

Summary
Dendritic cells (DCs) play critical roles in initiating and regulating innate immunity as well as adaptive immune responses. However, the role of conventional dendritic cells (cDCs) in concanavalin A (ConA)‐induced fulminant hepatitis is unknown. In this study, we demonstrated that depletion of cDCs using either CD11c‐diphtheria toxin receptor transgenic mice (DTR Tg) mice or anti‐CD11c antibody reduced the severity of liver injury significantly, indicating a detrimental role of cDCs in ConA‐induced hepatitis. We elucidated further the pathological role of cDCs as being the critical source of interleukin (IL)‐12, which induced the secretion of interferon (IFN)‐γ by natural killer (NK) T cells. Reconstitution of cDCs‐depleted mice with IL‐12 restored ConA‐induced hepatitis significantly. Furthermore, we determined that NK T cells were the target of DC‐derived IL‐12, and NK T cells contributed to liver inflammation and injury through production of IFN‐γ. In summary, our study demonstrated a novel function of cDCs in mediating ConA‐induced hepatitis through regulating IFN‐γ secretion of NK T cells in an IL‐12‐dependent fashion. Targeting cDCs might provide potentially therapeutic applications in treating autoimmune related liver diseases.
Keywords: cDCs, IFN‐γ, IL‐12, hepatitis
Introduction
Liver diseases, including viral hepatitis, alcoholic liver disease and autoimmune hepatitis, affect millions of people worldwide. The high risk of hepatitis is due in part to the lack of complete understanding of its pathogenesis. T cell‐mediated immune responses play critical roles in a series of liver diseases, such as viral infections 1, autoimmune hepatitis (AIH) 2 and alcoholic‐related liver diseases 3. Concanavalin A (ConA)‐induced hepatitis is a well‐established model for investigating T cell‐dependent hepatitis in mice, which involves a series of hepatic cells and proinflammatory cytokines. Interferon (IFN)‐γ has been known to play an important role in ConA‐induced hepatitis 4, 5; in addition, CD4+ T cells and natural killer (NK) T cells have been shown to be implicated in the pathogenesis 6, 7, 8.
As critical regulators of both innate and adaptive immunity, DCs play important roles in immune responses 9, 10, 11. DCs are heterogeneous, differing in origin, location, function and migratory pathways 12. Infections or inflammatory stimuli can also affect their function and generation 13. Conventional dendritic cells (cDCs) are a DC subset with a dendritic form and exhibit DC functions in steady state. Predendritic cells (pre‐DCs) are cells with the capacity to develop into DCs, including plasmacytoid dendritic cells (pDCs) and monocytes 13. cDCs account for 1% of hepatic non‐parenchymal cells (NPC). Due to the relative scarcity of hepatic DC in normal mice, investigations have been limited. To investigate the role of cDCs, a conditional diphtheria toxin (DT)‐based cDCs depletion was generated in mice. Transgenic mice expressed the primate DT receptor (DTR) under the control of the DC‐specific CD11c promoter. A single administration of DT to these mice allowed the specificity and timing of CD11chigh cDCs ablation in vivo 14. Hepatic DCs are poised to orchestrate immune responses in a way that maintains tolerance to self 15 and be activated to induce T cell activation, cytokine and chemokine‐mediated immune responses. DCs from healthy liver exhibit a decreased ability to capture antigens and stimulate T cells; they are also reported to be less immunogenic than their splenic counterparts 16 and produce increased interleukin (IL)‐10 17, which might help to maintain allograft survival after liver transplantation. Lines of evidence suggest that hepatic DCs play a regulatory role in murine liver disease models. Using CD11c–DTR Tg mice, Bamboat et al. reported that during ischaemia/reperfusion (I/R), production of IL‐10 by cDCs suppressed inflammatory monocyte function and reduced liver injury 18. cDC depletion in DTR mice also exacerbates acetaminophen (APAP) hepatotoxicity in a manner independent of neutrophils, NK cells or inflammatory mediators 19. However, whether or not cDCs are involved in ConA‐mediated hepatitis, especially the regulation of NK T or CD4+ T cells, has not been investigated.
IL‐12 is composed of two subunits, namely p35 and p40, and secreted by a series of haematopoietic cells, mainly antigen‐presenting cells (APCs) such as DCs and macrophages 20. It binds to its receptor composed of IL‐12Rβ1 and IL‐12Rβ2 chains expressed on activated T cells, NK, NK T cells and DCs. Physiological responses to IL‐12 are mediated mainly through Janus‐associated kinase 2 (Jak2), tyrosine kinase 2 (Tyk2) and signal transducer and activator of transcription (STAT), especially STAT‐4 20, 21. The central role of IL‐12 is to induce the T helper type 1 (Th1) response. IL‐12 can promote the differentiation of naive T cells towards Th1 cells that enable them to produce IFN‐γ 22. In response to α‐galactosylceramide (α‐GalCer), NK T cells expressed IFN‐γ, which also required direct contact with DC and IL‐12 production by DCs 23. In hepatitis, neutralizing IL‐12 could diminish IFN‐γ in serum and alanine aminotransferase (ALT) levels 24. Blockade of IL‐12 activity leads to loss of early IFN‐γ production IL‐12 in mice infected with Leishmania major 25. IL‐12‐induced IFN‐γ also plays a critical part in the control of tumour growth 26.
In this study, we investigated the role of cDCs in T cell‐mediated acute liver injury. Using a model of ConA‐induced fulminant hepatitis in transgenic CD11c–diphtheria toxin receptor (DTR) mice, we observed that upon DT treatment, DTR mice were highly protected against ConA‐induced hepatitis, and we elucidated further the underlying mechanisms of cDC‐derived IL‐12 in promoting IFN‐γ production by NK T cells. Our experiment showed that interventions of DC or IL‐12 may be a potential therapy for hepatitis.
