

Abstract
Nuclear-receptors are often overexpressed in tumours and can thereby be used as targets when designing novel selective chemotherapeutic agents. To date, many conjugates incorporating an estrogen receptor (ER) ligand have been synthesised in order to direct chemical agents to tissue sites containing ERs. A series of ER ligand conjugates were synthesised incorporating an antagonistic ER ligand scaffold based on endoxifen, covalently-bound via an amide linkage to a variety of combretastatin-based analogues, which may act as antimitotic agents. These novel endoxifen-combretastatin hybrid scaffold analogues were biochemically evaluated in order to determine their antiproliferative and cytotoxicity effects in both the ER-positive MCF-7 and the ER-negative MDA-MB-231 human breast cancer cell lines. ER competitive binding assays were carried out to assess the binding affinity of the lead conjugate 28 towards both the ERα and ERβ isoforms. In results from the NCI 60-cell line screen, the lead conjugate 28 displayed potent and highly selective antiproliferative activity towards the MCF-7 human cancer cell line (IC50 = 5 nM). In the ER-binding assays, the lead conjugate 28 demonstrated potent ER competitive binding in ERα (IC50 value: 0.9 nM) and ERβ (IC50 value: 4.7 nM). Preliminary biochemical results also demonstrate that the lead conjugate 28 may exhibit pure antagonism. This series makes an important addition to the class of ER antagonists and may have potential applications in anticancer therapy.
Keywords: estrogen receptor ligands, selective estrogen receptor modulators, tumour targeting, conjugates, tamoxifen, endoxifen, hormone-dependent breast cancer
1. Introduction
Estrogen receptors (ER), principally present as two main isoforms; ERα and ERβ, are found in abundance in female reproductive tissues such as the breast, uterus and ovary, while also found in bone, liver and brain tissue [1,2,3,4,5]. ERs can be overexpressed in tumour tissue and this provides a means to selectively target these tissues by both steroidal and non-steroidal ER ligands. ER ligands can be classified by their agonistic and antagonistic behaviour in the different ER-isoforms [3,4,5,6]. The term selective estrogen receptor subtype modulator (SERSM) refers to the observation that a large number of reported ER-ligands have varying degrees of agonist/antagonistic behaviour towards the ERα and ERβ isoforms at the different tissue sites [3,4,5,6,7]. This leads to a complex action [1] where the benefits of a ligand at one ER-tissue site may be lessened by the negative effects the same ligand exerts at another ER-tissue site. For many decades, tamoxifen has been prescribed worldwide for the treatment of hormone-dependent breast cancer, (Figure 1). Tamoxifen displays antagonistic behaviour in breast tumour tissue; however this drug also displays agonistic behaviour on uterine tissue, which can lead to an increased risk of developing uterine cancer in postmenopausal women [3,4,5]. The other main concern in the use of tamoxifen is increased incidence of blood clots [8]. Breast cancer is often hormone dependent in its early stages of development. However as the disease progresses, the tumours can become less hormone dependent and difficult to treat [9,10]. For an effective treatment of hormone-dependent breast cancer, one goal would be to design an ER-ligand with no noticeable agonistic effects, thus displaying pure antagonistic properties.
Figure 1.

Estradiol, Tamoxifen, 4-Hydroxytamoxifen, Endoxifen and Combretastatin A-4.
Various strategies attempt to improve the selectivity of chemotherapeutic agents by specifically targeting cancer cells and tumour environments [11,12]. Conjugates have been designed containing multiple pharmacophore elements or ligands, individually separated by a linker group, which aim to exert a synergistic and improved selective action on the target disease [13]. To date, a number of ER-targeting conjugates have been reported which attempt to exploit the high affinity and receptor selectivity of estrogen receptor ligands to deliver cytotoxic drugs to tumour cells [14,15,16,17,18]. In our investigation, antagonistic ER-ligands are key structural components utilised as the conjugate’s targeting mechanism. In the present study, the ER-targeting antagonist endoxifen is linked via a covalent amide bond to a Combretastatin A-4 analogue—itself a possible antimitotic agent. We now investigate if the introduction of steric hindrance provided by the Combretastatin CA-4 amide fragment, would enhance the ER antagonistic effects of the endoxifen conjugate in the ER positive MCF-7 cells, possibly by interferance with Helix-12. It is hypothesised that the combination of an antagonistic ER-ligand and the Combretastatin CA-4 related acrylic acid antimitotic agent may produce a selective antiproliferative action on ER-dependent cancers.
2. Experimental Section
2.1. Chemistry
All reagents were commercially available and were used without further purification unless otherwise indicated [19]. Tetrahydrofuran (THF) was distilled immediately prior to use from Na/Benzophenone under a slight positive pressure of nitrogen, toluene was dried by distillation from sodium and stored on activated molecular sieves (4 Å) and dichloromethane was dried by distillation from calcium hydride prior to use. Uncorrected melting points were measured on a Gallenkamp apparatus. Infra-red (IR) spectra were recorded as thin film on NaCl plates, or as potassium bromide discs on a Perkin Elmer FT-IR Specrtum 100 spectrometer (Perkin Elmer, Waltham, MA, USA). 1H, 13C and 19F nuclear magnetic resonance (NMR) spectra were recorded at 27 °C on a Brucker Avance DPX 400 spectrometer (400.13 MHz, 1H; 100.61 MHz, 13C; 376.47 MHz, 19F) (Brucker, Billerica, MA, USA) at 20 °C in either CDCl3 (internal standard tetramethylsilane (TMS)) or CD3OD by Dr. John O’Brien and Dr. Manuel Ruether in the School of Chemistry, Trinity College Dublin. For CDCl3, 1H-NMR spectra were assigned relative to the TMS peak at 0.00 δ and 13C-NMR spectra were assigned relative to the middle CDCl3 triplet at 77.00 ppm. For CD3OD, 1H and 13C-NMR spectra were assigned relative to the centre peaks of the CD3OD multiplets at 3.30 δ and 49.00 ppm respectively. 19F-NMR spectra were not calibrated. Electrospray ionisation mass spectrometry (ESI-MS) was performed in the positive ion mode on a liquid chromatography time-of-flight (TOF) mass spectrometer (Micromass LCT, Waters Ltd., Manchester, UK), equipped with electrospray ionization (ES) interface operated in the positive ion mode at the High Resolution Mass Spectrometry Laboratory by Dr. Martin Feeney in the School of Chemistry, Trinity College and a Micromass spectrometer (E.I. Mode) by Dr. Dilip Rai at the Centre for Synthesis and Chemical Biology, University College Dublin. Mass measurement accuracies of <±5 ppm were obtained. Low resolution mass spectra (LRMS) were acquired on a Hewlett-Packard 5973 MSD GC-MS system (Hewlett-Packard, Palo Alto, CA, USA) in electron impact (EI) mode. Rf values are quoted for thin layer chromatography on silica gel Merck F-254 plates, unless otherwise stated. Compounds were visually detected with UV at 254 and 366 nm. Flash column chromatography was carried out on Merck Kieselgel 60 (particle size 0.040–0.063 mm), Aldrich aluminium oxide, (activated, neutral, Brockmann I, 50 mesh) or Aldrich aluminium oxide, (activated, acidic, Brockmann I, 50 mesh). All products isolated were homogenous on TLC. Analytical high-performance liquid chromatography (HPLC) to determine the purity of the final compounds was performed using a Waters 2487 Dual Wavelength Absorbance detector, a Waters 1525 binary HPLC pump, a Waters In-Line Degasser AF and a Waters 717 plus Autosampler (Waters Corporation, Milford, MA, USA). The column used was a Varian Pursuit XRs C18 reverse phase 150 × 4.6 mm chromatography column (Agilent, Santa Clara, CA, USA). Samples were detected using a wavelength of 254 nm. All samples were analyzed using acetonitrile (70%): water (30%) over 10 min and a flow rate of 1 mL/min. Combretastatin A-4 (CA4) 26 was prepared as previously reported [20]. The acrylic acids 13 [21], 24 [21], 15 [22], 16 [23], 21 [22], 23 [24], 14 [25] and 25 [26] were prepared as previously reported.
2.1.1. 4-{1-[4-(tert-Butyldimethylsilanyloxy)phenyl]-2-phenylbut-1-enylphenol 5
Zinc dust (5.85 g, 90.0 mmol) was weighed out and transferred to a three-necked round-bottomed flask containing dry THF (100 mL). Titanium tetrachloride (8.55 g, 4.94 mL, 45 mmol), was carefully added via syringe to the mixture and then refluxed for 2 h under darkness and a nitrogen environment. The benzophenone 2 [27] (3.285 g, 10 mmol) and propiophenone 4a (4.03 g, 4.00 mL, 30 mmol), were dissolved in dry THF (40 mL). This mixture was carefully added to the refluxing mixture in the round-bottomed flask via syringe. The mixture was then refluxed for a further 3 h. Afterwards, the mixture was allowed to cool then diluted with ethyl acetate (150 mL) and washed with 10% potassium carbonate solution (60 mL). After filtration, the organic layer was separated out and the aqueous layer was extracted with ethyl acetate (100 mL × 3). The combined organic layers were washed with 10% potassium carbonate solution (40 mL), water (50 mL) and brine (50 mL) then dried over anhydrous sodium sulfate, filtered and evaporated to dryness in vacuo to yield crude product. The material was purified via flash chromatography on silica gel (hexane:diethyl ether = 6:1) to afford an isomeric product mixture 5 (4.01 g, 93%, E/Z = 1.2:1) as a brown oil. 1H-NMR (400 MHz, CDCl3): δ 0.13 (s, 0.51 × 6H, SiCH3), 0.25 (s, 0.49 × 6H, SiCH3), 0.94–1.03 (m, 12H, SiC(CH3)3, CH3), 2.49–2.54 (q, 2H, J = 7.6 Hz, CH2), 4.78 (bs, 0.5H, OH), 5.05 (bs, 0.5H, OH), 6.49–6.52 (m, 2H, ArH), 6.68 (t, 2H, J = 8.5 Hz, ArH), 6.83–6.85 (m, 2H, ArH), 7.11–7.19 (m, 7H, ArH). 13C-NMR (100 MHz, CDCl3): δ −4.92, −4.82, −4.80, 13.17, 13.20, 17.74, 25.23, 27.21, 113.74, 114.44, 118.50, 119.08, 125.40, 126.21, 126.41, 126.68, 126.93, 127.19, 127.29, 127.37, 127.37, 129.26, 130.08, 130.35, 131.38, 131.70, 135.62, 135.94, 140.64, 142.11, 152.84, 153.05, 153.68. IR: νmax (KBr) cm−1: 3560.4, 2967.6, 1738.9, 1598.4, 1463.1, 1445.1, 1251.1, 1115.8, 1072.3, 896.1, 739.1, 703.2, 655.0. HRMS (EI): Found 453.2220 (M + Na)+, C28H34O2NaSi requires 453.2226.
