

Summary
Receptor interacting protein kinase 3 (RIPK3) induces necroptosis, a type of regulated necrosis, through its kinase domain and RIP homotypic interaction motif (RHIM). In addition, RIPK3 has been shown to regulate NLRP3 inflammasome and NF-κB activation. However, the relative contribution of these signaling pathways to RIPK3-dependent inflammation in distinct immune effectors is unknown. To interrogate these questions, we generated RIPK3-GFP reporter mice. We found that colonic CD11c+CD11b+CD14+ mononuclear phagocytes (MNPs) expressed the highest level of RIPK3 in the lamina propria. Consequently, deletion of the RIPK3 RHIM in CD11c+ cells alone was sufficient to impair DSS-induced IL-23 and IL-1β expression, leading to severe intestinal inflammation. In contrast, mice expressing kinase inactive RIPK3 were not hypersensitive to DSS. Thus, a key physiological function of RIPK3 is to promote reparative cytokine expression through intestinal CD11c+ MNPs in a kinase- and necroptosis-independent manner.
Graphical Abstract
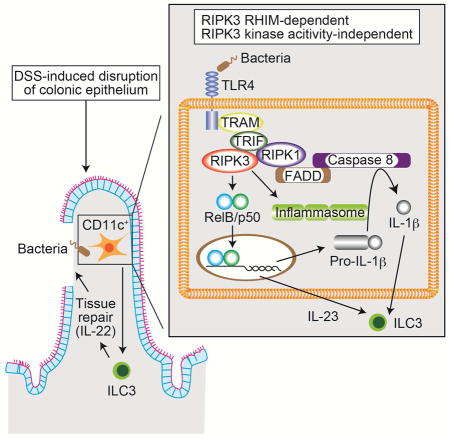
Indroduction
Necroptosis is a type of regulated necrosis induced by several cell surface immune receptors such as tumor necrosis factor (TNF) receptor (TNFR), toll like receptor (TLR) 3 (TLR3), TLR4, interferon receptor, and T cell receptor (Chan et al., 2015). Receptor interacting protein kinase 3 (RIPK3) is a cytosolic master regulator of necroptosis (Moriwaki and Chan, 2013). RIPK3 has an active serine/threonine kinase domain at the N-terminus, and a unique protein-protein interaction domain called the RIP homotypic interaction motif (RHIM) at the C-terminus. Both kinase activity and RHIM are indispensable for necroptosis (Cho et al., 2009). RIPK3 interacts with other RHIM-containing proteins such as RIPK1 (Cho et al., 2009; He et al., 2009), Toll/interleukin-1 (IL-1) receptor domain-containing adaptor protein inducing interferon β (TRIF) (He et al., 2011) or DNA-dependent activator of interferon regulatory factor (DAI) (Upton et al., 2012). RHIM-RHIM interaction leads to amyloid-like conformational change and enhancement of RIPK3 kinase activity (Li et al., 2012). Activated RIPK3 phosphorylates the downstream adaptor MLKL (Sun et al., 2012), which triggers oligomerization and translocation of MLKL to the plasma membrane (Cai et al., 2014; Chen et al., 2014; Dondelinger et al., 2014; Wang et al., 2014).
Besides necroptosis, recent emerging evidence shows that RIPK3 also has necroptosis-independent functions (Moriwaki and Chan, 2014). For example, RIPK3 stimulates NF-κB-dependent Il23p19 (Il23a) expression and pro-IL-1β processing downstream of TLR4 in bone marrow (BM)-derived DCs (BMDCs) (Moriwaki et al., 2014). Similar RIPK3-dependent processing of pro-IL-1β has also been observed in BM-derived macrophages (BMDMs) treated with Smac mimetics and/or caspase inhibitors (Kang et al., 2015; Kang et al., 2013; Lawlor et al., 2015; Vince et al., 2012). However, the relative contribution of necroptosis-dependent and independent effects of RIPK3 in physiological inflammation has not been clearly elucidated.
Germline Ripk3-deficient mice have been used extensively to investigate the pathophysiological roles of RIPK3 (Chan et al., 2014). The resolution of inflammation in Ripk3−/− mice has often been attributed to as a consequence of blocking necroptosis. Strikingly, RIPK3 has recently been shown to have paradoxical function in promoting injury-induced tissue repair (Godwin et al., 2015). In mouse model of chemical-induced intestinal injury, RIPK3 facilitates expression of IL-23 and IL-1β, which in turn stimulates expression of the tissue repair cytokine IL-22. Thus, Ripk3−/− mice developed sustained injury and inflammation (Moriwaki et al., 2016; Moriwaki et al., 2014). Based on these results, we asked if RIPK3 promotes cytokine-induced tissue repair mainly through DCs. To achieve our goal, we generated “knock-in” mice expressing a RIPK3-GFP fusion reporter as well as mice with tissue-specific deletion of the essential RHIM (Ripk3ΔR/ΔR). Using the RIPK3-GFP reporter mice, we found that RIPK3 was highly expressed in intestinal CD11c+CD11b+CD14+CD103− mononuclear phagocytes (MNPs). Deletion of RIPK3 RHIM in CD11c+ cells fully recapitulated the severe intestinal inflammation observed in germline Ripk3−/− mice. In contrast, kinase inactive RIPK3 knock-in mice did not exhibit hypersensitivity in response to chemical-induced colitis. Collectively, these results reveal cell type-specific functions of RIPK3 in inflammation as well as the importance of RHIM-RHIM mediated interaction in biological responses beyond necroptosis.
Results
Generation of RIPK3-GFP reporter mice
RIPK3 expression is often induced in tissues and cells during inflammation (Gautheron et al., 2014; Moriwaki et al., 2014; Vitner et al., 2014). To determine the physiological expression pattern of RIPK3 in quiescent and inflamed states, we generated RIPK3-GFP reporter mice in which the Ripk3 gene was fused in-frame with eGfp sequence at the end of the RIPK3 translated sequence (Fig. S1A). Proper targeting of the Ripk3 locus was confirmed by Southern blot analysis (Fig. S1A–C). We confirmed that RIPK3-GFP and wild type RIPK3 were expressed at similar levels in Ripk3-gfpfl/+ MEFs (Fig. 1A). The percentage and number of T and B cells in the thymus and lymph node were normal in Ripk3+/+ and Ripk3-gfpfl/fl mice (Fig. S1D–E and data not shown). In addition, the composition of bone marrow cells was unchanged in Ripk3-gfpfl/fl mice (Fig. S1F). Hence, the RIPK3-GFP fusion protein faithfully reports in vivo expression of RIPK3 without disrupting RIPK3 function.
