection of a first graft from the same donors. Ten mice per group received busulphan/cyclophosphamide as described in the Materials and methods, but only one of these subgroups subsequently received autologous marrow cells (5 × 106 T‐depleted cells intravenously). Two equivalent groups of 10 naive mice were treated in the same way. All animals continued to receive rapamycin for 21 days beginning 5 days after marrow infusion. Data in Fig. 1(a) shows skin graft survival (pooled across two independent experiments: total 20 mice per group) for this study.
Figure 1.
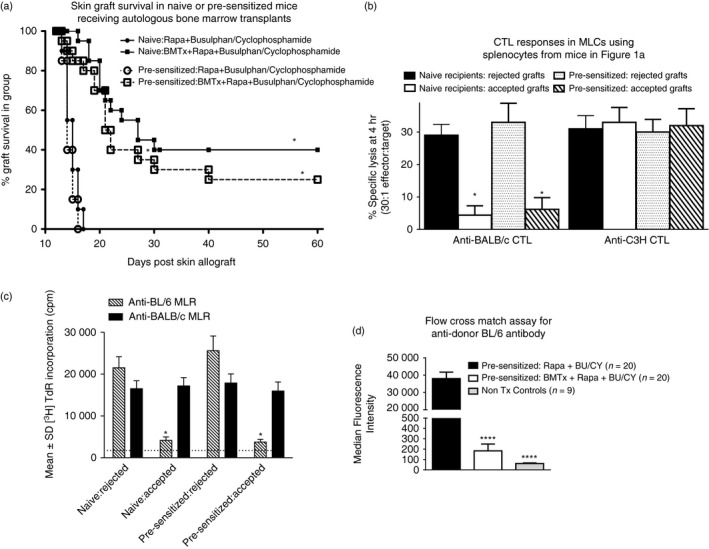
Long‐term skin allograft survival following autologous bone marrow transplantation (BMTx) of naive and pre‐sensitized mice – see text for more details. (a) Ten mice/group naive (●) or pre‐sensitized (○) C3H mice (the latter having rejected a first BL/6 graft 21 days earlier) received BL/6 skin grafts and a sequence of rapamycin treatment, busulphan/cyclophosphamide for 6 days, followed by a further 21 days of rapamycin treatment beginning 5 days later. Two groups of 10 naive (▄) or pre‐sensitized (□) mice also received autologous T‐cell‐depleted C3H bone marrow cells (5 × 106 per recipient) 2 days after completion of the course of busulphan/cyclophosphamide, and began 21 days of rapamycin 3 days later. Graft survival is shown for all four groups, using data pooled from two independent studies (20 mice/group). *P < 0·05 (Mann–Whitney U‐test) for equivalent recipients with/without bone marrow. (b) Attenuation of cytotoxic T lymphocyte (CTL) responses (51Cr‐release assays performed at day 5 using labelled EL4 target cells) in mixed lymphocyte co‐cultures (MLC) from splenocytes of skin graft recipients shown in (a). CTL in control cultures stimulated with third party cells (BALB/c) were assayed using P815 tumour target cells. Data are pooled within groups in which mice had accepted/rejected skin grafts. *P < 0·02 compared with equivalent recipients with rejected grafts. (c) Attenuation of mixed lymphocyte reaction (MLR) proliferative responses ([3 H]TdR‐incorporation assays) at 72 hr using splenocytes of skin graft recipients shown in (a), stimulated with either BL/6 or BALB/c (third‐party) splenocytes. Once again data are pooled within groups in which mice had accepted/rejected skin grafts. *P < 0·02 compared with equivalent recipients with rejected grafts. The broken line indicates [3 H]TdR incorporation in unstimulated cells. (d) Anti‐donor (BL/6) IgG levels in pre‐sensitized mice treated with rapamycin and busulphan/cyclophosphamide with (middle bar) and without (far left hand bar) BMTx (n = 20 each). Serum alloantibodies were also quantified for non‐transplanted C3H mice (far right hand bar) as controls (n = 9). Data are reported as MFI (median fluorescence intensity; mean ± SD). ****P < 0·0001 versus pre‐sensitized mice receiving rapamycin but not BMTx.
Importantly, skin graft survival following autologous marrow transplantation was essentially equivalent in both naive and pre‐sensitized mice, and in each case, was significantly superior to that seen in mice not receiving marrow (P < 0·02 in Fig. 1a). When splenocytes from these mice were tested individually in MLC for induction of donor‐specific (versus third‐party) CTL, marked attenuation of donor‐specific cytotoxicity was seen in both naive and pre‐sensitized mice with graft acceptance (grouped data shown in Fig. 1b) compared with animals rejecting their grafts. Third‐party CTL induction was equivalent across all groups of mice (naive and pre‐sensitized, with/without graft acceptance). Similar attenuation of proliferation in mixed lymphocyte reaction (MLR) cultures, assessed by [3H]TdR incorporation, was seen in both naive and pre‐sensitized mice with prolonged graft acceptance (Fig. 1c).
Anti‐donor immunoglobulin may play a role in graft rejection in pre‐sensitized mice, so we also compared the titres of anti‐BL/6 IgG in various groups of mice. Data in Fig. 1(d) show the results of a flow cross‐match assay for individual mice broken down into three categories. The categories are non‐transplanted (Non‐Tx) control mice as well as, pre‐sensitized mice treated with rapamycin and busulphan/cyclophosphamide, with and without, bone marrow transplant. Serum was obtained from age‐matched non‐transplanted naive C3H mice. For mice receiving autologous bone marrow transplants, serum was obtained at 60 days post transplant, and at 21 days following graft rejection in the pre‐sensitized group that did not receive bone marrow transplants. Following bone marrow transplantation, anti‐BL/6 titres (reported as median fluorescence intensities) were significantly (P < 0·0001) diminished in pre‐sensitized mice compared with pre‐sensitized mice not receiving bone marrow. Hence, enhanced graft survival in pre‐sensitized mice was associated both with attenuation of MLC responses, and marked reduction of donor‐specific IgG in recipients with surviving grafts.
Evidence for an important role for Treg cells in prolonged graft survival in pre‐sensitized mice
In our previous study using autologous bone marrow transplantation in naive mice we reported that in mice with extended allograft survival, functional Treg cells of both host and donor bone marrow origin could be documented in vitro by antigen‐specific suppression of fresh MLC.2 To investigate whether this was also the case in pre‐sensitized mice we performed studies analogous to those shown in Fig. 1, but using CD45.2 BL/6 (not C3H) mice as recipients, with autologous marrow from CD45.1 BL/6 mice. Two independent studies, each with groups of 10 naive and 10 pre‐sensitized mice, were used. A total of eight long‐term graft survivors were available in the naive group, and seven in the pre‐sensitized group at 60 days post skin grafting. Individual spleen preparations were made for all animals, and CD4+‐enriched cells were obtained using an aliquot (70%) of each individual spleen cell pool. The MLC were initiated using 3 × 106 responder cells pooled from four naive mice, mixed with either 1 × 106 whole spleens (Fig. 2a), or CD4+‐enriched cells from MACS columns (Fig. 2b), for each graft recipient. In addition, for each graft recipient cell preparation, cells were further treated in vitro with rabbit complement alone, or complement with anti‐CD45.1 or anti‐CD45.2 mAbs. All MLC were stimulated with irradiated BALB/c or C3H cells (third‐party) and assayed at 5 days for CTL specific for the stimulating antigen. Grouped data for these studies are shown in Fig. 2.
