

Abstract
Emerging evidence indicates that long noncoding RNAs (lncRNAs) are actively involved in a number of developmental and tumorigenic processes. Here, we describe the first successful use of spherical nucleic acids (SNAs) as an effective nanoparticle platform for regulating lncRNAs in cells; specifically, for the targeted knockdown of the nuclear-retained metastasis associated lung adenocarcinoma transcript 1 (Malat1), a key oncogenic lncRNA involved in metastasis of several cancers. Utilizing the liposomal spherical nucleic acid (LSNA) constructs, we first explored the delivery of antisense oligonucleotides to the nucleus. We show a dose-dependent inhibition of Malat1 upon LSNA treatment as well as the consequent up-regulation of tumor suppressor mRNA associated with Malat1 knockdown. These findings reveal the biologic and therapeutic potential of a LSNA-based antisense strategy in targeting disease-associated, nuclear-retained lncRNAs.
Keywords: DNA nanoparticle, spherical nucleic acids, gene regulation, nuclear targeting, liposomal SNA
Graphical Abstract
The regulation of nuclear-retained RNAs with liposomal spherical nucleic acids (LSNAs) expands the abilities of these unique nanoconstructs for intracellular nucleic acid delivery. RNAs of the nucleus were previously inaccessible for gene regulation by SNA constructs; this work demonstrates the delivery of ASOs to the nucleus and the subsequent regulation of the nuclear long non-coding RNA, Malat1.
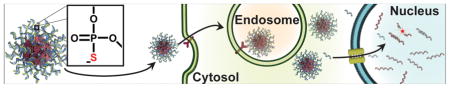
1. Introduction
Spherical nucleic acids (SNAs) have emerged as a new class of regulatory nucleic acids typically constructed by chemically arranging oligonucleotides (DNA or RNA) on a nanoparticle core in a densely packed and highly oriented manner.[1] The spherical, three-dimensional arrangement of oligonucleotides defining the SNA architecture is responsible for several unique properties that make them different when compared to conventional, particle-free antisense oligonucleotides and siRNAs and attractive for biological and medical applications[2–6]. For example, SNAs are taken up rapidly by cells despite their negative charge and do not require the use of ancillary transfection reagents (polymers or viral vectors).[7] In addition, they exhibit nuclease resistance,[8] low immunogenicity and toxicity,[9, 10] and are highly effective as gene regulation and immunomodulation agents.[3, 7, 11–16] Although the prototypical SNA structure consists of oligonucleotides arranged around a Au nanoparticle core[1, 8, 17], many new forms have also been introduced, including hollow architectures[13], micelles with organic cores[18], versions based upon other inorganic core materials, such as silica[19] and infinite coordination polymers[16], protein core constructs[20], and versions derived from sub-100 nm liposomal cores (LSNAs).[21–24] The liposomal architectures, which are made from 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) small unilamellar vesicles (SUVs) and tocopherol-terminated oligonucleotides, are extremely interesting from a therapeutics standpoint, since they can be made from materials that are known to be biocompatible and already a part of a variety of FDA-approved drugs. In addition, they define a new class of SNA that is held together via non-covalent bonds and, in principle, more dynamic than the prototypical Au nanoparticle SNAs (SNA-AuNP).
In general, most SNAs studied to date enter cells via a scavenger receptor- and caveolin-mediated endocytotic process, which highlights the importance of the nucleic acid shell architecture. They are internalized via early endosomes, which mature into late endosomes and then a fraction of the SNAs escape the endosome and are responsible for antisense or siRNA activity. However, SNA-AuNPs, because of their relatively large size, do not enter the nucleus, even when coupled to nuclear localization peptides.[25] Indeed, although small particles (< ~40 kDa) are able to passively diffuse through the nuclear pore complex,[26] structures greater than 25 nm in diameter and over 500 kDa typically do not enter the nucleus, which precludes most SNA architectures.[26] In contrast, it is not clear, whether or not the non-covalent LSNA architecture once internalized remains fully intact upon endocytosis, and we believe that its dynamic structure could lead to release of the tocopherol-modified oligonucleotides and the ability for such structures to achieve gene regulation within the nucleus. Further, phosphorothioate (PS) oligonucleotides have been shown to localize within the nucleus upon cellular entry via a Ras-related nuclear (RAN) protein-mediated pathway.[27–30] PS oligonucleotides have a sulfur atom in place of a non-bridging oxygen of traditional phosphodiester (PO) oligonucleotides. We hypothesize that incorporating these chemically modified antisense oligonucleotides (ASOs) into LSNAs will allow for the regulation of RNAs within the nucleus. Herein, we evaluate this hypothesis in the context of the antisense regulation of Malat1, a nuclear-retained, oncogenic, long non-coding RNA (lncRNA).