Fig. 1. RIPK3 is highly expressed in colonic CD11c+CD11b+CD103− MNPs, See also Figure S1–2.

(A) RIPK3-GFP and RIPK3 expression in MEFs of the indicated genotypes were examined by Western blot. (B and C) RIPK3 expression level in various subsets of immune cells in (B) spleen and (C) colonic lamina propria of Ripk3-gfpfl/fl reporter mice was determined by measuring the GFP fluorescence intensity using flow cytometer (n=2). CD19+ and CD3+ cells were defined as B and T cells, respectively. CD3−CD19−CD11c+ cells were defined as DCs, except for colonic CD3−CD19−CD11c+CD11b+ cells, which were defined as MNPs. CD3−CD19−CD11c−CD11b+ cells were defined as macrophages (Mϕ), except for the CD3−CD19−CD11c−CD11bhiF4/80lo cells, which were defined as monocytes (Mo). (D) Spleen was harvested from Ripk3+/+ (dashed line) and Ripk3-gfpfl/fl reporter mice (solid line) 15 hours after intraperitoneal injection of LPS (50 ug/mouse). Representative histogram of GFP fluorescence intensity in CD11bhiF4/80lo monocytes is shown. (E) Mean fluorescence intensity (MFI) of the GFP fluorescence in various colonic lamina propria mononuclear cells from DSS-treated Ripk3-gfpfl/fl reporter mice is shown (n=5–6). (F) Representative histograms of the GFP fluorescence intensity in CD44hi or CD44lo splenic CD3+CD4+ or CD3+CD8+ T cells are shown. (G) Splenocytes were stimulated with either anti-CD3 and anti-CD28 antibodies or concanavalin A (Con A) for 3 days. Representative FACS plots in CD3+CD4+ or CD3+CD8+ T cells were shown. Results shown are mean ± SEM. Asterisks: p < 0.05.
Real-time PCR analysis using various mouse tissues showed the highest expression of the Ripk3 transcript in the spleen (Fig. S2A), suggesting that RIPK3 is highly expressed in immune cells. Indeed, strong GFP fluorescence signal was detected in various immune cells in the spleen (Fig. 1B, S2B). In particular, splenic CD11c−CD11bhighF4/80low monocytes expressed RIPK3 at the highest level (Swirski et al., 2009) (Fig. 1B). In contrast, CD11c+CD11b+CD103− MNPs, but not CD11c−CD11b+ macrophages, showed the highest RIPK3 expression in the colon (Fig. 1C, Fig. S2C). These results indicate that RIPK3 expression pattern in immune cells is highly variable in different cell types and tissues.
We next examined how inflammation might affect RIPK3 expression. To this end, we first injected LPS intraperitoneally and analyzed splenocytes for RIPK3 expression. While RIPK3 expression in CD11c−CD11bhighF4/80low monocytes was further enhanced (Fig. 1D), no significant change in GFP fluorescence was observed in other cell types (Fig. S2D). Moreover, RIPK3 protein expression in multiple tissues were unchanged despite clear induction of mature IL-1β (Fig. S2E). We previously showed that RIPK3 expression was increased in immune cells in the lamina propria of DSS-treated mice (Moriwaki et al., 2014). Consistent with our previous results, RIPK3-GFP expression in colonic CD3+ T cells, but not other immune cells, was significantly increased upon DSS treatment (Fig. 1E). In addition, higher RIPK3 expression was found in splenic CD44hi effector/memory T cells compared with CD44lo naïve T cells (Fig. 1F). In vitro activation of T cells by anti-CD3 and anti-CD28 antibodies or concanavalin A (Con A) also enhanced RIPK3 expression (Fig. 1G and S2F). These results indicate that RIPK3 expression is highly inducible in T cells.
The RIPK3 RHIM is critical for RIPK3-mediated cell death
RIPK3 has two distinct functional domains: the kinase domain and the RHIM. Studies in knock-in mice expressing kinase inactive RIPK3 (Ripk3K51A/K51A and Ripk3D161N/D161N) showed that RIPK3 kinase activity is crucial for necroptosis (Mandal et al., 2014; Newton et al., 2014). In contrast, the physiological function of the RHIM has not been fully explored. To examine the physiological importance of the RHIM, we generated mice specifically lacking RIPK3 RHIM at the C-terminus using Cre-loxP mediated recombination (Fig. S3A). We crossed the Ripk3-gfpfl/fl reporter mice with Sox2-Cre deletor mice to generate mice with germline deletion of the RHIM (Ripk3ΔR/ΔR) (Fig. S3B). Loss of GFP fluorescence signal from all immune cell subsets from Ripk3ΔR/ΔR mice was confirmed by flow cytometry (Fig. 2A). Since the stop codon of the Ripk3 gene was removed in the Ripk3ΔR allele, we performed 3′-rapid amplification of cDNA ends (RACE) experiment using total RNA from Ripk3ΔR/ΔR mouse embryonic fibroblast (MEFs) to determine the C-terminal amino acid sequence of the RIPK3-ΔRHIM protein. The result revealed two possible mRNA transcripts generated from the Ripk3ΔR allele (Fig. S3C). DNA sequencing confirmed that the both transcripts are expected to encode truncated and shorter RIPK3 that lack the RHIM (Fig. S3D). A widely used rabbit polyclonal antibody that recognizes the C-terminal end of mouse RIPK3 revealed loss of full-length RIPK3 expression from Ripk3ΔR/ΔR cells and tissues (Fig. 2B–C, S3E, middle panel, RIPK3-P). In contrast, the anti-RIPK3 antibody clone 1G6.1.4 (RIPK3-G) recognized both wild type RIPK3 and the RIPK3-ΔRHIM proteins that were absent in Ripk3−/− cells (Fig. 2B–C, S3E, top panel, RIPK3-G) (Newton et al., 2014). It should be noted that the RIPK3-G antibody detected two bands, both of which were specific for RIPK3 (Newton et al. 2014), and that a single band of RIPK3-ΔRHIM protein was detected between these two WT bands (Fig. 2B–C). This suggests that WT RIPK3 has two isoforms with different C-terminal sequences. Similar to mice expressing kinase inactive RIPK3 (Mandal et al., 2014), RIPK3-ΔRHIM was expressed at lower level compared with wild type RIPK3 (Fig. 2B–C, top panel). This suggests that the RHIM and kinase activity are both required to stabilize RIPK3 protein expression.
Fig. 2. The RIPK3 RHIM is essential for necroptosis, See also Figure S3.