2.1.2. 4-{1,2-Bis-[4-(tert-butyldimethylsilanyloxy)phenyl]but-1-enyl}phenol 6
According to the general McMurry reaction method above with zinc dust (4.91 g, 75.1 mmol), titanium tetrachloride (7.12 g, 4.12 mL, 37.5 mmol), the benzophenone 2 [27] (2.74 g, 8.34 mmol) and the silylated propiophenone 4b [28] (6.62 g, 25.0 mmol), the isomeric product mixture 6 was afforded (4.44 g, 95%, E/Z = 1.4:1) as a brown oil. 1H-NMR (400 MHz, CDCl3): δ 0.15 (s, 3H, CH3), 0.20 (s, 6H, CH3), 0.26 (s, 3H, CH3), 0.96–1.04 (m, 21H, CH3), 2.48 (q, 2H, J = 7.5 Hz, CH2), 6.50 (dd, 2H, J = 12.6 Hz, 8.6 Hz, ArH), 6.68 (t, 2H, J = 8.5 Hz, ArH), 6.74 (dd, 2H, J = 8.5 Hz, 5.0 Hz, ArH), 6.83 (dd, 2H, J = 11.5 Hz, 8.5 Hz, ArH), 6.98 (dd, 2H, J = 8.5 Hz, 4.0 Hz, ArH), 7.12 (dd, 2H, J = 8.6 Hz, 4.5 Hz, ArH), OH not observed. 13C-NMR (100 MHz, CDCl3): δ −4.88 (CH3), −4.88, −4.84, −4.80, 13.26, 17.75, 17.77, 17.81, 17.84, 25.23, 25.26, 25.29. 25.40, 28.34, 28.46, 113.72, 114.46, 118.43, 119.05, 119.10, 119.16, 130.15, 130.25, 130.37, 131.49, 131.73, 135.11, 135.21, 135.72, 136.08, 136.23, 136.54, 136.87, 140.19, 140.22, 152.82, 152.92, 153.27, 153.69, 153.72. IR: νmax (KBr) cm−1: 3400.3, 2957.5, 2930.6, 2858.5, 1604.3, 1507.6, 1255.2, 1167.5, 914.9, 838.6, 804.1, 780.6. HRMS (EI): Found 583.3015 (M + Na)+, C34H48O3NaSi2 requires 583.3040.
2.1.3. (4-{1-[4-(2-Bromoethoxy)phenyl]-2-phenylbut-1-enyl}phenoxy)-tert-butyldimethylsilane 7
The phenolic triarylethylene 5 (3.16 g, 7.34 mmol), was dissolved in 1,2-dibromoethane (69.0 g, 32.0 mL, 367 mmol), with stirring. Tetrabutylammonium hydrogen sulfate (2.24 g, 6.61 mmol) was added, followed by 1 M sodium hydroxide solution (30 mL). The biphasic mixture was stirred vigorously at room temperature for 16 h. The reaction mixture was worked up via the addition of dichloromethane (100 mL) and sodium bicarbonate solution (100 mL). The aqueous layer was extracted with dichloromethane (100 mL). The organic layers were combined, dried over sodium sulfate and concentrated under in vacuo to yield crude product. The material was purified via flash chromatography on silica gel (hexane:diethyl ether = 40:1) to afford the product 7 (2.05 g, 52%, E/Z = 1.2:1) as a yellow oil. 1H-NMR (400 MHz, CDCl3): δ 0.12 (s, 0.55 × 6H, SiCH3), 0.25 (s, 0.45 × 6H, SiCH3), 0.94–1.02 (m, 12H, SiC(CH3)3, CH3), 2.47–2.53 (m, 2H, CH2), 3.58 (t, 0.45 × 2H, J = 6.0 Hz, NCH2), 3.69 (t, 0.55 × 2H, J = 6.5 Hz, NCH2), 4.18 (t, 0.45 × 2H, J = 6.5 Hz, OCH2), 4.34 (t, 0.55 × 2H, J = 6.0 Hz, OCH2), 6.49–6.58 (m, 2H, ArH), 6.71–6.93 (m, 4H, ArH), 7.09–7.21 (m, 7H, ArH). 13C-NMR (100 MHz, CDCl3): δ −4.92, −4.81, 13.16, 13.21, 13.70, 25.21, 25.23, 28.45, 28.73, 28.79, 31.15, 67.35, 113.00, 113.76, 118.52, 119.10, 125.48, 127.30, 127.40, 129.25, 130.09, 130.26, 131.37, 131.61, 135.88, 136.56, 140.74, 142.04, 155.45, 155.45, 156.28. IR: νmax (KBr) cm−1: 3436.4, 2957.9, 2930.4, 2858.5, 1604.9, 1507.3, 1472.3, 1254.7, 1168.3, 916.2, 838.8, 804.2, 780.3. HRMS (EI): Found 559.1630 (M + Na)+, C30H37O2BrSiNa requires 559.1644.
2.1.4. [2-(4-{1,2-Bis-[4-(tert-butyldimethylsilanyloxy)phenyl]but-1-enyl}phenoxy)ethyl] bromide 8
According to the general alkylation method above with the phenol 6 (4.78 g, 8.51 mmol), 1,2-dibromoethane (80.0 g, 37.0 mL, 426 mmol), tetrabutylammonium hydrogen sulfate (2.60 g, 7.66 mmol) and 1 M sodium hydroxide solution (30 mL), the product 8 was afforded (3.07 g, 54%, E/Z = 1.4:1) as a yellow oil. 1H-NMR (400 MHz, CDCl3): δ 0.20–0.30 (m, 12H, CH3), 0.99–1.08 (m, 21H, CH3), 2.52–2.54 (m, 2H, CH2), 3.60 (t, 0.41 × 2H, J = 6.3 Hz, CH2), 3.68 (t, 0.59 × 2H, J = 6.0 Hz, CH2), 4.21 (t, 0.41 × 2H, J = 6.5 Hz, CH2), 4.34 (t, 0.59 × 2H, J = 6.3 Hz, CH2), 6.55–7.24 (m, 12H, ArH). 13C-NMR (100 MHz, CDCl3): δ −4.80, −4.77, 13.33, 13.36, 17.79, 17.85, 17.88, 25.31, 25.33, 25.36, 28.41, 28.54, 28.76, 28.83, 67.19, 67.37, 76.36, 76.68, 77.00, 130.19, 130.29, 130.35, 131.53, 131.70, 135.05, 135.15, 136.16, 136.41, 136.46, 136.78, 136.88, 140.38, 140.46, 153.03, 153.39, 153.82, 155.43, 156.28. IR: νmax (KBr) cm−1: 3436.4, 2957.9, 2930.4, 2858.5, 1604.9, 1507.3, 1472.3, 1254.7, 1168.3, 916.2, 838.8, 804.2, 780.3. HRMS (EI): Found 689.2450 (M + Na)+, C36H51O3BrSi2Na requires 689.2458.
2.1.5. [2-(4-{1-[4-(tert-Butyldimethylsilanyloxy)phenyl]-2-phenylbut-1-enyl}phenoxy)ethyl]methylamine 9
Methylamine (in a 20 molar equivalent excess), was dissolved in anhydrous tetrahydrofuran (20 mL) together with the bromide 7 (0.54 g, 1.00 mmol) and sealed in a high-pressure tube. The reaction is heated to 60 °C while stirring for 48–72 h. After this time the reaction vessel was cooled. The reaction is worked up via the addition of a sodium carbonate/sodium hydrogencarbonate pH 10 buffer solution (50 mL) and the organics were extracted with dichloromethane (3 × 50 mL). The organic phases were combined, dried over sodium sulfate and the solvent evaporated in vacuo to afford a crude product, which is then purified via flash chromatography (dichloromethane:methanol) to afford the product 9 as a brown oil (0.38 g, 78%, E/Z = 1.3:1) [16]. 1H-NMR (400 MHz, CDCl3): δ 0.13 (s, 0.57 × 6H, Si(CH3)2), 0.26 (s, 0.43 × 6H, Si(CH3)2), 0.95–1.03 (m, 12H, (CH3)3), 2.45–2.53 (m, 5H, NCH3, CH2), 2.89 (s, 0.43 × 2H, CH2), 2.99 (s, 0.57 × 2H, CH2), 3.36 (s, 1H, NH), 3.95 (t, 0.43 × 2H, J = 5.0 Hz, CH2), 4.11 (t, 0.43 × 2H, J = 5.0 Hz, CH2), 6.50–7.20 (m, 13H, ArH). 13C-NMR (100 MHz, CDCl3): δ −4.91, −4.80, 13.21, 13.24, 17.75, 25.24, 25.26, 28.47, 28.60, 35.39, 35.26, 49.47, 50.03, 50.12, 65.73, 65.99, 112.80, 113.54, 118.54, 119.12, 125.44, 125.48, 127.33, 127.41, 129.25, 130.12, 130.20, 131.43, 131.57, 135.68, 136.02, 136.06, 136.35, 137.44, 137.53, 140.59, 140.65, 142.11, 142.19, 153.06, 153.84, 156.10, 156.94. IR: νmax (KBr) cm−1: 3340.9, 2956.9, 2930.0, 2857.0, 1605.5, 1507.3, 1462.7, 1253.0, 1170.6, 1100.7, 1044.8, 914.4, 836.2, 805.0, 779.9, 700.0. HRMS (EI): Found 488.2980 (M + H)+, C31H42NO2Si requires 488.2985.
2.1.6. [2-(4-{1,2-Bis-[4-(tert-butyldimethylsilanyloxy)phenyl]but-1-enyl}phenoxy)ethyl]methylamine 10
According to the general amination method above with the bromide 8 (0.67 g, 1.00 mmol) and methylamine (in a 20 molar equivalent excess), the product 10 was afforded as a brown oil (0.47 g, 76%, E/Z = 1.4:1). 1H-NMR (400 MHz, CDCl3): δ 0.14 (s, 6H, CH3), 0.19 (s, 6H, CH3), 0.95–1.00 (m, 21H, CH3), 2.48 (q, 2H, J = 7.0 Hz, CH2), 2.55 (s, 3H, CH3), 2.80 (s, 1H, NH), 3.03 (s, 2H, NCH2), 4.13 (s, 2H, OCH2), 6.50 (d, 2H, J = 8.0 Hz, ArH), 6.66 (d, 2H, J = 6.5 Hz, ArH), 6.72 (d, 2H, J = 8.0 Hz, ArH), 6.90 (d, 2H, J = 7.0 Hz, ArH), 6.97 (d, 2H, J = 8.0 Hz, ArH), 7.17 (d, 2H, J = 7.0 Hz, ArH). 13C-NMR (100 MHz, CDCl3): δ −4.88, −4.84, 13.29, 17.74, 17.80, 25.24, 25.29, 28.35, 35.61, 50.22, 66.18, 113.51, 118.40, 119.03, 130.22, 130.25, 131.48, 135.06, 136.19, 136.24, 136.94, 140.22, 152.94, 153.30, 156.91. IR: νmax (KBr) cm−1: 3401.3, 2956.7, 2930.4, 2857.6, 1606.1, 1508.0, 1471.8, 1253.9, 1169.7, 915.2, 835.6, 804.7, 779.5. HRMS (EI): Found 618.3785 (M + H)+, C37H56NO3Si2 requires 618.3799.