2. Results and Discussion
2.1. DNA backbone dictates DNA localization to the nucleus using LSNAs
To investigate the ability of LSNAs to target the nuclear-retained lncRNA Malat1, we first explored the Lipofectamine delivery of free PS ASOs to the nucleus. To test their nuclear localization, we transfected a human adenocarcinomic cell line (A549 cells) with Cy5-labeled ASOs (see Table S1 for a list of ASO sequences) and examined the intracellular fluorescence by confocal microscopy (Figure S1). From this experiment, we observed Cy5 fluorescence co-localized with the DAPI fluorescence in the nucleus confirming previous reports of PS oligos entering the nucleus.[27–29]
To assess if ASO delivery via LSNAs also leads to the ability to regulate gene expression in the nucleus, we synthesized both PS and PO LSNAs by functionalizing DOPC SUVs with tocopherol-terminated (5′ terminus), Cy5-labeled (3′ terminus) ASOs. Loading of the oligonucleotides on the DOPC SUV was confirmed by gel electrophoresis with the observation of a large shift in gel displacement due to the size increase upon ASO functionalization (Figure S2). In addition, from this we observed that no detectable free oligos were present following LSNA purification. Using dynamic light scattering, we measured the average size of the LSNAs to be 51.6 ± 1.7 nm and calculated the average loading to be 70 ± 9 DNA strands per particle (see Table S2 for particle characterization). A549 cells were then treated with Cy5- PO or PS LSNAs for 16 h (overnight), and the nuclear localization of the Cy5-labeled oligo was examined by confocal microscopy. The mean fluorescence intensity (MFI) for Cy5 was measured at the center of the nucleus (Scheme 1) for Malat1 targeting, PO (Malat1-PO-LSNAs) and PS LSNAs (Malat1-PS-LSNAs), and a non-targeting PS LSNA (NT-PS-LSNAs) (Figure 1A). A concentration-dependent increase in the Cy5 MFI was observed for both Malat1-PS-LSNAs and NT-PS-LSNAs with a significant increase in Cy5 ASO localization within the nucleus when compared to the Malat1-PO-LSNAs (Figure 1B). In addition, to determine if the increase in nuclear fluorescence was due to differences in cellular uptake of either LSNA, we performed flow cytometry on A549 cells treated with either Malat1-PO- or PS- LSNAs, significantly, saw no measurable difference in cellular fluorescence (Figure S3). This is consistent with the conclusion that the chemically-modified PS LSNAs are uptaken in similar quantities compared to PO LSNAs but trafficked differently within the cell. We suspect that the dynamic nature of the LSNA frees the oligonucleotides from the particle upon cellular entry, allowing it to be taken up within the nucleus. The concentration-dependent increase in nuclear localization of Cy5 suggests that PS LSNAs are able to deliver ASOs to the nucleus. However, it is possible that the Cy5 fluorophore is cleaved from the ASO within the cell and enters the nucleus separate from the oligo. To determine if the complete ASO, as opposed to just the fluorophore, enters the nucleus, we examined the gene regulatory ability of PS LSNAs in targeting the nuclear-retained lncRNA, Malat1.
Scheme 1.

The Cy5 mean fluorescence intensity (MFI) was quantified at the center of the nucleus in A549 cells. Z-stacks were generated with optical sections of 0.3 μm throughout the cell. When measuring the MFI intensity with the nucleus (yellow circle), we used the peaks in the DAPI MFI vs. Z-Displacement graph to identify the top and bottom of the nucleus (green line). The center of the nucleus was defined as the midpoint between these two edges (yellow line) where the Cy5 MFI was recorded.
Figure 1.

(A) Representative images of Cy5-labeled LSNA uptake within the A549 cells. The DAPI and MFI intensity was measured near the center of the nucleus (yellow circle) and was used to examine the MFI through the cell in the Z-direction. (B) Mean fluorescence intensity for Cy5 at the center of the nucleus in A549 cells; *** - p<0.001; * - p<0.05; scale bar = 20 μm.