Materials and methods
Mice
C57BL/6J (B6) wild‐type (wt) mice were purchased from the Academy of Military Medical Science (Beijing, China). B6.FVB‐Tg [integrin subunit alpha X (Itgax)–DTR/enhanced green fluorescent protein (EGFP)] and 57Lan/J (B6 CD11c–DTR) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). CD1d–/– mice were kindly provided by Professor Li Bai (University of Science and Technology of China, Hefei, China). All experimental mice were maintained in a specific pathogen‐free facility at Nankai University (Tianjin, China) and used when they were 6–8 weeks old. All experiments were performed in accordance with guidelines for animal care, created by the Nankai University Experimental Animal Ethics Committee.
Reagents
ConA (type IV) was purchased from Sigma‐Aldrich (St Louis, MO, USA). Recombinant mouse (rm) granulocyte–macrophage colony‐stimulating factor (GM‐CSF) and rmIL‐4 were purchased from PeproTech (London, UK). Purified IL‐12 mAb (clone C17.8) was purchased from Sungene Biotech (Tianjin, China). rm IL‐12 was purchased from Biolegend (San Diego, CA, USA). rm IL‐2 was purchased from R&D Systems (Minneapolis, MN, USA). Fluorescein isothiocyanate (FITC)‐conjugated anti‐mouse T cell receptor (TCR)‐β (clone H57‐597), phycoerythrin (PE)‐conjugated CD69 (clone H1.2F3), natural killer group 2, member D (NKG2D) (clone CX5), CD11b (clone M1/70) and lymphocyte antigen 6 complex, locus C1 (Ly6c) (clone HK1.4) were purchased from BioLegend. PE‐conjugated IFN‐γ (clone XMG1.2), major histocompatibility complex (MHC) class II clone M5/114.15.2), FITC‐conjugated anti‐mouse CD4 (clone RM4‐5) and peridinin chlorophyll (PerCP)‐conjugated CD11c were purchased from Sungene Biotech. APC‐conjugated NK1.1 (clone PK136), CD11c (clone N418), Ly6g (clone IA8), IL‐12 and anti‐Gr‐1 (clone RB6‐8C5) were purchased from BioLegend.
T cell‐mediated hepatitis model
To induce hepatitis, we injected wt and DTR mice by intravenous injection (i.v.) with 10 mg/kg ConA from our laboratory, as described previously 7. The dose was increased to 25 mg/kg body weight to study the survival rate.
Assay for serum transaminase activity
Mice blood samples were obtained at different time‐points after ConA injection, maintained at room temperature for 2 h, followed by centrifugation at 2516 g for 8 min to separate the serum. Serum ALT activities were measured using an ALT test kit (Rong Sheng Biotech, Shanghai, China), according to the manufacturer's protocol.
Liver mononuclear cell preparation
Liver mononuclear cells were isolated by centrifugation in a Percoll gradient, as described previously 7.
Flow cytometry
For analysis of surface markers, cells were incubated in the dark for 15 min with antibodies. For IFN‐γ staining, cells were stimulated with 50 ng/ml phorbol myristate acetate (PMA) (Sigma‐Aldrich, St Louis, MO, USA), 0·5 μM ionomycin calcium salt (Sigma), in the presence of GolgiPlug (BD Biosciences, San Jose, CA, USA) for 4 h. Cells were first stained with antibodies in the dark for 15 min against surface markers, then fixed and permeabilized in BD Cytofix/Cytoperm solution (BD Biosciences), according to the manufacturer's protocol. All staining samples were acquired by BD fluorescence activated cell sorter (FACS)Calibur (BD Biosciences) and analysed by FlowJo software (TreeStar, Inc., Ashland, OR, USA).
Cytokine reconstitution and neutralization
A total recombinant (400 ng) of IL‐12 (BioLegend) in 200 μl phosphate‐buffered saline (PBS) was injected intraperitoneally (i.p.) into mice in 24 h and 2 h before ConA injections. PBS was given as control. For IL‐12 neutralization, a total of 1 mg anti mouse‐IL‐12 (clone C17.8; Sungene Biotech) was injected intraperitoneally (i.v.) 1 h before ConA treatment.
In‐vivo cell depletion
For depletion of cDCs, mice were i.p. injected with DT (4 ng/g body weight) 24 h before ConA treatment. In our preliminary experiments, the depletion of cDCs was more than 90% and lasted for 36 h. For depletion of NK1.1+ cells, mice were i.p. injected with 200 μg purified anti‐mouse NK1.1 mAb (Biolegend (clone PK136)) 24 h before ConA injections; for depletion of CD11c+ cells, mice were i.p. injected with 200 μg purified anti‐mouse CD11c mAb (clone N418) in 24 h and 2 h before ConA injections. Depletion efficiency was confirmed by flow cytometry.
Adoptive transfer of liver MNCs
The recipient mice were treated with anti‐NK1.1 monoclonal antibody (mAb) 72 and 48 h before adoptive transfer to deplete NK and NK T cells while not affecting the transferred mononuclear cells (MNCs) 27. Under isoflurane anaesthesia, hepatic MNCs (5 × 106 cells) suspended in 30 μl PBS were injected into the lateral left lobe of the liver using a 1‐ml insulin syringe (BD Biosciences, Franklin Lakes, New Jersey, USA), as described previously 28.
Enzyme‐linked immunosorbent assay (ELISA)
Mouse IL‐12, IFN‐γ, tumour necrosis factor (TNF)‐α, IL‐4, IL‐17 and IL‐10 ELISA kits were purchased from (BioLegend) and ELISA was performed according to the manufacturer's protocol.
