

Summary
Mesenchymal stromal cells (MSC) have emerged as promising cell therapies for multiple conditions based on demonstrations of their potent immunomodulatory and regenerative capacities in models of inflammatory disease. Understanding the effects of MSC on T cells has dominated the majority of work carried out in this field to date; recently, however, a number of studies have shown that the therapeutic effect of MSC requires the presence of macrophages. It is timely to review the mechanisms and manner by which MSC modulate macrophage populations in order to design more effective MSC therapies and clinical studies. A complex cross‐talk exists through which MSC and macrophages communicate, a communication that is not controlled exclusively by MSC. Here, we examine the evidence that suggests that MSC not only respond to inflammatory macrophages and adjust their secretome accordingly, but also that macrophages respond to encounters with MSC, creating a feedback loop which contributes to the immune regulation observed following MSC therapy. Future studies examining the effects of MSC on macrophages should consider the antagonistic role that macrophages play in this exchange.
Keywords: inflammation, macrophage, mesenchymal stem cells
Introduction
Adult progenitor cells such as mesenchymal stromal cells (MSC) and multi‐potent adult progenitor cells (MAPC) are heterogeneous populations present in a range of adult tissues, often derived from bone marrow (BM) or adipose tissue (AT) for experimental use 1. While both MSC and MAPC can protect and repair damaged tissues 2, 3, 4, it is their immunomodulatory action that has garnered most attention over the past decade, with a large number of studies demonstrating that MSC can suppress inflammation and adaptive immunity 5, 6, 7, 8. Understandably, the focus of MSC research has centred on interactions between MSC and T cells; however, there is now substantial evidence to suggest that MSC‐derived soluble factors also suppress activation and maturation of innate immune cells, while skewing early innate reactions towards an anti‐inflammatory phenotype. Studies of adaptive immune modulation have shown that MSC suppress proliferation and activation of proinflammatory T cells preferentially, while promoting anti‐inflammatory regulatory T cells (Treg) simultaneously 6, 9, 10. MAPC and MSC that have been activated or licensed by inflammatory signals such as interferon (IFN)‐γ suppress the proliferation of T helper type 1 (Th1) cells via production of indoleamine 2,3‐dioxygenase (IDO) 11, while prostaglandin E2 (PGE2) and programmed death ligand 1 (PDL‐1) are required for the suppression of Th17 activity 5, 9, 12, 13, 14. The combination of these effects, with suppression of the inflammatory storm, make MSC and similar therapies important candidates for intervention against chronic immune pathologies.
The features of the MSC interaction with innate immune cells are becoming clear. MSC‐derived PGE2, the products of IDO and tumour necrosis factor‐inducible gene 6 (TSG‐6) promote the conversion of monocytes and proinflammatory macrophages into anti‐inflammatory populations producing interleukin (IL)‐10 15, 16, 17, 18, 19, 20. Proliferation, IFN‐γ production and the cytotoxic action of natural killer (NK) cells is inhibited during co‐culture with MSC, with IDO, PGE2 and human leucocyte antigen G5 (HLA‐G5) playing important roles 21, 22, 23, 24, 25. MSC also suppress adaptive immunity indirectly through shaping the response patterns of dendritic cells (DC). Initially these MSC–DC interactions appeared bewildering; however, the mechanisms by which MSC direct DC towards a regulatory phenotype through IL‐6 secretion and Notch signalling have been clarified recently and characterized by a number of teams 8, 26, 27, 28, 29.
The studies summarized above suggest that MSC action during cell therapy is complex, and involves more than shaping adaptive immunity: MSC therapy is not a simple form of global immune suppression that can be reduced to, or replaced by, a single soluble factor. A complex interaction occurs whereby MSC seem to be licensed or primed by an inflammatory milieu and respond by producing anti‐inflammatory soluble factors and surface molecules (Fig. 1), suggesting that the early MSC–innate interaction might be central to successful design of effective therapies. For example, IDO, which converts tryptophan to kynurenine, is not produced by MSC under basal conditions, but is induced in human MSC by exposure to IFN‐γ 20. Similarly, TNF‐α induces cyclooxygenase 2 (COX‐2) expression by murine BM‐MSC 30, and IL‐1β has been shown to prime human MAPC to generate PGE2 5. It is also likely that an intricate communication exists with cells of the immune system requiring direct cell–cell contact involving signals such as Notch‐Jagged or PDL‐1 for MSC to support Treg and regulatory DC populations. The implication from these studies is that suppression of innate immune processes contributes to the beneficial effects seen when MSC are deployed therapeutically in conditions where an inflammatory/cytokine storm is prominent.
Figure 1.
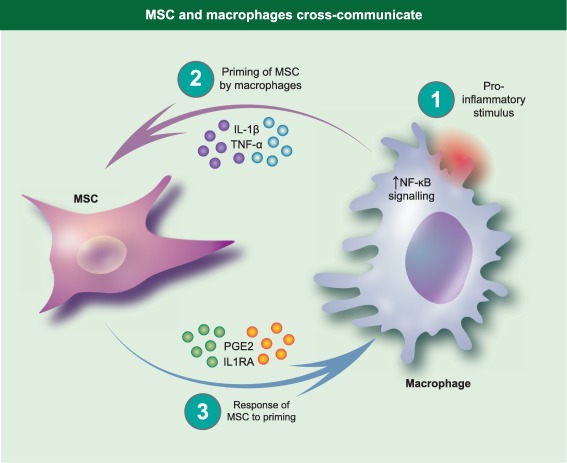
Mesenchymal stromal cells (MSC) and macrophages cross‐communicate. Macrophages are activated by stimuli to produce proinflammatory factors. This creates a feedback loop whereby proinflammatory cytokines produced by macrophages stimulate MSC to produce prostaglandin E2 (PGE2) and interleukin (IL)−1RA, among other immune modulators.
Clinical trials using MSC and MAPC have been justified and performed targeting a range of ischaemic and inflammatory conditions including chronic obstructive pulmonary disease (COPD), graft‐versus‐host disease (GVHD) and Crohn's disease, with varying levels of success (www.clinicaltrials.gov) 31, 32, 33. While much has been done to understand MSC biology, it is evident from the clinical trial data that mechanistic understanding lags behind the phenomenological observations of MSC efficacy. The gap in our understanding is perhaps most evident with regard to the cross‐talk between MSC and macrophages, although a number of interesting studies have addressed this issue recently. Multiple and varied mechanisms are emerging by which MSC modulate macrophage populations; however, the priority, redundancy and species specificity of these interactions is still unclear. The remainder of this paper concentrates on the mechanisms by which MSC promote anti‐inflammatory macrophage populations, with a particular focus on the ways in which macrophages and MSC interact.