2.2. Phosphorothioate-modified LSNAs regulate Malat1 expression in the nucleus
A549 cells were cultured on cover glass slides and treated with PS and PO Malat1 targeting LSNAs for 16 h. Using fluorescence in situ hybridization (FISH) and confocal microscopy, we examined the localization and expression of Malat1 (Figure 2A). This confirmed the intra-nuclear localization of Malat1 in A549 cells and further supports previous reports that Malat1 is a nuclear-retained long non-coding RNA.[31–33] Upon measuring the MFI of the FISH probes for Malat1, we observed a significant decrease in Malat1 expression in cells treated with Malat1-PS-LSNAs compared to those treated with NT-PS-LSNAs (Figure 2B). This suggests that upon chemically modifying the ASOs on the surface of the LSNAs, we gain the ability to deliver active ASOs within the nucleus and regulate the expression of the nuclear RNAs. To further validate these results, we next examined Malat1 RNA expression levels using quantitative real time polymerase chain reaction (qRT-PCR).
Figure 2.

(A) Fluorescence in situ hybridization was used to examine expression of Malat1 and confirm knockdown within the nucleus. Scale bar = 20 μm. (B) The mean fluorescence intensity (MFI) of Malat1 was quantified to examine the reduction in Malat1 expression with Malat1-PS-LSNA treatment. A significant decrease in MFI is seen with Malat1-PS-LSNAs; *** - p<0.001.
A549 cells were again treated with LSNAs overnight (16 h). RNA extracts were harvested 48 h after treatment initiation, and Malat1 expression levels were quantified using qRT-PCR. This revealed a concentration-dependent down regulation in Malat1 expression (Figure 3A) and further demonstrates the PS LSNAs effectiveness in targeting the nuclear-retained lncRNA Malat1. Furthermore, we examined the gene regulatory ability of Malat1-PO-LSNAs, and saw no decrease in gene expression (Figure S4). This supports the conclusion that the chemically modified PS backbone is an enabling entity in targeting nuclear RNAs using LSNAs.
Figure 3.

Malat1 mRNA expression levels 48 h after treatment with Malat1-PS-LSNAs. (A) A concentration-dependent down regulation of Malat1 is seen with treatment. Free PS ASOs delivered with Lipofectamine RNAiMax at 0.1 μM were used as a positive (+) control. (B) Malat1-PS-LSNAs were used to treat A549 cells following a siRNA treatment to knockdown the nuclear trafficking protein RAN. A significant decrease in Malat1 gene knockdown was observed with RAN protein knockdown while no difference was observed with non-targeting (Scr) siRNA treatment; *** - p<0.001.
Since, mechanistically, PS oligonucleotides are trafficked to the nucleus via a RAN-mediated pathway,[30] we can inhibit nuclear uptake of PS ASOs delivered by LSNAs with the use of RAN-targeting siRNA. In a typical experiment, A549 cells were pre-treated with RAN targeting siRNA (see Figure S5 for RAN protein knockdown) and subsequently treated with Malat1-PS-LSNAs. This resulted in a significant decrease in the ability of the LSNAs to regulate Malat1 while no significant change was observed when pretreating with a non-targeting siRNA (Figure 3B). This suggests that PS ASOs of LSNAs are partially taken up in the nucleus via a RAN-mediated pathway; afterwards, they are able to participate in the antisense gene regulation of Malat1.
Within the nucleus, Malat1 associates with serine arginine (SR) proteins of nuclear speckles, modulating their phosphorylation[32] and ultimately regulating alternative splicing within the cell.[32, 34, 35] Malat1 knockdown has previously been shown to induce the up-regulation of the tumor suppressor, interferon-induced protein with tetratricopeptide repeats 2 (IFIT2).[35–37] To further investigate the efficacy of Malat1-PS-LSNAs, we next performed a loss-of-function analysis by examining IFIT2 mRNA expression levels after Malat1-PS-LSNA treatment. This showed a significant and concentration-dependent increase in IFIT2 mRNA levels compared to when the NT-PS-LSNAs were used (Figure 4A). This illustrates the functional ability of LSNAs through the upregulation of the downstream tumor suppressor, IFIT2, and further demonstrates the power of these constructs as biological tools and therapeutics. Gutshner et al. have established a loss-of-function model for Malat1 using zinc finger nucleases to integrate RNA destabilizing elements into the human genome, but this approach requires extensive work on molecular cloning and cell selection, along with the risk of random integration into other genomic loci.[34] Notably, here we show that treatment of cultured cells with PS-LSNAs effectively silenced Malat1 up to 90%.