Real‐time polymerase chain reaction (PCR)
Total RNA of liver monocular cells or sorted cells was extracted by TRIzol reagent (Invitrogen, Carlsbad, CA, USA). RNA was reverse‐transcribed using Tiangen reverse transcription reagent (Sungene Biotech). RNA expression was quantified by real‐time PCR. SYBR PreMix HotMaster Taq (Sungene Biotech) was used in real‐time PCR on a Mastercyclerep Realplex (Eppendorf, Hamburg, Germany). Results represent the relative expression level normalized against glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). Mouse genes were amplified by the following primers: T‐bet, 5′‐TTTCATTTGGGAAGCTAAAG‐3′ (forward), 5′‐GGCTGGTACTTGTGGAGAGA‐3′ (reverse); IL‐12p40, 5′‐ATGTGGAATGGCGTCTC‐3′ (forward), 5′‐GTCTCCTCGGCAGTTGG‐3′ (reverse); GAPDH, 5′‐TTGAGGTCAATGAAGGGGTC‐3′ (forward), 5′‐TCGTCCCGTAGACAAAATGG‐3′ (reverse). IFN‐γ, 5′‐CACAGTCATTGAAAGCCTAGA‐3′ (forward), 5′‐TTGCCAGTTCCTCCAGATAT‐3′ (reverse); FasL, 5′‐CAG CTT CAG ATG CAA GTG AGT GG‐3′ (forward), 5′‐CAA GGA CAG AAC TCT GAC GCT GAC‐3′ (reverse); IL‐23p19, 5′‐ACTCAGCCAACTCCTCC‐3′, 5′‐TCCTTGCCCTTCACG‐3′ (reverse); IL‐17a, 5′‐CACCGCAATGAAGACC‐3′ (forward), 5′‐CGAAGCAGTTTGGGAC3‐3′ (reverse) and 5′‐TTGCCAGTTCCTCCAGATAT‐3′ (reverse); IL‐12Rβ1, 5′‐TGAGTGCTCCTGGCAGTATG‐3′ (forward), 5′‐TATGGTTCGGAGGGACAAAG‐3′ (reverse). The results were analysed by Realplex software (version 5.01 for Windows. *P < 0·05, **P < 0·01 and ***P < 0·001 were used to show statistical significance throughout the text, figures and legends.
Results
Liver cDC depletion alleviates ConA‐induced fulminant hepatitis
To investigate the role of cDCs in hepatitis, age‐ and sex‐matched B6 wt and CD11c–DTR Tg mice were used. As expected, based on prior studies 14, 18, 19, 29, a single injection of DT depleted approximately 90% of hepatic cDCs in CD11c–DTR mice (Fig. 1a). Meanwhile, DT did not show any toxic effect to wt mice (data not shown). We then challenged sex‐ and age‐matched DT‐treated B6 wt and DTR mice with lethal doses of ConA (25 mg/kg) and survival rates of mice were observed and recorded. Surprisingly, in comparison with B6 wt mice, all DTR mice survived for 72 h (Fig. 1b), indicating an important role of cDCs in ConA‐induced liver damage. We then used lower concentrations of ConA in our experimental system to elucidate the mechanisms as titrated previously in our laboratory. Therefore, unless mentioned specifically, we used a low dose (10 mg/kg) of ConA for experiments in this study. Less severe hepatitis appeared in DTR mice than those in wt mice, as indicated by the serum ALT level (Fig. 1c). This was evidenced further by liver histopathology at 12 h after treatment of ConA. Significantly large areas of necrosis in liver tissues were observed in wt mice, while no visible histological changes were found in DTR mice (Fig. 1e). To define further the role of DCs in ConA‐induced hepatitis, B6 wt mice were administered anti‐CD11c antibodies, as described in Materials and methods. In our preliminary studies, this approach resulted in 95% of depletion of CD11c+ cells and lasted for 72 h. Depletion of CD11c+ DCs reduced hepatitis serum ALT significantly (Fig. 1d). Histological evaluation of the liver tissues after 12 h of ConA was consistent with serum ALT results, and wt mice displayed larger percentages of hepatocyte necrosis than those of CD11c cell‐depleted mice (Fig. 1e,f). These results demonstrate collectively a proinflammation role of cDCs in the ConA‐induced hepatitis model.
Figure 1.