Characterizing macrophage phenotypes
The measurement of the effect of MSC on the inflammatory signature of isolated macrophage populations has been an important initial topic for study. Macrophages and their monocytic precursors are professional phagocytes responsible for the clearance of pathogens and apoptotic cells. Macrophages can further acquire specialized roles based on their anatomical location. For example, the predominant function of alveolar macrophages is the elimination of particulates, allergens and microbes from the alveolar surfaces of the lung, while microglia in the brain are responsible for tissue remodelling and homeostasis 34. These issues of macrophage diversity give rise to problems of interpretability and questions of how applicable data from macrophage cell lines, or from different tissues, can be broadly interpreted and applied.
Regardless of their anatomical location, macrophages are categorized typically by immunologists into two populations: the M1 and M2 subsets. M1 macrophages are considered to be ‘classically activated’, in that they are activated by Toll‐like receptor (TLR) ligands and IFN‐γ, whereas M2 macrophages are ‘alternatively activated’ by IL‐4 and IL‐13 35. M1 macrophages are considered to mediate defence against pathogens and secrete proinflammatory cytokines and inducible nitric oxide synthase (iNOS), while M2 macrophages are a regulatory‐like population associated with production of IL‐10. Tumour‐associated macrophages (TAMs) and myeloid‐derived suppressor cells (MDSC) share many of the immunosuppressive features of M2 macrophages 34.
Differentiating between M1 and M2 macrophages (especially in vivo) can be challenging, and there is plasticity between the two states. Signal transducer and activator of transcription 6 (STAT‐6) phosphorylation has been a useful distinguishing marker, as it is phosphorylated in M2 but not M1 cells. Conversely, in M1 cells STAT‐3 and STAT‐1 are phosphorylated, often accompanied by activation of the IFN‐γ signalling pathway 34. The most commonly used method of M1/M2 identification at population level is the comparative expression of IL‐10 and TNF‐α measured by either mRNA or enzyme‐linked immunosorbent assay (ELISA) 19, 36, 37. These technological difficulties in characterizing M1/M2 patterns of macrophage response compound the differences seen between studies on macrophages from different anatomical locations. Nevertheless, in attempts to overcome these potentially confounding issues, some groups have measured the regulatory capacity of macrophages following cell therapy through introduction of MSC‐influenced macrophages into different functional assays which have helped to advance and clarify our understanding 38, 39.
An array of studies have demonstrated the capacity for MSC to modulate inflammatory M1 macrophages and promote anti‐inflammatory M2 macrophages 15, 37, 40. The recent progress made in the field of immunometabolism has been reflected in the MSC field, with Selleri et al. 39 reporting the capacity of umbilical cord MSC (UC‐MSC) to promote an M2 phenotype in a lactate‐dependent manner. The M2 gene expression signature was confirmed by expression of typical macrophage and M2 gene transcripts such as CD14, CD16, CD68 and IL‐10. Furthermore, these MSC influenced monocytes showed a greater capacity to skew activated T cells towards a Th2 phenotype than monocytes differentiated in the absence of MSC. While no role was attributed to IDO or IL‐6 in this study, inhibition of lactate production by MSC resulted in lower expression of CD14, CD16 and CD163 and higher expression of CD1a. In the presence of UC‐MSC, monocytes undergoing differentiation into DC show decreased mitochondrial mass and increased spare respiratory capacity, indicating that MSC‐derived lactate shifts the metabolic programming of monocytes undergoing differentiation towards the M2 phenotype, rather than DC.
MSC‐derived soluble factors promote M2 macrophages
MSC‐derived soluble factors can promote the conversion of monocytes or M1 macrophages into an M2‐, IL‐10‐producing population 15, 19, 20. The role of PGE2 in this context has been highlighted most recently by Chiossone et al. 15, who reported that in the presence of MSC, monocytes driven to differentiate into macrophages by macrophage colony‐stimulating factor (M‐CSF) adopted an alternative phenotype compared to those differentiated in the absence of MSC (Fig. 2). Macrophages generated in the presence of MSC expressed higher levels of CD14, CD16, MHCII, CD11b, CD209, CD163 and CD206, an effect that was lost when PGE2 production was inhibited using a COX‐2 inhibitor (Table 1). Interestingly, this population of MSC‐influenced macrophages were unlike IL‐4‐driven M2 cells, as macrophages cultured with MSC during differentiation produced less IL‐10 and more IL‐1β and TGF‐β than traditionally activated M2 cells.
Figure 2.
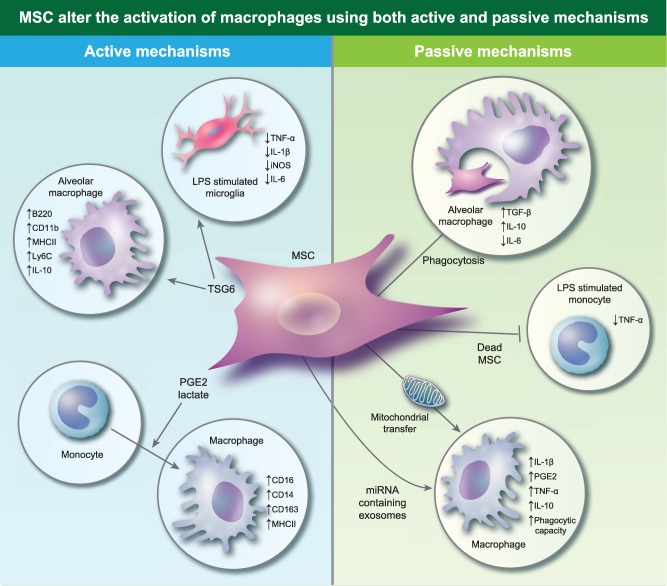
Mesenchymal stromal cells (MSC) alter the activation of macrophages using both active and passive mechanisms. Active mechanisms include the production of soluble factors such as tumour necrosis factor‐inducible gene 6 (TSG‐6), produce prostaglandin E2 (PGE2) and lactate which promote macrophages with an anti‐inflammatory profile. MSC can also passively affect the profile of macrophages, by being phagocytosed by macrophages. Dead or effete MSC suppress tumour necrosis factor (TNF)‐α production by monocytes. MSC also produce exosomes loaded with miRNA which down‐regulate Toll‐like receptor (TLR) signalling in macrophages, making them permissive to the uptake of MSC‐derived mitochondria. Exosomes up‐regulate nuclear factor kappa B (NF‐κB) signalling in macrophages, while the uptake of mitochondria increases their phagocytic capacity.