Figure 4.

A) IFIT2 mRNA expression with treatment of Malat1-PS-LSNAs. A significant up-regulation was seen with Malat1-PS-LSNAs. (B) Cell viability was examined in A549 cells treated with PO and PS LSNAs (1 μM by DNA). Curcumin (50 μM), an apoptotic inducing agent, was used as a positive control (+). No change in viability was observed 24 hours after treatment; ** - p<0.01.
To ensure the LSNAs remain non-toxic with the incorporation of a PS backbone, we next employed a cell viability assay. Cell viability was measured 24 h after treatment with both PO and PS LSNAs (Figure 4B) and no cytotoxic response was observed under either set of conditions. Thus, the PS LSNA remains non-toxic making it a viable candidate as a gene regulatory construct for nuclear-retained RNAs.
3. Conclusion
Importantly, this work shows, for the first time, that PS forms of SNAs can be used to effectively deliver ASOs that can traffic to the nucleus and interact with target lncRNA in a sequence-specific manner and subsequently effect gene knockdown. This is both a fundamentally and technologically important observation. First, PS SNAs, with gold cores, are difficult to make because of the reactivity between the sulfur-rich oligonucleotides and the gold cores; therefore, the realization of the easily prepared PS LSNAs points to an advantage of the liposomal core. Second, the observation that lncRNAs in the nucleus can be effectively knocked down with the PS LSNAs suggests that at least this form of SNA can escape, in full or in part, the endosome to an extent that it can effectively reach and bind target in the nucleus. Third, the data suggest that the non-covalently assembled LSNAs are dynamic architectures, and while the intact SNA architecture is critical for facilitating uptake, once inside the cell, the particle slowly releases the ASOs that comprise it in order to interact with nucleic acids in the nucleus. SNAs made from covalent chemistry have never been able to enter the nucleus, presumably due in part to their large size and relatively static structure.
Taken together, this work introduces the concept of using PS LSNAs for targeting RNAs in the nucleus that are not addressable with conventional PO oligonucleotides. Furthermore, it underscores the importance of chemical tunability with respect to controlling SNA trafficking within the cell, thereby emphasizing the need to fully examine the role of other chemical modifications (i.e., morpholino-modified backbones and fluorinated nucleosides) on SNA properties and function.
4. Experimental Section
Cell Culture
A549 (lung carcinoma) cells were obtained from American Tissue Culture Collection (ATCC) and cultured in F-12K Medium supplemented with 10% fetal bovine serum (FBS) and 1% Pen Strep (Invitrogen). Cells were maintained at 37°C and 5% CO2 and seeded 1 day prior to LSNA treatment at 30–40% confluency. Lipofectamine RNAiMax (3 μL/mL) was used for cellular transfection of both ASOs and siRNAs in Optimem (serum-free media from Invitrogen). All particle treatments were done in Optimem overnight (16 h) followed by a subsequent media change with serum containing media.
Oligonucleotide Synthesis
All antisense oligonucleotides were generated using a Mermaid 12 Synthesizer. Universal CPGs and 2′-O-Methyl RNA phosphoramidites were obtained from ChemGenes while all other phosphoramidites and reagents were purchased from Glen Research and synthesized per manufacturers recommendations with DCI activator. Following solid phase synthesis, oligos were cleaved from the support using aqueous ammonium hydroxide (28–30%, Sigma Aldrich) at room temperature for 16–20 h. Successfully synthesized nucleic acids were purified by reverse phase high pressure liquid chromatography (RP-HPLC) using a Microsorb C18 column (Varian ProStar Model No. 210) using a gradient of triethylammonium acetate (TEAA) with 3% acetonitrile (ACN) to pure ACN. Subsequently, purified oligos were lyophilized and stored at −80°C. To ensure proper synthesis, the molecular weight of the complete oligonucleotide was measured by matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) using Autoflex III Smartbeam (Bruker).
Liposomal Spherical Nucleic Acids
Liposomal spherical nucleic acids were generated as previously described.[21] DOPC in chloroform was obtained from Avanti Polar Lipids. Solvent was evaporated off and DOPC was suspended in 1X HEPES buffer saline (pH = 7.4). The solution was then probe sonicated for 45 minutes yielding small unilamellar vesicles (SUVs). To remove impurities, ultracentrifugation was conducted and the supernatant was extruded 10 times through a 50-nm pore membrane and then 10 times through a 30-nm pore membrane. The concentration of lipid (or phosphorus) was determined using ICP-OES against a standard curve for phosphorus. The liposome core was functionalized with ASOs and the number of strands loaded was calculated using previously reported methods.[21] Following functionalization with oligonucleotides the concentration of nucleic acid was determined by measuring the absorbance at 260 nm.