Liver conventional dendritic cell (cDC) depletion alleviates concanavalin A (ConA)‐induced fulminant hepatitis. (a) Administration of diphtheria toxin (DT) depletes major histocompatibility complex (MHC) class II+CD11chigh conventional dendritic cell populations from CD11c‐diphtheria toxin receptor transgenic mice (DTR Tg) mice. Sex and age‐matched C57BL/6 wild‐type and CD11c–DTR Tg mice were treated with intraperitoneal (i.p.) injections of DT (4 ng/g body weight); 24 h before ConA injections, liver cells were isolated and used for flow cytometry analysis. Results from one of the representative experiments are shown. (b) Complete resistance against lethal dose of ConA in the absence of cDCs. Sex‐ and age‐matched C57BL/6 wild‐type (wt) and CD11c–DTR Tg mice were treated with DT as described above, followed by a lethal dose of ConA (25 mg/kg). Survival rate was monitored and recorded for 72 h. Data shown are the combined results from three experiments (n = 10 per each group). (c) Depletion of cDCs reduced the severity of ConA‐induced hepatitis significantly. Sex and age‐matched B6 wt and DTR mice (n = 5 or 7 for each group) were treated with DT as above, then injected intravenously with subdoses of ConA (10 mg/kg). Serum samples were obtained at different time‐points post‐ConA injection to be used for measuring the level of alanine aminotransferase (ALT). Results from one typical experiment of many repeating ones are shown. (d) Similar results were obtained when mice were treated with anti‐CD11c antibodies. Sex‐ and age‐matched B6 wt and CD11c–DTR Tg mice (n = 5 or 7 for each group) were treated with DT as above; some wt mice were also treated with anti‐CD11c antibodies to deplete CD11c+ cells. Three groups of mice were injected intravenously with ConA (10 mg/kg). Serum samples were obtained at different time‐points post‐ConA injection to be used for measuring the level of ALT. Results from one representative experiment are shown. (e) Histology analysis showed similar results. Liver tissues at 12 h post‐ConA treatment were fixed and embedded in paraffin for haematoxylin and eosin staining and one representative tissue staining is shown. N = necrosis area; scale bars = 200 μm. (f) The percentage of necrosis is calculated and shown; n = 3 mice/group. *P < 0·05; **P < 0·01; ***P < 0·001. [Colour figure can be viewed at wileyonlinelibrary.com]
Characterization of cDCs in the liver during ConA‐induced hepatitis
To define the characterization of cDCs (CD11chighMHC class II+) after hepatic injury, liver NPCs were isolated from liver at different time points post ConA treatment (0, 2, 6, 12 and 18 h). We showed that during the early stages of ConA induced hepatitis, the absolute number and percentage of cDCs decreased; however, the number increased from the time‐points of 6 h and later (Fig. 2a). Interestingly, a significant correlation was observed between the frequency of cDCs in the liver and ALT levels (Fig. 2b). Consistent with previous work by Bamboat et al. 18, the remaining cDCs in the liver were mature, with increased expressions of CD40, CD80 and CD86 (Fig. 2c). These suggested collectively that there is a significant correlation between cDCs and ConA‐induced hepatitis.
Figure 2.

Characterization of conventional dendritic cells (cDCs) in the liver during concanavalin A (ConA)‐induced hepatitis. C57BL/6 wild‐type (wt) mice were treated with ConA as described above. Mice were killed 0, 6, 12 and 18 h after ConA injections and hepatic mononuclear cells (MNCs) were collected. (a) Hepatic cDCs percentage decreased after ConA and began to recover at 6 h. Liver cDCs [CD11chighmajor histocompatibility complex II (MHC class II+)] percentages of different time‐points were assessed by flow cytometry and are shown. (b) There was a correlation of alanine aminotransferase (ALT) level and hepatic cDCs percentage or numbers after ConA. Serum was collected at different time‐points to measure the level of ALT. The correlation of ALT (black) and hepatic cDCs percentages or numbers (red) are shown. (c) The maturation level of hepatic cDCs increased after ConA. The relative fold of mean fluorescence intensity (MFI) of CD80, CD86 and CD40 expressed by cDCs after ConA was detected by flow cytometry. Data show the combined results from two experiments with similar results. *P < 0·05; **P < 0·01; ***P< 0·001. [Colour figure can be viewed at wileyonlinelibrary.com]
cDCs deficiency reduces the inflammatory cytokine storms upon ConA treatment
Various cytokines are involved in the pathogenesis of ConA‐induced hepatitis. To define the mechanisms of decreased liver injury in CD11c–DTR Tg mice, the inflammation cytokine levels were analysed in serum samples collected at different time‐points post‐ConA challenge. A remarkable cytokine storm was observed in wt mice upon ConA treatment. Notably, compared to wt mice, extremely decreased levels of IFN‐γ, TNF‐α, IL‐4 and IL‐12 in DTR mice were observed during 2–12 h upon ConA administration (Fig. 3). Interestingly, levels of IL‐10 and IL‐17 showed no significant changes upon cDC depletion. These results demonstrate collectively the role of cDCs in regulating inflammatory cytokine production in ConA‐mediated hepatitis.
Figure 3.
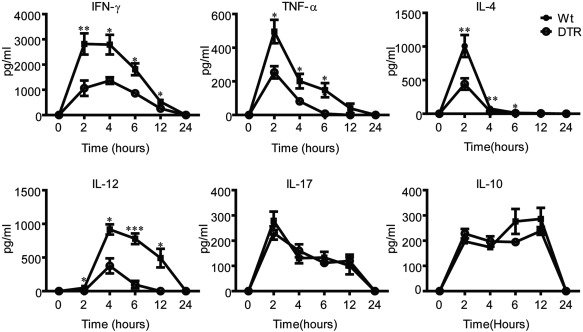
Conventional dendritic cells (cDCs) deficiency reduces the inflammatory cytokine storms upon concanavalin A (ConA) treatment. Serum interferon (IFN)‐γ and interleukin (IL)‐12 levels decreased in DTR mice after ConA injections. Sex and age‐matched C57BL/6 wild‐type (wt) and CD11c‐diphtheria toxin receptor transgenic (DTR Tg) mice (n = 6–10) were treated with diphtheria toxin (DT) as above, then injected intravenously with 10 mg/kg ConA. Blood serum was collected 0, 2, 4, 6, 12 and 24 h after ConA injections. The serum levels of IFN‐γ, tumour necrosis factor (TNF)‐α, IL‐4, IL‐12, IL‐10 and IL‐17 in C57BL/6 wt and CD11c–DTR Tg mice were detected by enzyme‐linked immunosorbent assay (ELISA). Data show the combined results from two experiments with similar results. *P < 0·05; **P < 0·01; ***P < 0·001.