Table 1.
Cross‐talk between macrophages and MSC in model systems
| Disease model/assay system | Effect of MSC on macrophages | Effect of macrophages on MSC | Reference |
|---|---|---|---|
| Monocyte to Macrophage M‐CSF driven differentiation | MSC‐derived PGE2 skews polarization of monocytes towards an M2‐like population | n.a. | 15 |
| Co‐culture of human MSC and naive lung cells and administration of MSC to naive mice | MSC promoted the polarization of lung macrophages towards an anti‐inflammatory population in a TSG‐6‐dependent manner | MSC co‐cultured in transwell inserts with lung cells were shown to have more than a twofold increase in gene transcripts for TSG‐6, TGF‐β, IL‐1β and COX‐2; however, this was not a purified macrophage population | 38 |
| Murine BM‐MSC were cultured with LPS‐stimulated microglia | MSC suppressed production of TNF‐α, IL‐1β, IL‐6 and iNOS by microglia in a TSG‐6‐dependent fashion | n.a. | 42 |
| Murine BM‐MSC and macrophages in an LPS‐stimulated co‐culture | MSC promoted the production of IL‐10 by macrophages in a PGE2‐dependent manner | Macrophage derived TNF‐α and iNOS were required for the production of PGE2 by MSC | 19 |
| Human MAPC cultured with human monocytes or human monocyte‐conditioned media | n.a. | MAPC cultured in the presence of monocytes or conditioned medium increased production of PGE2 in an IL‐1β‐dependent manner | 5 |
| Murine Macrophages were stimulated with silica or LPS in the presence of conditioned media from murine MSC‐conditioned media | TNF‐α secretion by stimulated macrophages was decreased by MSC‐conditioned media in an IL‐1RA‐dependent fashion | n.a. | 36 |
| Murine macrophages were stimulated with LPS in the presence of murine BM‐MSC | MSC decreased TNF‐α production and increased IL‐10 production by LPS‐stimulated macrophages in an IL‐1RA‐dependent manner | n.a. | 40 |
| Co‐culture of human MSC with mouse and human macrophages or treatment of mouse macrophages with human MSC‐derived exosomes | MSC secrete miRNA containing exosomes that down‐regulate TLR signalling; these exosomes also increase transcripts of cytokines associated with NF‐κB signalling such as IL‐1β, PGE2, TNF and IL‐10 | Macrophages engulf mitochondria which are shuttled to the cell membrane of MSC during mitophagy. This improves the bioenergetics of macrophages | 44 |
MSC = mesenchymal stromal cells; M‐CSF = macrophage colony‐stimulating factor; BM‐MSC = bone marrow MSC; LPS = lipopolysaccharide; MAPC = multi‐potent adult progenitor cells; PGE2 = prostaglandin E2; TSG‐6 = tumour necrosis factor‐inducible gene 6; IL = interleukin; iNOS = inducible nitric oxide synthase; TNF = tumour necrosis factor; TGF = transforming growth factor; NF‐κb = nuclear factor kappa B; COX‐2 = cyclooxygenase 2; n.a. = not applicable.
A role for TSG‐6 from MSC on macrophages has also been investigated. In a xenogeneic administration model, Ko et al. 38 reported that human BM‐MSC elevated murine pulmonary cell populations expressing major histocompatibility complex (MHC)‐II with a significant increase in the frequency of B220+ CD11b+ cells compared to controls. Characterization of this population showed high expression of IL‐10, F4/80 and Ly6C (Table 1). Furthermore, the B220+ CD11b+ population had greater capacity than B220–CD11b+ cells to suppress CD3/CD28‐driven T cell proliferation and could prolong the survival of corneal allografts in vivo, whereas B220–CD11b+ cells could not. The promotion of this anti‐inflammatory population required the production of TSG‐6 by MSC in both in‐vitro and in‐vivo studies 38 (Fig. 2). Similarly, in a murine model of dextran sodium sulphate (DSS)‐induced colitis, the therapeutic efficacy of intraperitoneally administered murine BM‐MSC is dependent upon the production of TSG‐6 41. In this model MSC form aggregates with immune cells in the peritoneal cavity, suggesting a close interaction. Confocal analysis of these aggregates confirmed the presence of macrophages using CD68 as a marker, while flow cytometry was used to confirm the presence of F4/80‐expressing cells. Furthermore, mRNA analysis of the aggregates showed high expression of arginase II, CC chemokine ligand 22 (CCL22), haem oxygenase 1 (HO‐1) and low expression of TNF‐α and IL‐12, suggesting that the macrophages adopt an M2 phenotype in these structures. MSC‐derived TSG‐6 has also been shown to regulate microglial cells (Table 1); in this case, murine BM‐MSC suppress TNF‐α, IL‐1β, iNOS and IL‐6 production by lipopolysaccharide (LPS)‐stimulated BV2 microglial cells in a TSG‐6‐dependent manner (Fig. 2) 42. These data, from different systems and using different measurement approaches, indicate that MSC‐derived TSG‐6 is a probable key player in the programming of macrophages in cell therapies, although additional knock‐down approaches may be needed to show definitively if this is a requirement or a redundant influence.
M1 macrophages respond to MSC
While soluble factors produced by MSC are considered to dominate their immunomodulatory effects, some groups have taken an alternative approach to understanding the effects of MSC on monocytes and macrophages. Recent studies have suggested that the MSC/macrophage relationship is collaborative, in that phagocytes also respond to the presence of MSC which may contribute to their anti‐inflammatory effect. For example, heat‐inactivated human AD‐MSC are unable to suppress T cell proliferation or induce regulatory B cells in vitro; however, they have the capacity to suppress TNF‐α production by LPS‐stimulated monocytes, despite being unable to produce anti‐inflammatory mediators (Fig. 2) 43. This work demonstrates that the effects of MSC on monocytes and macrophages are due not only to soluble factor‐mediated anti‐inflammatory activity by MSC, but that the response of the host cells to MSC may contribute to their regulatory effect. In support of this hypothesis, a murine model of house dust mite‐induced asthma was used to demonstrate that murine BM‐MSC are phagocytosed by some lung macrophages. Interestingly, macrophages characterized as F4/80+CD11c+ which phagocytosed MSC expressed higher mRNA levels of TGF‐β and IL‐10 and lower mRNA levels of IL‐6 than cells that did not engulf MSC (Fig. 2) 37.