2.1.7. 4-{1-[4-(2-Methylaminoethoxy)phenyl]-2-phenylbut-1-enyl}phenol 11 (Endoxifen)
The silyl ether amine 9 (0.12 g, 0.25 mmol), was dissolved in a minimum amount (~5 mL) of THF while stirred under nitrogen. An equimolar quantity of TBAF was added, relative to the number of silyl protecting groups present and the mixture was allowed stir for 16–24 h. The reaction was monitored via TLC (dichloromethane:methanol). The solvent was evaporated to dryness. The residue was redissolved in dichloromethane (~30 mL) and then washed with a quantity of 10% HCl solution (~20 mL). The organic phase was dried over sodium sulphate and evaporated to dryness in vacuo. The residue was purified via flash chromatography on silica gel (DCM:MeOH) to afford the product as an isomeric mixture of product 11 [29] as a brown oil (85 mg, 93%, E/Z = 1.1:1). 1H-NMR (400 MHz, CDCl3): δ 0.95 (t, 3H, J = 7.5 Hz, CH3), 2.48–2.56 (m, 6H, NCH3, CH3), 2.94 (s, 0.52 × 2H, CH2), 3.04 (s, 0.48 × 2H, CH2), 3.96 (t, 0.52 × 2H, J = 4.8 Hz, CH2), 4.12 (t, 0.48 × 2H, J = 4.8 Hz, CH2), 4.92 (s, 2H, NH, OH), 6.43–7.20 (m, 13H, ArH). 13C-NMR (100 MHz, CDCl3): δ 13.25, 28.56, 28.63, 49.42, 57.90, 64.66, 112.81, 113.58, 114.12, 114.82, 125.42, 127.36, 127.39, 129.27, 130.17, 130.20, 131.54, 131.58, 134.32, 134.67, 136.07, 136.52, 137.48, 140.20, 140.41, 142.23, 142.26, 154.29, 155.22, 155.62, 156.49. IR: νmax (KBr) cm−1: 3391.6, 3188.4, 2956.7, 2929.8, 2870.4, 1606.2, 1507.7 (C=C), 1462.0, 1238.8, 1170.4, 1036.6, 835.9, 770.8, 699.9. HRMS (EI): Found 374.2116 (M + H)+, C25H28NO2 requires 374.2120.
2.1.8. 4-(1-(4-(2-(Methylamino)ethoxy)phenyl)-2-(4-hydroxyphenyl)but-1-enyl)phenol 12
According to the silyl ether deprotection method above with amine 10 (0.15 g, 0.24 mmol), an isomeric mixture of product 12 was afforded as a brown oil (87 mg, 92%, E/Z = 1:1.2). 1H-NMR (400 MHz, CDCl3): δ 0.89–0.94 (m, 6H, CH3), 2.39–2.49 (m, 2H, CH2), 2.65–2.70 (m, 3H, NCH3), 3.21–3.34 (m, 3H, OH, NCH2), 4.08–4.25 (m, 2H, OCH2), 6.42–7.14 (m, 12H, ArH). 13C-NMR (100 MHz, CDCl3): δ 12.21, 27.90, 27.97, 32.65, 32.71, 48.23, 48.30, 63.28, 63.62, 112.60, 113.17, 113.26, 113.30, 113.41, 113.85, 113.98, 114.01, 129.75, 129.87, 130.07, 130.23, 131.19, 131.22, 131.28, 133.06, 134.71, 136.76, 136.90, 137.01, 139.88, 140.22, 154.75, 154.78, 155.29, 155.51. IR: νmax (KBr) cm−1: 3391.3, 3289.9, 2960.8, 2927.5, 2870.1, 1607.7, 1509.4, 1461.6, 1371.3, 1236.1, 1169.8, 1102.2, 1035.4, 832.0. HRMS (EI): Found 390.2057 (M + H)+, C25H28NO3 requires 386.2069.
2.1.9. (E)-3-(3-Amino-4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)acrylic acid 16
The nitro compound 21 [22], (0.40 g, 1.02 mmol) was dissolved in 19.0 mL glacial acetic acid while zinc powder (3.84 g, 0.06 mmol) was added to the mixture. The reaction was stirred at room temperature for 3 h, then filtered through Celite. The filtrate was diluted with DCM (50 mL) and washed with 1 M sodium hydroxide solution (3 × 250 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to afford yellow crystals 16 (0.25 g, 67%, m.p. 199–202 °C) [23]. 1H-NMR (400 MHz, d-DMSO): δ 3.69–3.72 (m, 12H, OCH3), 3.78 (t, 2H, NH2, J = 9.3 Hz), 6.28 (d, 1H, J = 8.5 Hz), 6.44 (s, 2H), 6.51 (s, 1H), 6.66 (d, 1H, J = 8.5 Hz), 6.75–6.79 (m, 1H), 7.54 (s, 1H, C=CH). 13C-NMR (100 MHz, d-DMSO): δ 55.27 (OCH3), 55.91 (OCH3), 60.10 (OCH3), 103.11, 106.77, 110.09, 116.20, 119.19, 126.87, 129.94, 132.38, 136.85, 137.17, 139.72, 147.39, 152.87, 152.99 (COOH). IR: νmax (KBr) cm−1: 3437.06 (w), 3357.48, 2939.23, 1672.37, 1587.20, 1505.99, 1439.78, 1411.72, 1268.86, 1238.95, 1171.02, 1123.26 (s), 1028.15. HRMS (EI): Found 360.1394 (M + H)+, C19H22NO6 requires 360.1369.
2.1.10. General Method for Synthesis of Acrylic Acids 17–20, 22
A mixture of the appropriate benzaldehyde (1 equivalent), the appropriate phenylacetic acid (~0.50 g, 1 equivalent), acetic anhydride (2 mL) and triethylamine (1 mL) were heated under reflux for 3 h. After acidification with concentrated hydrochloric acid (~5 mL), the resulting solid was filtered off and recrystallised to yield the appropriate acrylic acid.
2.1.11. (E)-3-(4-Bromophenyl)-2-(3,4,5-trimethoxyphenyl)acrylic Acid 17
4-Bromobenzaldehyde (0.41 g, 2.21 mmol) and 3,4,5-trimethoxyphenylacetic acid (0.50 g, 2.21 mmol) were reacted following the general method above. Recrystallisation from ethanol yielded the acrylic acid 17 as fine yellow needles (0.36 g, 41%, m.p. 227–230 °C) [22]. 1H-NMR (400 MHz, d-DMSO): δ 3.67 (s, 6H, OCH3), 3.71 (s, 3H, OCH3), 6.45 (s, 2H, ArH), 7.06 (d, 2H, J = 8.5Hz), 7.46 (d, 2H, J = 8.5 Hz), 7.69 (s, 1H, C=CH), 12.76 (s, 1H, COOH). 13C-NMR (100 MHz, d-DMSO): δ 55.94 (OCH3), 60.15 (OCH3), 106.60, 122.46 (C-Br), 131.32, 133.73, 134.07, 137.13, 137.54, 153.11, 168.15 (COOH). IR: νmax (KBr) cm−1: 3435.94 (w), 2936.34, 1667.10, 1582.78, 1505.97, 1465.88, 1453.56, 1411.97, 1309.07, 1287.21, 1240.39, 1132.43, 1008.37. HRMS (EI): Found 415.0170 (M + Na)+, C18H17O5NaBr requires 415.0157.
2.1.12. (E)-3-(4-Fluorophenyl)-2-(3,4,5-trimethoxyphenyl)acrylic acid 18
4-Fluorobenzaldehyde (0.27 g, 2.21 mmol) and 3,4,5-trimethoxyphenylacetic acid (0.50 g, 2.21 mmol) were reacted following the general method above. Recrystallisation from ethanol yielded the acrylic acid 18 as fine yellow needles (0.43 g, 59%, m.p. 211–213 °C). 1H-NMR (400 MHz, d-DMSO): δ 3.67 (s, 6H, OCH3), 3.71 (s, 3H, OCH3), 6.46 (s, 2H, ArH), 7.08–7.19 (m, 4H, ArH), 7.73 (s, 1H, C=CH), 12.52 (s, 1H, COOH). 13C-NMR (100 MHz, d-DMSO): δ 55.91 (OCH3), 60.13 (OCH3), 106.59, 115.24, 115.46, 131.01, 131.53, 132.39, 132.48, 132.98, 137.02, 137.64, 153.16, 168.27 (COOH). 19F-NMR (100 MHz, d-DMSO): δ −111.64 IR: νmax (KBr) cm−1: 3436.06 (w), 2942.89, 2832.08, 1665.92, 1597.65, 1582.99, 1507.68, 1458.71, 1411.77, 1306.89, 1294.76, 1240.29, 1219.25, 1130.42, 1005.88, 837.04. HRMS (EI): Found 355.0989 (M + Na)+, C18H17O5FNa requires 355.0958.
2.1.13. (E)-3-p-Tolyl-2-(3,4,5-trimethoxyphenyl)acrylic acid 19
p-Tolualdehyde (0.27 g, 2.21 mmol) and 3,4,5-trimethoxyphenylacetic acid (0.50 g, 2.21 mmol) were reacted following the general method above. Recrystallisation from ethanol yielded the acrylic acid 19 as fine yellow needles (0.27 g, 37%, m.p. 190–193 °C). 1H-NMR (400 MHz, d-DMSO): δ 2.24 (s, 3H, CH3), 3.67 (s, 6H, OCH3), 3.71 (s, 3H, OCH3), 6.45 (s, 2H, ArH), 7.03 (2xd, 4H, 8 Hz, ArH), 7.69 (s, 1H, C=CH), 12.61 (s, 1H, COOH). 13C-NMR (100 MHz, d-DMSO): δ 20.87 (CH3), 55.92 (OCH3), 60.14 (OCH3), 106.63, 128.96, 130.26, 131.56, 132.01, 132.22, 136.95, 138.91, 139.04, 153.09, 168.40 (COOH). IR: νmax (KBr) cm−1: 3436.54 (w), 2939.20, 1670.98 (s), 1581.01, 1505.34, 1412.52, 1294.34, 1241.50, 1185.30, 1127.19 (s), 1000.57. HRMS (EI): Found 351.1198 (M + Na)+, C19H20O5Na requires 351.1208.
