

Summary
Intestinal mucositis is a serious complication of chemotherapy that leads to significant morbidity that may require dose or drug adjustments. Specific mitigating strategies for mucositis are unavailable, due partly to an incomplete understanding of the pathogenic mechanisms. We have previously shown an effect of properdin, a positive regulator of complement activation, in models of colitis. Here we use properdin‐deficient (PKO) mice to interrogate the role of properdin and complement in small intestinal mucositis. Mucositis was induced by five daily injections of 5‐fluorouracil (5‐FU) in wild‐type (WT), PKO, interleukin (IL)‐10–/– and properdin/IL‐10–/– double knock‐out (DKO) mice. At the time of euthanasia their jejunum was collected for histology, immunohistochemistry and cytokine and complement activation measurements. Complement became activated in mice receiving 5‐FU, indicated by increased intestinal levels of C3a and C5a. Compared to WT, PKO mice experienced significantly less mucositis, despite C3a levels as high as inflamed WT mice and slightly less C5a. Conversely, PKO mice had higher intestinal levels of IL‐10. IL‐10 expression was mainly by epithelial cells in both uninflamed and inflamed PKO mice. IL‐10–/– mice proved to be highly susceptible to mucositis and DKO mice were equally susceptible, demonstrating that a lack of properdin does not protect mice lacking IL‐10. We interpret our findings to indicate that, to a significant extent, the inflammation of mucositis is properdin‐dependent but complement activation‐independent. Additionally, the benefit achieved in the absence of properdin is associated with increased IL‐10 levels, and IL‐10 is important in limiting mucositis.
Keywords: anaphylatoxin, chemotherapy, mucositis, properdin, 5‐fluorouracil
Introduction
Gastrointestinal mucositis is a frequent and severe side effect of chemotherapy in cancer patients. Depending on the dose and type of chemotherapy, between 50 and 80% of the patients suffer from mucositis that results in vomiting, severe diarrhoea and abdominal pain 1, 2. In particular, small intestinal mucositis leads to reductions in the drug dosage and delayed treatment that risks compromising the effectiveness of chemotherapy 3, 4. At the histological level, small intestinal mucositis is characterized by epithelial cell death, villi shortening, crypt damage and leucocyte infiltration 4. Moreover, intestinal mucositis predisposes to infections that can lead to further significant morbidity and mortality 5, 6. Despite research into possible drugs to directly mitigate mucositis none are available as yet in the clinic, so there remains an urgent need to understand more clearly the underlying pathogenic mechanisms to focus the effort 7.
Recent studies using animal models have implicated both the innate and adaptive immune systems mechanistically in the inflammation underlying chemotherapy‐induced mucositis 8, 9, 10. Complement is a crucial component of the innate immune system that can lead to inflammation, but can also modulate the development of the emerging adaptive immune response 11, 12, yet a role for complement, particularly the pattern recognition receptors of the lectin and alternative pathways, has not been explored directly in mucositis. Our understanding is limited to a report where mRNA for proteins belonging to the classical and alternative pathway were found up‐regulated in intestines of rats subjected to irinotecan‐induced mucositis 13.
Properdin, a protein of the alternative pathway, is a positive regulator of complement that can initiate and amplify ongoing complement activation 14. Because the loss of properdin abolishes terminal complement activation in multiple models of intestinal injury 15, 16, we thought to use properdin‐deficient mice to explore the contribution of complement to 5‐fluorouracil (5‐FU)‐induced mucositis. Surprisingly, loss of properdin did not abolish terminal complement activation but was associated with reduced pathology compared to 5‐FU inflamed wild‐type (WT) controls. We further demonstrate that the protection in the absence of properdin is probably dependent upon interleukin (IL)‐10. Overall, we establish a previously unappreciated complement activation‐independent, proinflammatory role of properdin in this model of intestinal mucositis.
Materials and methods
Mice
Age‐ and sex‐matched WT, properdin knock‐out (PKO), IL‐10–/– (Jackson Laboratories, Bar Harbour, ME, USA) and IL‐10/properdin double gene knock‐out (DKO, developed locally) mice, all on the C57BL/6 background and of both sexes, were used. Mice had free access to food and water and were housed on woodchip bedding under specific pathogen‐free conditions on a 12‐h dark/light cycle. All animals used were Helicobacter‐negative, assayed by polymerase chain reaction (PCR) on stool DNA preparations. Helicobacter triggers colitis in susceptible strains of mice, including IL‐10 gene knock‐out animals 17. The numbers of mice used in each experiment is indicated in each figure. The experiments were undertaken with the approval of the University Committee on Laboratory Animals, Dalhousie University, who in turn adjudicate the standards of the Canadian Council on Animal Care.