(A) Representative histograms of the GFP fluorescence in splenocytes of mice with indicated genotype are shown. (B–C) Western blotting of (B) BMDCs and (C) ConA-activated T cell blasts from mice of the indicated genotypes was performed using two distinct anti-RIPK3 antibodies, RIPK3-P and RIPK3-G. The wild type RIPK3 and RIPK3-ΔRHIM bands are indicated. * = non-specific signals. (D–G) Necroptosis was induced by various stimuli in (D) MEFs, (E–F) BMDCs, and (G) BMDMs from Ripk3+/+ and Ripk3ΔR/ΔR mice. N, no stimulation; T, 100 ng/ml TNF; L, 100 ng/ml LPS; P, 20 μg/ml poly(I:C). (H) Fadd+/−Ripk3ΔR/ΔR mice were intercrossed, and the number of offspring with each genotype is shown. (I) A representative picture of the spleen and lymph node at 13 week-old mice of the indicated genotypes is shown. (J) Splenocytes from 13 week-old mice of the indicated genotypes were analyzed by flow cytometer. Results shown are mean ± SEM of triplicates.
To evaluate the impact of the RHIM on necroptosis, we treated Ripk3ΔR/ΔR MEFs, with TNF, the pan-caspase inhibitor z-VAD-fmk (zVAD), and cycloheximide (CHX). In contrast to wild type MEFs, Ripk3ΔR/ΔR MEFs were resistant to TNF-induced necroptosis (Fig. 2D). Ripk3ΔR/ΔR BMDCs were also resistant to necroptosis induced by LPS and zVAD-fmk (Fig. 2E) and zVAD-fmk and the Smac mimetic BV6 (Fig. 2F), which induces autocrine TNF production (McComb et al., 2012). In addition, Ripk3ΔR/ΔR BMDMs were resistant to necroptosis induced by TNF, LPS, or polyIC in the presence of the pan-caspase inhibitor zVAD-fmk (Fig. 2G). Similar resistance to necroptosis was observed in Ripk3−/− 3T3 cells reconstituted with HA-tagged RIPK3-ΔRHIM proteins (Fig. S3F). By contrast, Ripk3ΔR/ΔR BMDMs were equally sensitive to apoptosis induced by TNF, LPS or poly(I:C) (Fig. S3G). Moreover, Ripk3ΔR/ΔR MEFs were resistant to apoptosis induced by the RIPK3 kinase inhibitor GSK’843 (Fig. S3H), which causes caspase 8 activation through RHIM-mediated formation of the ripoptosome (Mandal et al., 2014; Moriwaki and Chan, 2016). Furthermore, Ripk3ΔR/ΔR BMDCs were resistant to LPS and CHX-induced caspase 3/8 activation and apoptosis (Fig. S3I and S3J) (Moriwaki et al., 2015). These results indicate that the RIPK3 RHIM is crucial for RIPK3-mediated necroptosis and apoptosis.
Caspase 8−/− and Fadd−/− mice suffer from embryonic lethality due to excessive necroptosis. This defect was completely rescued by inactivation of RIPK3 (Kaiser et al., 2011; Oberst et al., 2011). We set up intercrosses of Fadd+/−Ripk3ΔR/ΔR mice and found that Fadd−/−Ripk3ΔR/ΔR mice were born at a Mendelian ratio (Fig. 2H). Moreover, Fadd−/−Ripk3ΔR/ΔR mice developed splenomegaly and lymphadenopathy (Fig. 2I), and exhibited an expansion of CD3+B220+CD4−CD8− T cells in the spleen (Fig. 2J). These phenotypes are characteristic of lpr and gld mice and the human autoimmune lymphoproliferative syndrome (ALPS) (Lenardo et al., 1999). Hence, the RIPK3 RHIM is essential for necroptosis induced by various physiological stimuli and deletion of the RHIM is functionally equivalent to the Ripk3 null allele.
The RHIM is essential for RIPK3-dependent cytokine production
We previously reported that RIPK3 promotes NF-κB and inflammasome activation in BMDCs and that these necroptosis-independent signaling functions are crucial for cytokine production downstream of TLR4 (Moriwaki et al., 2014). In contrast to BMDCs treated with RIPK3 kinase inhibitor (Moriwaki et al., 2014), LPS-induced Il23p19 expression was reduced in Ripk3ΔR/ΔR BMDCs (Fig. 3A). TNF expression was also significantly diminished in LPS-treated Ripk3ΔR/ΔR BMDCs (Fig. 3B). The RelB-p50 heterodimer potently induces Il23p19 expression by BMDCs in response to TLR stimulation (Shih et al., 2012). This led us to test if the RIPK3 RHIM is required for optimal RelB-p50 activation and cytokine expression (Moriwaki et al., 2014). Indeed, LPS-induced nuclear translocation of RelB and p50 was similarly impaired in Ripk3ΔR/ΔR and Ripk3−/− BMDCs (Fig. 3C).
Fig. 3. The RIPK3 RHIM is required for cytokine production in BMDCs.

Ripk3+/+, Ripk3−/−, and Ripk3ΔR/ΔR BMDCs were stimulated with LPS for 6 hours (A, B, D, and G), 2 hours (C), or 1 hour (E, F, and H). Where indicated, the cells were pretreated with GSK’843 for 1 hour (F–H). Il23p19 expression was determined by Q-PCR (n=3) (A). TNF (B) and IL-1β (D and G) secretion were determined by ELISA (n=3–8). Caspase 1 activity was quantified using fluorescent substrate YVAD-AFC (n=2) (F). Nuclear extracts (C) or whole cell lysates (E and H) were subjected to Western blotting. Results shown are mean ± SEM. Asterisks: p < 0.05.
In monocytes and DCs, LPS alone is sufficient to stimulate mature IL-1β secretion without a second inflammasome activation signal (He et al., 2013; Moriwaki et al., 2015). This response requires an intact RIPK3, but not its kinase activity (Moriwaki et al., 2015). LPS-induced IL-1β secretion was blunted in Ripk3ΔR/ΔR BMDCs (Fig. 3D), indicating that unlike the kinase activity, the RHIM is essential for this function. Mature IL-1β secretion is achieved by cleavage of pro-IL-1β by caspase 1 or caspase 8 (Latz et al., 2013). The activation of both caspases was equally impaired in Ripk3ΔR/ΔR and Ripk3−/− BMDCs (Fig. 3E and 3F). RIPK3 stimulates caspase 8-mediated cleavage of pro-IL-1β through a ripoptosome-like complex (Kang et al., 2015; Lawlor et al., 2015; Moriwaki et al., 2015). Assembly of this complex is enhanced by addition of GSK’843, which causes a conformational change of RIPK3 to promote interaction with RIPK1, FADD and caspase 8 (Moriwaki et al., 2015). GSK’843 enhanced LPS-induced caspase 8 activation and IL-1β secretion in Ripk3+/+ BMDCs (Fig. 3G and H). In contrast, GSK’843 failed to stimulate IL-1β secretion or caspase 8 activation in Ripk3ΔR/ΔR BMDCs (Fig. 3G and H). Hence, unlike its kinase activity, the RIPK3 RHIM is essential for optimal LPS-induced NF-κB and inflammasome activation.