cDC deficiency leads to decreased IL‐12 levels in DTR mice
To test the hypothesis that cDCs might be an important source of IL‐12, liver lymphocytes were isolated from wt and DTR mice at 2 h post‐ConA treatment and an array of cytokines were analysed by real‐time PCR. Consistently, reduced expressions of IL‐12 p40, IL‐12 receptor β and T‐bet were found in DTR mice; no significant changes were observed between these groups of mice for IL‐17 (Fig. 4a). To define further the cellular source of IL‐12, hepatic CD11c+ cells, CD11c–CD11b+ cells and CD11c–CD11b– cells were sorted from ConA‐challenged wt and DTR mice for IL‐12 p40 expression analysis. Upon DT treatment, CD11c+ cells from DTR mice decreased the expression level of IL‐12 p40 significantly (Fig. 4b). To test our hypothesis further, we directly examined cytokine expression of cDCs upon ConA treatment. Indeed, expression levels of IL‐12 p35 and p40 in cDCs was increased significantly, whereas no dramatic differences were seen in the expression levels of IL‐4 and IL‐10 (Fig. 4c). These results indicated that cDCs were the main source of IL‐12 in this model of acute liver injury.
Figure 4.

Conventional dendritic cells (cDCs) deficiency leads to decreased interleukin (IL)‐12 levels in diphtheria toxin receptor (DTR) mice. (a) Liver lymphocytes of CD11c–DTR transgenic (Tg) mice expressed significantly lower levels of IL‐12 and IL‐12 receptor (IL‐12r). Sex‐ and age‐matched C57BL/6 wild‐type (wt) and CD11c–DTR Tg mice (n = 6–8) were treated with diphtheria toxin (DT) and ConA as above, and liver mononuclear cells (MNCs) were isolated; gene expression was analysed by real‐time polymerase chain reaction (PCR). (b) CD11c+ cells in DTR mice, other than other cell subsets, expressed lower levels of IL‐12 than wt after ConA. Sex‐ and age‐matched B6 wt and DTR mice were treated with DT as mentioned above, then treated with ConA and killed 2 h after injection. CD11c+, CD11c–CD11b+ and CD11c–CD11b– cells from the two groups of mice were sorted for RNA isolation (10 mice pooled together per group) and gene expression of IL‐12 p40 was analysed by real‐time PCR. (c) The expression of IL‐12 in hepatic cDCs increased after ConA. C57BL/6 wt mice were treated with ConA or phosphate‐buffered saline (PBS) and killed 2 h after injections; cDCs were sorted (20 mice pooled together per group) for real‐time PCR analysis. Data are representative of two independent experiments. *P < 0·05; **P < 0·01; ***P < 0·001.
Essential role of IL‐12 in mediating IFN‐γ production of NK T cells in hepatitis
To test the role of IL‐12 in the pathogenesis of acute liver injury, sex‐ and age‐matched B6 wt and DTR mice were treated with DT as mentioned above, then these mice were treated with i.p. injections of either 400 ng rmIL‐12 or PBS followed by ConA injection. Liver lymphocytes were prepared 2 h post‐ConA treatment. IFN‐γ secretion by NK T and CD4+ T cells was analysed by flow cytometry. Upon DT treatment, numbers of IFN‐γ‐producing NK T cells decreased significantly, however, without affecting those of CD4+ T cells (Fig. 5a). Administration of IL‐12 recovered the IFN‐γ secretion fully only by NK T cells, but not from CD4+ T cells (Fig. 5a,b). We next tested further the effect of IL‐12 in ConA‐induced hepatitis; sex‐ and age‐matched B6 wt were treated with or without anti‐IL‐12 and DTR mice were treated with DT as above, followed with IL‐12 only, or IL‐12 plus anti‐NK1.1 antibodies. NK1.1 antibody depleted NK and NK T cells efficiently (Fig. 5c). Treatment with anti‐IL‐12 impaired ConA‐induced hepatitis significantly (Fig. 5d), confirming the role of IL‐12 in the pathogenesis of acute liver injury. Interestingly, administration of IL‐12 to DT‐treated DTR mice reconstitute the ConA‐induced hepatitis significantly, whereas depletion of NK T cells using NK1.1 antibodies neutralized the phenotype, indicating strongly the effect of IL‐12 on NK T cells (Fig. 5d). These results were confirmed further by liver histopathology sections (Fig. 5e,f). NK1.1 antibody also depleted NK cells (Fig. 5c); to exclude the possibility that NK cells in the liver are involved in this reaction, CD1d–/– mice were used. DTR mice were treated with DT, IL‐12 and anti‐NK1.1 antibody, then liver MNCs from wt mice or CD1d–/– mice were transferred to these mice. Hepatic MNCs from wt but not CD1d–/– mice could mediate liver injury in these mice, shown by significantly increased serum ALT levels (Fig. 5g) and bigger necrosis areas (Fig. 5h,i), suggesting that NK T cells were the target of IL‐12 in hepatitis.
Figure 5.