Dynamic Light Scattering
LSNAs were analyzed in triplicates using a Malvern Zetasizer Instrument as previously described by Banga et al.[21]
RNA expression analysis by qRT-PCR
To quantify gene expression, total RNA was extracted from cells plated in 96-well plates using the RNeasy 96 well plate kit (Qiagen) per the manufacturer’s protocol. RNA was subsequently reverse transcribed to generate cDNA using the High-Capacity cDNA reverse Transcription Kit (Applied Biosystems). cDNA was mixed with Roche’s Lightcycler 480 Probe Master Mix along with probes and primers (per manufacturer’s protocol). GAPDH was used as a housekeeping gene with the primers and probes generated in house using the following sequences: Forward - 5′-CAA GGT CAT CCA TGA CAA CTT TG -3′, Reverse - 5′-GGG CCA TCC ACA GTC TTC T -3′, Probe – 5′ - HEX – ACC ACA GTC CAT GCC ATC ACT GCC A - BHQ1. All other primers/probes were obtained from Life Technologies. qRT-PCR was performed on a Roche Lightcycler 480 and the relative abundance of each mRNA transcript was normalized to GAPDH expression and compared to untreated cells.
Fluorescence In Situ Hybridization (FISH)
FISH was performed with the RNAScope kit using Human Malat1 probes both of which were obtained from Advanced Cell Diagnostics. The manufacturer’s protocol was followed for FISH staining and images acquired on a confocal microscope as stated below.
Imaging and Analysis
All imaging was done with a Leica SP5 confocal or a Zeiss LSM 800 microscope for FISH and nuclear uptake, respectively. For nuclear uptake of ASOs, cells were plated on 3.5 cm, cover glass bottom, tissue culture dishes (World Precision Instruments). Following treatment, cells were washed with 1X PBS and fixed for 30 minutes in 4% paraformaldehyde and subsequently incubated with DAPI for 15 minutes (in PBS). The cells were then washed in 1X PBS and remained in PBS throughout imaging. A 60X objective was used for imaging. To quantify nuclear fluorescence, optical sections were taken in the Z-direction every 0.3 μm. The mean fluorescence intensity was quantified using the measurement tool of Zeiss’ Zen Blue imaging software by drawing a circular with a diameter of 2 μm. Peaks in nuclear fluorescence were used to determine the top and the bottom (or “edges”) of the nucleus. The center of the nucleus was considered the halfway point between these two edges. The MFI was recorded for Cy5 as a Cy5 MFI in the Z-plane at the center. The MFI was also quantified for Malat1 following the application of FISH probes using a similar method on the Leica SP5 using Leica Application Suite software. In this case, optical sections were taken every 0.25 μm. The MFI was quantified throughout the nucleus (n=12 planes, total z-distance spanning 3.0 μm) using a circular measurement tool 6 μm in diameter.
Agarose Gel Electrophoresis
Cy5-labeled PO and PS LSNAs were analyzed using 1% agarose gels run at 100 V for 30 min in 1X TBE buffer. Gels were imaged using a FluorChem Q gel imager.
Western Blotting
Protein analysis was employed to examine expression of RAN following siRNA treatment. Proteins were separated by SDS-PAGE gel electrophoresis. Gels were transferred using Invitrogen’s iBlot2 and stained with anti-RAN (Abcam) and anti-Actin (cell signaling) antibodies using Invitrogen’s iBind at a 1:100 dilution. Anti-rabbit secondary antibodies were obtained from Licor and used at a 1:2000 dilution. Licor’s gel imager was used to measure and quantify protein expression.
Flow Cytometry
A549 cells were cultured in 96 well plates in serum containing media. Once ~70–80% confluent, cells were treated with either LSNAs and collected for cytometric analysis by first washing with 1X PBS (3 times), trypsinizing following by fixing with 4% PFA (in 1X PBS). Flow cytometry was then conducted using a Becton Dickinson LSR II to measure the fluorescence (excitation 561 nm, emission 685–735 nm) of 5,000 single cell events per sample (in triplicates).