2.1.14. (E)-3-(4-Methylsulfanylphenyl)-2-(3,4,5-trimethoxyphenyl)acrylic acid 20
4-Methylthiobenzaldehyde (0.34 g, 2.21 mmol), 3,4,5-trimethoxyphenylacetic acid (0.50 g, 2.21 mmol) reacted following the general method above. Recrystallisation from ethanol yielded the acrylic acid 20 as fine yellow needles (0.38 g, 48%, m.p. 194–196 °C). 1H-NMR (400 MHz, d-DMSO): δ 2.43 (s, 3H, SCH3), 3.68 (s, 6H, OCH3), 3.72 (s, 3H, OCH3), 6.47 (s, 2H, ArH), 7.08 (2xd, 4H, J = 8.5 Hz, ArH), 7.68 (s, 1H, C=CH), 12.64 (s, 1H, COOH). 13C-NMR (100 MHz, d-DMSO): δ 14.01 (SCH3), 55.93 (OCH3), 60.15 (OCH3), 106.50, 125.02, 130.56, 130.71, 132.01, 132.13, 136.95, 138.43, 140.36, 153.17, 168.36 (COOH). IR: νmax (KBr) cm−1: 3435.88 (w), 2939.66, 1667.30, 1587.76, 1506.19, 1410.67, 1286.01, 1240.91, 1125.30, 1087.58, 1001.09. HRMS (EI): Found 383.0940 (M + Na)+, C19H20O5NaS requires 383.0929.
2.1.15. (E)-3-Naphthalen-2-yl-2-(3,4,5-trimethoxyphenyl)acrylic acid 22
β-Naphthaldehyde (0.35 g, 2.21 mmol), 3,4,5-trimethoxyphenylacetic acid (0.50 g, 2.21 mmol) were reacted following the general method above. Recrystallisation from ethanol yielded the acrylic acid 22 as fine yellow needles (0.41 g, 51%, m.p. 238–240 °C). 1H-NMR (400 MHz, d-DMSO): δ 3.65 (s, 6H, OCH3), 3.73 (s, 3H, OCH3), 6.52 (s, 2H, ArH), 7.02 (dd, 1H, J = 1.5 Hz, 8.5 Hz, ArH), 7.50 (d, 2H, J = 2 Hz, ArH), 7.52–7.90 (m, 6H ArH, CH=), 12.73 (s, 1H, COOH). 13C-NMR (100 MHz, d-DMSO): δ 55.97 (OCH3), 60.20 (OCH3), 106.99, 126.18, 126.56, 127.18, 127.40, 127.46, 128.25, 131.36, 131.75, 132.16, 132.58, 132.80, 133.43, 137.20, 138.88, 153.08, 168.34 (COOH). IR: νmax (KBr) cm−1: 3435.50, 2937.78, 1664.26, 1581.88, 1504.78, 1454.14, 1411.09, 1287.35, 1239.16, 1127.39, 1005.19. HRMS (EI): Found 387.1222 (M + Na)+, C22H20O5Na requires 387.1208.
2.1.16. General Method for Synthesis of Endoxifen-Acrylic Acid Conjugates 27–46
A mixture of the required acrylic acid (1 equivalent (eq.), 0.15 mmol), DCC (1 eq., 0.15 mmol, 0.03 g) and HOBt (1 eq., 0.15 mmol, 0.02 g) were suspended in 3 mL of anhydrous DCM and stirred for 10 min under a nitrogen atmosphere. The required silyl-protected endoxifen analogue, 9 (0.08 g, 0.15 mmol, 1 eq.) or 10 (0.10 g, 0.15 mmol, 1 eq.), was dissolved in 3 mL of anhydrous DCM and slowly added to the mixture via syringe. Reaction was allowed stir for 24–48 h. Reaction was monitored via TLC (DCM:MeOH, 4:1). The reaction mixture was diluted to 15 mL with anhydrous DCM and filtered to remove DCU. The filtrate was evaporated to dryness under reduced pressure. The residue was dissolved in 3 mL anhydrous THF and stirred under a nitrogen atmosphere. A solution of 0.1 M TBAF (2 equivalents) was added to the mixture and allowed stir for 24 h. The mixture was evaporated to dryness under reduced pressure. The residue was dissolved in DCM and washed with 10% HCl solution. The resulting organic phase was dried over sodium sulfate and evaporated to dryness under vacuum. The residue was purified via flash chromatography on silica gel (DCM:MeOH, 20:1) to yield a E/Z isomeric mixture of the products.
2.1.17. (E)-3-(3-Hydroxy-4-methoxyphenyl)-N-(2-{4-[(E/Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-enyl]phenoxy}ethyl)-N-methyl-2-(3,4,5-trimethoxyphenyl) acrylamide 27
The acrylic acid analogue 13 was reacted with the endoxifen derivative 9, following the general method above. The product 27 was afforded as a brown oil (103 mg, 94%), then changed to a semi-solid resin. 1H-NMR (400 Hz, CDCl3): δ 0.90–0.96 (m, 3H, CH3), 2.47–2.52 (m, 3H, CH3), 3.04–3.24 (m, 3H, NCH3), 3.44–4.35 (m, 16H, OCH3, CH2,, NCH2, OCH2), 6.40–7.20 (m, 19H, ArH), OH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.17, 13.20, 24.45, 25.12, 28.62, 33.44, 48.72, 55.44, 55.57, 55.67, 60.50, 65.71, 105.58, 109.76, 112.65, 113.40, 113.93, 114.64, 115.01, 121.52, 125.46, 127.37, 127.39, 128.07, 129.25, 129.58, 130.17, 130.23, 131.55, 131.59, 135.20, 137.33, 140.44, 142.12, 144.68, 145.96, 145.98, 152.83, 153.72, 154.66, 156.62. IR: νmax (KBr) cm−1: 3420.3, 3376.8, 3270.5, 2930.8, 2850.2, 1626.4, 1606.8, 1580.6, 1507.9, 1462.9, 1410.8, 1274.7, 1238.4, 1169.3, 1126.3, 1026.8, 901.1, 834.9, 762.8, 701.4. HRMS (EI): Found 738.3043 (M + Na)+, C44H45NO8Na requires 738.3043.
2.1.18. (E)-N-(2-{4-[(E/Z)-1,2-Bis-(4-hydroxyphenyl)but-1-enyl]-phenoxy}ethyl)-3-(3-hydroxy-4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide 28
The acrylic acid analogue 13 was reacted with the endoxifen derivative 10, following the general method above,. The product 28 was afforded as a brown oil (104 mg, 92%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.90–0.96 (m, 3H, CH3), 2.40–2.50 (m, 3H, CH3), 3.04–3.25 (m, 3H, NCH3), 3.53–4.30 (m, 16H, OCH3, CH2,, NCH2, OCH2), 6.40–7.20 (m, 18H, ArH), 2xOH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.28, 25.11, 33.42, 48.67, 55.46, 55.68, 60.55, 66.29, 105.32, 105.47, 112.61, 113.37, 113.99, 114.49, 114.53, 114.65, 128.44, 128.73, 128.88, 130.15, 130.29, 130.48, 131.52, 131.59, 133.59, 134.94, 137.49, 140.06, 152.72, 152.81, 153.12, 153.74, 154.17, 154.66, 156.88. IR: νmax (KBr) cm−1: 3428.6, 3376.8, 3270.5, 2932.4, 2869.6, 1607.9, 1580.6, 1509.6, 1462.9, 1410.0, 1272.0, 1238.1, 1169.2, 1125.6, 1050.2, 1023.7, 901.0, 833.5. HRMS (EI): Found 732.3162 (M + H)+, C44H46NO9 requires 732.3173.
2.1.19. (E)-N-(2-{4-[(E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl]phenoxy}-ethyl)-3-(4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide 29
The acrylic acid analogue 14 was reacted with the endoxifen derivative 9, following the general method above. The product 29 was afforded as a brown oil (97 mg, 90%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.91–0.95 (m, 3H, CH3), 2.48–2.50 (m, 2H, CH2), 3.06–3.21 (m, 3H, NCH3), 3.50–3.91 (m, 14H, OCH3, CH2,), 4.10 (m, 0.30 × 2H, OCH2), 4.26–4.33 (m, 0.70 × 2H, OCH2), 5.68 (s, 1H, OH), 6.51–7.17 (m, 19H, ArH). 13C-NMR (100 MHz, CDCl3): δ 13.17, 13.20, 24.45, 25.12, 28.55, 28.62, 33.44, 48.72, 53.00, 55.44, 55.57, 55.67, 60.50, 105.58, 109.76, 112.65, 113.40, 113.93, 114.64, 115.01, 121.52, 125.46, 127.37, 127.39, 128.07, 129.25, 129.58, 130.17, 130.23, 131.55, 131.59, 134.78, 135.20, 137.33, 140.44, 142.12, 144.68, 145.96, 145.98, 152.83, 153.72, 154.66, 156.62. IR: νmax (KBr) cm−1: 3468.6, 3372.0, 3327.3, 2929.7, 2850.8, 1626.3, 1606.0, 1579.0, 1509.4, 1463.2, 1411.3, 1310.6, 1243.3, 1174.1, 1127.6, 1030.3, 829.4. HRMS (EI): Found 722.3073 (M + Na)+, C44H45NO7Na requires 722.3094.
2.1.20. (E)-N-(2-{4-[(E/Z)-1,2-Bis-(4-hydroxyphenyl)but-1-enyl]phenoxy}ethyl)-3-(4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide 30
The acrylic acid analogue 14 was reacted with the endoxifen derivative 10, following the general method above. The product 30 was afforded as a brown oil (104 mg, 94%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.91–0.94 (m, 3H, CH3), 2.42–2.46 (m, 2H, CH2), 3.06–3.22 (m, 3H, NCH3), 3.53–4.24 (m, 16H, OCH3, CH2,), 5.74–5.80 (m, 2H, OH), 6.42–7.12 (m, 18H, ArH). 13C-NMR (100 MHz, CDCl3): δ 13.26, 28.30, 28.40, 38.75, 47.38, 55.45, 55.50, 55.56, 60.54, 60.58, 65.87, 105.36, 105.54, 109.80, 112.63, 113.41, 113.93, 114.40, 114.57, 115.08, 121.54, 127.92, 129.84, 130.21, 130.37, 131.55, 133.78, 134.78, 135.11, 136.66, 144.54, 144.62, 146.05, 152.77, 152.87, 153.46, 153.79, 153.92, 154.24. IR: νmax (KBr) cm−1: 3327.3, 2929.5, 2850.8, 1626.3, 1607.1, 1579.7, 1510.3, 1449.5, 1310.9, 1243.3, 1172.5, 1127.2, 1046.6, 892.6, 829.4. HRMS (EI): Found 738.3076 (M + Na)+, C44H45NO8Na requires 738.3043.