5‐FU‐induced small intestinal mucositis
Mucositis was induced by administering 5‐FU once daily beginning on day 1 to either days 3 or 5. PBS‐injected mice were treated as vehicle controls. The study began with optimizing the dose of 5‐FU, examining 50, 100 and 200 mg/kg, delivered intraperitoneally. The animals' weight was recorded daily and any mice that reached a loss of 20% were killed immediately and were excluded from the data analyses. In some experiments, 5‐FU‐injected WT mice were gavaged orally with the selective C5a receptor (C5aR1) antagonist PMX205 15 (200 μg/mouse) or water (control) daily from days 1 to 5. One day after the final injection of 5‐FU, a stool specimen was collected and a rectal bleeding score was assigned based on stool consistency and whether blood was detectable, with the scale ranging from 0 (normal stool with no blood) to 4 (diarrhoea with blood). The mice were then anaesthetized and blood was withdrawn by cardiac puncture and serum was isolated. The mice were then killed by cervical dislocation, their jejunum was excised, flushed with cold phosphate‐buffered saline (PBS), opened longitudinally and divided into two parts. One longitudinal part was used for histology and the other part was homogenized.
Histopathology
The longitudinal strip of jejunum was fixed in buffered formalin overnight, rehydrated in 70% v/v ethanol, embedded in paraffin and 4‐μm sections were stained using haematoxylin and eosin. A pathologist blinded to the treatments determined the inflammation score summed from the following parameters: villous shortening (0 = normal, 1 = mild blunting, 2 = marked blunting), apoptosis (0 = none, 1 = focal, 2 = diffuse), epithelial damage (0 = normal, 1 = reactive epithelial change, 2 = superficial erosion, 3 = ulceration), inflammation (0 = none, 1 = mild, 2 = moderate, 3 = severe) and crypt loss (0 = none, 1 = focal, 2 = diffuse).
Enzyme‐linked immunosorbent assay (ELISA) measurements
The remaining longitudinal strip of the jejunum was homogenized in 50 mM HEPES buffer supplemented with soy trypsin inhibitor (100 µg/ml) and centrifuged at 16,000 g for 30 min at 4ºC, after which the supernatants were stored at −80°C until examined by ELISA. Interferon (IFN)‐γ, IL‐1β and IL‐10 concentrations were measured using ELISA kits from eBiosciences (San Diego, CA, USA). C3a and C5a levels were assessed using reagents from BD Pharmingen (Mississauga, ON, Canada). IL‐4 and IL‐6 were measured using ELISA kits from Peprotech (Dollard, QC, Canada). All the protocols were performed according to the manufacturer's instructions.
Immunohistochemistry
Four‐μm‐thick paraffin embedded sections were deparaffinized in xylene and graded ethanol washes. To detect neutrophils, antigen retrieval was performed by heat in 10 mM sodium citrate buffer (pH = 6·0) and the slides incubated with rat anti‐mouse Ly6G (1 : 4000; BD Pharmingen, San Jose, CA, USA). For the detection of IL‐10, antigen retrieval was performed by heating in Tris‐ethylenediamine tetraacetic acid (EDTA) buffer (pH = 6·0) and sections were incubated subsequently with a blocking buffer [2% goat serum, 1% bovine serum albumin (BSA), 0·1% Triton X‐100, 0·05% Tween 20 and 0·05% sodium azide in PBS] for 1 h at room temperature (RT) to block non‐specific binding sites. Tissue sections were then incubated with rat monoclonal anti‐mouse IL‐10 antibody (clone JES5‐2A5; Abcam, Cambridge, MA, USA) as the primary antibody. Goat anti‐rat immunoglobulin (Ig)G secondary antibody was used to detect the primary antibodies. Isotype‐specific control monoclonal antibodies and tissues from IL‐10–/– mice were used to confirm the specificity of the staining. Positively stained cells were counted in the remaining intact villi only.
Statistical analyses
Statistical analyses were performed using GraphPad Prism version 5 (La Jolla, CA, USA). Parametric data are shown as mean ± standard deviation and were compared using two‐tailed t‐test (two groups) or one‐way analysis of variance (anova) with Tukey's multiple comparison post‐test (more than two groups). Body weights were compared using two‐way anova with Bonferroni post‐hoc tests. Inflammation scores are shown in scatter‐plots with the median as a line. Differences between groups were tested using the non‐parametric Mann–Whitney U‐test. The threshold for declaring a significant difference was defined as P ≤ 0·05.
Results
Titration of 5‐FU‐induced mucositis in mice
In a pilot experiment to determine a dose of 5‐FU for the study, WT mice were injected intraperitoneally with 50, 100 or 200 mg/kg/day or PBS for 5 days then killed 24 h later (day 6). All mice receiving 5‐FU lost weight during the period of the experiment. The average maximum body weight loss in mice receiving 50 and 100 mg/kg/day 5‐FU was 10 and 19%, respectively (Fig. 1a). Mice treated with 200 mg/kg lost close to 20% body weight by day 5 and were killed; this dose was not considered further, nor were mice which reached this unintended end‐point used in any further data sets.