The RHIM of RIPK3 mediates protection against DSS-induced colitis
DSS induces intestinal epithelial cell injury and the resultant inflammation exhibits certain features resembling acute colitis. We previously showed that RIPK3-mediated expression of the cytokines IL-23, IL-1β, and IL-22 has a crucial role in the resolution of intestinal injury (Moriwaki et al., 2014). To determine whether this tissue repair function of RIPK3 requires an intact RHIM, we treated Ripk3ΔR/ΔR mice with DSS for 7 days, followed by recovery in normal drinking water for another 7 days. When compared to Ripk3+/+ littermates, Ripk3ΔR/ΔR mice suffered from more severe body weight loss (Fig. 4A). In addition, colon length after DSS treatment was significantly reduced in Ripk3ΔR/ΔR mice (Fig. 4B). In contrast, Ripk3kd/kd (K51A) kinase inactive mice developed similar intestinal inflammation compared to co-housed Ripk3+/− controls, in which RIPK3 protein is expressed at the level similar to RIPK3 K51A protein, as determined by body weight loss and colon length on day 15 (Fig. 4C and D). Since the kinase activity of RIPK3 is essential for necroptosis, these results indicate that RIPK3 mediates reparative inflammation in the intestine through RHIM-dependent but necroptosis- and kinase-independent mechanisms.
Fig. 4. The RIPK3 RHIM, but not its kinase activity, mediates protection against DSS.

Ripk3ΔR/ΔR (n=10) and Ripk3+/+ littermates (n=16) (A–B), or Ripk3kd/kd (n=17) and Ripk3+/− co-housed mice (n=14) (C–D) were treated with 3% DSS for 7 days, followed by regular water for an additional 7 days. (A and C) Body weight was measured daily. (B and D) Colon length was measured on day 15. The inset in C shows equal RIPK3 protein expression between Ripk3kd/kd and Ripk3+/− colon. Results shown are mean ± SEM. Asterisks: p < 0.05.
RIPK3 promotes injury-induced cytokine expression by CD11c+ cells
Radiation bone marrow chimera experiments revealed that RIPK3 expression in hematopoietic cells was important for protection against DSS-induced colitis (Moriwaki et al., 2014). Adoptive transfer of LPS-treated Ripk3+/+ BMDCs, but not Ripk3−/− BMDCs, rescued production of the tissue repair-associated cytokines IL-22, IL-23 and IL-1β. Because RIPK3 promotes optimal cytokine expression in BMDCs, we reasoned that DC-like cells are responsible for the protective effect of RIPK3 in DSS-induced colitis and tissue repair. To test our hypothesis, we generated DC-specific Ripk3ΔR/ΔR (CD11c:Ripk3-gfpfl/fl) mice by crossing the Ripk3-gfpfl/fl reporter mice with CD11c (Itgax)-Cre transgenic mice. Flow cytometry confirmed that the GFP fluorescence signal was lost in CD11c+ cells (Fig. 5A). Consistent with the reported weak activity of the CD11c-Cre transgene in T and B cells (Caton et al., 2007), we also observed slight reduction in GFP signals in these cells. When the CD11c:Ripk3-gfpfl/fl mice were challenged with DSS, they exhibited much more severe body weight loss and shortening of the colon than Ripk3-gfpfl/fl littermates (Fig. 5B–C). Similar to Ripk3−/− mice (Moriwaki et al., 2014), CD11c:Ripk3-gfpfl/fl mice produced significantly reduced IL-22, a critical promoter of tissue repair, in the colon after DSS treatment than littermate controls (Fig. 5D). In addition, expression of IL-23 and IL-1β, which stimulate IL-22 production by type 3 innate lymphoid cells (Lee et al., 2013), was also significantly suppressed in the colon of DSS-treated CD11c:Ripk3-gfpfl/fl mice (Fig. 5D). By contrast, TNF expression, which was normal in DSS-treated Ripk3−/− mice (Moriwaki et al., 2014), was also unaffected in CD11c:Ripk3-gfpfl/fl mice (Fig. S4A). The number of T cells, B cells, CD11c+ and CD11b+ mononuclear phagocytes in the intestinal lamina propria was similar in Ripk3-gfpfl/fl and CD11c:Ripk3-gfpfl/fl mice (Fig. S4B–C). In addition, the percentage of type 3 innate lymphoid cells and RIPK3 in these cells was also unchanged in CD11c:Ripk3-gfpfl/fl mice (Fig. S4D–E). These results are consistent with those observed in germline Ripk3−/− mice and indicate that RIPK3 promotes optimal expression of repair-associated cytokines by CD11c+ cells in a cell-intrinsic manner.
Fig. 5. CD11c+ cell-specific deletion of the RIPK3 RHIM confers protection against DSS, See also Figure S4.

(A) Representative histograms of the GFP fluorescence intensity in colonic lamina propria mononuclear cells from mice of the indicated genotypes are shown. (B and C) Ripk3-gfpfl/fl (n=11) and CD11c:Ripk3-gfpfl/fl littermates (n=16) were treated with 3% DSS for 7 days, followed by regular water for additional 7 days. Body weight was measured daily (B). Colon length on day 15 is shown (C). (D) Expression of Il22, Il23p19, and Il1b in colon from the mice treated with DSS for 7 days was determined by Q-PCR (n=3–10). Results shown are mean ± SEM. Asterisks: p < 0.05.