Essential role of interleukin (IL)‐12 in mediating interferon (IFN)‐γ production of natural killer T cells (NKT cells) in hepatitis. (a) The ability of NKT cells to produce IFN‐γ after concanavalin A (ConA) treatment was abolished in CD11c‐diphtheria toxin receptor transgenic (DTR Tg) mice, which can be restored by IL‐12 reconstitution. Sex and age‐matched C57BL/6 wild‐type (wt) and CD11c–DTR Tg mice were treated with diphtheria toxin (DT) as mentioned above, then these mice (n = 5–8) were treated with intraperitoneal (i.p.) injections of either 400 ng recombinant mouse (rm)IL‐12 or phosphate‐buffered saline (PBS) followed by ConA injection. Liver lymphocytes were prepared 2 h after ConA for intracellular cytokine staining. NKT cells [gated on T cell receptor (TCR) β+ NK1.1+] and CD4+ T cells (CD4+ NK1.1–) were analysed for IFN‐γ production by flow cytometry. Statistical analysis of IFN‐γ‐secreting NK T and CD4+ T cells of wt, DTR and IL‐12‐treated DTR mice are shown. (b) One representative staining result is shown. (c) NK1.1 antibody depleted NK and NK T cells effectively. The liver mononuclear cells (MNCs) of NK1.1 antibody‐ or PBS‐treated mice were analysed by flow cytometry, NKT cells (gated on CD3+ NK1.1+) and NK cells (gated on CD3–NK1.1+) were depleted in NK1.1 antibody‐treated mice. (d,e) IL‐12 played a proinflammation role in hepatitis, which was dependent upon NK1.1+ cells. Sex‐ and age‐matched B6 wt were treated with or without anti‐IL‐12, and DTR mice were treated with diphtheria toxin (DT) as above, followed with IL‐12 only or IL‐12 plus anti‐NK1.1 antibodies. Alanine aminotransferase (ALT) values of five groups of mice were measured at different time‐points after ConA injections. (f) Similar results were obtained by histology analysis. Five groups of mice were killed 12 h after ConA, liver tissues were collected, fixed and subjected to haematoxylin and eosin (H&E) staining. The percentage of necrosis is calculated and shown and one representative tissue staining is shown. N = necrosis area; scale bars = 200 μm; n = 3 mice/group. (g) NKT cells were the target of IL‐12 in hepatitis. Sex and age‐matched DTR mice were treated with DT, IL‐12 and anti‐NK1.1 antibodies as mentioned above. Liver MNCs from wt mice or CD1d–/– mice were transferred to these mice as described in the Materials and methods section. PBS was given as control. These mice were injected intravenously with ConA (10 mg/kg). ALT values of three groups of mice were measured at different time‐points after ConA injections. (h,i) Similar results were obtained by histology analysis. Three groups of mice were killed 12 h after ConA, liver tissues were collected, fixed and subjected to haematoxylin and eosin (H&E) staining. The percentage of necrosis is calculated and shown and one representative tissue staining is shown. N = necrosis area; scale bars, 200 μm; n = 3 mice/group. *P < 0·05; **P < 0·01; ***P< 0·001; n.s. = not significant. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
Acute liver failure is a devastating liver disease with significant mortality and without an effective therapeutic approach. ConA‐induced fulminant hepatitis is a well‐used model for studying the pathophysiology mechanisms of acute liver injury 6. DCs have been well defined in orchestrating both innate and adaptive immunity 30, 31; however, the role of DC in ConA‐induced hepatitis has been elusive. In this report we demonstrated for the first time, to our knowledge, that DCs, especially cDCs, played a detrimental role in ConA‐induced liver injury through IL‐12 production which, in turn, regulates NK T cell function, especially the production of IFN‐γ.
One of the key findings in our study is establishing for the first time, to our knowledge, a unique role of cDCs in ConA‐induced hepatitis. DCs are not only the primary APCs, but also regulate immune response through secreting an array of inflammatory cytokines 32, 33, 34, 35, 36. However, the role of DCs in acute liver injury has not yet been reported. Using either CD11c–DTR Tg mice or anti‐CD11c antibodies to deplete CD11c+ DCs, we demonstrated collectively that DCs played a detrimental role in promoting ConA‐induced liver injury (Fig. 1). DCs have many subsets, with divergent functions 30, 31, 36, 37, 38. Administration of DT depletes CD11chighMHC class II+ conventional DCs (cDCs) selectively (Fig. 1a), indicating a predominant role of this subset in increasing the susceptibility of ConA‐induced acute hepatitis. It has to be emphasized that the role of other subsets of DCs, such as pDC, which produce predominantly IFN‐α 39, 40, remains to be studied further.
We next addressed the question concerning how cDCs mediated the ConA‐induced liver injury. We hypothesized that these cDCs would act through producing cytokines. Indeed, our study established firmly that it was IL‐12 deriving from cDCs that played a critical role in mediating the detrimental role in ConA‐induced acute hepatitis. Our conclusion was supported by results obtained from multiple approaches. First, both systemic IL‐12 level and liver tissue IL‐12 gene expression increased significantly upon ConA treatment. In contrast, upon DT treatment, the expression level of IL‐12 reduced dramatically (Figs 3 and 4a), implying that cDCs are the main source of IL‐12. Secondly, neutralizing IL‐12 with anti‐IL‐12 antibody ameliorated ConA‐induced inflammation (Fig. 5c). Finally, administration of recombinant IL‐12 into DT‐treated DTR Tg mice showed that these mice regained susceptibility (Fig. 5c). Our results thus define a critical role of cDCs in providing the source of IL‐12 in ConA‐induced hepatitis.
One of the essential functions of IL‐12 is inducing IFN‐γ production by a few cell types, including CD4+ T cells, NK and T cells 22, 41, 42. We wished to determine the source of IFN‐γ induced by IL‐12 in the ConA‐induced hepatitis model. Although our previous studies revealed that IFN‐γ produced by either CD4+ T cells or NK T cells was involved in the pathogenesis of ConA‐induced liver injury depending the immune environment 7, 8, interestingly, we provided solid evidence that DC‐derived IL‐12 triggered only NK T cells producing IFN‐γ, without affecting the function of CD4+ T cells (Fig. 5a,b). This conclusion was supported with results obtained from in‐vivo studies, with either reconstitution with IL‐12 in DTR mice or depletion of NK T cells in IL‐12 treated DTR mice (Fig. 5d–f). In addition, adoptively transferred liver MNCs from wt but not CD1d–/– mice to IL‐12 and anti‐NK1.1 antibody‐treated DTR mice confirmed further that NK T cells are the target of IL‐12 in hepatitis, excluding the possibility that NK cells are involved in the reaction (Fig. 5g–i). We do not yet understand fully the underlying mechanisms of why DC‐derived IL‐12 in this special circumstance acts on only NK T cells. One possibility is the differential threshold of activation between CD4+ T cells and NK T cells, with a better response of NK T cells to lower concentrations of IL‐12. Other possibilities might include the response to other ConA or ConA‐induced signalling pathways. Further studies are needed to clarify the matter and it would lead to clearer understanding of the pathogenesis of this model.