Figure 1.
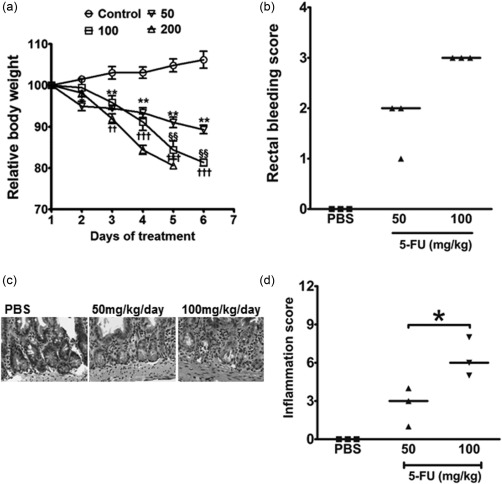
5‐Fluorouracil (5‐FU) titration and clinical illness and histopathological phenotypes in wild‐type (WT) mice. Mice were administered 5‐FU daily, to day 5, and were then killed 24 h after the final injection. (a) Each animal's weight was measured daily and is reported as the average percentage of the weight from the start of the treatments. Data are shown as mean ± standard error of the mean (s.e.m.) (n = 3 mice/group). *,†Comparison of 50 and 100 mg/kg groups with no 5‐FU controls on the same day, respectively. §Indicative of a difference between 50 and 100 mg/kg groups. *P < 0·05; **P < 0·01; ††P < 0·01; †††P < 0·001 and §§P < 0·01. (b) Rectal bleeding scores. Each dot represents a single mouse and the line represents the median score. Any animal whose weight loss reached 20% was not included in further data collection or analyses. (c) Representative images of the jejunum prepared from phosphate‐buffered saline (PBS)‐ or 5‐FU‐treated mice. Jejunum of mice receiving 5‐FU at a dose of 100 mg/kg demonstrated severe villus height shortening, epithelial damage, crypt loss and inflammation. (d) The total inflammation scores were calculated based on criteria outlined in the methods. Each dot represents a single mouse and the line represents the median inflammation score. *P < 0·05 comparing the 50 versus 100 mg/kg groups.
As another macroscopic measure of the treated animals' phenotype, the stool consistency was evaluated on day 6. PBS‐injected WT mice had firm faecal pellets and no rectal bleeding. Treatment with 5‐FU resulted in soft stool pellets and blood in the stool, with the rectal bleeding score higher in the 100 mg/kg group compared to PBS controls (Fig. 1b). Microscopic examination of the small intestines of mice treated with 100 mg/kg 5‐FU revealed extensive epithelial damage, crypt loss and villi shortening, and an inflammation score statistically higher in the 100 mg/kg 5‐FU treatment group compared to the 50 mg/kg group and/or PBS‐treated controls (Fig. 1c,d). Based on these clinical and histopathological responses the 100 mg/kg 5‐FU dose was chosen for experiments including the PKO mice.
Genetic deficiency of properdin ameliorates 5‐FU‐induced intestinal mucositis
To evaluate whether properdin and complement play a role in our model of mucositis we compared the response of WT and PKO mice to the 100 mg/kg 5‐FU treatment regimen. As shown in Fig. 2a, 5‐FU treatment led to comparable body weight losses in the two genotypes. Despite weight loss, PKO mice had significantly less rectal bleeding compared to the WT mice (Fig. 2b). Consistent with the bleeding score data, 5‐FU inflamed PKO mice also experienced significantly lower inflammation scores compared to inflamed WT mice (Fig. 2c,d). It is noteworthy that, despite the lower inflammation score in PKO mice, the numbers of neutrophils infiltrating the small intestine was not different between the strains (Fig. 2e). In addition to neutrophil infiltration, a reduction in goblet cell numbers is another hallmark of intestinal mucositis 18, 19. We stained tissue sections with periodic acid‐Schiff stain to enumerate goblet cells. The two untreated mouse strains had similar numbers of small intestinal goblet cells, and while both experienced a reduction in numbers with 5‐FU treatment (Fig. 2f), inflamed PKO mice registered significantly higher numbers compared to inflamed WT controls. Thus, the preservation of goblet cell numbers in PKO mice was associated with the evidence for reduced inflammation. Overall, these results indicate that PKO mice have an attenuated inflammatory phenotype in 5‐FU‐induced small intestinal mucositis.
Figure 2.