The role of colonic CX3CR1+ MNPs in RIPK3-dependent cytokine expression
Colonic CD11c+ cells are subdivided into two main populations: CD11c+CD11b+CD103− and CD11c+CD11b−CD103+ cells (Fig. S2C) (Denning et al., 2011). While CD11c+CD11b−CD103+ cells are considered to be conventional DCs developed from common DC precursors, CD11c+CD11b+CD103− cells are developed from monocytes and express both DC and macrophage markers (Cerovic et al., 2014). Within the CD11c+CD11b+CD103− MNPs, the chemokine receptor CX3CR1 was reported to mark a population of cells that is the main source of IL-23 and IL-1β during intestinal inflammation (Longman et al., 2014). We therefore tested the role of RIPK3 in cytokine production by the CX3CR1+ MNPs. Since there is no reliable CX3CR1 antibody available for flow cytometry, we used CD14 as a marker to test RIPK3 expression in this population. Consistent with a previous report (Diehl et al., 2013), we confirmed by using Cx3cr1gfp/+ reporter mice that the CD11c+MHC-II+CD14+ MNPs were indeed excusively positive for CX3CR1 expression (Fig. S5A). Interestingly, these CD11c+CD11b+CD103−CD14+ MNPs also expressed high levels of RIPK3 compared to CD11c+CD11b−CD103+ DCs (Fig. 6A). We isolated CX3CR1+ MNPs and CD103+ DCs from colonic lamina propria of untreated Ripk3+/+Cx3cr1gfp/+ and Ripk3−/−Cx3cr1gfp/+ mice or the mice challenged with DSS for 7 days and compared their cytokine expression profile (Fig. S5B). Consistent with a published report (Longman et al., 2014), Il23p19 and Il1b expression was induced in CX3CR1+ MNPs in response to DSS (Fig. 6B). In contrast, the cytokine expression was undetectable in CD103+ DCs (Fig. 6B). Strikingly, the expression of Il23p19 was significantly reduced in CX3CR1+ MNPs isolated from Ripk3−/−Cx3cr1gfp/+ mice compared with those from Ripk3+/+Cx3cr1gfp/+ littermates. The expression of Il1b was also reduced in CX3CR1+ MNPs isolated from Ripk3−/−Cx3cr1gfp/+ mice, although the difference was not statistically significant. To determine if CX3CR1+ MNPs are responsible for the impaired cytokine expression in CD11c:Ripk3-gfpfl/fl mice, we isolated CD11c+CD11b+CD14+ MNPs from DSS-treated Ripk3-gfpfl/fl and CD11c:Ripk3-gfpfl/fl mice. Indeed, the cells from CD11c:Ripk3-gfpfl/fl mice also exhibited reduction in Il1b and Il23p19 expression (Fig. S5C), although the difference was not statistically significant. Collectively, these results suggest that CX3CR1+ MNPs in the lamina propria contributes to injury-induced cytokine expression.
Fig. 6. RIPK3 promotes cytokine production by colonic CX3CR1+ MNPs, See also Figure S5.

(A) Mononuclear cells from untreated or DSS-treated Ripk3-gfpfl/fl and Ripk3+/+ mice were analyzed for RIPK3-GFP mean fluorescence intensity (MFI). CD14+ MNPs and CD103+ DCs were defined as CD45+CD3−CD19−CD11c+MHC-II+CD14+CD103− (for CD14+ MNPs) and CD45+CD3−CD19−CD11c+MHC-II+CD14−CD103+ (for CD103+ DCs) respectively (n=5–6). (B) CD45+CD11c+MHC-II+CD11b+CX3CR1+ MNPs and CD45+CD11c+MHC-II+CD11b−CD103+ DCs were FACS-sorted from mice treated with DSS for 7 days and subjected to RNA extraction and Q-PCR analysis (n=2–6). Results shown are mean ± SEM. Asterisks: p < 0.05.
Discussion
RIPK3 is a key signal adaptor for necroptosis. Recently, RIPK3 has also been shown to promote activation of the NLRP3 inflammasome, NF-κB, and apoptosis. The role of RIPK3 in necroptosis-independent signaling is especially prominent in innate immune sentinels such as macrophages and DCs. However, tools that allow examination of RIPK3 functions in distinct cell populations have been lacking. In this study, we report two mouse models that address this deficiency. Using the RIPK3-GFP reporter mice, we found that RIPK3 expression is dynamically regulated in different immune subsets. For example, RIPK3 expression was increased in macrophages in response to LPS stimulation. In contrast, RIPK3 expression was strongly induced in T cells during intestinal inflammation. T cell receptor-dependent induction of RIPK3 expression suggests that RIPK3 may regulate T cell responses and functions. Indeed, previous studies showed that Fadd−/−Ripk3−/− and Casp8−/−Ripk3−/− mice developed a lpr/gld-like autoimmune disease (Dillon et al., 2012; Kaiser et al., 2011; Oberst et al., 2011), indicating that RIPK3 cooperates with FADD/Caspase 8 to enforce peripheral T cell tolerance. Whether RIPK3 regulates other functions in T cells is unknown at present.
In contrast to T cells, RIPK3 has a key role in cytokine expression by DCs and macrophages. RIPK3 expression in CD11c+ DCs and CD11b+ macrophages was not dramatically altered during LPS- or DSS-induced inflammation, although its expression in these innate immune effectors in different anatomical locations are highly variable. For instance, CD11c+MHC-II+CD11b+ MNPs in the colonic lamina propria highly expressed RIPK3 compared to those in spleen and showed the highest RIPK3 expression among various immune cell subsets. These cells have been implicated to be major producers of IL-23 and IL-1β, which in turn stimulate IL-22 production by type 3 innate lymphoid cells (Aychek et al., 2015; Longman et al., 2014; Manta et al., 2013). Depletion of this population or knockout of CX3CR1 exacerbated experimental colitis (Longman et al., 2014; Medina-Contreras et al., 2011). Although some of the effect might be due to the reduced expression of RIPK3-ΔRHIM protein, deletion of the RIPK3 RHIM in CD11c+ cells alone was sufficient to recapitulate the severe colitis observed in germline Ripk3−/− mice. This observation strongly suggests that the RHIM is essential for necroptosis-dependent and independent functions of RIPK3. Ripk3−/− CX3CR1+ MNPs and CX3CR1+ MNPs from CD11c:Ripk3-gfpfl/fl mice produced reduced levels of Il-1b, Il23p19 and Il22, although the differences were not always statistically significiant. Thus, there may be other immune effectors within the lamina propria that require RIPK3 to achieve optimal cytokine expression.