Two exciting studies have also indicated that other mechanisms may have a significant impact upon MSC–macrophage interaction. Phinney et al. described a process by which macrophages engulf the mitochondria shuttled to the cell membrane by MSC undergoing mitophagy (Table 1). During this process, MSC secrete simultaneously miRNA‐containing exosomes which, upon uptake by macrophages, down‐regulate TLR signalling, rendering macrophages receptive to these MSC‐derived mitochondria. These exosomes also increase transcripts of cytokines associated with nuclear factor kappa B (NF‐κB) signalling such as IL‐1β, PGE2, TNF and IL‐10 when compared with silica‐activated macrophages (Fig. 2). Production of these cytokines by macrophages following MSC treatment may therefore be responsible for the stimulation of MSC (Fig. 1) 44. Jackson et al. have also reported the transfer of MSC mitochondria to macrophages in the lung; here the transfer is via tunnelling nanotube‐like structures 45. Notably, both the above studies show that macrophage bioenergetics and phagocytic capacities are enhanced following the uptake of MSC mitochondria, which may be beneficial in increasing microbial clearance in conditions such as pneumonia and sepsis.
Cross‐talk between MSC and macrophages
The importance of cross‐talk between MSC and macrophages was highlighted in 2009, when it was shown that murine BM‐MSC reprogramme CD11b+ monocytes/macrophages to produce IL‐10 in a murine model of sepsis (Table 1). In this study, PGE2 secretion by MSC was required to increase IL‐10 production by monocytes/macrophages; however, the generation of PGE2 by MSC in this case was dependent upon TNF‐α and iNOS signalling from monocytes/macrophages 19. In the human context it has been shown that human BM‐MAPC require priming by monocyte‐derived IL‐1β to produce PGE2 5 (Table 1). The importance of IL‐1 signalling is supported by the fact that human BM‐MSC require the IL‐1 signalling pathway in order to produce PGE2 and TSG‐6 46. Interestingly, MSC require IFN‐γ stimulation to suppress Th1 cell activation and proliferation (a negative feedback loop) 7, 30, and so it is very likely that MSC maintain a similar cross‐talk with macrophages via IL‐1β. This is supported in a recent study by Ko et al., where human MSC directed lung macrophages towards an anti‐inflammatory phenotype (Fig. 2). In this case, MSC co‐cultured in a transwell system with mouse lung cells increased expression of gene transcripts for TSG‐6, TGF‐β, COX‐2 and IL‐1β; however, it is unclear if macrophages alone caused this, as the lung cells represented a mixed population of cell types (Table 1) 38.
The importance of IL‐1 in MSC biology has been known for some time. In 2007 it was reported that IL‐1RA produced by MSC blocked TNF‐α production by activated macrophages 36 (Table 1). IL‐1RA production has been shown to be up‐regulated in murine BM‐MSC by simultaneous IFN‐γ and IL‐1β stimulation, and is an important contributor to the effects of MSC on macrophage activation 40 (Fig. 1). CD11b+ monocytes cultured alone, with MSC or with IL‐1RA knock‐out MSC were driven to differentiate into macrophages in the presence of M‐CSF, and then activated with LPS. Macrophages which differentiated in the presence of wild‐type MSC produced lower levels of TNF‐α and higher levels of IL‐10 than those cultured in the presence of IL‐1RA knock‐down MSC 40 (Table 1). Similarly, in a model of mitogen‐driven liver injury, murine MSC promoted an M2 population of macrophages in the lung. This was shown by confocal microscopic analysis of double staining for F4/80 and IL‐10, combined with mRNA and immunohistochemical analyses of iNOS and Arg1 expression in lung tissue. MSC therapy was associated with an increase in IL‐1RA mRNA expression in the lung: knock‐down of IL‐1RA in MSC reduced their ability to ameliorate liver injury. Furthermore, IL‐1RA knock‐down MSC did not exhibit the same capacity as control MSC to increase IL‐10 or Arg1 mRNA expression in the lung, or to decrease iNOS mRNA levels 47.
Human MSC‐, murine MSC‐ and human MSC‐derived exosomes increase IL‐1β transcripts in murine macrophages. Interestingly, however, only murine MSC increased expression of IL‐1 receptor 1, indicating that the cross‐talk between MSC and macrophages is probably species‐restricted 44 and cautions against over‐extrapolation from xenogeneic or humanized mouse studies. Nevertheless, it has been reported that human BM‐MSC require activation to achieve efficacy in a murine model of LPS‐induced acute respiratory distress syndrome (ARDS) 48. In this case, activation of MSC by incubation with serum from ARDS patients for 16 h prior to administration is associated with an increase in IL‐10 and IL‐1RA production by MSC. Animals treated with activated human MSC exhibited higher levels of IL‐10 and lower levels of IL‐1β in bronchoalveolar lavage (BAL) and plasma than those treated with non‐activated MSC. It is possible that the improved efficacy of MSC therein could be due to increased IL1RA expression, leading to polarization of M2 macrophages. Taken together, these studies suggest that macrophage derived IL‐1β may stimulate MSC to secrete IL‐1RA and PGE2, and that this process may contribute to the reciprocal cross‐talk between these cell populations (Fig. 1).
MSC and macrophages: effects at secondary sites
While recent years have seen progress in clarifying the manner by which MSC and macrophages interact, the underlying mechanisms behind the phenomena observed in inflammatory diseases are less clear. The current candidates for cell therapy (including MSC and MAPC) suppress inflammation effectively in murine models of inflammatory disease 4, 7, 13, 41, 49, 50, 51, 52. Efficacy is linked to migration of MSC to target organs, suppression of T cell proliferation, induction of Treg, cytoprotection of damaged tissue and suppression of inflammation. A role for PGE2 has been demonstrated in MSC‐ and MAPC‐mediated suppression of pathology in murine models of GVHD 53, 54, while in murine models of colitis TSG‐6 production is required for efficacy of BM‐MSC 41. Similarly, preclinical studies using MSC in pulmonary disorders have provided promising results 2, 4, 26, 55, 56, 57, 58, 59, 60, with MSC therapy being associated with expansion of Treg, promotion of anti‐inflammatory macrophages, suppression of Th2‐ and Th17‐associated cytokines and enhanced microbial clearance by macrophages in ARDS 45, 48, 61.