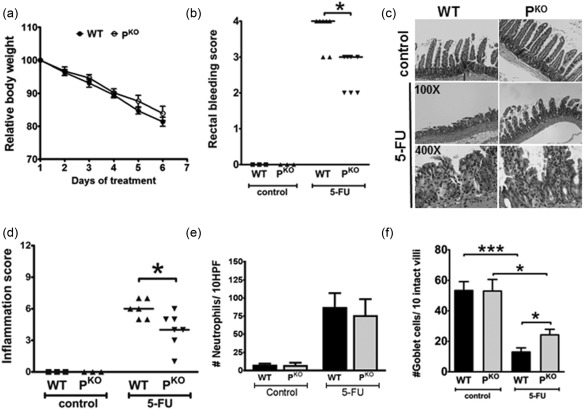
Properdin deficiency protects mice from 5‐fluorouracil (5‐FU)‐induced mucositis. (a) Weight loss during 5‐FU treatments. Data is the mean percentage of each animal's weight at the start of the experiment ± standard error of the mean (s.e.m.) (n = 6 mice/group). (b) Inflamed PKO mice registered significantly less rectal bleeding and (c,d) intestinal inflammation, compared to wild‐type (WT) mice. Each dot represents a single mouse and the line represents the median score. *P < 0·05 versus inflamed WT mice. (e) Neutrophil infiltration into the 5‐FU‐treated mouse intestine. Similar numbers of neutrophils were detected in the two strains. (f) Periodic acid‐Schiff (PAS)‐positive cells/10 intact villi/section and averaged for all mice within a treatment group. The data are the mean ± s.e.m. of four or five mice.
Reduced mucositis in properdin‐deficient mice is C5a‐independent
Because properdin is a positive regulator of complement activation we measured complement activation products, C3a and C5a, in the intestinal homogenates of treated mice. C3a and C5a were detectable at comparable levels in uninflamed mice of both strains (Fig. 3a,b). 5‐FU‐treated WT mice possessed significantly higher levels of C3a and C5a compared to uninflamed controls, confirming that complement is activated during mucositis (Fig. 3a,b). Interestingly, in inflamed PKO mice the increase in C3a levels reached statistical significance but the level of C5a did not, suggesting that perhaps reduced levels of C5a were mechanistically responsible for the lower inflammation in PKO mice (Fig. 3b). Also, considering that we discovered recently that properdin impacts the inflammation in murine infectious colitis in a C5a‐dependent mechanism 15, we sought to elucidate whether C5a is inflammatory in mucositis. We gavaged 5‐FU‐inflamed WT mice with the selective C5aR1 antagonist, PMX205. As shown in Fig. 3c,d, PMX205 treatments did not protect the mice from mucositis, suggesting that the apparent lower levels of C5a in PKO mice is not contributing mechanistically to the protection. Together, these data suggest that while properdin is involved in driving mucositis, it is independent of complement activation.
Figure 3.
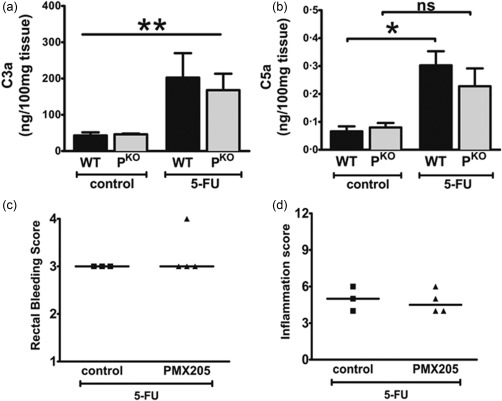
Reduced inflammation in PKO mice is independent of complement activation. Levels of (a) C3a and (b) C5a measured by enzyme‐linked immunosorbent assay (ELISA) in tissue homogenates of uninflamed and inflamed mice. Data are shown as mean ± standard error of the mean (s.e.m.) (n = three to 10 mice/group). *P < 0·05 and **P < 0·01 versus control, n.s. = not significant. Control refers to mice that did not receive 5‐fluorouracil (5‐FU). To investigate the role of C5a during mucositis, 5‐FU inflamed wild‐type (WT) mice were gavaged with PMX205 or water. All mice received 5‐FU; control refers to mice which received water in their gavage. (c) Inflammation and (d) rectal bleeding score of control or mice receiving PMX205. Each dot represents a single mouse and the line indicates the median score.
Reduced mucositis in PKO mice is associated with higher IL‐10 levels
To decipher further the mechanism behind the protection observed in PKO mice, we measured a number of relevant pro‐ and anti‐inflammatory cytokines in intestinal homogenates. IFN‐γ, IL‐6, IL‐17A, IL‐12 and IL‐13 were all below the limit of detection in all groups of mice. IL‐1β, while detectable, was neither increased nor statistically different between the inflamed groups (Fig. 4a). With regard to IL‐10, 5‐FU‐inflamed WT mice demonstrated significantly reduced levels compared to uninflamed mice. Conversely, IL‐10 levels were not significantly different comparing inflamed and control PKO mice (Fig. 4b).