2.1.21. (E)-N-(2-{4-[(E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl]phenoxy}ethyl)-2-(4-methoxyphenyl)-N-methyl-3-(3,4,5-trimethoxyphenyl) acrylamide 31
The acrylic acid analogue 23 was reacted with the endoxifen derivative 9, following the general method above. The product 31 was afforded as a brown oil (99 mg, 92%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.91–0.95 (m, 3H, CH3), 2.49–2.51 (m, 2H, CH2), 3.08 (s, 1.5H, NCH3), 3.17 (s, 1.5H, NCH3), 3.57–3.88 (m, 14H, OCH3, NCH2,), 4.09 (m, 0.35 × 2H, OCH2), 4.25 (m, 0.33 × 2H, OCH2), 4.39 (d, 0.32 × 2H, J = 8.0 Hz, OCH2), 6.39–7.31 (m, 20H, ArH), OH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.15, 13.19, 24.45, 25.14, 33.44, 54.84, 55.33, 60.42, 106.20, 106.23, 113.72, 113.98, 114.69, 125.43, 127.36, 129.23, 129.91, 129.94, 130.12, 130.19, 130.41, 131.49, 131.52, 152.27, 152.30. IR: νmax (KBr) cm−1: 3430.0, 3327.5, 2929.6, 2850.8, 1626.4, 1579.1, 1509.0, 1462.7, 1244.2, 1174.2, 1127.0, 835.5, 641.0. HRMS (EI): Found 722.3099 (M + Na)+, C44H45NO7Na requires 722.3094.
2.1.22. (E)-N-(2-{4-[(E/Z)-1,2-Bis-(4-hydroxyphenyl)but-1-enyl]phenoxy}ethyl)-2-(4-methoxyphenyl)-N-methyl-3-(3,4,5-trimethoxyphenyl)acrylamide 32
The acrylic acid analogue 23 was reacted with the endoxifen derivative 10, following the general method above. The product 32 was afforded as a brown oil (99 mg, 90%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.90–0.93 (m, 3H, CH3), 2.44–2.45 (m, 2H, CH2), 3.09 (s, 1.5H, NCH3), 3.16 (s, 1.5H, NCH3), 3.57–3.86 (m, 14H, OCH3, NCH2,), 4.08 (m, 0.31 × 2H, OCH2), 4.24 (m, 0.28 × 2H, OCH2), 4.39 (d, 0.41 × 2H, J = 8.0 Hz, OCH2), 6.39–7.28 (m, 19H, ArH), 2xOH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.22, 24.43, 25.12, 25.20, 33.41, 55.34, 60.42, 106.25, 112.79, 113.51, 113.76, 113.98, 114.47, 114.66, 129.92, 130.14, 130.20, 130.29, 130.39, 130.78, 130.89, 131.54, 137.18, 152.27, 154.61, 156.82, 158.97. IR: νmax (KBr) cm−1: 3327.7, 2929.5, 2850.7, 1625.8, 1579.3, 1510.5, 1449.9, 1244.1, 1171.7, 1126.7, 1033.0, 834.6, 641.1. HRMS (EI): Found 738.3065 (M + Na)+, C44H45NO8Na requires 738.3043.
2.1.23. (E)-3-Benzo[1,3]dioxol-5-yl-N-(2-{4-[(E/Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-enyl]phenoxy}ethyl)-2-(4-methoxyphenyl)-N-methylacrylamide 33
The acrylic acid analogue 25 was reacted with the endoxifen derivative 9, following the general method above. The product 33 was afforded as a brown oil (94 mg, 93%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.92–0.96 (m, 3H, CH3), 2.49–2.51 (m, 2H, CH2), 3.04–3.13 (m, 3H, NCH3), 3.47–4.24 (m, 7H, OCH3, CH2,), 5.91 (s, 2H, O2CH2), 6.46–7.23 (m, 21H, ArH), OH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.16, 13.19, 24.37, 25.05, 28.54, 28.60, 33.24, 48.97, 54.75, 100.57, 107.69, 108.61, 112.77, 113.52, 113.83, 113.96, 114.68, 123.61, 125.43, 127.36, 129.06, 129.25, 129.69, 129.73, 130.15, 130.20, 131.54, 140.42, 142.25, 146.71, 146.86, 146.89, 153.75, 154.71. IR: νmax (KBr) cm−1: 3435.8, 3323.7, 2929.1, 2850.7, 1626.2, 1575.7, 1509.7, 1243.6, 1036.2, 835.0, 630.8. HRMS (EI): Found 676.2667 (M + Na)+, C42H39NO6Na requires 676.2675.
2.1.24. (E)-3-Benzo[1,3]dioxol-5-yl-N-(2-{4-[(E/Z)-1,2-bis-(4-hydroxyphenyl)but-1-enyl]phenoxy}ethyl)-2-(4-methoxyphenyl)-N-methylacrylamide 34
The acrylic acid analogue 25 was reacted with the endoxifen derivative 10, following the general method above. The product 34 was afforded as a brown oil (95 mg, 92%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.91–0.94 (m, 3H, CH3), 2.44–2.46 (m, 3H, CH2), 3.05–3.12 (m, 3H, NCH3), 3.48–4.53 (m, 7H, OCH3, CH2,), 5.90 (m, 2H, OCH2O), 6.45–7.26 (m, 20H, ArH), OH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.26, 13.25, 24.42, 25.10, 28.42, 33.38, 48.76, 54.76, 54.80, 100.57, 107.73, 108.62, 109.21, 112.79, 113.52, 113.84, 113.99, 114.50, 114.66, 128.99, 129.73, 130.17, 130.22, 130.33, 130.51, 131.56, 140.04, 146.74, 146.84, 146.87, 154.53, 156.93. IR: νmax (KBr) cm−1: 3412.2, 2929.5, 2850.2, 1606.9, 1510.6, 1488.3, 1444.2, 1242.9, 1172.0, 1035.9, 930.8, 833.3. HRMS (EI): Found 692.2648 (M + Na)+, C42H39NO7Na requires 692.2624.
2.1.25. (E)-N-(2-{4-[(E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl]phenoxy}ethyl)-N-methyl-3-naphthalen-2-yl-2-(3,4,5-trimethoxyphenyl) acrylamide 35
The acrylic acid analogue 22 was reacted with the endoxifen derivative 9, following the general method above. The product 35 was afforded as a brown oil (101 mg, 91%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.91–0.95 (m, 3H, CH3), 2.48–2.50 (m, 2H, CH2), 3.11–3.27 (m, 3H, NCH3), 3.50–4.35 (m, 13H, OCH3, CH2,), 6.45–7.77 (m, 23H, ArH), OH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.18, 13.23, 24.45, 25.13, 33.45, 48.72, 53.01, 55.48, 55.61, 60.52, 105.32, 112.65, 113.41, 113.95, 114.66, 125.46, 125.78, 125.80, 125.97, 126.27, 126.93, 126.95, 127.17, 127.37, 127.39, 127.57, 128.87, 129.24, 130.16, 130.27, 131.55, 131.62, 132.34, 132.73, 134.75, 135.09, 136.93, 137.33, 140.44, 140.59, 142.08, 152.85, 153.80, 154.75, 156.63, 171.43, 171.50. IR: νmax (KBr) cm−1: 3425.1, 3327.0, 2929.1, 2850.6, 1625.9, 1579.0, 1507.3, 1449.4, 1410.0, 1310.4, 1242.0, 1170.2, 1127.2, 905.1, 833.8, 701.1, 641.0. HRMS (EI): Found 742.3143 (M + Na)+, C47H45NO6Na requires 742.3145.
2.1.26. (E)-N-(2-{4-[(E/Z)-1,2-Bis-(4-hydroxyphenyl)but-1-enyl]phenoxy}ethyl)-N-methyl-3-naphthalen-2-yl-2-(3,4,5-trimethoxyphenyl)acrylamide 36
The acrylic acid analogue 22 was reacted with the endoxifen derivative 10, following the general method above. The product 36 was afforded as a brown oil (98 mg, 87%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.90–0.92 (m, 3H, CH3), 2.42–2.44 (m, 2H, CH2), 3.11–3.26 (m, 3H, NCH3), 3.44–4.54 (m, 13H, OCH3, CH2,), 6.45–7.77 (m, 22H, ArH), 2xOH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.26, 24.43, 25.09, 33.39, 48.75, 55.41, 55.47, 55.56, 60.53, 60.57, 105.43, 105.57, 106.40, 112.62, 113.38, 114.00, 114.49, 114.54, 114.65, 125.82, 126.01, 126.24, 126.52, 126.97, 127.08, 127.17, 127.58, 128.03, 128.83, 128.89, 129.59, 129.85, 130.17, 130.27, 130.32, 131.17, 131.54, 132.37, 132.73, 133.63, 136.52, 140.07, 152.79, 152.88, 152.96, 153.71, 154.06, 154.14, 154.61, 155.67, 156.93, 171.69. IR: νmax (KBr) cm−1: 3430.0, 3327.7, 2929.5, 2850.7, 1626.0, 1578.4, 1507.4, 1311.2, 1243.1, 1126.8, 641.0. HRMS (EI): Found 758.3123 (M + Na)+, C47H45NO7Na requires 758.3094.
2.1.27. (E)-N-(2-{4-[(E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl]phenoxy}ethyl)-N-methyl-3-p-tolyl-2-(3,4,5-trimethoxyphenyl)acrylamide 37
The acrylic acid analogue 19 was reacted with the endoxifen derivative 9, following the general method above. The product 37 was afforded as a brown oil (98 mg, 93%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.91–0.94 (m, 6H, CH3), 2.31 (s, 3H, CH3), 2.49–2.51 (m, 2H, CH2), 3.08–3.24 (m, 3H, NCH3), 3.56–3.91 (m, 12H, OCH3, CH2,), 4.11 (m, 0.35 × 2H, OCH2), 4.27 (m, 0.30 × 2H, OCH2), 4.47 (d, 0.35 × 2H, J = 8.0 Hz, OCH2), 6.43–7.20 (m, 20H, ArH), OH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.19, 13.23, 20.83, 24.45, 25.12, 25.22, 33.43, 33.60, 38.59, 48.65, 55.47, 55.60, 60.50, 105.49, 112.62, 113.37, 113.98, 114.68, 125.45, 127.37, 127.39, 128.42, 128.44, 128.91, 129.26, 130.14, 130.25, 131.53, 131.62, 131.87, 135.79, 137.43, 142.25, 152.79, 153.99, 154.93, 156.78. IR: νmax (KBr) cm−1: 3425.1, 3327.1, 2929.2, 2850.7, 1626.1, 1579.8, 1507.5, 1449.4, 1411.5, 1310.5, 1242.3, 1170.2, 1127.5, 892.5, 834.3, 641.0. HRMS (EI): Found 706.3118 (M + Na)+, C44H45NO6Na requires 706.3145.