Figure 2.
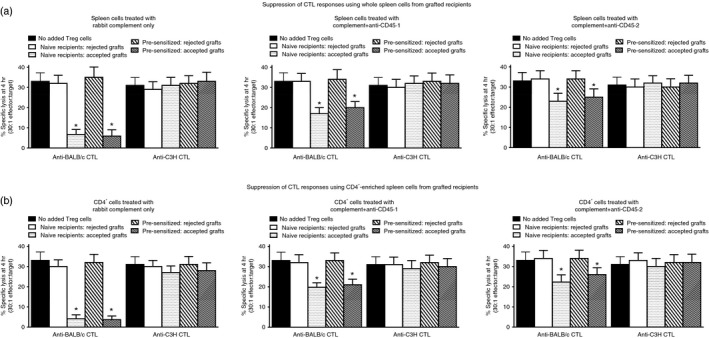
Attenuation of cytotoxic T lymphocyte (CTL) responses (51Cr‐release assays performed at day 5 using labelled P815 target cells) in mixed lymphocyte co‐cultures (MLC) using splenocytes pooled from naive BL/6 mice (data to far left in each panel for anti‐BALB/c and anti‐C3H responses) following addition of whole spleen cells (a) or CD4+‐enriched splenocytes (b) of skin graft recipients shown in Fig. 1. CTL in cultures stimulated with third‐party cells (C3H) were assayed using C3H Concanavalin A blasts. Data were obtained using individual spleen preparations from all mice shown in Fig. 1, and were then pooled within groups in which mice had accepted/rejected skin grafts (see Fig. 1). In each set of upper/lower panels the added cells from grafted recipients were treated with either rabbit complement alone, or anti‐CD45.1/anti‐CD45.2 with rabbit complement. *P < 0·05 compared with equivalent recipients with rejected grafts.
It is apparent from these data that both whole spleen cells and, more especially, CD4+‐enriched splenocytes from both naive and pre‐sensitized mice with surviving grafts, were able to attenuate, in an antigen‐specific manner, CTL induction from a pool of naive responder spleen cells (see data with cells treated with complement only – these responses were equivalent to those seen using untreated splenocytes; data not shown for clarity). Interestingly, treatment with anti‐CD45.1 led to significant loss of suppression, implying existence of a regulatory population derived from the infused marrow. In addition, reduction in suppression also followed treatment with anti‐CD45.2 mAb, implying the existence of a regulatory population derived from the original recipient's cells. Similar data were observed in the reverse scenario (CD45.1 BL/6 recipients of CD45.2 BL/6 marrow; data not shown). In a subsequent study (see Supplementary material, Fig. S1a), combined treatment with anti‐CD45.1 and anti‐CD45.2 antibody with complement completely abolished all suppression seen by the addition of splenocytes from grafted mice to naive spleen cell cultures. Further analysis revealed that not only was the combination of anti‐CD45.1 and anti‐CD45.2 able to abolish all measured suppression from cells of grafted mice, so too did a depleting anti‐Thy‐1.2 + (see Supplementary material, Fig. S1b). These latter data imply that non‐T cells are probably not responsible for the regulatory activity measured in splenocytes from grafted mice when they were sacrificed (see also Discussion).
Expansion of CD4+ Treg cells using mAbs to TNFRSF25 to augment regulation of alloimmunity
We had previously reported that sera from rabbits immunized with a peptide derived from TNFRSF25, reported to be expressed preferentially on Treg cells, could augment allograft survival in vivo in mice also receiving autologous marrow transplantation.2, 13 Data in Fig. 3 show results from screening rat hybridomas prepared following fusion of rat spleen cells from animals immunized against the same peptide. In Fig. 3(a) data are shown from an ELISA screen using wells coated with Treg cells and fixed with glutaraldehyde, whereas Fig. 3(b) shows results from FACS analysis using similar Treg cells. Based upon these data we selected three mAbs for more exhaustive study (1B4, 2D4 and 7C3) as well as a control mAb (2D6). Typical data from staining subsets of activated lymphocytes by one of these (2D4) are shown in the Supplementary material (Fig. S2). Immunoglobulin from each was added (2 μg/ml final concentration) to 72‐hr cultures of Treg cells induced from CD4+ cells (MACS‐columns) by incubation on anti‐CD3‐coated plates with medium also containing 2 μg/ml anti‐CD28 and 10 ng/ml TGF‐β. After 48 hr (5 days post induction of Treg cells) cells were harvested, counted and Treg cells were added (7·5 × 105, 2·5 × 105, 0·8 × 105) in fresh MLC with 2 × 106 spleen responder cells (syngeneic with the cells used to induce Treg cells) and 1 × 106 irradiated stimulator cells. The CTL were assayed in standard fashion at day 5. Data from one such assay (of four) are shown in Fig. 3(c). The MLR proliferation cultures receiving these same Treg cells were assessed by [3H]TdR incorporation (Fig. 3d).
Figure 3.