Figure 4.
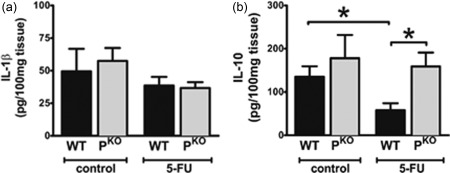
Reduced inflammation in PKO mice is associated with higher interleukin (IL)‐10 levels. Tissue homogenates from the jejunum of control [did not receive 5‐fluorouracil (5‐FU)] or 5‐FU treated mice were examined for (a) IL‐1β and (b) IL‐10.
As the level of IL‐10 was similar between uninflamed and inflamed PKO mice and uninflamed WT mice, we used an immunohistological approach to identify the cellular source of IL‐10, particularly to determine whether the source changes when the animals received 5‐FU. Tissue sections stained with an anti‐IL‐10 antibody revealed that epithelial cells were the predominant source of IL‐10 in healthy animals of both strains (Fig. 5a). Importantly, and consistent with the ELISA data and the idea that resident cells remained the principal source of IL‐10, the number of epithelial cells positive for IL‐10 was significantly higher in 5‐FU‐inflamed PKO mice compared to inflamed WT (Fig. 5b). Taken together, lower mucositis in PKO mice is associated with higher numbers of epithelial cells preserved in the intestine synthesizing IL‐10. This finding can be explained by a lack of inflammation in the PKO strain receiving 5‐FU, leaving more of the epithelium intact and, importantly, implicating properdin in triggering the inflammation.
Figure 5.
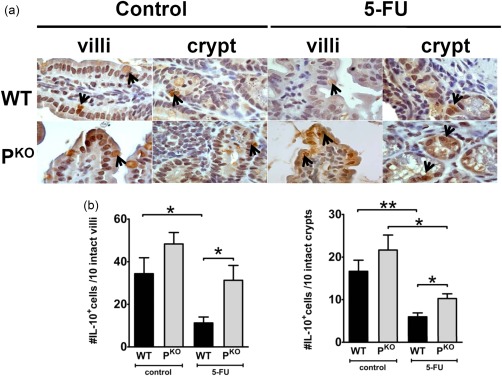
Interleukin (IL)‐10 expression is detected in the epithelium. “Control” in the figure refers to mice which did not receive 5‐fluorouracil (5‐FU). (a) Immunohistochemical staining for IL‐10 shows that epithelial cell expression was reduced in inflamed wild‐type (WT) but not PKO mice. Isotype matched antibody or anti‐IL‐10 used on sections from IL‐10–/– mice did not demonstrate any staining (not shown). (b) Quantification of IL‐10 positive epithelial cells/10 crypts or villi (shown separately). Cells were counted from intact villi/crypts only. Data are shown as mean ± standard error of the mean (s.e.m.) (n = four or five mice/group). *P < 0·05 and **P < 0·01.
Protection from mucositis is IL‐10‐dependent in properdin‐deficient mice
IL‐10 is a potent anti‐inflammatory cytokine in multiple models of intestinal inflammation but a role for IL‐10 specifically during 5‐FU‐induced mucositis has not been reported. Consequently, we conducted experiments to explore the properdin/IL‐10 relationship in our model. If properdin and IL‐10 are linked mechanistically, a lack of properdin will not protect IL‐10–/– mice from mucositis and properdin/IL‐10 double knock‐out mice (DKO) will presumably experience similar mucositis as IL‐10–/– mice. In preliminary experiments treating IL‐10–/– mice with 5‐FU the animals reached the weight loss end‐point early and had to be euthanized, indicating that this strain is considerably more sensitive than WT to 5‐FU (data not shown). Consequently, the 5‐FU regimen was shortened to 3 days and the mice killed 24 h later (day 4). Despite the shorter treatment regimen, significantly higher weight loss was observed in both IL‐10–/– and DKO mice compared to WT controls (Fig. 6a). When combined with IL‐10 deficiency the protective effect of properdin deficiency was lost, as the inflammation in DKO mice was comparable to 5‐FU‐treated IL‐10–/– and higher than WT mice (Fig. 6b,c). This outcome indicates that a lack of properdin fails to protect DKO animals from mucositis, and points to the importance of IL‐10 in protecting the intestine from mucositis.
Figure 6.