In the context of the rat model of type 2 diabetes, for example, intravenously administered UC‐MSC can alleviate insulin resistance, increase the numbers of CD163+ and Arg‐1+ cells in the stromovascular fraction of adipose tissue and decrease numbers of CD11c+ cells. Expression of proinflammatory cytokines was lower in the stromal vascular fraction (SVF) of MSC‐treated mice, while Arg1, CD206 and CD163 expression was higher, suggesting that MSC promote the M2 phenotype at this site, despite no MSC being detected in either adipose tissue or the pancreas. Interestingly, this study observed an increase in Arg1+ cells in the liver following MSC therapy, which suggests M2 polarization at other sites 62. This is supported by a model of corneal allotransplantation, which showed that intravenously administered MSC were ineffective when lung monocytes and macrophages were depleted 38. Furthermore, in a murine model of cardiac allotransplantation, intravenously administered rat MAPC induced Treg in a process that required MDSC 52, while a similar allograft model has also been used to show that intravenously administered murine AD‐MSC stimulated MDSC to induce Th17, which were consequently converted to Treg 13. Thus, the presence of monocytes/macrophages are required for MSC‐ and MAPC‐mediated suppression of allograft rejection in these instances. Therefore, polarization of macrophages at sites of MSC distribution in intravenous cell therapy may be required for the systemic response to MSC.
The capacity for both tissue repair and immunomodulation have led to MSC being seen as a potential cell therapy for asthma 26, 55, 56, 57, 58. In these studies, the therapeutic efficacy of MSC has been attributed to reduction of airway inflammation, expansion of Treg in vivo and suppression of Th2‐ and Th17‐associated cytokines. However, in 2013 Mathias et al. 63 showed that depletion of alveolar macrophages abrogated the therapeutic effects of MSC in a murine model of ovalbumin (OVA)‐induced asthma – a surprising effect, given that the pathology is in the conducting airways. In this study lung macrophages from MSC‐treated mice showed no increase in mRNA levels of typical M2 markers such as Arg1, Chi313 or IL‐10 compared to untreated mice. Despite this, overall IL‐10 protein levels were higher in the lung homogenates of MSC‐treated mice compared to untreated mice, or MSC‐treated mice in which alveolar macrophages were depleted. Therefore, this study suggests that MSC treatment increases IL‐10 production by cell populations other than macrophages in the asthmatic lung (however, this IL‐10 production requires the presence of alveolar macrophages). Similarly, in a murine model of Der F‐induced asthma, human MSC improved lung function, inhibited inflammation and decreased Th2 and Th17 cytokines, as expected. However, this study also showed that MSC were phagocytosed by some lung macrophages. The macrophages which phagocytosed MSC expressed higher mRNA levels of TGF‐β and IL‐10 and lower mRNA levels of IL‐6 than macrophages that did not phagocytose MSC 37. While both studies suggest a role for anti‐inflammatory macrophages in the therapeutic efficacy of MSC for asthma, it would be beneficial to characterize M2 populations more thoroughly and to study further localization in the lung to gain insight into the interactions occurring.
In the case of ARDS, it is not only the immunomodulatory and regenerative capacities of MSC which are useful, but also their ability to improve microbial clearance by macrophages. Anti‐microbial effects of MSC in a murine model of Eschericia coli‐induced pneumonia through the production of anti‐microbial peptides was first reported in 2012 64. More recently a number of studies not only support this observation, but also implicate a role for macrophages in MSC‐mediated bacterial clearance. Both intravenous and intratracheal administration of human MSC reduced the severity of E. coli‐induced pneumonia through production of the anti‐microbial peptide LL‐37 and their ability to enhance the phagocytic capacity of host monocytes and macrophages 65. Similarly, enhanced macrophage phagocytosis in ARDS was due to mitochondrial transfer from MSC to macrophages in the same model 45. Phase I trials have demonstrated a safety profile for the use of MSC in ARDS, while two Phase IIa trials are currently under way to determine the efficacy of MSC in this condition 66.
Both BM‐MSC and AD‐MSC administered intravenously and intratracheally have shown efficacy in a murine model of elastase‐induced emphysema 4, 59. However, while BM‐MSC and AD‐MSC reduced the number of M1 macrophages in the lung, intratracheally administered MSC from either tissue source did not 59. Furthermore, only BM‐MSC had the capacity to increase the number of M2 macrophages in the lung, as characterized by Arg1 expression in the tissue. Interestingly, intravenously administered BM‐MSC which localize to the lung vasculature were superior at promoting M2 macrophages compared to intratracheally administered BM‐MSC. Similarly, rat BM‐MSC suppressed numbers of CD68+COX‐2+ macrophages which were associated with inflammation in lung tissue in a cigarette smoke‐induced model of emphysema. Furthermore, this study demonstrated that BM‐MSC increased IL‐10 production by CD68+ macrophages in BAL 60. Together these data suggest a complex cross‐talk between macrophages in the alveoli, circulation and with therapeutic MSC.
While these preclinical studies suggest that MSC are a promising therapy for a range of disorders, clinical trials are needed urgently to confirm unambiguously the efficacy of MSC for these conditions. In 2004, the first report of successful MSC therapy in a paediatric case of steroid‐refractory GVHD highlighted the potential of MSC as an alternative treatment for inflammatory diseases in humans 67. Since then a large number of Phase I trials have proved the safety profile of MSC and MAPC for GVHD and other conditions 32, 68, 69. Response rates to MSC generally occur in 50–60% of GVHD patients, but even in this well‐studied condition for MSC therapy it is difficult to make efficacy comparisons between trials and groups 31, 69, 70. These trials show that there is still much to do to elucidate the exact mechanisms of action of MSC, and failure to consider issues of dose, timing, route and viability at the preclinical stage risks suboptimally designed clinical studies or studies in which efficacy will not be evident, despite an effective product.
Discussion
While research during the past decade has improved our understanding of MSC mechanisms of action, it is clear that much is left to be uncovered. The majority of studies to date have shown that MSC mediate their immunomodulation through the production of soluble factors in response to inflammatory stimuli 5, 6, 71. These soluble factors suppress proliferation and activity of T cells while promoting Treg, regulatory DC and M2 populations in a myriad of inflammatory diseases 38, 54. Despite the poor capacity of MSC to migrate to specific regions of inflammation and injury, it is believed that these trophic factors promote a tolerant immune state which persists long after MSC have been cleared from the system 72.