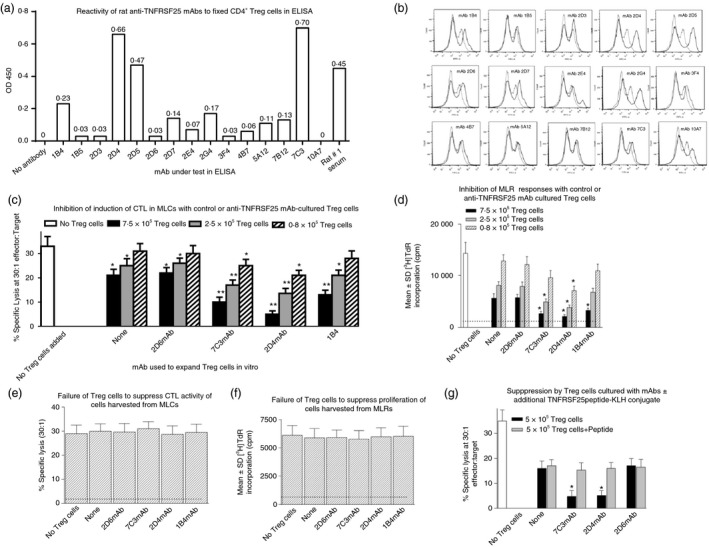
Characterization of monoclonal antibodies (mAbs) to TNFRSF25 and use of mAbs to expand regulatory T (Treg) cells induced in vitro. Screening of rat mAbs to TNFRSF25 in ELISA (a) or FACS (b) using Treg cells induced over 5 days in vitro from BALB/c CD4+ cells (MACS column enrichment) incubated on anti‐CD3 coated plates in medium containing 2 μg/ml anti‐CD28 and 10 ng/ml transforming growth factor‐β (TGF‐β). All mAb supernatants were tested at 2 μg/ml concentration. Data are representative for one of three similar studies for each assay. (c) Augmented activity of Treg cells induced in vitro from CD4+‐enriched splenocytes cultured on anti‐CD3 coated plates with (anti‐CD28 + TGF‐β) to suppress cytotoxic T lymphocyte (CTL) induction in naive splenocytes syngeneic to those producing Treg cells, when hybridoma supernatants (50% final concentration) were added for the last 48 hr of Treg induction in vitro. Data to far‐left show CTL responses in cultures with no Treg cells added. Other groups represent Treg cells from cultures with no mAbs, or with either of the four mAbs shown. Following addition of the latter, Treg numbers harvested at 5 days of culture were increased by a mean of (80 ± 25%) relative to control mAb, with no reproducible difference for any mAb. Data show mean ± SD of lysis for three different Treg quantities included in mixed lymphocyte co‐cultures (MLC). *P < 0·02, **P < 0·05, compared with equivalent cultures with untreated Treg cells. (d) Augmented ability of Treg cells induced as in (c) to suppress proliferation (measured as [3 H]TdR incorporation at 72 hr of culture) in allo‐stimulated naive splenocytes syngeneic to those producing Treg cells, when hybridoma supernatants (2 μg/ml final concentration) were added for the last 48 hr of Treg induction in vitro. Data to far‐left show [3 H]TdR incorporation in cultures with no Treg cells added. Other groups represent Treg cells from cultures with no mAbs, or with either of the three mAbs shown. Data show mean ± SD for cultures with two different Treg numbers. *P < 0·05 compared with equivalent cultures with untreated Treg cells (no mAb). The broken line indicates [3 H]TdR incorporation in unstimulated cells. (e) Treg cells induced in cultures as shown in Fig. 3(c) do not directly suppress activity of CTL in 51Cr‐release assay. Treg cells were harvested from cultures of CD4+‐enriched splenocytes (±mAbs shown) and 3 × 105 Treg cells added in duplicate to 1·5 × 105 CTL harvested from 5‐day MLC of fresh BL/6 splenocytes and irradiated BALB/c splenocytes. After 6 hr in culture, 5 × 103 51Cr‐labelled P815 target cells were added and lytic activity was measured from duplicate wells at 4 hr. The broken line shows mean lytic activity from Treg cells added alone (in all groups lytic activity measured was < 2%). (f) As for panel (e) except that 4 × 105 Treg cells were added to 2 × 105 CD4+‐enriched cells harvested from 72 hr mixed lymphocyte reaction (MLR) cultures (BL/6 stimulated with irradiated BALB/c). After 6 hr, 1 μCi [3 H]TdR was added to cultures, with all cultures harvested to assess [3 H]TdR incorporation 12 hr later. The broken line represents mean proliferation in cultures of Treg cells alone (in all cases counts/min were < 1000). (g) Loss of augmented suppression activity in Treg cells induced in vitro in the presence of hybridoma supernatants (2 μg/ml final concentration) added for the last 48 hr of Treg induction in vitro when TNFRSF25 peptide‐keyhole limpet haemocyanin was also added as a competitive inhibitor. As before Treg cells were induced from CD4+‐enriched splenocytes cultured on anti‐CD3‐coated plates with (anti‐CD28 + TGF‐β), and used to suppress CTL induction in allo‐stimulated naive splenocytes syngeneic to those producing Treg cells. Data to far‐left show CTL in cultures with no Treg cells added. Other groups represent Treg cells from cultures with no mAbs, or with one of three mAbs (2D6 represents a negative control mAb), with/without peptide. Data show mean ± SD for all cultures. *P < 0·05 compared with equivalent cultures with untreated Treg cells (no mAb).
Consistent with data from screening the various hybridomas, all three of the chosen mAbs led to augmented Treg cell functional activity as defined in these assays. All three mAbs were observed to produce similar numerical increases in Treg cells isolated from the cultures (80 ± 25%), with no reproducible difference among the mAbs tested, although two mAbs (2D4 and 7C3) showed most expansion of functional Treg activity. We found no functional Treg cells induced in cultures of CD4+ cells incubated only with anti‐TNFRSF25 mAbs (unpublished results). In control studies, adding Treg cells from cultures analogous to those shown in Fig. 3(c) to either effector CTL for 6 hr before assay with 51Cr targets (Fig. 3e), or to proliferating cells in MLC 6 hr before addition of [3H]TdR (Fig. 3f), led to no attenuation of responses. Hence, the Treg cells induced in the presence/absence of the mAbs were not themselves directly cytotoxic to CTL (or proliferating cells in MLRs).
In a further study (Fig. 3g) we performed a similar expansion of Treg cells but in addition to adding mAbs at 72 hr to cultures used to induce Treg cells, we also added purified peptide–keyhole limpet haemocyanin conjugate (20 μg/ml) as a competitive inhibitor to the mAbs. This completely abolished any augmentation of Treg functional activity achieved by addition of either 2D4 or 7C3.
Use of mAbs to TNFRSF25 to expand Treg cells in vivo
Twenty‐one naive (Fig. 4a) and pre‐sensitized (Fig. 4b) BL/6 mice were grafted with BALB/c skin, followed by treatment as described for Fig. 1, with all mice receiving autologous bone marrow transplants 2 days after completion of busulphan/cyclophosphamide treatment. Beginning 2 days after bone marrow infusion (day 15 post engraftment), for a total of eight injections at 96‐hr intervals, seven mice in each group received either control rat immunoglobulin (10 μg/mouse purified from hybridoma 2D6), or 10 μg/mouse of either 2D4 or 7C3 mAbs (see Fig. 3a). Mice continued to receive rapamycin until 21 days after bone marrow infusion. Grafts were inspected daily and rejection was recorded by an individual blinded to the groups. Survival data to 60 days are shown in Fig. 4. All mice were killed 60 days post transplant and splenocytes were harvested. There was no difference in absolute cell numbers recovered per spleen, or in frequencies of spleen cells that were CD4+ (15 ± 3·8% across all groups) or CD8+ (6·5 ± 1·9% across all groups) as determined by FACS. Splenocytes were used directly for MLC (induction of CTL‐Fig. 5a; MLR proliferation‐ Fig. 5b), and, following CD4+ enrichment, for analysis of Treg cells able to attenuate the induction of CTL (Fig. 5c) or MLR proliferation (Fig. 5d) from a fresh pool of naive responder splenocytes. Data pooled from one of two such studies are shown in these figures.