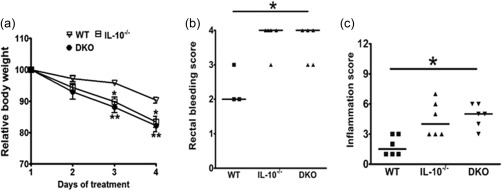
PKO mice lacking interleukin (IL)‐10 are not protected against intestinal mucositis. To assess if IL‐10 contributes to the protection observed in PKO mice against mucositis, PKO mice were crossed with IL‐10–/– and double knock‐out (DKO) offspring were subjected to 5‐fluorouracil (5‐FU) mucositis. Wild‐type (WT), IL‐10–/– and DKO were administered 5‐FU from days 1 to 3 and killed 24 h later. All the mice in the figure received 5‐FU. (a) Weight loss during 5‐FU treatments. Data are shown as mean ± standard error of the mean (s.e.m.) (n = four mice/group) and (b) rectal bleed and (c) inflammation scores of the mice. Each dot represents a single mouse and the line represents the median score. *P < 0·05; **P < 0·02 versus WT control.
Discussion
Complement is understood to be inflammatory, yet the history of investigation into complement and mucositis is scant. Bowen et al. surveyed changes in gene expression in the intestines of rats subjected to a single injection of irinotecan leading to mucositis and found that levels of mRNA for C1q and C2 of the classical pathway and factor D of the alternative pathway were increased considerably 13. Whether the increase in mRNA of complement proteins was a direct product of damaged epithelial cells, infiltrating leucocytes or triggered indirectly by the inflammation (many complement proteins are acute phase reactants) or whether the complement was involved mechanistically in the pathogenesis of mucositis were not assessed. Here we report that complement indeed becomes activated during mucositis but the mechanism by which the genetic deficiency of properdin protects against mucositis is complement activation‐independent. We determined that C5a is not driving mucositis, as the C5aR1 antagonist failed to prevent inflammation; however, other split complement molecules, such as C3a, remain to be assessed.
Complement is a network of approximately 30 proteins that co‐ordinate a cascade of enzymatic reactions ultimately generating opsonins, the anaphylatoxins and the membrane attack complex (MAC). The common understanding was that these activation products presumably impact positively the phenotype of inflammatory diseases, not limited to the intestines. However, a new understanding is emerging that these proteins, particularly the proximal activators in the cascade, have activities independent of complement activation. For example, mannose binding lectins (MBL), initiators of the lectin pathway, can modulate lipolysaccharide (LPS)‐mediated secretion of IL‐8, IL‐6 and monocyte chemotactic protein 1 (MCP‐1) without activating complement 20. MBL binds the extracellular domain of Toll‐like receptor (TLR)‐4 diminishing binding of LPS 21. Properdin was shown recently to facilitate removal of apoptotic cells in a complement activation‐independent manner 22. Here we demonstrate that the lack of properdin did not impact complement activation significantly, apart from slightly lower C5a levels, yet protected mice from 5‐FU injury. As C5a is a potent inflammatory product of complement activation, we thought to test directly the impact of C5a in the model. We determined that C5a was not driving the inflammation by administering a C5aR1 antagonist, PMX205, which failed to protect WT mice from mucositis. It is noteworthy that we showed previously that PMX205 reduced DSS‐induced colitis significantly, indicating that perhaps complement/C5a has different impacts on the large versus small intestine 23. Additionally, the finding that C5a does not influence mucositis only strengthens our conclusion that properdin is a positive regulator of mucositis in a complement activation‐independent manner. How properdin contributes positively to inflammation consequential to the cell injury caused by 5‐FU remains to be discovered.
IL‐10 has profound anti‐inflammatory effects in the gastrointestinal tract, demonstrated clearly by the fact that a lack of IL‐10 or the IL‐10 receptor indisputably predisposes to colitis, both in experimental animal models and humans. In cancer patients, IL‐10 levels were found to correlate negatively with the degree of mucositis 24 and LPS/IFN‐γ‐stimulated monocytes from patients with high‐grade mucositis produced less IL‐10 compared to patients without mucositis 24. These observations may mean that individuals who make less IL‐10 experience worse mucositis or, alternatively, that events related to mucositis may suppress IL‐10 production actively. We observed a significant reduction in the number of IL‐10‐producing cells and the soluble IL‐10 levels in WT mice with mucositis. Using mice deficient in IL‐10 we can address further whether homeostatic levels of IL‐10 provide any initial barrier to mucositis 25. We show further that IL‐10 in the small intestine is detectable mainly in epithelial cells, including in PKO mice (proving that this strain is not IL‐10‐deficient), suggesting that local constitutive expression of IL‐10 plays a role in limiting the cell injury directly due to 5‐FU, and that the lack of properdin interrupts the subsequent inflammatory response. When both molecules are absent, the extent of cell injury overwhelms the properdin‐dependent process or provokes an alternate mechanism leading to inflammation and severe mucositis. We are currently investigating how IL‐10 may directly prevent the apoptosis triggered by 5‐FU in gut epithelium. Ultimately, the fact that DKO mice were affected as severely by 5‐FU as IL‐10–/– mice links these two molecules mechanistically in mucositis.