Recently, attention has shifted to the additional mechanisms by which MSC therapy may help promote a tolerant immune state. These studies have highlighted a second facet of MSC‐mediated immunosuppression mediated by host immune cells responding to the presence of MSC and altering their immune profile accordingly. Macrophages phagocytose MSC and alter their proinflammatory signature following contact with dead MSC, suggesting that there is a complex cross‐talk between MSC and macrophages that is not explained simply by the production of anti‐inflammatory mediators by MSC 37, 43. Understanding this cross‐talk may help resolve one of the paradoxes of MSC therapy. MSC delivered therapeutically have a short half‐life following intravenous administration in animal models, and yet therapy has profound long‐term effects. Intravenously administered MSC accumulate initially in the lung vasculature 73, despite a short survival time; MSC in this niche would encounter most of the circulating monocyte populations rapidly, and these could potentially interact. Furthermore, other studies and our own unpublished work have shown that MSC can be detected in distal organs and at sites of inflammation in the hours following administration 72. A hallmark of innate immune cells is the rapidity of their activation; therefore, it is feasible for MSC to interact with large numbers of monocytes, macrophages or other cells of the innate immune system prior to being cleared. Clearly, new experimental methodologies such as whole animal cryovisualization will be needed to link cell distribution and cell–cell interaction during MSC therapies to resolve the paradox of poor in‐vivo survival with high efficacy 54, 74. Furthermore, it is known that MSC derived exosomes promote NF‐κB signalling and subsequent transcription of proinflammatory cytokines by macrophages 44, suggesting that MSC localized in the lung may also be shedding exosomes for action in distant tissues. While the cytokines that macrophages produce in response to MSC may be responsible for the activation or ‘licensing’ of MSC, these induce important MSC‐derived anti‐inflammatory signals that, in turn, modulate macrophages (Fig. 1). Further studies investigating the response of innate immune cells to the presence of MSC in vivo may therefore provide new insight into the exact events which occur following MSC administration, and so may improve future prospects for the efficacy of intravenously administered MSC in clinical trials.
Disclosure
None declared.
Acknowledgements
F. C. is supported through an Irish Research Council Enterprise Partnership Schemes (IRCEPS). The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (FP7/2007‐2013) under REA grant agreement no PCIG11‐GA‐2012‐321697 and from the financial support of Science Foundation Ireland (SFI) under grant number 13/SIRG/2172 through a Starting Investigator Research Grant awarded to K. E.
References
- 1. Caplan A. Why are MSCs therapeutic? New data: new insight. J Pathol 2009; 217:318–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cahill EF, Kennelly H, Carty F, Mahon BP, English K. Hepatocyte growth factor is required for mesenchymal stromal cell protection against bleomycin‐induced pulmonary fibrosis. Stem Cells Transl Med 2016; 5:1307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lehman N, Cutrone R, Raber A et al Development of a surrogate angiogenic potency assay for clinical‐grade stem cell production. Cytotherapy 2012; 14:994–1004. [DOI] [PubMed] [Google Scholar]
- 4. Kennelly H, Mahon BP, English K. Human mesenchymal stromal cells exert HGF dependent cytoprotective effects in a human relevant pre‐clinical model of COPD. Sci Rep 2016; 6:38207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reading JL, Vaes B, Hull C et al Suppression of IL‐7‐dependent effector T‐cell expansion by multipotent adult progenitor cells and PGE2. Mol Ther 2015; 23:1783–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reading JL, Yang JHM, Sabbah S et al Clinical‐grade multipotent adult progenitor cells durably control pathogenic T cell responses in human models of transplantation and autoimmunity. J Immunol 2013; 190:4542–52. [DOI] [PubMed] [Google Scholar]
- 7. Tobin LM, Healy ME, English K, Mahon BP. Human mesenchymal stem cells suppress donor CD4+ T cell proliferation and reduce pathology in a humanized mouse model of acute graft‐versus‐host disease. Clin Exp Immunol 2013; 172:333–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. English K, Barry FP, Mahon BP. Murine mesenchymal stem cells suppress dendritic cell migration, maturation and antigen presentation. Immunol Lett 2008; 115:50–8. [DOI] [PubMed] [Google Scholar]
- 9. Ghannam S, Pène J, Torcy‐Moquet G, Jorgensen C, Yssel H. Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J Immunol 2010; 185:302–12. [DOI] [PubMed] [Google Scholar]
- 10. Luz‐Crawford P, Kurte M, Bravo‐Alegría J et al Mesenchymal stem cells generate a CD4+CD25+Foxp3+ regulatory T cell population during the differentiation process of Th1 and Th17 cells. Stem Cell Res Ther 2013; 4:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chinnadurai R, Copland IB, Ng S et al Mesenchymal stromal cells derived from Crohn's patients deploy indoleamine 2,3‐dioxygenase‐mediated immune suppression, independent of autophagy. Mol Ther 2015; 23:1248–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Duffy MM, Pindjakova J, Hanley SA et al Mesenchymal stem cell inhibition of T‐helper 17 cell‐ differentiation is triggered by cell–cell contact and mediated by prostaglandin E2 via the EP4 receptor. Eur J Immunol 2011; 41:2840–51. [DOI] [PubMed] [Google Scholar]
- 13. Obermajer N, Popp FC, Soeder Y et al Conversion of Th17 into IL‐17A(neg) regulatory T cells: a novel mechanism in prolonged allograft survival promoted by mesenchymal stem cell‐supported minimized immunosuppressive therapy. J Immunol 2014; 193:4988–99. [DOI] [PubMed] [Google Scholar]
- 14. Luz‐Crawford P, Noël D, Fernandez X et al Mesenchymal stem cells repress Th17 molecular program through the PD‐1 pathway. PLOS ONE 2012; 7:e45272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chiossone L, Conte R, Spaggiari GM et al Mesenchymal stromal cells induce peculiar alternatively activated macrophages capable of dampening both innate and adaptive immune responses. Stem Cells 2016; 34:1909–21. [DOI] [PubMed] [Google Scholar]