Figure 4.
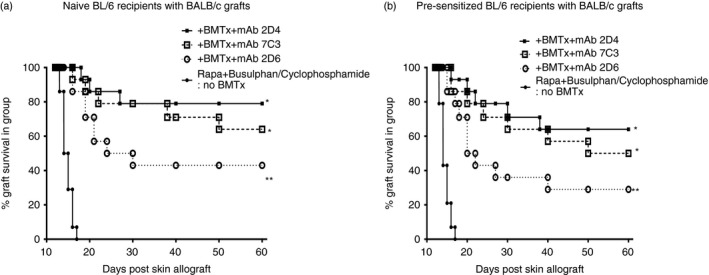
Graft survival in naive (a) or pre‐sensitized (b) mice receiving autologous bone marrow transplantation (BMTx) following busulphan/cyclophosphamide and, in addition, serial injections (20 μg/mouse) of control rat immunoglobulin [monoclonal antibody (mAb) 2D6], or of mAbs (2D4; 7C3) at 96‐hr intervals (eight injections) beginning 2 days after BMTx. Data are pooled from two studies, with seven mice/group in each individual study. **P < 0·05 compared with no BMTx control, and *P < 0·05 compared with the control mAb (2D6) ‐treated group, using Mann–Whitney U‐test. See also Fig. 1 for comparison.
Figure 5.
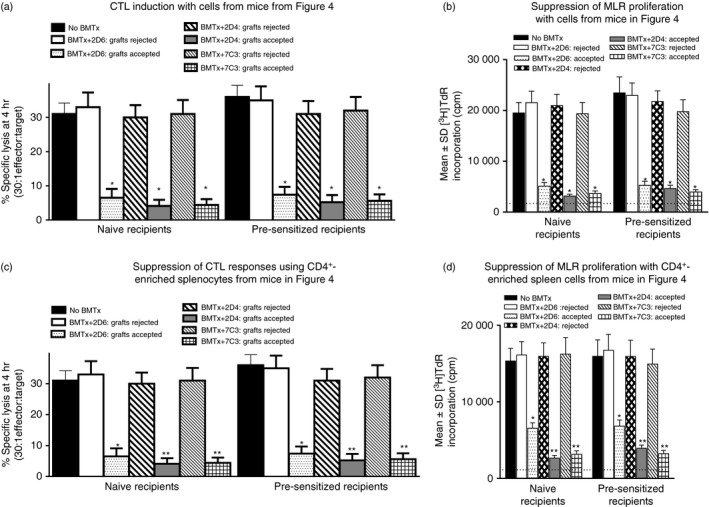
Decreased cytotoxic T lymphocyte (CTL) induction/proliferation in mixed lymphocyte co‐cultures (MLC) (a,b) and augmented CD4+ regulatory T (Treg) function (c,d) in splenocytes harvested at day 60 post skin grafting from mice shown in Fig. 4. Naive or pre‐sensitized mice had received autologous marrow transplants followed by serial injections (eight times at 96‐hr intervals) with 20 μg/mouse of the hybridomas shown. MLC and CD4+ Treg assays were performed as described in Fig. 1. Only data for anti‐BALB/c responses are shown to retain clarity. No perturbation of third‐party (anti‐C3H) responses was seen. CTL activity in control cells in (c) (no Treg cells added) was 34 ± 4·6% specific lysis (30 : 1 effector : target). *P < 0·05 compared with group receiving same mAb but with rejected grafts; **P < 0·05 compared with groups receiving control 2D6 mAb.
There are several points worthy of note in these data. First, infusion of 2D4 and 7C3 mAbs in vivo into mice receiving autologous bone marrow transplantation as per the protocol outlined in Fig. 1, substantially increased graft survival at d60 (approximately twofold), in both naive and pre‐sensitized mice, compared with that seen in Fig. 1, or in Fig. 4 with control immunoglobulin (2D6). There was a trend to a greater survival advantage in naive compared with pre‐sensitized mice (compare equivalent groups in Fig. 4a,b). As before, when we compared CTL induction or MLR proliferation in splenocytes from all mice shown in Fig. 4, animals with rejected grafts showed CTL responses equivalent to mice receiving no bone marrow transplantation, whereas all mice with surviving grafts, regardless of the mAb injected, showed decreased CTL induction and MLR proliferation (Fig. 5a,b). However, when equivalent numbers of CD4+‐enriched cells from splenocytes of these mice were compared for their ability to attenuate CTL induction or MLR proliferation in a fresh MLC with naive responder splenocytes, cells from mice receiving 2D4 or 7C3 mAb, with accepted grafts, showed the most marked suppressive capacity (Fig. 5c,d), although Treg cells were demonstrable even in mice receiving autologous bone marrow transplantation but only control mAb (2D6).
In a subsequent study, we transplanted pre‐sensitized BL/6 mice as before with BALB/c skin using the same protocol as described in Fig. 4, infusing again eight injections of 10 μg/mouse of the mAbs 2D4, 2D6 and 7C3 to separate groups of eight mice per group. Control mice received no mAb immunoglobulin infusions. At 60 days post transplant, mice were killed and CD4+ cells were isolated on MACS columns from splenocytes of three individuals/group with surviving grafts. Cells were stained in FACS with anti‐CD4 and Foxp3 mAbs (Fig. 6a), and 3 × 105 cells from each preparation assayed in triplicate for the suppression of CTL induction from fresh BL/6 splenocytes stimulated with either BALB/c or C3H irradiated splenocytes (Fig. 6b). CTL were assayed as before at 5 days of culture. These data show that enhanced suppression by CD4+ cells from mice receiving 2D4 or 7C3 was paralleled by an approximately twofold increase in CD4+ Foxp3+ cells isolated from those mice.
Figure 6.