Supplementary Material
Acknowledgments
This research was supported by the National Cancer Institute of the National Institutes of Health under Award Number U54CA199091. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This material is also based upon work supported by the NTU-NU Institute for NanoMedicine located at the International Institute for Nanotechnology, Northwestern University, USA and the Nanyang Technological University, Singapore. A.J.S. acknowledges the NSF Graduate Research Fellowship. L.H. was a Howard Hughes Medical Institute International Student Research Fellow and acknowledges a Ryan Fellowship from Northwestern University. Confocal imaging was performed at the Biological Imaging Facility (BIF) at Northwestern University.
Footnotes
Supporting Information is available from the Wiley Online Library.
Contributor Information
Anthony J. Sprangers, Department of Biomedical Engineering, International Institute for Nanotechnology, Northwestern University, Evanston, IL 60208, USA
Dr. Liangliang Hao, Interdisciplinary Biological Sciences, International Institute for Nanotechnology, Northwestern University, Evanston, IL 60208, USA
Resham J. Banga, Department of Chemical and Biological Engineering, International Institute for Nanotechnology, Northwestern University, Evanston, IL 60208, USA
Prof. Chad A. Mirkin, Department of Biomedical Engineering, International Institute for Nanotechnology, Northwestern University, Evanston, IL 60208, USA. Department of Chemical and Biological Engineering, International Institute for Nanotechnology, Northwestern University, Evanston, IL 60208, USA
References
- 1.Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. Nature. 1996;(382):607–9. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- 2.Alhasan AH, Kim DY, Daniel WL, Watson E, Meeks JJ, Thaxton CS, Mirkin CA. Analytical chemistry. 2012;84(9):4153–4160. doi: 10.1021/ac3004055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen SA, Day ES, Ko CH, Hurley LA, Luciano JP, Kouri FM, Merkel TJ, Luthi AJ, Patel PC, Cutler JI. Science translational medicine. 2013;5(209):209ra152. doi: 10.1126/scitranslmed.3006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Randeria PS, Seeger MA, Wang XQ, Wilson H, Shipp D, Mirkin CA, Paller AS. Proceedings of the National Academy of Sciences. 2015;112(18):5573–5578. doi: 10.1073/pnas.1505951112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Briley WE, Bondy MH, Randeria PS, Dupper TJ, Mirkin CA. Proceedings of the National Academy of Sciences. 2015;112(31):9591–9595. doi: 10.1073/pnas.1510581112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seferos DS, Giljohann DA, Hill HD, Prigodich AE, Mirkin CA. Journal of the American Chemical Society. 2007;129(50):15477–15479. doi: 10.1021/ja0776529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AK, Han MS, Mirkin CA. Science. 2006;312(5776):1027–1030. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- 8.Seferos DS, Prigodich AE, Giljohann DA, Patel PC, Mirkin CA. Nano letters. 2008;9(1):308–311. doi: 10.1021/nl802958f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massich MD, Giljohann DA, Schmucker AL, Patel PC, Mirkin CA. ACS nano. 2010;4(10):5641–5646. doi: 10.1021/nn102228s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massich MD, Giljohann DA, Seferos DS, Ludlow LE, Horvath CM, Mirkin CA. Molecular pharmaceutics. 2009;6(6):1934–1940. doi: 10.1021/mp900172m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng D, Giljohann DA, Chen DL, Massich MD, Wang XQ, Iordanov H, Mirkin CA, Paller AS. Proceedings of the National Academy of Sciences. 2012;109(30):11975–11980. doi: 10.1073/pnas.1118425109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim JH, Yeom JH, Ko JJ, Han MS, Lee K, Na SY, Bae J. Journal of biotechnology. 2011;155(3):287–292. doi: 10.1016/j.jbiotec.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 13.Cutler JI, Zhang K, Zheng D, Auyeung E, Prigodich AE, Mirkin CA. Journal of the American Chemical Society. 2011;133(24):9254–9257. doi: 10.1021/ja203375n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giljohann DA, Seferos DS, Prigodich AE, Patel PC, Mirkin CA. Journal of the American Chemical Society. 