RIPK3 is widely believed to induce inflammation through necroptosis-associated release of damage-associated molecular patterns (Kaczmarek et al., 2013). This current dogma is mostly based on studies from mice lacking caspase 8 or FADD, or multiple cellular inhibitor of apoptosis proteins (cIAPs), all of which are strong inhibitors of RIPK3 pro-necroptotic function (Chan et al., 2015). It is therefore striking that unlike Ripk3ΔR/ΔR or Cd11c-Cre:Ripk3-gfpfl/fl mice, Ripk3kd/kd mice did not develop more severe colitis. Thus, in injury-induced cytokine expression and tissue repair, RIPK3 functions exclusively as an inducer of cytokine expression rather than as a cell death adaptor. A necroptosis-independent role for RIPK3 in NLRP3 inflammasome activation has been observed in in vitro studies with BMDMs and BMDCs (Kang et al., 2015; Lawlor et al., 2015; Moriwaki et al., 2015; Vince et al., 2012). However, as in the case of necroptosis, maximal RIPK3-mediated inflammasome activation also requires FADD, caspase 8 or cIAPs inhibition. Thus, our result demonstrates a necroptosis-independent role for RIPK3 in physiological inflammation without pharmacological or genetic manipulation of FADD, caspase 8 or cIAPs. Our results also reveal that the non-necroptotic functions of RIPK3 may be confined to specific cell compartments. In this scenario, its function can only be revealed using tissue-specific inactivation approaches.
We previously reported that RIPK3-RIPK1-FADD-caspase 8 complex was formed upon LPS stimulation in BMDCs. Assembly of this ripoptosome-like complex also requires another RHIM-containing adaptor TRIF (Moriwaki et al., 2015). Results from this present study indicate that an intact RHIM is not only crucial for necroptosis, but also for RIPK3-dependent ripoptosome assembly and pro-IL-1β processing. However, this function of RIPK3 does not require its pro-necroptotic kinase activity. In contrast, the mechanism by which RIPK3 promotes RelB-p50 activation is less understood. RIPK3 has been implicated to shuttle between the cytoplasm and nucleus (Yang et al., 2004; Yoon et al., 2016), suggesting that RIPK3 may directly engage the transcriptional machinery under certain conditions.
Experimental Procedures
Mice
To generate Ripk3-gfpfl/fl reporter mice, a targeting construct in which eGfp sequence was inserted at the end of coding region of the Ripk3 gene was created. The neomycine resistance gene flanked by FRT sites was inserted in intron 9 for positive selection. For negative selection, thymidine kinase was added outside the 5′ homology arm. LoxP sites were also inserted before the neomycine cassette and after eGFP sequence. Embryonic stem (ES) cells were transfected with the targeting construct and subsequently selected by neomycine in transgenic animal core at University of Massachusetts Medical School (UMMS). ES clones in which proper recombination occurs were selected by Southern blot analysis with probe 1 located in the 5′ homology arm and probe 2 located outside the 3′ homology arm and subsequently used for injection into albino C57BL/6 blastocysts to generate chimeric mice. Germline transmission of the transgene was confirmed by Southern blot analysis. The neomycine cassette was removed by crossing the Ripk3-gfpflox−neo/+ mice with the flippase transgenic mice (Gt(ROSA)26Sortm1(Flp1)Dym, kindly provided from S. Jones in UMMS). To generate mice constitutively deficient for the Ripk3 RHIM, Ripk3-gfpflox−neo/+ mice were crossed with Sox2-Cre transgenic mice (kindly provided from S. Jones in UMMS). The resultant Ripk3fl/+ and Ripk3ΔRHIM/+ mice were backcrossed with C57BL/6 mice for 8–10 generations. To delete the RHIM specifically in CD11c+ cells, Ripk3-gfpfl/fl mice were crossed with CD11c (Itgax)-Cre transgenic mice obtained from the Jackson Laboratory (Bar Harbor, Maine). Ripk3−/− mice were obtained from Genentech. Fadd−/− mice were described before (Zhang et al., 2011). Ripk3 K51A kinase dead knock-in (Ripk3kd/kd) mice were generated in GlaxoSmithKline (Mandal et al., 2014). Cx3cr1gfp/gfp reporter mice were obtained from the Jackson Laboratory. All animal experiments were approved by the institutional animal care and use committee.
DSS treatment
Female mice (9–12 week old) were treated with 3% DSS (MP Biomedicals, molecular mass 36,000–50,000 Da) for 7 days. The DSS water was replaced with fresh DSS water on days 3 and 5 and with regular water on day 8. Body weight was monitored for 15 days and the weight at the beginning of the experiments was normalized as 100%. To obtain Ripk3+/− control mice with a minimum difference of genetic background, Ripk3kd/+ mice were crossed with Ripk3−/− mice. The resultant Ripk3kd/− or Ripk3+/− mice were intercrossed to obtain Ripk3kd/kd or Ripk3+/− mice. After weaning, these mice were co-housed to minimize the influence of intestinal microflora. All experiments, except for the ones using Ripk3kd/kd mice, were performed using littermates. To minimize the differences in intestinal microbial environment among cages, bedding materials from different cages were mixed once a week after weaning.
Flow cytometry
Colon was harvested from untreated or DSS-treated mice. After removing feces with PBS, the colon was longitudinally opened and cut into three pieces. The tissues were washed with PBS and subsequently incubated in 1 mM DTT for 10 min at RT. Then, the tissues were shaken in 10 mM HEPES buffer containing 30 mM EDTA for 10 min at 37 °C (225 rpm). After washing, the tissues were digested with 0.5 mg/ml collagenase IV (Sigma) and 150 μg/ml DNase I (Sigma) for 90 min at 37 °C. Tissues were broken by vigorously shaking, filtered, and subjected to Percoll density gradient separation. LPMCs at the interphase of the two Percoll solutions were collected and subjected to flow cytometric analysis. To isolate splenocytes, spleen was incubated with 2 mg/ml collagenase D solution (10 mM HEPES pH 7.4, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2) for 30 min at 37 °C. After gliding the spleen, red blood cells were lysed with ACK lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA). Splenocytes were filtered and subjected to flow cytometric analysis. Prior to incubation with primary antibodies, cells were incubated with anti-Fc receptor 2.4G2 antibody for 10 min. PE-labeled anti-CD3 (145-2C11), PerCP-Cy5.5-labeled anti-CD11b (M1/70), Pacific blue-labeled anti-F4/80 (BM8), Pacific blue-labeled anti-CD45.2 (104), biotin-labeled CD103 (2E7), PerCP-Cy5.5-labeled anti-CD8b (YTS/56.7.7), and PE-labeled anti-MHCII I-Ab (AF6-120.1) antibodies were obtained from BioLegend. PE-Cy7-labeled CD19 (eBio10-3), PE-Cy7-labeled CD44 (IM7), PE-Cy7-labeled anti-CD14 (Sa2-8) antibodies, and APC-eFluor780-labeled streptavidin were obtained from eBioscience. APC-labeled anti-CD11c (HL3), APC-Cy7-labeled anti-CD8a (53-6.7), APC-labeled anti-CD3 (145-2C11), APC-Cy7-labeled anti-CD4 (GK1.5), FITC-labeled anti-CD19 (1D3), PE-labeled anti-B220 (RA3-6B2), Alexa Fluor 647-labeled CCR6 (140706), PE-labeled Ter119 (TER-119) and Pacific blue-labeled anti-CD4 (RM4-5) antibodies were obtained from BD Biosciences. Cells were analyzed by LSRII (BD Biosciences). For cell sorting, FACS Aria II (BD Biosciences) was used.