2.1.28. (E)-N-(2-{4-[(E/Z)-1,2-Bis-(4-hydroxyphenyl)but-1-enyl]phenoxy}ethyl)-N-methyl-3-p-tolyl-2-(3,4,5-trimethoxyphenyl)acrylamide 38
The acrylic acid analogue 19 was reacted with the endoxifen derivative 10, following the general method above. The product 38 was afforded as a brown oil (99 mg, 92%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.90–0.93 (m, 3H, CH3), 2.32 (s, 3H, CH3), 2.44–2.46 (m, 2H, CH2), 3.08–3.23 (m, 3H, NCH3), 3.49–3.92 (m, 12H, OCH3, CH2,), 4.08 (m, 0.31 × 2H, OCH2), 4.25 (m, 0.27 × 2H, OCH2), 4.51 (d, 0.42 × 2H, J = 8.0 Hz, OCH2), 6.42–7.14 (m, 19H, ArH), 2xOH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.28, 20.83, 24.44, 25.11, 33.42, 48.67, 55.46, 55.68, 60.55, 105.32, 105.47, 112.61, 113.37, 113.99, 114.49, 114.53, 114.65, 128.44, 128.73, 128.88, 130.15, 130.29, 130.48, 131.52, 131.59, 133.59, 134.94, 137.49, 140.06, 152.72, 152.81, 153.12, 153.74, 154.17, 154.66, 156.88. IR: νmax (KBr) cm−1: 3428.8, 3328.5, 2929.5, 2850.8, 1625.9, 1580.6, 1509.4, 1449.8, 1411.3, 1310.5, 1241.6, 1169.9, 1127.4, 832.9. HRMS (EI): Found 722.3099 (M + Na)+, C44H45NO7Na requires 722.3094.
2.1.29. (E)-N-(2-{4-[(E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl]phenoxy}ethyl)-N-methyl-3-(4-methylsulfanylphenyl)-2-(3,4,5-trimethoxyphenyl) acrylamide 39
The acrylic acid analogue 20 was reacted with the endoxifen derivative 9, following the general method above. The product 39 was afforded as a brown oil (99 mg, 90%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.91–0.93 (m, 3H, CH3), 2.45–2.50 (m, 5H, CH2), 3.07–3.22 (m, 3H, NCH3), 3.57–3.89 (m, 11H, OCH3, CH2), 4.10 (m, 0.33 × 2H, OCH2), 4.26 (m, 0.35 × 2H, OCH2), 4.48 (d, 0.32 × 2H, J = 8.0 Hz, OCH2), 6.50–7.18 (m, 20H, ArH), OH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.20, 13.24, 14.95, 24..45, 25.13, 25.23, 33.43, 48.64, 55.53, 55.82, 60.51, 105.40, 112.64, 113.38, 113.98, 114.69, 125.21, 125.45, 127.37, 129.24, 129.39, 130.13, 130.25, 131.52, 131.62, 136.16, 137.38, 138.18, 138.24, 152.92, 154.02, 154.95, 156.79. IR: νmax (KBr) cm−1: 3326.9, 2929.2, 2850.8, 1626.1, 1579.2, 1507.6, 1449.4, 1409.1, 1312.0, 1242.1, 1170.1, 1127.3, 1050.0, 1005.7, 892.5, 834.0, 701.8. HRMS (EI): Found 738.2897 (M + Na)+, C44H45NO6NaS requires 738.2865.
2.1.30. (E)-N-(2-{4-[(E/Z)-1,2-Bis-(4-hydroxyphenyl)but-1-enyl]phenoxy}ethyl)-N-methyl-3-(4-methylsulfanylphenyl)-2-(3,4,5-trimethoxyphenyl)acrylamide 40
The acrylic acid analogue 20 was reacted with the endoxifen derivative 10, following the general method above. The product 40 was afforded as a brown oil (103 mg, 91%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.90–0.93 (m, 3H, CH3), 2.42–2.45 (m, 5H, CH2), 3.07–3.22 (m, 3H, NCH3), 3.52–3.89 (m, 11H, OCH3, CH2,), 4.08 (m, 0.27 × 2H, OCH2), 4.25 (m, 0.28 × 2H, OCH2), 4.50 (d, 0.45 × 2H, J = 8.0 Hz, OCH2), 6.48–7.11 (m, 19H, ArH), 2xOH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.28, 14.91, 24..43, 25.10, 33.40, 48.68, 55.52, 60.56, 105.23, 105.37, 112.61, 113.37, 113.99, 114.52, 114.65, 125.20, 129.41, 130.15, 130.30, 131.53, 133.58, 152.92, 153.74, 154.09, 154.17, 154.65, 156.89. IR: νmax (KBr) cm−1: 3376.8, 3327.7, 2929.4, 2850.9, 1626.0, 1607.1, 1580.3, 1509.0, 1449.7, 1409.1, 1312.3, 1241.6, 1170.0, 1127.3, 892.8, 833.2. HRMS (EI): Found 754.2819 (M + Na)+, C44H45NO7NaS requires 754.2814.
2.1.31. (E)-3-(4-Bromophenyl)-N-(2-{4-[(E/Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-enyl]phenoxy}ethyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide 41
The acrylic acid analogue 17 was reacted with the endoxifen derivative 9, following the general method above. The product 41 was afforded as a brown oil (105 mg, 91%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.91–0.94 (m, 3H, CH3), 2.47–2.50 (m, 2H, CH2), 3.05–3.21 (m, 3H, NCH3), 3.56–4.47 (m, 13H, OCH3, CH2), 6.42–7.35 (m, 20H, ArH), OH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.18, 13.26, 24.44, 25.12, 25.22, 33.43, 48.66, 55.52, 60.52, 105.35, 112.63, 113.38, 113.98, 114.68, 121.42, 121.44, 125.46, 127.38, 129.24, 129.26, 130.13, 130.27, 130.53, 130.89, 130.91, 131.52, 131.62, 133.81, 137.35, 137.59, 140.37, 142.10, 142.25, 152.96, 154.02, 154.94, 156.78, 162.19. IR: νmax (KBr) cm−1: 3327.0, 2929.3, 2850.8, 1626.2, 1579.0, 1507.5, 1449.4, 1409.0, 1311.9, 1242.4, 1170.5, 1127.9, 1009.7, 892.5, 834.3. HRMS (EI): Found 770.2120 (M + Na)+, C43H42BrNO6Na requires 770.2094.
2.1.32. (E)-N-(2-{4-[(E/Z)-1,2-Bis-(4-hydroxyphenyl)but-1-enyl]phenoxy}ethyl)-3-(4-bromophenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide 42
The acrylic acid analogue 17 was reacted with the endoxifen derivative 10, following the general method above. The product 42 was afforded as a brown oil (106 mg, 90%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.90–0.93 (m, 3H, CH3), 2.38–2.48 (m, 2H, CH2), 3.08–3.20 (m, 3H, NCH3), 3.51–4.51 (m, 13H, OCH3, CH2,), 6.41–7.35 (m, 19H, ArH), 2xOH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.28, 24.43, 25.10, 33.41, 48.70, 55.46, 55.51, 55.62, 60.57, 105.19, 105.32, 112.59, 113.36, 114.00, 114.47, 114.52, 114.66, 121.48, 129.41, 129.79, 129.95, 130.15, 130.30, 130.53, 130.91, 131.52, 131.60, 133.59, 133.73, 135.29, 136.55, 137.15, 137.49, 152.88, 152.96, 153.02, 153.73, 154.08, 154.15, 154.64, 156.89, 171.26. IR: νmax (KBr) cm−1: 3327.2, 2929.2, 2850.8, 1626.4, 1508.9, 1508.9, 1449.3, 1409.2, 1311.9, 1242.9, 1169.9, 1127.9, 1009.9, 892.5, 833.9, 641.2. HRMS (EI): Found 786.2071 (M + Na)+, C43H42BrNO7Na requires 786.2043.
2.1.33. (E)-3-(3-Fluoro-4-methoxyphenyl)-N-(2-{4-[(E/Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-enyl]phenoxy}ethyl)-N-methyl-2-(3,4,5-trimethoxyphenyl) acrylamide 43
The acrylic acid analogue 15 was reacted with the endoxifen derivative 9, following the general method above. The product 43 was afforded as a brown oil (103 mg, 93%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.91–0.93 (m, 3H, CH3), 2.47–2.50 (m, 2H, CH2), 3.06–3.21 (m, 3H, NCH3), 3.60–3.86 (m, 14H, OCH3, CH2,), 4.10 (m, 0.34 × 2H, OCH2), 4.25 (m, 0.27 × 2H, OCH2), 4.43 (d, 0.35 × 2H, J = 8.0 Hz, OCH2), 6.43–7.19 (m, 19H, ArH), OH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.17, 13.21, 24.44, 25.11, 28.55, 28.63, 33.42, 48.66, 53.02, 55.58, 55.69, 60.53, 105.41, 112.26, 112.59, 113.37, 113.95, 114.66, 116.19, 116.38, 125.45, 125.55, 127.36, 129.23, 130.13, 130.24, 131.52, 131.60, 134.58, 135.94, 137.29, 140.39, 142.10, 146.82, 146.93, 150.04, 152.46, 152.97, 153.94, 154.88, 156.75, 171.31. IR: νmax (KBr) cm−1: 3327.6, 2929.2, 2850.8, 1626.4, 1578.4, 1509.0, 1437.1, 1311.2, 1272.5, 1242.7, 1127.2, 892.8, 834.9. HRMS (EI): Found 740.2983 (M + Na)+, C44H44NO7NaF requires 740.3000.
2.1.34. (E)-N-(2-{4-[(E/Z)-1,2-Bis-(4-hydroxyphenyl)but-1-enyl]phenoxy}ethyl)-3-(3-fluoro-4-methoxyphenyl)-N-methyl-2-(3,4,5-trimethoxyphenyl)acrylamide 44
The acrylic acid analogue 15 was reacted with the endoxifen derivative 10, following the general method above. The product 44 was afforded as a brown oil (99 mg, 88%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.90–0.93 (m, 3H, CH3), 2.41–2.45 (m, 2H, CH2), 3.07–3.21 (m, 3H, NCH3), 3.48–4.48 (m, 16H, OCH3, CH2,), 6.44–7.14 (m, 19H, ArH), 2xOH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.26, 24.43, 25.10, 33.41, 48.69, 55.58, 55.71, 60.56, 60.59, 105.24, 105.38, 112.29, 112.60, 113.36, 113.98, 114.51, 114.65, 116.21, 116.34, 125.59, 127.66, 129.70, 130.15, 130.26, 131.52, 133.61, 146.87, 146.98, 150.03, 152.93, 153.02, 153.71, 154.07, 154.14, 154.63, 156.85. IR: νmax (KBr) cm−1: 3376.8, 3328.3, 2930.5, 2850.8, 1608.1, 1581.4, 1511.1, 1463.1, 1410.9, 1302.2, 1274.2, 1239.5, 1169.9, 1126.8, 1050.7, 1025.9, 902.0, 832.9. HRMS (EI): Found 756.2915 (M + Na)+, C44H44NO8NaF requires 756.2949.