- 16. Choi H, Lee RH, Bazhanov N, Oh JY, Prockop DJ. Anti‐inflammatory protein TSG‐6 secreted by activated MSCs attenuates zymosan‐induced mouse peritonitis by decreasing TLR2/NF‐κB signaling in resident macrophages. Blood 2011; 118:330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim J, Hematti P. Mesenchymal stem cell‐educated macrophages: a novel type of alternatively activated macrophages. Exp Hematol 2009; 37:1445–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maggini J, Mirkin G, Bognanni I et al Mouse bone marrow‐derived mesenchymal stromal cells turn activated macrophages into a regulatory‐like profile. PLOS ONE 2010; 5:e9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Németh K, Leelahavanichkul A, Yuen PST et al Bone marrow stromal cells attenuate sepsis via prostaglandin E2‐dependent reprogramming of host macrophages to increase their interleukin‐10 production. Nat Med 2009; 15:42–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. François M, Romieu‐Mourez R, Li M, Galipeau J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3‐dioxygenase and bystander m2 macrophage differentiation. Mol Ther 2012; 20:187–95. [DOI] [PubMed] [Google Scholar]
- 21. Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005; 105:1815–22. [DOI] [PubMed] [Google Scholar]
- 22. Noone C, Kihm A, English K, O'Dea S, Mahon BP. IFN‐γ stimulated human umbilical‐tissue‐derived cells potently suppress NK activation and resist NK‐mediated cytotoxicity in vitro . Stem Cells Dev 2013; 22:3003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Selmani Z, Naji A, Zidi I et al Human leukocyte antigen‐G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem Cells 2008; 26:212–22. [DOI] [PubMed] [Google Scholar]
- 24. Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2, 3‐dioxygenase and prostaglandin E2. Blood 2008; 111:1327–33. [DOI] [PubMed] [Google Scholar]
- 25. Spaggiari GM, Capobianco A, Becchetti S, Mingari MC, Moretta L. Mesenchymal stem cell–natural killer cell interactions: evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL‐2‐induced NK‐cell proliferation. Blood 2006; 107:1484–90. [DOI] [PubMed] [Google Scholar]
- 26. Cahill EF, Tobin LM, Carty F, Mahon BP, English K. Jagged‐1 is required for the expansion of CD4+ CD25+ FoxP3+ regulatory T cells and tolerogenic dendritic cells by murine mesenchymal stromal cells. Stem Cell Res Ther 2015; 6:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chiesa S, Morbelli S, Morando S et al Mesenchymal stem cells impair in vivo T‐cell priming by dendritic cells. Proc Natl Acad Sci 2011; 108:17384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li FR, Wang XG, Deng CY, Qi H, Ren LL, Zhou HX. Immune modulation of co‐transplantation mesenchymal stem cells with islet on T and dendritic cells. Clin Exp Immunol 2010; 161:357–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Favaro E, Carpanetto A, Caorsi C et al Human mesenchymal stem cells and derived extracellular vesicles induce regulatory dendritic cells in type 1 diabetic patients. Diabetologia 2016; 59:325–33. [DOI] [PubMed] [Google Scholar]
- 30. English K, Barry FP, Field‐Corbett CP, Mahon BP. IFN‐γ and TNF‐α differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol Lett 2007; 110:91–100. [DOI] [PubMed] [Google Scholar]
- 31. Weiss DJ, Casaburi R, Flannery R, LeRoux‐Williams M, Tashkin DP. A placebo‐controlled, randomized trial of mesenchymal stem cells in COPD. Chest 2013; 143:1590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Galipeau J. The mesenchymal stromal cells dilemma – does a negative phase III trial of random donor mesenchymal stromal cells in steroid‐resistant graft‐versus‐host disease represent a death knell or a bump in the road? Cytotherapy 2013; 15:2–8. [DOI] [PubMed] [Google Scholar]
- 33. Panés J, García‐Olmo D, Van Assche G et al Expanded allogeneic adipose‐derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn's disease: a phase 3 randomised, double‐blind controlled trial. Lancet 2016; 6736:1–10. [DOI] [PubMed] [Google Scholar]
- 34. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011; 11:723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mills EL, O'Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti‐inflammatory signal. Eur J Immunol 2016; 46:13–21. [DOI] [PubMed] [Google Scholar]
- 36. Ortiz LA, Dutreil M, Fattman C et al Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proc Natl Acad Sci USA 2007; 104:11002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Braza F, Dirou S, Forest V, Sauzeau V, Hassoun D. Mesenchymal stem cells induce suppressive macrophages through phagocytosis in a mouse model of asthma. Stem Cells 2016; 34:1836–45. [DOI] [PubMed] [Google Scholar]
- 38. Ko JH, Lee HJ, Jeong HJ et al Mesenchymal stem/stromal cells precondition lung monocytes/macrophages to produce tolerance against allo‐ and autoimmunity in the eye. Proc Natl Acad Sci 2016; 113:158–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Selleri S, Bifsha P, Civini S et al Human mesenchymal stromal cell‐secreted lactate induces M2‐ macrophage differentiation by metabolic reprogramming. Oncotarget 2016; 7:30193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luz‐Crawford P, Djouad F, Toupet K et al Mesenchymal stem cell‐derived interleukin 1 receptor antagonist promotes macrophage polarization and inhibits B cell differentiation. Stem Cells 2016; 483–92. [DOI] [PubMed] [Google Scholar]
- 41. Sala E, Genua M, Petti L et al Mesenchymal stem cells reduce colitis in mice via release of TSG6, independently of their localization to the intestine. Gastroenterology 2015; 149:163–76. [DOI] [PubMed] [Google Scholar]
- 42. Liu Y, Zhang R, Yan K et al Mesenchymal stem cells inhibit lipopolysaccharide‐induced inflammatory responses of BV2 microglial cells through TSG‐6. J Neuroinflammation 2014; 11:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luk F, de Witte S, Korevaar SS et al Inactivated mesenchymal stem cells maintain immunomodulatory capacity. Stem Cells Dev 2016; 25:1342–54. [DOI] [PubMed] [Google Scholar]
- 44. Phinney DG, Di Giuseppe M, Njah J et al Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat Commun 2015; 6:8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jackson MV, Morrison TJ, Doherty DF et al Mitochondrial transfer via tunneling nanotubes is an important mechanism by which mesenchymal stem cells enhance macrophage phagocytosis in the in vitro and in vivo models of ARDS. Stem Cells 2016; 34:2210–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bartosh TJ, Ylostalo JH, Bazhanov N, Kuhlman J, Prockop DJ. Dynamic compaction of human mesenchymal stem/precursor cells (MSC) into spheres self‐activates caspase‐dependent IL1 signaling to enhance secretion of modulators of inflammation an immunity (PGE2, TSG6 and STC1). Stem Cells 2013; 100:130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee KC, Lin HC, Huang YH, Hung SC. Allo‐transplantation of mesenchymal stem cells attenuates hepatic injury through IL1Ra dependent macrophage switch in a mouse model of liver disease. J Hepatol 2015; 63:1405–12. [DOI] [PubMed] [Google Scholar]