Therefore, our results established the detrimental role of cDCs in T cell‐dependent hepatitis. cDCs are the critical source of IL‐12, and IL‐12 can target NK T cells to regulate their IFN‐γ production, a mechanism different from other liver injury models mentioned above. Our finding could also shed light upon developing novel therapeutic approaches targeting cDCs in hepatitis.
Disclosure
None.
Author contributions
J. W. conceived the project, designed and performed all experiments, analysed data and wrote the manuscript. X. C., J. Z., Q. L., X. Q. and L. W. performed animal experiments and analysed data. Z. Y. and H. Z. helped to perform the cell cultures. J. W. and H. Z. helped conceive the project. L. B. kindly gave us CD1d–/– mice. Z. W., L. Z. and Z. H. supervised and coordinated this research. Z. Y. helped conceive the project, wrote the manuscript, mentored and supervised its participants.
Acknowledgements
This work was supported by the Key Program of the National Natural Science Foundation of China (Grant no. 31230025), National High Technology Research and Development Program of China (863 Program, Grant no. SS2014AA021601), Tianjin Technology Research Funding (08QTPTJC28400) to Z. Y. and grants from the National Natural Science Foundation of China (31270926) to Z. H.
References
- 1. Kazmierczak J, Caraballo Cortes K, Bukowska‐Osko I, Radkowski M. Virus‐specific cellular response in hepatitis C virus infection. Arch Immunol Ther Exp (Warsz) 2016; 64:101–10.] [DOI] [PubMed] [Google Scholar]
- 2. Czaja AJ. Current concepts in autoimmune hepatitis. Ann Hepatol 2005; 4:6–24. [PubMed] [Google Scholar]
- 3. McVicker BL, Thiele GM, Tuma DJ, Casey CA. Hepatocyte‐mediated cytotoxicity and host defense mechanisms in the alcohol‐injured liver. Hepatol Int 2014; 8:432–8. [DOI] [PubMed] [Google Scholar]
- 4. Tagawa Y, Sekikawa K, Iwakura Y. Suppression of concanavalin A‐induced hepatitis in IFN‐gamma(–/–) mice, but not in TNF‐alpha(–/–) mice: role for IFN‐gamma in activating apoptosis of hepatocytes. J Immunol 1997; 159:1418–28. [PubMed] [Google Scholar]
- 5. Jaruga B, Hong F, Kim WH, Gao B. IFN‐gamma/STAT1 acts as a proinflammatory signal in T cell‐mediated hepatitis via induction of multiple chemokines and adhesion molecules: a critical role of IRF‐1. Am J Physiol Gastrointest Liver Physiol 2004; 287:G1044–52. [DOI] [PubMed] [Google Scholar]
- 6. Wang HX, Liu M, Weng SY et al Immune mechanisms of concanavalin A model of autoimmune hepatitis. World J Gastroenterol 2012; 18:119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao N, Hao J, Ni Y et al Vgamma4 gammadelta T cell‐derived IL‐17A negatively regulates NKT cell function in Con A‐induced fulminant hepatitis. J Immunol 2011; 187:5007–14. [DOI] [PubMed] [Google Scholar]
- 8. Zhang S, Liang R, Luo W et al High susceptibility to liver injury in IL‐27 p28 conditional knockout mice involves intrinsic interferon‐gamma dysregulation of CD4+ T cells. Hepatology 2013; 57:1620–31. [DOI] [PubMed] [Google Scholar]
- 9. Huarte E, Cubillos‐Ruiz JR, Nesbeth YC et al Depletion of dendritic cells delays ovarian cancer progression by boosting antitumor immunity. Cancer Res 2008; 68:7684–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuwajima S, Sato T, Ishida K, Tada H, Tezuka H, Ohteki T. Interleukin 15–dependent crosstalk between conventional and plasmacytoid dendritic cells is essential for CpG‐induced immune activation. Nat Immunol 2006; 7:740–6. [DOI] [PubMed] [Google Scholar]
- 11. Boscardin SB, Hafalla JC, Masilamani RF et al Antigen targeting to dendritic cells elicits long‐lived T cell help for antibody responses. J Exp Med 2006; 203:599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol 2002; 2:151–61. [DOI] [PubMed] [Google Scholar]
- 13. Shortman K, Naik SH. Steady‐state and inflammatory dendritic‐cell development. Nat Rev Immunol 2007; 7:19–30. [DOI] [PubMed] [Google Scholar]
- 14. Jung S, Unutmaz D, Wong P et al In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell‐associated antigens. Immunity 2002; 17:211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morelli AE, Thomson AW. Dendritic cells: regulators of alloimmunity and opportunities for tolerance induction. Immunol Rev 2003; 196:125–46. [DOI] [PubMed] [Google Scholar]
- 16. Pillarisetty V, Shah A, Bleier J, DeMatteo R. Liver dendritic cells are less immunogenic than spleen dendritic cells because of differences in subtype composition. J Immunol 2004; 172:1009–17. [DOI] [PubMed] [Google Scholar]
- 17. Thomson AW, Knolle PA. Antigen‐presenting cell function in the tolerogenic liver environment. Nat Rev Immunol 2010; 10:753–66. [DOI] [PubMed] [Google Scholar]