Knowing constitutive gut IL‐10 levels are not elevated in PKO mice is important because there are precedents indicating that the lack of properdin may result in increased IL‐10. In one example, PKO mice with zymosan‐induced peritonitis demonstrated higher levels of IL‐10 26. Properdin is composed of thrombospondin repeats type I and it possibly has activities in mucositis similar to other members of this protein family 27. In this regard, thrombospondin I (also contains type I repeats), regulates negatively the production of IL‐10 from dendritic cells through interactions with CD47 and CD36 28. It remains to be determined in our model whether properdin uses these or other receptors to influence production of IL‐10 in the small intestine and inflammation, but currently our understanding is that properdin deficiency interrupted inflammation, leaving a greater number of epithelial cells intact and making IL‐10.
Although our data support the conclusion that PKO mice have normal levels of IL‐10, it is possible that IL‐10–/– mice may have abnormal levels of properdin, where high properdin levels would presumably make the mice more susceptible to mucositis. While we did not measure properdin levels in the IL‐10–/– strain, we interpret our earlier experiments exploiting this strain's susceptibility to colitis to infer that there is no obvious reason to believe they have high levels of properdin 16.
Despite incomplete knowledge of the mechanisms, our findings identify properdin as a potential target for ablating mucositis, particularly considering the lack of impact properdin has on complement activation. Complement is required for controlling the escape of microbes from inflamed intestines 29, and this will presumably be intact in patients in which properdin levels are reduced intentionally. Properdin is present in very low levels in plasma (4–25 μg/ml), so purposely reducing levels may be easily achievable 30.
Our study is the first experimental evidence, to our knowledge, for a mechanistic role of a complement protein during small intestinal mucositis, although not acting through the presumed role of supporting complement activation. We demonstrate that the protective effects of properdin deficiency is linked to IL‐10. In addition to further investigations into the mechanisms of properdin dependent mucositis, experiments are now needed to assess the impact of properdin on tumour progression and chemotherapeutic efficacy against tumours before reducing levels could be applied to mitigate mucositis in humans.
Disclosure
The authors have no financial conflicts of interest.
Acknowledgements
We thank Hana James for expert technical assistance. The project was supported by the Beatrice Hunter Cancer Research Institute (BHCRI). U. J. was supported by a trainee award from the BHCRI with funds provided by the Cancer Research Training Program as part of the Terry Fox Foundation Strategic Health Research Training Program in Cancer Research at CIHR and CIBC Graduate Scholarship in Medical Research.
References
- 1. Keefe DM, Cummins AG, Dale BM, Kotasek D, Robb TA, Sage RE. Effect of high‐dose chemotherapy on intestinal permeability in humans. Clin Sci 1997; 92:385–9. [DOI] [PubMed] [Google Scholar]
- 2. Benson AB 3rd, Ajani JA, Catalano RB et al Recommended guidelines for the treatment of cancer treatment‐induced diarrhea. J Clin Oncol 2004; 22:2918–26. [DOI] [PubMed] [Google Scholar]
- 3. Sonis ST. The pathobiology of mucositis. Nat Rev Cancer 2004; 4:277–84. [DOI] [PubMed] [Google Scholar]
- 4. Sonis ST, Elting LS, Keefe D et al Perspectives on cancer therapy‐induced mucosal injury: pathogenesis, measurement, epidemiology, and consequences for patients. Cancer 2004; 100:1995–2025. [DOI] [PubMed] [Google Scholar]
- 5. Elting LS, Cooksley C, Chambers M, Cantor SB, Manzullo E, Rubenstein EB. The burdens of cancer therapy. Clinical and economic outcomes of chemotherapy‐induced mucositis. Cancer 2003; 98:1531–9. [DOI] [PubMed] [Google Scholar]
- 6. Ruescher TJ, Sodeifi A, Scrivani SJ, Kaban LB, Sonis ST. The impact of mucositis on alpha‐hemolytic streptococcal infection in patients undergoing autologous bone marrow transplantation for hematologic malignancies. Cancer 1998; 82:2275–81. [PubMed] [Google Scholar]
- 7. Gibson RJ, Keefe DM, Lalla RV et al Systematic review of agents for the management of gastrointestinal mucositis in cancer patients. Suppl Care Cancer 2013; 21:313–26. [DOI] [PubMed] [Google Scholar]
- 8. Frank M, Hennenberg EM, Eyking A et al TLR signaling modulates side effects of anticancer therapy in the small intestine. J Immunol 2015; 194:1983–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaczmarek A, Brinkman BM, Heyndrickx L, Vandenabeele P, Krysko DV. Severity of doxorubicin‐induced small intestinal mucositis is regulated by the TLR‐2 and TLR‐9 pathways. J Pathol 2012; 226:598–608. [DOI] [PubMed] [Google Scholar]