- 48. Bustos ML, Huleihel L, Meyer EM et al Activation of human mesenchymal stem cells impacts their therapeutic abilities in lung injury by increasing interleukin (IL)‐10 and IL‐1RN levels. Stem Cells Transl Med 2013; 2:884–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gonzalez‐Rey E, Anderson P, González M, Rico L, Büscher D, Delgado M. Human adult stem cells derived from adipose tissue protect against experimental colitis and sepsis. Gut 2009; 58:929–39. [DOI] [PubMed] [Google Scholar]
- 50. González MA, Gonzalez‐Rey E, Rico L, Büscher D, Delgado M. Adipose‐derived mesenchymal stem cells alleviate experimental colitis by inhibiting inflammatory and autoimmune responses. Gastroenterology 2009; 136:978–89. [DOI] [PubMed] [Google Scholar]
- 51. Eggenhofer E, Steinmann JF, Renner P et al Mesenchymal stem cells together with mycophenolate mofetil inhibit antigen presenting cell and T cell infiltration into allogeneic heart grafts. Transpl Immunol 2011; 24:157–63. [DOI] [PubMed] [Google Scholar]
- 52. Eggenhofer E, Popp FC, Mendicino M et al Heart grafts tolerized through third party multipotent adult progenitor cells can be retransplanted to secondary hosts with no immunosuppression. Stem Cells Transl Med 2013; 2:595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Highfill SL, Kelly RM, Shaughnessy MJO et al Multipotent adult progenitor cells can suppress graft‐versus‐host disease via prostaglandin E 2 synthesis and only if localized to sites of allopriming. Blood 2009; 114:693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Auletta JJ, Eid SK, Wuttisarnwattana P et al Human mesenchymal stromal cells attenuate graft‐versus‐host disease and maintain graft‐versus‐leukemia activity following experimental allogeneic bone marrow transplantation. Stem Cells 2015; 33:601–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kavanagh H, Mahon BP. Allogeneic mesenchymal stem cells prevent allergic airway inflammation by inducing murine regulatory T cells. Allergy 2011; 66:523–31. [DOI] [PubMed] [Google Scholar]
- 56. Cruz FF, Borg ZD, Goodwin M et al Freshly thawed and continuously cultured human bone marrow‐derived mesenchymal stromal cells comparably ameliorate allergic airways inflammation in immunocompetent mice. Stem Cells Transl Med 2015; 4:615–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lathrop MJ, Brooks EM, Bonenfant NR et al Mesenchymal stromal cells mediate Aspergillus hyphal extract‐induced allergic airway inflammation by inhibition of the Th17 signaling pathway. Stem Cells Transl Med 2014; 3:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cruz FF, Borg ZD, Goodwin M et al CD11b+ and Sca‐1 cells exert the main beneficial effects of systemically administered bone marrow‐derived mononuclear cells in a murine model of mixed Th2/Th17 allergic airway inflammation. Stem Cells Transl Med 2016; 5:488–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Antunes MA, Abreu SC, Cruz FF et al Effects of different mesenchymal stromal cell sources and delivery routes in experimental emphysema. Respir Res 2014; 15:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gu W, Song L, Li X‐M, Wang D, Guo X‐J, Xu W‐G. Mesenchymal stem cells alleviate airway inflammation and emphysema in COPD through down‐regulation of cyclooxygenase‐2 via p38 and ERK MAPK pathways. Sci Rep 2015; 5:8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Asmussen S, Ito H, Traber DL et al Human mesenchymal stem cells reduce the severity of acute lung injury in a sheep model of bacterial pneumonia. Thorax 2014; 69:819–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xie Z, Hao H, Tong C et al Human umbilical cord‐derived mesenchymal stem cells elicit macrophages into an anti‐inflammatory phenotype to alleviate insulin resistance in type 2 diabetic rats. Stem Cells 2016; 34:627–39. [DOI] [PubMed] [Google Scholar]
- 63. Mathias LJ, Khong SML, Spyroglou L et al Alveolar macrophages are critical for the inhibition of allergic asthma by mesenchymal stromal cells. J Immunol 2013; 191:5914–24. [DOI] [PubMed] [Google Scholar]
- 64. Gupta N, Krasnodembskaya A, Kapetanaki M et al Mesenchymal stem cells enhance survival and bacterial clearance in murine Escherichia coli pneumonia. Thorax 2012; 67:533–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Devaney J, Horie S, Masterson C et al Human mesenchymal stromal cells decrease the severity of acute lung injury induced by E. coli in the rat. Thorax 2015; 70:625–35. [DOI] [PubMed] [Google Scholar]
- 66. Millar JE, Fraser JF, McAuley DF. Mesenchymal stromal cells and the acute respiratory distress syndrome (ARDS): challenges for clinical application. Thorax 2015; 70:611–2. [DOI] [PubMed] [Google Scholar]
- 67. Le Blanc K, Rasmusson I, Sundberg B, Hassan M, Uzunel M. Treatment of severe acute graft‐versus‐host disease with third party haploidentical mesenchymal stem cells. Lancet 2004; 363:1439–42. [DOI] [PubMed] [Google Scholar]
- 68. Kuci Z, Bonig H, Kreyenberg H et al Mesenchymal stromal cells generated from pooled mononuclear cells of multiple bone marrow donors as a rescue therapy for children with severe steroid‐refractory graft versus host disease: a multicenter survey. Haematologica 2016; 101:985–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kurtzberg J, Prockop S, Teira P et al Allogeneic human mesenchymal stem cell therapy (Remestemcel‐L, Prochymal) as a rescue agent for severe refractory acute graft‐versus‐host disease in pediatric patients. Biol Blood Marrow Transplant 2014; 20:229–35. [DOI] [PubMed] [Google Scholar]
- 70. Ball LM, Bernardo ME, Roelofs H et al Multiple infusions of mesenchymal stromal cells induce sustained remission in children with steroid‐refractory, grade III‐IV acute graft‐versus‐host disease. Br J Haematol 2013; 163:501–9. [DOI] [PubMed] [Google Scholar]
- 71. Krampera M, Cosmi L, Angeli R et al Role for interferon‐gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells 2006; 24:386–98. [DOI] [PubMed] [Google Scholar]
- 72. Eggenhofer E, Luk F, Dahlke MH, Hoogduijn MJ. The life and fate of mesenchymal stem cells. Front Immunol 2014; 5:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Schrepfer S, Deuse T, Reichenspurner H, Fischbein MP, Robbins RC, Pelletier MP. Stem cell transplantation: the lung barrier. Transplant Proc 2007; 39:573–6. [DOI] [PubMed] [Google Scholar]
- 74. Roy D, Steyer GJ, Gargesha M, Stone ME, Wilson DL. 3D cryo‐imaging: a very high‐resolution view of the whole mouse. Anat Rec 2009; 292:342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]