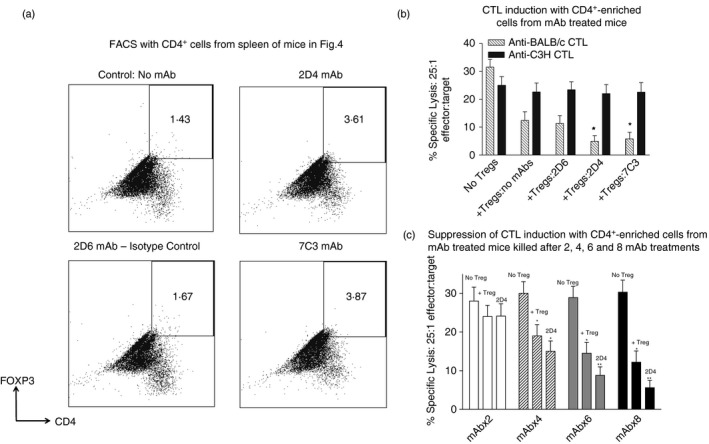
Expansion of regulatory T (Treg) cells in vivo using monoclonal antibodies (mAbs) to TNFRSF25. (a) Flow cytometric plots of MACS‐purified CD4+ cells from individual spleens of pre‐immunized BL/6 mice receiving BALB/c skin grafts and mAbs as per the protocol outlined in Fig. 4, with surviving grafts at 60 days post transplant. Control mice received no mAb immunoglobulin infusions. CD4+ cells were stained in FACS with anti‐ Foxp3 mAbs. Data show an approximately twofold enrichment of CD4+ Foxp3+ cells in spleens from 2D4 and 7C3 mAb‐treated mice versus control mAb (2D6) and untreated mice. Plots are representative of three mice per group. (b) Augmented CD4+ Treg function in CD4+ MACS‐enriched BL/6 splenocytes from mAb‐treated mice or mice with PBS only (no mAbs). 3 × 105 cells of each individual CD4+ preparation was assayed in triplicate for suppression of proliferation from fresh BL/6 splenocytes stimulated with either BALB/c or C3H (third‐party) irradiated splenocytes. Cytotoxic T lymphocytes (CTL) were assayed as before at 5 days of culture. *P < 0·05 compared with mice receiving no mAbs. (c) Kinetics of evolution of functional Treg activity in CD4+‐enriched splenocytes from transplanted mice as in Fig. 6(b), but killed 10 days after two, four, six and eight injections of mAb. Splenocytes were pooled from three mice/group. There was no perturbation of the anti‐C3H response at any time shown (not shown; see Fig. 6b). *P < 0·05 compared with group with no Treg cells; **P < 0·05 compared with Treg cells from mice receiving no mAbs.
In a preliminary study, we assayed the evolution of enhanced Treg functional activity over time in mice receiving 2D4 mAb as in Figs 4, 5, 6. These data are shown in Fig. 6(c), for mice receiving two, four, six or eight injections of mAb, with the four different groups each killed 10 days after the last antibody injection. These data indicate that significant functional activity of CD4+ Treg cells is evident only ~16 days after bone marrow transplantation, with no significant effect of Treg cells from mAb‐treated mice at this time, but Treg activity increases thereafter and is then augmented by mAb administration.
mAbs to 2D4 and 7C3 expand human CD4+ Treg cells as well as mouse CD4+ Treg cells
As the peptide used for immunization to produce the anti‐TNFRSF25 mAbs 2D4 and 7C3 was common to human/mouse TNFRSF25, we compared the ability of these same mAbs to expand human and mouse Treg cells induced over 5 days on anti‐CD3‐coated dishes, with anti‐CD28/TGF‐β added in vitro. As above, mAbs were added at 50% final concentration (supernatant of hybridoma cultures) over the last 48 hr of culture. Once more, expansion of cell numbers relative to control cultures (receiving 2D6 mAbs) was seen for both mouse (81 ± 23%) and human (68 ± 20%) cultures. Treg cells were harvested at 5 days and added at different numbers (1·5 × 105 and 5 × 105) in MLC with responder cells (2·5 × 106) syngeneic with the Treg donor, and irradiated allogeneic stimulator cells. CTL were assayed in standard 51Cr release assays using 72‐hr stimulator Concanavalin A‐activated blasts as targets. Data are shown in the Supplementary material (Fig. S3; one of two such studies). It is apparent that both 2D4 and 7C3 augmented, on a per cell basis, functional Treg activity in both human and mouse cultures of CD4+ cells incubated with anti‐CD3/anti‐CD28 and TGF‐β.
Discussion
Transplantation is recognized as being the treatment of choice for end‐stage organ failure, although limitations still remain associated with the need for long‐term immunosuppressive therapy in recipients. Development of reproducible mechanisms to produce graft‐specific tolerance in recipients has yet to be achieved, although a number of novel strategies have been described and tested in the past several years, as our knowledge of the biology of regulation of immunity improves. We ourselves recently described a novel approach, using autologous bone marrow transplantation following allografting, which led to a high success rate of skin graft acceptance in rodents, with withdrawal of all immunosuppression in recipients. Graft survival was associated with existence of Treg cells of both recipient and donor bone marrow origin.2
An auxiliary daunting problem for the transplant field is the increased rejection seen when grafts are attempted into recipients with pre‐formed anti‐MHC antibodies, a scenario that is linked to an enhanced risk of antibody‐mediated rejection, as well as the anticipated cellular rejection seen even in naive recipients. However, other groups have established that B‐cell‐mediated chronic allograft rejection can occur even in the absence of antibody production, through mechanism(s) potentially related to B‐cell antigen presentation and support of T‐cell immune responses.17 Over the past decade a number of attempts have been made to improve transplant success among this pre‐sensitized group, using protocols that have been referred to as ‘desensitization’. Included among these are the use of B‐cell‐depleting agents (rituximab), intravenous immunoglobulin and plasmapheresis.25, 26
We have asked whether the autologous bone marrow infusion protocol used so successfully in naive recipients to enhance allograft survival could be adapted to pre‐sensitized recipients. Our data argue that indeed skin allograft survival is significantly prolonged even in pre‐sensitized mice following autologous bone marrow infusions, and that the long‐term graft survivors show both enhanced Treg cells, of both host and donor marrow origin (Fig. 2) and significantly reduced donor‐specific antibody to the sensitizing antigens compared with control mice not receiving this bone marrow. That there is no generalized dysfunction in B cells in these recipients is evident from independent data (RMG‐unpublished) showing equivalent levels of mouse immunoglobulin in LPS‐stimulated cultures from all groups of mice (naive and pre‐sensitized, with/without autologous marrow transplants) as assessed by ELISA using 72‐hr culture supernatants. The significant reduction of alloantibodies observed in the bone marrow transplant group may reflect the ability of newly emerging cells from the transferred bone marrow to ‘outcompete’ long‐lived host bone marrow plasma cells. The bone marrow is a niche for immunoglobulin‐secreting long‐lived plasma cells capable of providing long‐term humoral protection and putatively supported in situ by eosinophils that produce plasma cell survival factors.27, 28 However, we found that sensitized mice receiving busulphan/cyclophosphamide and rapamycin without bone marrow transplantation maintain high donor‐specific antibody serum levels, unlike the markedly lower levels (> 100‐fold) seen in similar recipients post bone marrow transplantation, suggesting that there may be a population post bone marrow transplantation capable either of suppressing immunoglobulin production by surviving host cells (e.g. Breg cells; see below), or attenuating (and/or competing for) their survival per se. In a study performed on 27 patients who underwent autologous bone marrow transplants for leukaemia, CD23+ B cells were depressed in blood for 3 months and in the bone marrow for 6 months post transplant, indicating an immature state.29 A clinical study in B‐cell non‐Hodgkin's lymphoma patients after autologous bone marrow transplants demonstrated impaired in vitro proliferative responses to anti‐immunoglobulin, Epstein–Barr virus and IL‐2, as well as, reduced IgG levels for 12 months post transplant.30 These studies were early indications of altered B‐cell function and phenotype after autologous bone marrow transplant. These results are corroborated by observations in patients receiving autologous haematopoietic stem cell transplants for autoimmune disease. In a study of seven patients with systemic lupus erythematosus receiving autologous haematopoietic stem cell transplants, a reduction of memory (IgD−) B cells was observed post transplant. In six of the seven patients, there was essentially a complete ablation of anti‐dsDNA antibodies and a significant reduction in antinuclear antibodies.31 These data suggest that the newly emerging B‐cell repertoire consists primarily of naive B cells contributing to self‐tolerance in autoimmune patients receiving autologous haematopoietic stem cell transplants.