2.1.35. (E)-3-(3-Amino-4-methoxyphenyl)-N-(2-{4-[(E/Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-enyl]phenoxy}ethyl)-N-methyl-2-(3,4,5-trimethoxyphenyl) acrylamide 45
The acrylic acid analogue 16 was reacted with the endoxifen derivative 9, following the general method above. The product 45 was afforded as a brown oil (100 mg, 91%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.91–0.95 (m, 3H, CH3), 2.47–2.50 (m, 2H, CH2), 3.06–3.22 (m, 3H, NCH3), 3.51–3.88 (m, 16H, OCH3, CH2,, NH2), 4.10 (m, 0.32 × 2H, OCH2), 4.26 (m, 0.29 × 2H, OCH2), 4.51 (d, 0.39 × 2H, J = 8.0 Hz, OCH2), 6.39–7.18 (m, 19H, ArH), OH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.19, 13.23, 24.46, 25.13, 25.21, 28.64, 33.44, 48.65, 55.01, 55.57, 60.50, 105.53, 109.39, 112.64, 113.40, 113.97, 114.67, 115.18, 120.17, 125.44, 127.37, 129.09, 129.26, 130.14, 130.24, 131.53, 131.60, 135.09, 142.14, 142.24, 146.81, 152.82, 153.95, 154.90, 156.67. IR: νmax (KBr) cm−1: 3327.2, 2929.4, 2850.7, 1625.9, 1579.2, 1508.9, 1449.3, 1411.2, 1310.6, 1242.1, 1169.5, 1126.1, 1046.3, 892.7, 833.9. HRMS (EI): Found 737.3238 (M + Na)+, C44H46N2O7Na requires 737.3203.
2.1.36. (E)-3-(3-Amino-4-methoxyphenyl)-N-(2-{4-[(E/Z)-1,2-bis-(4-hydroxyphenyl)but-1-enyl]phenoxy}ethyl)-N-methyl-2-(3,4,5-trimethoxyphenyl) acrylamide 46
The acrylic acid analogue 16 was reacted with the endoxifen derivative 10, following the general method above. The product 46 was afforded as a brown oil (106 mg, 94%), then changes to a semi-solid resin. 1H-NMR (400 MHz, CDCl3): δ 0.91–0.94 (m, 3H, CH3), 2.44–2.46 (m, 2H, CH2), 3.08–3.22 (m, 3H, NCH3), 3.51–4.34 (m, 18H, OCH3, CH2, NH2), 6.49–7.12 (m, 18H, ArH), 2xOH not observed. 13C-NMR (100 MHz, CDCl3): δ 13.27, 24.46, 25.13, 29.26, 33.44, 48.70, 55.03, 55.26, 55.56, 55.82, 60.55, 105.42, 106.23, 109.40, 113.39, 113.93, 114.36, 114.44, 114.51, 114.61, 130.17, 130.24, 130.31, 131.53, 133.62, 152.82, 153.85, 154.57, 156.61. IR: νmax (KBr) cm−1: 3327.3, 2929.2, 2850.7, 1625.8, 1578.6, 1510.6, 1436.5, 1311.1, 1242.9, 1169.1, 1126.2, 892.6, 833.8. HRMS (EI): Found 753.3172 (M + Na)+, C44H46N2O8Na requires 753.3152.
2.1.37. (E)-2-(4-Methoxyphenyl)-1-(piperidin-1-yl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one 47
To a suspension of 23 (1.45 mmol, 0.5 g) in anhydrous dichloromethane (20 mL), 2-chloro-1-methylpyridinium iodide (4.35 mmol 1.11 g) was added and the reaction mixture was stirred for 5 min at room temperature. To this was added piperidine (1.45 mmol, 0.12 g, 0.14 mL) followed by the addition of triethylamine (7.25 mmol, 0.73 g, 1.01 mL). The reaction mixture was stirred at room temperature for 1 h and was then diluted with 10% hydrochloric acid (10 mL), washed with 10% sodium hydroxide (10 mL), water and brine and dried over sodium sulphate. The crude product was purified by column chromatography (dichloromethane:ethyl acetate, 1:1) to afford the product as a yellow powder, 48% (0.287 g), Mp 106–108 °C, HPLC 95%. IR: νmax (KBr) cm−1: 2993, 2948, 2865, 2835, 1615, 1599, 1577, 1505, 1453, 1432, 1329, 1281, 1238, 1125, 1021, 989, 904, 845, 833, 718, 657. 1H NMR (400 MHz, CDCl3) δ 1.32 (br. s., 2 H, CH2), 1.57 (br. s., 4 H, CH2), 3.46 (br. s., 2 H, CH2), 3.54 (d, J = 2.49 Hz, 2 H, CH2), 3.58 (s, 6 H, CH3), 3.76 (s, 3 H, CH3), 3.78 (s, 3 H, CH3), 6.35 (s, 2 H, Ar-H), 6.50 (s, 1 H, CH), 6.81 (d, J = 8.71 Hz, 2 H, Ar-H), 7.27 (d, J = 8.71 Hz, 2 H, Ar-H). 13C NMR (101 MHz, CDCl3) δ 24.51 (3 × CH2), 55.24 (2 × CH2), 55.73 (3 × CH3), 60.82 (CH3), 106.55 (2 × CH), 114.00 (2 × CH), 127.99 (C), 128.29 (C), 130.21 (2 × CH), 131.02 (C), 137.00 (CH), 137.47 (C), 152.66 (2C), 159.22 (C), 170.09 (C=O). HRMS (EI): Found 434.1929 (M + Na)+, C24H29NO5 requires 434.1944.
2.1.38. (E)-2-(4-methoxyphenyl)-1-(pyrrolidin-1-yl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one 48
The acrylic acid 23 (1.45 mmol 0.5 g) was reacted with 2-chloro-1-methylpyridinium iodide (4.35 mmol 1.11 g), pyrrolidine (1.45 mmol, 0.1 g, 0.12 mL) and triethylamine (7.25 mmol, 0.73 g, 1.01 mL) in anhydrous dichloromethane (20 mL) as described for compound 47 above. The crude product was purified by column chromatography (dichloromethane: ethyl acetate,1:1) to afford the product as a yellow oil, 44% (0.256 g); HPLC; 91%. IR: νmax (KBr) cm−1: 3463, 2937, 2877, 2837, 1605, 1578, 1505, 1450, 1416, 1327, 1239, 1121, 1005, 913, 872, 836, 770. 1H NMR (400 MHz, CDCl3) δ 1.80–1.89 (m, 4 H, CH2), 3.26 (t, J = 6.22 Hz, 2 H, CH2), 3.55 (t, J = 6.43 Hz, 2 H, CH2), 3.61 (s, 6 H, CH3), 3.80 (s, 3 H, CH3), 3.82 (s, 3 H, CH3), 6.37 (s, 2 H, Ar-H), 6.74 (s, 1 H, CH), 6.83–6.88 (m, 2 H, Ar-H), 7.27–7.30 (m, 2 H, Ar-H). 13C NMR (101 MHz, CDCl3) δ 24.27 (CH2), 26.13 (CH2), 46.03 (CH2), 48.16 (CH2), 55.27 (CH3), 55.72 (2 × CH3), 60.83 (CH3), 106.73 (2 × CH), 114.04 (2 × CH), 127.86 (C), 130.34 (2 × CH), 130.63 (C), 137.58 (CH), 137.88 (C), 152.63 (2 × C), 159.21 (C), 170.03 (C=O). HRMS (EI): Found 420.1794 (M + Na)+, C23H27NO5 requires 420.1787.
2.1.39. (E)-3-(3-hydroxy-4-methoxyphenyl)-1-(piperidin-1-yl)-2-(3,4,5-trimethoxyphenyl)prop-2-en-1-one 49
The acrylic acid 13 (1.45 mmol 0.5 g) was reacted with 2-chloro-1-methylpyridinium iodide (4.35 mmol 1.11 g), piperidine (1.38 mmol, 0.12 g, 0.13 mL) and triethylamine (7.25 mmol, 0.73 g, 1.01 mL) in anhydrous dichloromethane (20 mL) as described for compound 47 above. The crude product was purified by column chromatography (dichloromethane: ethyl acetate,1:1) to afford the product as a yellow oil, 6.5% (0.038 g), HPLC 92%. IR: νmax (KBr) cm−1: 3209, 2998, 2936, 2854, 2250, 1605, 1578, 1505, 1440, 1256, 1234, 1120, 1024, 999, 907, 852, 803, 772, 725. 1H NMR (400 MHz, CDCl3) δ 1.32 (br. s., 2 H, CH2), 1.55 (br. s., 4 H, CH2), 3.43 (br. s., 2 H, CH2),3.54 (br. s., 2 H, CH2), 3.65 (s, 6 H, CH3), 3.78 (s, 3 H, CH3), 3.79 (s, 3 H, CH3), 6.46 (s, 1 H, CH), 6.51 (s, 2 H, Ar-H), 6.60 (d, J = 0.83 Hz, 2 H, Ar-H), 6.72 (s, 1 H,Ar-H). 13C NMR (101 MHz, CDCl3) δ 24.55 (3 × CH2), 55.88 (C), 56.14 (CH3), 60.93 (2 × CH3), 106.09 (2 × CH), 110.16 (CH), 115.42 (CH), 121.82 (CH), 128.77 (2 × C), 131.03 (C), 136.11 (CH), 137.82 (C), 145.10 (2 × C), 153.23 (2 × C), 170.09 (C=O). HRMS (EI): Found 450.1898 (M + Na)+, C24H29NO6 requires 450.1893.
2.1.40. (E)-3-(4-Methoxy-3-nitrophenyl)-1-(piperidin-1-yl)-2-(3,4,5-trimethoxyphenyl)prop-2-en-1-one 50
The acrylic acid 21 (1.45 mmol 0.5 g) was reacted with 2-chloro-1-methylpyridinium iodide (4.35 mmol 1.11 g), piperidine (1.38 mmol, 0.12 g, 0.13 mL) and triethylamine (7.25 mmol, 0.73 g, 1.01 mL) in anhydrous dichloromethane (20 mL) as described for compound 47 above. The crude product was purified by column chromatography (ethyl acetate:methanol, 9:1) to afford the product as a yellow oil, 31% (0.177 g), HPLC 67%. IR: νmax (KBr) cm−1: 2937, 2854, 1614, 1578, 1528, 1503, 1440, 1411, 1279, 1234, 1122, 999, 915, 827, 732, 675. 1H NMR (400 MHz, CDCl3) δ 1.40 (br. s., 2 H, CH2), 1.63 (br. s., 4 H, CH2), 3.48 (br. s., 2 H, CH2), 3.63 (br. s., 2 H, CH2), 3.73 (s, 6 H, CH3), 3.88 (s, 3 H, CH3), 3.93 (s, 3 H, CH3), 6.55 (s, 1 H, CH), 6.56 (s, 2 H, Ar-H), 6.89 (d, J = 8.71 Hz, 1 H, Ar-H), 7.29 (d, J = 2.49 Hz, 1 H, Ar-H), 7.70 (d, J = 2.07 Hz, 1 H, Ar-H). 13C NMR (101 MHz, CDCl3) δ 24.47 (3 × CH2), 56.25 (2 × CH2), 56.57 (3 × CH3), 61.02 (CH3), 105.78 (2 × CH), 113.05 (CH), 126.00 (CH), 126.53 (C), 128.07 (C), 130.13 (C), 134.91 (CH), 138.35 (CH), 138.92 (C), 152.13 (C), 153.64 (2 × C), 169.16 (C=O). HRMS (EI): Found 457.1974 (M + H)+, C24H28N2O7 requires 457.1975.