- 10. Guabiraba R, Besnard AG, Menezes GB et al IL‐33 targeting attenuates intestinal mucositis and enhances effective tumor chemotherapy in mice. Mucos Immunol 2014; 7:1079–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res 2010; 20:34–50. [DOI] [PubMed] [Google Scholar]
- 12. Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J. The role of the anaphylatoxins in health and disease. Mol Immunol 2009; 46:2753–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bowen JM, Gibson RJ, Tsykin A, Stringer AM, Logan RM, Keefe DM. Gene expression analysis of multiple gastrointestinal regions reveals activation of common cell regulatory pathways following cytotoxic chemotherapy. Intern J Cancer 2007; 121:1847–56. [DOI] [PubMed] [Google Scholar]
- 14. Harboe M, Mollnes TE. The alternative complement pathway revisited. J Cell Mol Med 2008; 12:1074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jain U, Cao Q, Thomas NA et al Properdin provides protection from Citrobacter rodentium‐induced intestinal inflammation in a C5a/IL‐6‐dependent manner. J Immunol 2015; 194:3414–21. [DOI] [PubMed] [Google Scholar]
- 16. Jain U, Midgen CA, Schwaeble WJ, Stover CM, Stadnyk AW. Properdin regulation of complement activation affects colitis in interleukin 10 gene‐deficient mice. Inflamm Bowel Dis 2015; 21:1519–28. [DOI] [PubMed] [Google Scholar]
- 17. Kullberg MC, Ward JM, Gorelick PL et al Helicobacter hepaticus triggers colitis in specific‐pathogen‐free interleukin‐10 (IL‐10)‐deficient mice through an IL‐12‐ and gamma interferon‐dependent mechanism. Inf Immun 1998; 66:5157–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stringer AM, Gibson RJ, Logan RM et al Gastrointestinal microflora and mucins may play a critical role in the development of 5‐fluorouracil‐induced gastrointestinal mucositis. Exp Biol Med 2009; 234:430–41. [DOI] [PubMed] [Google Scholar]
- 19. Saegusa Y, Ichikawa T, Iwai T et al Changes in the mucus barrier of the rat during 5‐fluorouracil‐induced gastrointestinal mucositis. Scand J Gastroenterol 2008; 43:59–65. [DOI] [PubMed] [Google Scholar]
- 20. Kang HJ, Lee SM, Lee HH et al Mannose‐binding lectin without the aid of its associated serine proteases alters lipopolysaccharide‐mediated cytokine/chemokine secretion from human endothelial cells. Immunology 2007; 122:335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang M, Chen Y, Zhang Y, Zhang L, Lu X, Chen Z. Mannan‐binding lectin directly interacts with Toll‐like receptor 4 and suppresses lipopolysaccharide‐induced inflammatory cytokine secretion from THP‐1 cells. Cell Mol Immunol 2011; 8:265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cortes C, Ohtola JA, Saggu G, Ferreira VP. Local release of properdin in the cellular microenvironment: role in pattern recognition and amplication of the alternative pathway of complement. Front Immunol 2013; 3:412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jain U, Woodruff T, Stadnyk AW. The C5a receptor antagonist PMX205 ameliorates experimentally induced colitis associated with increased IL‐4 and IL‐10. Br J Pharmacol 2013; 168:488–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schauer MC, Holzmann B, Peiper M, Friess H, Knoefel WT, Theisen J. Interleukin‐10 and −12 predict chemotherapy‐associated toxicity in esophageal adenocarcinoma. J Thor Oncol 2010; 5:1849–54. [DOI] [PubMed] [Google Scholar]
- 25. de Koning BA, van Dieren JM, Lindenbergh‐Kortleve DJ et al Contributions of mucosal immune cells to methotrexate‐induced mucositis. Int Immunol 2006; 18:941–9. [DOI] [PubMed] [Google Scholar]
- 26. Ivanovska ND, Dimitrova PA, Luckett JC, El‐Rachkidy Lonnen R, Schwaeble WJ, Stover CM. Properdin deficiency in murine models of nonseptic shock. J Immunol 2008; 180:6962–9. [DOI] [PubMed] [Google Scholar]
- 27. de Lau WB, Snel B, Clevers HC. The R‐spondin protein family. Genome Biol 2012; 13:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doyen V, Rubio M, Braun D et al Thrombospondin 1 is an autocrine negative regulator of human dendritic cell activation. J Exp Med 2003; 198:1277–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schepp‐Berglind J, Atkinson C, Elvington M, Qiao F, Mannon P, Tomlinson S. Complement‐dependent injury and protection in a murine model of acute dextran sulfate sodium‐induced colitis. J Immunol 2012; 188:6309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cortes C, Ohtola JA, Saggu G, Ferreira VP. Local release of properdin in the cellular microenvironment: role in pattern recognition and amplification of the alternative pathway of complement. Front Immunol 2012; 3:412. [DOI] [PMC free article] [PubMed] [Google Scholar]