T cell stimulation
A 24 well tissue culture plate was coated with 10 μg/ml anti-CD3 (145-2C11) and 5 μg/ml anti-CD28 (37.51) antibodies at 4 °C overnight. Splenocytes were plated onto the coated plate. For Con A stimulation, splenocytes were plated onto uncoated plate and stimulated with 5 μg/ml Con A. After 3 days, the cells were subjected to flow cytometric analysis.
Western blot
Cell lysates prepared using RIPA lysis buffer supplemented with protease (Roche) and phosphatase inhibitors (Sigma) were subjected to SDS-PAGE. Nuclear extracts were prepared as described before (Moriwaki et al., 2014). After transferring proteins to nitrocellulose membrane, immunoblot analysis was performed with the following antibodies: Anti-RIPK3 (2283, Prosci, RIPK3-P), anti-mouse RIPK3 (Genentech clone 1G6.1.4, RIPK3-G), anti-HSP90 (68/Hsp90, BD Biosciences), anti-HA, anti-RelB (C1E4, Cell Signaling), anti-p50 (H-119, Santa Cruz), anti-BRG1 (kindly provided by A. Imbalzano), anti-caspase 8 (1G12, Enzo Lifesciences), anti-pro-IL-1β (AF-401-NA, R&D systems), and anti-caspase 3 (46, Santa Cruz) antibodies.
Cell culture and stimulation
BM cells were cultured for 7 days in either RPMI media supplemented with 10 ng/ml GM-CSF and 5 ng/ml IL-4 to generate BMDCs or L929-conditioned media to generate BMDMs (Moriwaki et al., 2014). Ripk3+/+, Ripk3−/−, Ripk3ΔR/ΔR MEFs, Ripk3−/− 3T3, and 293T cells were cultured in DMEM media. Ten percent fetal calf serum, 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin were added to the media. Purified LPS from Invivogen was used for all experiments. In some experiments, cells were pretreated with z-VAD-fmk (Enzo Lifesciences), cycloheximide (Sigma), BV6 (kindly provided from D. Vucic in Genentech), and/or RIPK3 kinase inhibitor GSK2393843A (GSK’843, GlaxoSmithKline) for an hour prior to LPS, TNF, or PolyIC stimulation, unless otherwise stated. After stimulation, culture media and cells were used for cell death assay, ELISA, RNA, and protein analyses. Caspase 1 activity was measured by caspase 1 substrate YVAD-AFC (BioVision).
Quantitative PCR
Total RNA was extracted from cells and tissues using RNeasy Mini kit (Qiagen) and subjected to reverse transcription using Superscript III (Invitrogen). Real-time PCR analysis using iQ SYBR Green supermix (Bio-Rad laboratories) was performed on C1000 thermal cycler and CFX96 real-time system (Bio-Rad laboratories). The following primers were used: 5′-TTGAGGTGTCCAACTTCCAGCA-3′ and 5′-AGCCGGACATCTGTGTTGTTA-3′ for Il22, 5′-CCAGCGGGACATATGAATCT-3′ and 5′-AGGCTCCCCTTTGAAGATGT-3′ for Il23a (Il23p19), 5′-CCCAACTGGTACATCAGCAC-3′ and 5′-TCTGCTCATTCACGAAAAGG-3′ for Il1b, 5′-TGTCAAGTTATGGCCTACTGGTGCG-3′ and 5′-AACCATAGCCTTCACCTCCCAGGAT-3′ for Ripk3, and 5′-CAAACCCAGAATTGTTCTCCTT-3′ and 5′-ATGTGGTCTTCCTGAATCCCT-3′ for Tbp.
Cell death assay
Cell death was determined by CellTiter-Glo Luminescent Cell Viability Assay (Promega), CytoTox 96® Non-Radioactive Cytotoxicity Assay (Promega), or CellTiter 96 AQueous One Solution Cell Proliferation Assay (promega). All cell death assays were performed in triplicates.
3′-RACE PCR
DNA sequence of 3′ region of Ripk3ΔR mRNA was determined by 3′-RACE experiment (Scotto-Lavino et al., 2006). Total RNA was extracted from Ripk3ΔR/ΔR MEFs and reverse transcribed using Qtotal primer (5′-CCAGTGAGCAGAGTGACGAGGACTCGAGCTCAAGCTTTTTTTTTTTTTTTTT-3′) and Superscript III. First PCR was performed using Qouter primer (5′-CCAGTGAGCAGAGTGACG-3′) and gene specific primer (GSP) 1 (5′-TGCTGCTGTCTCCGAGGTAAAG-3′). PCR product was subjected to second PCR using Qinner primer (5′-ATCGGATCCGAGGACTCGAGCTCAAGC-3′) and GSP2 (5′-ATCGAATTCCAAAGGAATCAGGGAGATGGAAG-3′). Pfu Turbo DNA polymerase AD was used for PCR. PCR product was run on a 1% agarose gel and purified from the gel. The purified products were digested by EcoRI and HindIII and subsequently cloned into pUC19 vector. DNA sequence was determined by sequence analysis.
Plasmids, viruses, Transfections, and Transductions
Mouse wild type Ripk3 gene, and Ripk3ΔR short and long isoforms were cloned into a modified lentiviral tet-on pTRIPZ/Puro vector. HA tag was introduced at the amino terminus of RIPK3 by PCR cloning. The sequence of all the genes inserted was confirmed by sequence analysis. Ripk3−/− 3T3 cells were transduced by lentivirus generated in 293T cells with pTRIPZ, pMD2.G and psPAX2 vectors. After transduction, the cells were selected by 2 μg/ml puromycin.
Statistical analysis
P values were calculated using unpaired t test with Welch’s correction or two-way repeated measures ANOVA. P values lower than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank Stephen Jones (UMMS) for the flippase and Cre deletor mice, Anthony Imbalzano (UMMS) for the anti-BRG1 antibody, Domagoj Vucic (Genentech) for BV6, and Kim Newton and Vishva Dixit for the RIPK3-G antibody. This work is supported by NIH grant AI119030 (FKMC) and a Crohn’s & Colitis Foundation of America Senior Research Award (FKMC).