The development after bone marrow transplantation of so‐called Breg cells, a heterogeneous group of B lymphocytes with immunosuppressive functions, has also been of recent interest.18 In an islet allograft murine model induced TGF‐β‐producing Breg cells could promote graft survival by increasing the number of Treg cells.32 In a clinical study with patients receiving autologous bone marrow transplantation for systemic sclerosis a significant increase in Breg cells (defined as CD19+ CD24hi CD38hi) was seen at 6 and 12 months post transplant with no differences observed in levels of Foxp3+ CD25hi CD4+ Treg cells.33 These results were taken to imply an important role for Breg cells in promoting and maintaining immune tolerance. Data in the Supplementary material (Fig. S1b) show in contrast that all suppressive function was lost from splenocytes harvested from transplanted mice following depletion of Thy‐1.2+ T cells. This does not negate an effect of Breg cells in vivo at earlier times during induction of the tolerant state, or indeed in induction of Treg cells themselves. Indeed, data in Fig. 6(c) show that Treg functional activity was only demonstrable at ~16 days post transplantation, yet grafts were still not rejected at this time, implying the existence of alternative regulatory activity. Current studies are examining directly the functional activity of Breg cells harvested from these mice.
Based on our previous studies, which had initially documented a potential role for Treg cells in long‐term graft survival, we explored mechanism(s) that might expand Treg cells in vivo, in an attempt to improve graft outcome above the 35–40% of mice shown in initial studies (Fig. 1). We and others have suggested that antibodies made against TNFRSF25 may produce this effect,2, 13 although there is significant controversy in this area, precipitated by evidence that TNFRSF25 itself is expressed on both Treg cells and some effector T cells34 (see Supplementary material, Fig. S2). In the current studies, we produced several rat mAbs to a peptide of TNFRSF25 common to both the human and mouse molecules, and observed by FACS and ELISA (with plate‐bound cells) significant binding of several mAbs to enriched CD4+ Treg cells. The mAbs binding CD4+ Treg cells produced functional expansion of murine Treg cells in vitro, as assayed by an augmented ability to attenuate induction of CTL and proliferation in vitro, in the absence of any evidence for direct cytotoxicity to CD8+ CTL or CD4+ cells proliferating in MLC (Fig. 3). Note (see earlier text) that these mAbs did not induce Treg cells in vitro directly in the absence of anti‐CD3/CD28 activation. Moreover, again while there are reports that activation through TNFRSF25 can augment cytokine production,9 we found that the addition of TGF‐β in vitro was necessary to induce Treg cells even in the presence of mAbs, though we have not titrated TGF‐β requirements ± mAbs.
Infusion of these mAbs in vivo into naive or pre‐sensitized mice with skin allografts that subsequently received autologous bone marrow transplantation, improved (approximately twofold) long‐term graft survival in these recipients. The notion that unique epitopes of TNFRSF25 were being recognized for this effect to be pronounced, as was previously suggested,10 seems likely given the concurrent inability of other mAbs to produce any modification of allograft survival in vivo, or augment induction of Treg cells in vitro despite their ability to bind the immunizing peptide in ELISA (compare data for mAb 2D6 in Figs 3 and 4).
As the TNFRSF25 peptide used to produce the mAbs used in these studies was common to both human and mouse, in a final study we have also assayed whether the same mAbs as were used to augment mouse skin allograft survival might similarly expand functional human Treg cells in vitro. Data in Fig. 3 suggest that this is indeed the case, with an augmented (on a per cell basis) ability of Treg cells incubated with the relevant mAbs to suppress induction of human CTL in vitro.
In summary, our data provide several new and important findings. First, they suggest that an autologous marrow transplant protocol, previously used successfully to enhance graft survival in naive mice, accomplishes the same enhanced graft survival even in pre‐sensitized individuals. Second, in pre‐sensitized mice receiving autologous bone marrow transplantation, attenuation of anti‐donor IgG (> 100‐fold compared with identically treated recipients lacking marrow infusion) occurs, which may in itself reflect a process key to the prolongation of graft survival seen. Third, in association with improved graft survival, there is a concomitant expansion of Treg cells of both donor marrow and recipient origin, which can attenuate activation of naive CD8+ and CD4+ T cells, in the absence of direct cytotoxicity. Expansion of such Treg cells in vivo using defined mAbs to TNFRSF25 leads to even greater graft prolongation. The mAbs active in this in vivo assay were also able to produce functional expansion of CD4+ Treg cells of human origin in vitro, which could suppress the induction of human MLC reactivity in vitro. Taken together, our data imply a clinical utility to the application of these protocols in human transplantation.
Disclosures
The authors declare no commercial or financial conflict of interest.
Supporting information
Figure S1. (a) Attenuation of cytotoxic T lymphocyte responses (51Cr‐release assays performed at d5) in mixed lymphocyte co‐cultures using splenocytes pooled from naive BL/6 mice (data to far left in each panel for anti‐BALB/c and anti‐C3H responses) following addition of whole spleen cells of skin graft recipients akin to those shown in Fig. 1 . In all cases, only mice with surviving grafts were used as spleen donors for regulatory T (Treg) cell assays. Data were obtained using four individual spleen preparations in all groups of mice shown, and were then pooled within groups in which mice had accepted skin grafts. Cells added from grafted recipients were treated with either rabbit complement alone, or anti‐CD45.1/anti‐CD45.2 with rabbit complement, or with both antibodies (sequentially) and rabbit complement. *P < 0·05 compared with cultures using untreated Treg cells from naive or pre‐sensitized mice. (b) Data from study equivalent to (a), but cells from naive/immune mice were either untreated, or treated with a combination of anti‐CD45.1 and anti‐CD45.2 with complement, or anti‐Thy‐1.2 and complement before use as Treg cells in suppressor assays , performed in triplicate, with splenocytes from fresh naive mice with irradiated stimulator cells. Data show mean ± SD for cytotoxic T lymphocytes measured at day 5 of culture. *P < 0·05 compared with corresponding untreated Treg cells.