- 18. Bamboat ZM, Ocuin LM, Balachandran VP, Obaid H, Plitas G, DeMatteo RP. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL‐10 secretion. J Clin Invest 2010; 120:559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Connolly MK, Ayo D, Malhotra A et al Dendritic cell depletion exacerbates acetaminophen hepatotoxicity. Hepatology 2011; 54:959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gee K, Guzzo C, Che Mat NF, Ma W, Kumar A. The IL‐12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm Allergy Drug Targets 2009; 8:40–52. [DOI] [PubMed] [Google Scholar]
- 21. Morinobu A, Gadina M, Strober W et al STAT4 serine phosphorylation is critical for IL‐12‐induced IFN‐gamma production but not for cell proliferation. Proc Natl Acad Sci USA 2002; 99:12281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seder RA, Gazzinelli R, Sher A, Paul WE. Interleukin 12 acts directly on CD4+ T cells to enhance priming for interferon gamma production and diminishes interleukin 4 inhibition of such priming. Proc Natl Acad Sci USA 1993; 90:10188–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kitamura H, Iwakabe K, Yahata T et al The natural killer T (NKT) cell ligand alpha‐galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)‐12 production by dendritic cells and IL‐12 receptor expression on NKT cells. J Exp Med 1999; 189:1121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nicoletti F, Di Marco R, Zaccone P et al Murine concanavalin A‐induced hepatitis is prevented by interleukin 12 (IL‐12) antibody and exacerbated by exogenous IL‐12 through an interferon‐gamma‐dependent mechanism. Hepatology 2000; 32:728–33. [DOI] [PubMed] [Google Scholar]
- 25. Scharton‐Kersten T, Afonso LC, Wysocka M, Trinchieri G, Scott P. IL‐12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental leishmaniasis. J Immunol 1995; 154:5320–30. [PubMed] [Google Scholar]
- 26. Tugues S, Burkhard SH, Ohs I et al New insights into IL‐12‐mediated tumor suppression. Cell Death Differ 2015; 22:237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen Y, Wei H, Sun R, Dong Z, Zhang J, Tian Z. Increased susceptibility to liver injury in hepatitis B virus transgenic mice involves NKG2D–ligand interaction and natural killer cells. Hepatology 2007; 46:706–15. [DOI] [PubMed] [Google Scholar]
- 28. Takeda K, Hayakawa Y, Van Kaer L, Matsuda H, Yagita H, Okumura K. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci USA 2000; 97:5498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim JH, Choi JY, Kim SB et al CD11c(hi) dendritic cells regulate Ly‐6C(hi) monocyte differentiation to preserve immune‐privileged CNS in lethal neuroinflammation. Sci Rep 2015; 5:17548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hsu W, Shu SA, Gershwin E, Lian ZX. The current immune function of hepatic dendritic cells. Cell Mol Immunol 2007; 4:321–8. [PubMed] [Google Scholar]
- 31. Liu J, Cao X. Regulatory dendritic cells in autoimmunity: a comprehensive review. J Autoimmun 2015; 63:1–12. [DOI] [PubMed] [Google Scholar]
- 32. Pillarisetty VG, Katz SC, Bleier JI, Shah AB, Dematteo RP. Natural killer dendritic cells have both antigen presenting and lytic function and in response to CpG produce IFN‐gamma via autocrine IL‐12. J Immunol 2005; 174:2612–8. [DOI] [PubMed] [Google Scholar]
- 33. Theiner G, Rossner S, Dalpke A et al TLR9 cooperates with TLR4 to increase IL‐12 release by murine dendritic cells. Mol Immunol 2008; 45:244–52. [DOI] [PubMed] [Google Scholar]
- 34. Stober D, Schirmbeck R, Reimann J. IL‐12/IL‐18‐dependent IFN‐gamma release by murine dendritic cells. J Immunol 2001; 167:957–65. [DOI] [PubMed] [Google Scholar]
- 35. von Stebut E, Belkaid Y, Nguyen BV, Cushing M, Sacks DL, Udey MC. Leishmania major‐infected murine Langerhans cell‐like dendritic cells from susceptible mice release IL‐12 after infection and vaccinate against experimental cutaneous Leishmaniasis. Eur J Immunol 2000; 30:3498–506. [DOI] [PubMed] [Google Scholar]
- 36. Hochrein H, Shortman K, Vremec D, Scott B, Hertzog P, O'Keeffe M. Differential production of IL‐12, IFN‐alpha, and IFN‐gamma by mouse dendritic cell subsets. J Immunol 2001; 166:5448–55. [DOI] [PubMed] [Google Scholar]
- 37. Jung S. Good, bad and beautiful – the role of dendritic cells in autoimmunity. Autoimmun Rev 2004; 3:54–60. [DOI] [PubMed] [Google Scholar]
- 38. Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol 2013; 13:566–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu YJ. IPC: professional type 1 interferon‐producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol 2005; 23:275–306. [DOI] [PubMed] [Google Scholar]
- 40. Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol 2008; 8:594–606. [DOI] [PubMed] [Google Scholar]
- 41. Guilmot A, Bosse J, Carlier Y, Truyens C. Monocytes play an IL‐12‐dependent crucial role in driving cord blood NK cells to produce IFN‐g in response to Trypanosoma cruzi . PLOS Negl Trop Dis 2013; 7:e2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jaime‐Ramirez AC, Mundy‐Bosse BL, Kondadasula S et al IL‐12 enhances the antitumor actions of trastuzumab via NK cell IFN‐gamma production. J Immunol 2011; 186:3401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]