Footnotes
Author Contributions
KM and FKMC designed the project. KM and SB performed experiments. JB and PJG provided critical reagents. KM and FKMC wrote a paper. FKMC supervised the project.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aychek T, Mildner A, Yona S, Kim KW, Lampl N, Reich-Zeliger S, Boon L, Yogev N, Waisman A, Cua DJ, Jung S. IL-23-mediated mononuclear phagocyte crosstalk protects mice from Citrobacter rodentium-induced colon immunopathology. Nat Commun. 2015;6:6525. doi: 10.1038/ncomms7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8− dendritic cells in the spleen. J Exp Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerovic V, Bain CC, Mowat AM, Milling SW. Intestinal macrophages and dendritic cells: what’s the difference? Trends Immunol. 2014;35:270–277. doi: 10.1016/j.it.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Chan FK, Luz NF, Moriwaki K. Programmed Necrosis in the Cross Talk of Cell Death and Inflammation. Annu Rev Immunol. 2014 doi: 10.1146/annurev-immunol-032414-112248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol. 2015;33:79–106. doi: 10.1146/annurev-immunol-032414-112248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Zheng X, Chen P, Han J. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–121. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning TL, Norris BA, Medina-Contreras O, Manicassamy S, Geem D, Madan R, Karp CL, Pulendran B. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J Immunol. 2011;187:733–747. doi: 10.4049/jimmunol.1002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, Schwab SR, Littman DR. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature. 2013;494:116–120. doi: 10.1038/nature11809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, Green DR. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012;1:401–407. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–981. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, Kreggenwinkel K, Schneider AT, Bartneck M, Neumann UP, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med. 2014;6:1062–1074. doi: 10.15252/emmm.201403856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godwin A, Sharma A, Yang WL, Wang Z, Nicastro J, Coppa GF, Wang P. Receptor-Interacting Protein Kinase 3 Deficiency Delays Cutaneous Wound Healing. PLoS One. 2015;10:e0140514. doi: 10.1371/journal.pone.0140514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108:20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- He Y, Franchi L, Nunez G. TLR agonists stimulate Nlrp3-dependent IL-1beta production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J Immunol. 2013;190:334–339. doi: 10.4049/jimmunol.1202737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Fernandes-Alnemri T, Rogers C, Mayes L, Wang Y, Dillon C, Roback L, Kaiser W, Oberst A, Sagara J, et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun. 2015;6:7515. doi: 10.1038/ncomms8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kumagai Y, Jang MS, Kim JH, Yang BG, Lee EJ, Kim YM, Akira S, Jang MH. Intestinal Lin− c-Kit+ NKp46− CD4− population strongly produces IL-22 upon IL-1beta stimulation. J Immunol. 2013;190:5296–5305. doi: 10.4049/jimmunol.1201452. [DOI] [PubMed] [Google Scholar]
- Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, Zheng L. Mature T lymphocyte apoptosis--immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, Swaminath A, Bonneau R, Scherl EJ, Littman DR. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med. 2014;211:1571–1583. doi: 10.1084/jem.20140678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manta C, Heupel E, Radulovic K, Rossini V, Garbi N, Riedel CU, Niess JH. CX(3)CR1(+) macrophages support IL-22 production by innate lymphoid cells during infection with Citrobacter rodentium. Mucosal Immunol. 2013;6:177–188. doi: 10.1038/mi.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McComb S, Cheung HH, Korneluk RG, Wang S, Krishnan L, Sad S. cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ. 2012;19:1791–1801. doi: 10.1038/cdd.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Contreras O, Geem D, Laur O, Williams IR, Lira SA, Nusrat A, Parkos CA, Denning TL. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J Clin Invest. 2011;121:4787–4795. doi: 10.1172/JCI59150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki K, Balaji S, Chan FK. Border Security: The Role of RIPK3 in Epithelium Homeostasis. Frontiers in cell and developmental biology. 2016;4:70. doi: 10.3389/fcell.2016.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki K, Balaji S, McQuade T, Malhotra N, Kang J, Chan FK. The Necroptosis Adaptor RIPK3 Promotes Injury-Induced Cytokine Expression and Tissue Repair. Immunity. 2014;41:567–578. doi: 10.1016/j.immuni.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki K, Bertin J, Gough PJ, Chan FKM. A RIPK3-Caspase 8 complex mediates atypical pro-IL-1b processing. J Immunol. 2015;194:1938–1944. doi: 10.4049/jimmunol.1402167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27:1640–1649. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki K, Chan FK. Necrosis-dependent and independent signaling of the RIP kinases in inflammation. Cytokine Growth Factor Rev. 2014;25:167–174. doi: 10.1016/j.cytogfr.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki K, Chan FK. Regulation of RIPK3 and RHIM-dependent necroptosis by the proteasome. J Biol Chem. 2016 doi: 10.1074/jbc.M115.700997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotto-Lavino E, Du G, Frohman MA. 3′ end cDNA amplification using classic RACE. Nat Protoc. 2006;1:2742–2745. doi: 10.1038/nprot.2006.481. [DOI] [PubMed] [Google Scholar]
- Shih VF, Davis-Turak J, Macal M, Huang JQ, Ponomarenko J, Kearns JD, Yu T, Fagerlund R, Asagiri M, Zuniga EI, Hoffmann A. Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-kappaB pathways. Nat Immunol. 2012;13:1162–1170. doi: 10.1038/ni.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L, Mason K, Gross O, Ma S, Guarda G, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–227. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- Vitner EB, Salomon R, Farfel-Becker T, Meshcheriakova A, Ali M, Klein AD, Platt FM, Cox TM, Futerman AH. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat Med. 2014;20:204–208. doi: 10.1038/nm.3449. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Yang Y, Hu W, Feng S, Ma J, Wu M. RIP3 beta and RIP3 gamma, two novel splice variants of receptor-interacting protein 3 (RIP3), downregulate RIP3-induced apoptosis. Biochemical and biophysical research communications. 2005;332:181–187. doi: 10.1016/j.bbrc.2005.04.114. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ma J, Chen Y, Wu M. Nucleocytoplasmic shuttling of receptor-interacting protein 3 (RIP3): identification of novel nuclear export and import signals in RIP3. J Biol Chem. 2004;279:38820–38829. doi: 10.1074/jbc.M401663200. [DOI] [PubMed] [Google Scholar]
- Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016;23:253–260. doi: 10.1038/cdd.2015.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471:373–376. doi: 10.1038/nature09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.