Figure S2. Staining of 2D4 and control 2D6 monoclonal antibodies in activated lymph node and spleen lymphocyte subsets. 2x107 lymph node cells and 4x107 splenocytes from BL/6 mice were incubated with 4x107 irradiated splenocytes from BALB/c mice. Cells were harvested at Day 5 and stained with anti‐TNFRSF25 mAbs (2D4 and control 2D6) as well as anti‐mouse CD4 and CD8. Cells with no mAbs were used as no primary antibody controls. FITC anti‐mouse IgM was used to detect anti‐TNFRSF25 staining in activated lymph node and spleen CD4+ and CD8+ cell subsets. All stains were performed in duplicate.
Figure S3. Augmented ability of regulatory T (Treg) cells induced in vitro from CD4+‐enriched mouse splenocytes (left side of figure) or human peripheral blood lymphocytes (PBL) (right side of figure) cultured on anti‐CD3 coated plates with (anti‐CD28 + transforming growth factor‐β) to suppress cytotoxic T‐lymphocyte induction in naive splenocytes/PBL syngeneic to those producing Treg cells, when hybridoma supernatants (50% final concentration) were added for the last 48 hr of Treg induction in vitro (see also Fig. 3c). Data to left (white bars) in each set of mouse/human cultures show CTL responses in cultures with no Tregs added. Other groups represent Tregs from cultures with no mAbs, or with either of the 3 mAbs shown. Data show mean±SD of lysis for 2 different Treg numbers included in MLCs. *, ** indicates P<0.05 compared with cultures with no Tregs, or with control (2D6) mAb added respectively.
Acknowledgements
The stidy was supported by a research grant (to RMG) from CIHR and Heart & Stroke Foundation (Canada).
References
- 1. Crone CC, Marcangelo MJ, Shuster JL Jr. An approach to the patient with organ failure: transplantation and end‐of‐life treatment decisions. Med Clin North Am 2010; 94:1241–54, xii. [DOI] [PubMed] [Google Scholar]
- 2. Gorczynski R, Chen Z, Khatri I, Yu K. Long‐term tolerance and skin allograft survival in CD200tg mice after autologous marrow transplantation. Transplantation 2014; 98:1271–8. [DOI] [PubMed] [Google Scholar]
- 3. Fang L, Adkins B, Deyev V, Podack ER. Essential role of TNF receptor superfamily 25 (TNFRSF25) in the development of allergic lung inflammation. J Exp Med 2008; 205:1037–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bodmer JL, Burns K, Schneider P, Hofmann K, Steiner V, Thome M et al TRAMP, a novel apoptosis‐mediating receptor with sequence homology to tumor necrosis factor receptor 1 and Fas(Apo‐1/CD95). Immunity 1997; 6:79–88. [DOI] [PubMed] [Google Scholar]
- 5. Screaton GR, Xu XN, Olsen AL, Cowper AE, Tan R, McMichael AJ et al LARD: a new lymphoid‐specific death domain containing receptor regulated by alternative pre‐mRNA splicing. Proc Natl Acad Sci USA 1997; 94:4615–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tan KB, Harrop J, Reddy M, Young P, Terrett J, Emery J et al Characterization of a novel TNF‐like ligand and recently described TNF ligand and TNF receptor superfamily genes and their constitutive and inducible expression in hematopoietic and non‐hematopoietic cells. Gene 1997; 204:35–46. [DOI] [PubMed] [Google Scholar]
- 7. Meylan F, Davidson TS, Kahle E, Kinder M, Acharya K, Jankovic D et al The TNF‐family receptor DR3 is essential for diverse T cell‐mediated inflammatory diseases. Immunity 2008; 29:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Prehn JL, Thomas LS, Landers CJ, Yu QT, Michelsen KS, Targan SR. The T cell costimulator TL1A is induced by FcγR signaling in human monocytes and dendritic cells. J Immunol 2007; 178:4033–8. [DOI] [PubMed] [Google Scholar]
- 9. Papadakis KA, Zhu D, Prehn JL, Landers C, Avanesyan A, Lafkas G et al Dominant role for TL1A/DR3 pathway in IL‐12 plus IL‐18‐induced IFN‐γ production by peripheral blood and mucosal CCR9+ T lymphocytes. J Immunol 2005; 174:4985–90. [DOI] [PubMed] [Google Scholar]
- 10. Pappu BP, Borodovsky A, Zheng TS, Yang X, Wu P, Dong X et al TL1A‐DR3 interaction regulates Th17 cell function and Th17‐mediated autoimmune disease. J Exp Med 2008; 205:1049–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takedatsu H, Michelsen KS, Wei B, Landers CJ, Thomas LS, Dhall D et al TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T‐helper 1 and T‐helper 17 activation. Gastroenterology 2008; 135:552–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bull MJ, Williams AS, Mecklenburgh Z, Calder CJ, Twohig JP, Elford C et al The Death Receptor 3‐TNF‐like protein 1A pathway drives adverse bone pathology in inflammatory arthritis. J Exp Med 2008; 205:2457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schreiber TH, Wolf D, Tsai MS, Chirinos J, Deyev VV, Gonzalez L et al Therapeutic Treg expansion in mice by TNFRSF25 prevents allergic lung inflammation. J Clin Invest 2010; 120:3629–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Puttarajappa C, Shapiro R, Tan HP. Antibody‐mediated rejection in kidney transplantation: a review. J Transplant 2012; 2012:193724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lefaucheur C, Loupy A, Hill GS, Andrade J, Nochy D, Antoine C et al Preexisting donor‐specific HLA antibodies predict outcome in kidney transplantation. J Am Soc Nephrol 2010; 21:1398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zachary AA, Eng HS. Desensitization: achieving immune detente. Tissue Antigens 2011; 77:3–8. [DOI] [PubMed] [Google Scholar]
- 17. Zeng Q, Ng YH, Singh T, Jiang K, Sheriff KA, Ippolito R et al B cells mediate chronic allograft rejection independently of antibody production. J Clin Invest 2014; 124:1052–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goode I, Xu H, Ildstad ST. Regulatory B cells: the new “it” cell. Transplant Proc 2014; 46:3–8. [DOI] [PubMed] [Google Scholar]
- 19. Vo AA, Lukovsky M, Toyoda M, Wang J, Reinsmoen NL, Lai CH et al Rituximab and intravenous immune globulin for desensitization during renal transplantation. N Engl J Med 2008; 359:242–51. [DOI] [PubMed] [Google Scholar]
- 20. Everly MJ, Everly JJ, Susskind B, Brailey P, Arend LJ, Alloway RR et al Bortezomib provides effective therapy for antibody‐ and cell‐mediated acute rejection. Transplantation 2008; 86:1754–61. [DOI] [PubMed] [Google Scholar]