2009;131(6):2072–2073. doi: 10.1021/ja808719p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radovic-Moreno AF, Chernyak N, Mader CC, Nallagatla S, Kang RS, Hao L, Walker DA, Halo TL, Merkel TJ, Rische CH. Proceedings of the National Academy of Sciences. 2015;112(13):3892–3897. doi: 10.1073/pnas.1502850112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calabrese CM, Merkel TJ, Briley WE, Randeria PS, Narayan SP, Rouge JL, Walker DA, Scott AW, Mirkin CA. Angewandte Chemie International Edition. 2015;54(2):476–480. doi: 10.1002/anie.201407946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lytton-Jean AK, Gibbs-Davis JM, Long H, Schatz GC, Mirkin CA, Nguyen ST. Advanced Materials. 2009;21(6):706–709. doi: 10.1002/adma.200801724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang C, Hao L, Calabrese CM, Zhou Y, Choi CHJ, Xing H, Mirkin CA. Small. 2015;11(40):5360–5368. doi: 10.1002/smll.201501573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young KL, Scott AW, Hao L, Mirkin SE, Liu G, Mirkin CA. Nano letters. 2012;12(7):3867–3871. doi: 10.1021/nl3020846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brodin JD, Sprangers AJ, McMillan JR, Mirkin CA. Journal of the American Chemical Society. 2015;137(47):14838–14841. doi: 10.1021/jacs.5b09711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banga RJ, Chernyak N, Narayan SP, Nguyen ST, Mirkin CA. Journal of the American Chemical Society. 2014;136(28):9866–9869. doi: 10.1021/ja504845f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cutler JI, Zheng D, Xu X, Giljohann DA, Mirkin CA. Nano letters. 2010;10(4):1477–1480. doi: 10.1021/nl100477m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JS, Lytton-Jean AK, Hurst SJ, Mirkin CA. Nano letters. 2007;7(7):2112–2115. doi: 10.1021/nl071108g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seferos DS, Giljohann DA, Rosi NL, Mirkin CA. ChemBioChem. 2007;8(11):1230–1232. doi: 10.1002/cbic.200700262. [DOI] [PubMed] [Google Scholar]
- 25.Patel PC, Giljohann DA, Seferos DS, Mirkin CA. Proceedings of the National Academy of Sciences. 2008;105(45):17222–17226. doi: 10.1073/pnas.0801609105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peters R. Xenopus Protocols. Springer; 2006. Introduction to nucleocytoplasmic transport; pp. 235–258. [DOI] [PubMed] [Google Scholar]
- 27.Marcusson EG, Bhat B, Manoharan M, Bennett CF, Dean NM. Nucleic acids research. 1998;26(8):2016–2023. doi: 10.1093/nar/26.8.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beltinger C, Saragovi H, Smith R, LeSauteur L, Shah N, DeDionisio L, Christensen L, Raible A, Jarett L, Gewirtz A. Journal of Clinical Investigation. 1995;95(4):1814. doi: 10.1172/JCI117860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iversen PL, Zhu S, Meyer A, Zon G. Antisense research and development. 1992;2(3):211–222. doi: 10.1089/ard.1992.2.211. [DOI] [PubMed] [Google Scholar]
- 30.Liang X-h, Shen W, Sun H, Prakash TP, Crooke ST. Nucleic Acids Research. 2014;42(12):7819. doi: 10.1093/nar/gku484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyagawa R, Tano K, Mizuno R, Nakamura Y, Ijiri K, Rakwal R, Shibato J, Masuo Y, Mayeda A, Hirose T. Rna. 2012;18(4):738–751. doi: 10.1261/rna.028639.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA. Molecular cell. 2010;39(6):925–938. doi: 10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ip JY, Nakagawa S. Development, growth & differentiation. 2012;54(1):44–54. doi: 10.1111/j.1440-169X.2011.01303.x. [DOI] [PubMed] [Google Scholar]
- 34.Gutschner T, Hämmerle M, Eißmann M, Hsu J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Groß M. Cancer research. 2013;73(3):1180–1189. doi: 10.1158/0008-5472.CAN-12-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tripathi V, Shen Z, Chakraborty A, Giri S, Freier SM, Wu X, Zhang Y, Gorospe M, Prasanth SG, Lal A. PLoS genetics. 2013;9(3):e1003368. doi: 10.1371/journal.pgen.1003368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lai KC, Chang KW, Liu CJ, Kao SY, Lee TC. Molecular Cancer Research. 2008;6(9):1431–1439. doi: 10.1158/1541-7786.MCR-08-0141. [DOI] [PubMed] [Google Scholar]
- 37.Lai K, Liu C, Chang K, Lee T. Oncogene. 2013;32(32):3686–3697. doi: 10.1038/onc.2012.384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.