- 21. Jordan N, D'Cruz DP. Belimumab for the treatment of systemic lupus erythematosus. Expert Rev Clin Immunol 2015; 11:195–204. [DOI] [PubMed] [Google Scholar]
- 22. Kim I, Wu G, Chai NN, Klein AS, Jordan S. Anti‐interleukin 6 receptor antibodies attenuate antibody recall responses in a mouse model of allosensitization. Transplantation 2014; 98:1262–70. [DOI] [PubMed] [Google Scholar]
- 23. Gorczynski RM, Chen Z, Khatri I, Yu K. Graft‐infiltrating cells expressing a CD200 transgene prolong allogeneic skin graft survival in association with local increases in Foxp3+Treg and mast cells. Transpl Immunol 2011; 25:187–93. [DOI] [PubMed] [Google Scholar]
- 24. Xu H, Yan J, Huang Y, Chilton PM, Ding C, Schanie CL et al Costimulatory blockade of CD154‐CD40 in combination with T‐cell lymphodepletion results in prevention of allogeneic sensitization. Blood 2008; 111:3266–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abu Jawdeh BG . Cuffy MC, Alloway RR, Shields AR, Woodle ES. Desensitization in kidney transplantation: review and future perspectives. Clin Transplant 2014; 28:494–507. [DOI] [PubMed] [Google Scholar]
- 26. Zachary AA, Leffell MS. Desensitization for solid organ and hematopoietic stem cell transplantation. Immunol Rev 2014; 258:183–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Slifka MK, Matloubian M, Ahmed R. Bone marrow is a major site of long‐term antibody production after acute viral infection. J Virol 1995; 69:1895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chu VT, Frohlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S et al Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol 2011; 12:151–9. [DOI] [PubMed] [Google Scholar]
- 29. Bengtsson M, Smedmyr B, Festin R, Oberg G, Simonsson B, Totterman TH. B lymphocyte regeneration in marrow and blood after autologous bone marrow transplantation: increased numbers of B cells carrying activation and progression markers. Leuk Res 1989; 13:791–7. [DOI] [PubMed] [Google Scholar]
- 30. Pedrazzini A, Freedman AS, Andersen J, Heflin L, Anderson K, Takvorian T et al Anti‐B‐cell monoclonal antibody‐purged autologous bone marrow transplantation for B‐cell non‐Hodgkin's lymphoma: phenotypic reconstitution and B‐cell function. Blood 1989; 74:2203–11. [PubMed] [Google Scholar]
- 31. Alexander T, Thiel A, Rosen O, Massenkeil G, Sattler A, Kohler S et al Depletion of autoreactive immunologic memory followed by autologous hematopoietic stem cell transplantation in patients with refractory SLE induces long‐term remission through de novo generation of a juvenile and tolerant immune system. Blood 2009; 113:214–23. [DOI] [PubMed] [Google Scholar]
- 32. Lee KM, Stott RT, Zhao G, SooHoo J, Xiong W, Lian MM et al TGF‐β‐producing regulatory B cells induce regulatory T cells and promote transplantation tolerance. Eur J Immunol 2014; 44:1728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arruda LCM, Oliveira MC, Moraes DA, Covas DT, Voltarelli JC, Malmegrim KCR. THU0501 hematopoietic stem cell transplantation increases naive and regulatory B cells while decreasing memory B cells in systemic sclerosis patients. Ann Rheum Dis 2014; 73:356. [Google Scholar]
- 34. Taraban VY, Ferdinand JR, Al‐Shamkhani A. Expression of TNFRSF25 on conventional T cells and Tregs. J Clin Invest 2011; 121:463–4; author reply 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (a) Attenuation of cytotoxic T lymphocyte responses (51Cr‐release assays performed at d5) in mixed lymphocyte co‐cultures using splenocytes pooled from naive BL/6 mice (data to far left in each panel for anti‐BALB/c and anti‐C3H responses) following addition of whole spleen cells of skin graft recipients akin to those shown in Fig. 1 . In all cases, only mice with surviving grafts were used as spleen donors for regulatory T (Treg) cell assays. Data were obtained using four individual spleen preparations in all groups of mice shown, and were then pooled within groups in which mice had accepted skin grafts. Cells added from grafted recipients were treated with either rabbit complement alone, or anti‐CD45.1/anti‐CD45.2 with rabbit complement, or with both antibodies (sequentially) and rabbit complement. *P < 0·05 compared with cultures using untreated Treg cells from naive or pre‐sensitized mice. (b) Data from study equivalent to (a), but cells from naive/immune mice were either untreated, or treated with a combination of anti‐CD45.1 and anti‐CD45.2 with complement, or anti‐Thy‐1.2 and complement before use as Treg cells in suppressor assays , performed in triplicate, with splenocytes from fresh naive mice with irradiated stimulator cells. Data show mean ± SD for cytotoxic T lymphocytes measured at day 5 of culture. *P < 0·05 compared with corresponding untreated Treg cells.
Figure S2. Staining of 2D4 and control 2D6 monoclonal antibodies in activated lymph node and spleen lymphocyte subsets. 2x107 lymph node cells and 4x107 splenocytes from BL/6 mice were incubated with 4x107 irradiated splenocytes from BALB/c mice. Cells were harvested at Day 5 and stained with anti‐TNFRSF25 mAbs (2D4 and control 2D6) as well as anti‐mouse CD4 and CD8. Cells with no mAbs were used as no primary antibody controls. FITC anti‐mouse IgM was used to detect anti‐TNFRSF25 staining in activated lymph node and spleen CD4+ and CD8+ cell subsets. All stains were performed in duplicate.
Figure S3. Augmented ability of regulatory T (Treg) cells induced in vitro from CD4+‐enriched mouse splenocytes (left side of figure) or human peripheral blood lymphocytes (PBL) (right side of figure) cultured on anti‐CD3 coated plates with (anti‐CD28 + transforming growth factor‐β) to suppress cytotoxic T‐lymphocyte induction in naive splenocytes/PBL syngeneic to those producing Treg cells, when hybridoma supernatants (50% final concentration) were added for the last 48 hr of Treg induction in vitro (see also Fig. 3c). Data to left (white bars) in each set of mouse/human cultures show CTL responses in cultures with no Tregs added. Other groups represent Tregs from cultures with no mAbs, or with either of the 3 mAbs shown. Data show mean±SD of lysis for 2 different Treg numbers included in MLCs. *, ** indicates P<0.05 compared with cultures with no Tregs, or with control (2D6) mAb added respectively.