2.1.41. (E)-3-(4-Methoxy-3-nitrophenyl)-1-(pyrrolidin-1-yl)-2-(3,4,5-trimethoxyphenyl)prop-2-en-1-one 51
The acrylic acid 21 (1.45 mmol 0.5 g) was reacted with 2-chloro-1-methylpyridinium iodide (4.35 mmol 1.11 g), pyrrolidine (1.38 mmol, 0.12 g, 0.13 mL) and triethylamine (7.25 mmol, 0.73 g, 1.01 mL) in anhydrous dichloromethane (20 mL) as described for compound 47 above. The crude product was purified by column chromatography (dichloromethane:ethyl acetate, 1:1) to afford the product as a yellow oil, 52% (0.292 g). HPLC 98% and was used in the following reaction without further purification. IR: νmax (KBr) cm−1: 3463, 2971, 2942, 2878, 2841, 1613, 1578, 1528, 1503, 1410, 1353, 1236, 1122, 1087, 1005, 912, 829, 808, 751, 670. 1H NMR (400 MHz, CDCl3) δ 1.85–1.92 (m, 4 H, CH2), 3.30 (t, J = 6.22 Hz, 2 H, CH2), 3.57 (t, J = 6.43 Hz, 2 H, CH2), 3.74 (s, 6 H, CH3), 3.88 (s, 3 H, CH3), 3.93 (s, 3 H, CH3), 6.55 (s, 2 H, Ar-H), 6.73 (s, 1 H, CH), 6.89 (d, J = 9.12 Hz, 1 H, Ar-H), 7.25 (d, J = 2.07 Hz, 1 H, Ar-H), 7.68 (d, J = 2.07 Hz, 1 H, Ar-H). 13C NMR (101 MHz, CDCl3) δ 24.26 (2 × CH2), 26.13 (CH2), 46.12 (CH2), 48.11 (CH2), 56.29 (2 × CH3), 56.57 (CH3), 61.04 (CH3), 106.11 (2 × CH), 113.03 (CH), 126.62 (CH), 127.79 (C), 128.07 (C), 129.96 (C), 135.07 (CH), 138.34 (CH),139.77 (C), 152.22 (C), 153.70 (2 × C), 169.01 (C=O).
2.1.42. (E)-3-(4-Methoxyphenyl)-1-(pyrrolidin-1-yl)-2-(3,4,5-trimethoxyphenyl)prop-2-en-1-one 52
The acrylic acid 14 (1.45 mmol 0.5 g) was reacted with 2-chloro-1-methylpyridinium iodide (4.35 mmol 1.11 g), pyrrolidine (1.38 mmol, 0.12 g, 0.13 mL) and triethylamine (7.25 mmol, 0.73 g, 1.01 mL) in anhydrous dichloromethane (20 mL) as described for compound 47 above. The crude product was purified by column chromatography (dichloromethane:ethyl acetate, 1:1) to afford the product as a clear oil 37% (0.215 g), HPLC 96%. IR: νmax (KBr) cm−1: 3463, 2936, 2873, 2837, 1603, 1577, 1505, 1431, 1235, 1175, 1122, 1027, 1004, 828, 665. 1H NMR (400 MHz, CDCl3) δ 1.82–1.92 (m, 4 H, CH2), 3.31 (t, J = 6.22 Hz, 2 H, CH2), 3.56 (t, J = 6.43 Hz, 2 H, CH2), 3.71 (s, 6 H, CH3), 3.77 (s, 3 H, CH3), 3.87 (s, 3 H, CH3), 6.57 (s, 2 H, Ar-H), 6.70–6.75 (m, 2 H, Ar-H), 6.77 (s, 1 H, CH), 7.08 (m, J = 8.71 Hz, 2 H, Ar-H). 13C NMR (101 MHz, CDCl3) δ 24.30 (CH2), 26.14 (CH2), 46.02 (CH2), 48.17 (CH2), 55.19 (CH3), 56.12 (2 × CH3), 60.94 (CH3), 106.32 (2 × CH), 113.46 (2 × CH), 127.86 (C), 130.60 (C), 130.94 (2 × CH), 136.69 (CH) 137.65 (C), 153.31 (2C), 159.21 (C), 169.97 (C=O). HRMS (EI): Found 398.1974 (M + H), C23H28NO5 requires 398.1967.
2.1.43. (E)-3-(4-Methoxyphenyl)-1-(piperidin-1-yl)-2-(3,4,5-trimethoxyphenyl)prop-2-en-1-one 53
The acrylic acid 14 (1.45 mmol 0.5 g) was reacted with 2-chloro-1-methylpyridinium iodide (4.35 mmol 1.11 g), piperidine (1.38 mmol, 0.12 g, 0.13 mL) and triethylamine (7.25 mmol, 0.73 g, 1.01 mL) in anhydrous dichloromethane (20 mL) as described for compound 47 above. The reaction mixture was stirred at room temperature for 1 h and was then diluted with 10% hydrochloric acid (10 mL), washed with 10% sodium hydroxide (10 mL), water and brine and dried over sodium sulphate. The crude product was purified by column chromatography (dichloromethane: ethyl acetate,1:1) to afford the product as a pale yellow oil, 38% (0.225 g), HPLC 99%. IR: νmax (KBr) cm−1: 3490, 2996, 2854, 2838, 1619, 1605, 1577, 1506, 1433, 1410, 1234, 1122, 1004, 996, 827, 716, 704, 554. 1H NMR (400 MHz, CDCl3) δ 1.40 (br. s., 2 H, CH2), 1.64 (br. s., 4 H, CH2), 3.53 (br. s., 2 H, CH2), 3.56–3.68 (m, 2 H, CH2), 3.71 (s, 6 H, CH3), 3.77 (s, 3 H, CH3), 3.87 (s, 3 H, CH3), 6.57 (s, 2 H, Ar-H), 6.59 (s, 1 H, CH), 6.71–6.77 (m, 2 H, Ar-H), 7.11 (m, J = 8.71 Hz, 2 H,Ar-H). 13C NMR (101 MHz, CDCl3) δ 24.57 (3 × CH2), 55.22 (2 × CH2), 56.11 (3 × CH3), 60.94 (CH3), 105.99 (2 × CH), 113.51 (2 × CH), 127.88 (C), 128.82 (C), 130.80 (2 × CH), 131.25 (C), 135.69 (CH), 137.70 (C), 153.29 (2 × C), 159.15 (C), 170.16 (C=O). HRMS (EI): Found 412.2121 (M + H), C24H29NO5 requires 412.2124.
2.1.44. (E)-3-(3-Amino-4-methoxyphenyl)-1-(pyrrolidin-1-yl)-2-(3,4,5-trimethoxyphenyl)prop-2-en-1-one 54
The acrylic acid 51 (0.34 mmol, 0.150 g) was dissolved in a mixture of ethanol (5 mL) acetic acid (5 mL) and water (2 mL). Hydrochloric acid(1 drop) was added followed by iron powder (3.4 mmol, 0.19 g). The mixture was vigorously stirred under reflux for 12 h. After completion of the reaction, the mixture was filtered through Celite, diluted with NaHCO3 solution and extracted with dichloromethane. The organic layers were combined, washed with water and brine and dried over sodium sulphate. The crude product was purified by column chromatography (dichloromethane:ethyl acetate, 1:1) to afford the product as a yellow oil, 79% (0.111 g), HPLC 70%. IR: νmax (KBr) cm−1: 3464, 3360, 2938, 2877, 2836, 1578, 1504, 1410, 1230, 1121, 1003, 911, 797, 743, 705. 1H NMR (400 MHz, DMSO-d6) δ 1.74–1.80 (m, 4 H, CH 2) 3.33 (br. s., 2 H, CH2), 3.60 (s, 6 H, CH3), 3.65 (s, 3 H, CH3), 3.68 (s, 3 H, CH3) 4.59 (br. s., 2 H, NH2), 6.32 (dd, J = 8.29, 2.07 Hz, 1 H, Ar-H), 6.48 (d, J = 2.07 Hz, 1 H, Ar-H), 6.50 (s, 1 H, CH), 6.51 (s, 2 H, Ar-H), 6.63 (d, J = 8.29 Hz, 1 H, Ar-H). 13C NMR (101 MHz, DMSO-d6) δ 23.89 (CH2), 25.68 (CH2), 47.86 (2 × CH2), 55.25 (CH3), 55.82 (2 × CH3), 60.12 (CH3), 106.22 (2 × CH), 110.05 (CH), 114.52 (CH), 117.79 (CH), 127.71 (C), 130.23 (C), 131.18 (C), 135.53 (C),137.11 (CH), 137.15 (C), 146.25 (C), 152.76 (2 × C), 168.81 (C=O). HRMS (EI): Found 413.2078 (M + H), C23H28N2O5 requires 413.2076.
2.1.45. (E)-3-(3-Amino-4-methoxyphenyl)-1-(piperidin-1-yl)-2-(3,4,5-trimethoxyphenyl)prop-2-en-1-one 55
The acrylic acid 50 (0.33 mmol, 0.150 g) was dissolved in a mixture of ethanol (5 mL), acetic acid (5 mL) and water (2 mL). Hydrochloric acid(1 drop) was added followed by iron powder (3.4 mmol, 0.19 g) following the procedure described above. The crude product was purified by column chromatography (dichloromethane:ethyl acetate, 1:1) to afford the product as a yellow oil, 81%, (0.114 g). IR: νmax (KBr) cm−1: 3329, 2933, 1580, 1507, 1411, 1235, 1123, 1023, 773. 1H NMR (400 MHz, DMSO-d6) δ 1.38 (br. s., 6 H, CH2), 3.29 (s, 6 H, CH3), 3.46 (d, J = 4.98 Hz, 4 H, CH2), 3.64 (s, 3 H, CH3), 3.75 (s, 3 H, CH3), 4.61 (br. s., 2 H, NH2), 6.32–6.35 (m, 1 H, Ar-H), 6.45 (s, 1 H, CH), 6.49 (s, 2 H, Ar-H), 6.51 (s, 1 H, Ar-H), 6.64 (d, J = 8.29 Hz, 1 H, Ar-H). 13C NMR (101 MHz, DMSO-d6) δ 24.05 (3 × CH2), 55.67 (2 × CH2), 55.77 (3 × CH3), 60.10 (CH3), 105.71 (2 × CH), 110.09 (CH), 114.33 (CH), 117.59 (CH), 126.98 (C), 127.66 (C), 131.19 (C), 135.46 (C), 137.18 (CH), 137.27 (C), 146.17 (C), 152.92 (2 × C), 168.95 (C=O). HRMS (EI): found 427.2237 (M + H), C24H30N2O5 requires 427.2233.
2.1.46. Stability Study for Compounds 27, 31 and 32
Analytical high-performance liquid chromatography (HPLC) stability studies were performed using a Symmetry® column (C18, 5 µm, 4.6 × 150 mm), a Waters 2487 Dual Wavelength Absorbance detector, a Waters 1525 binary HPLC pump and a Waters 717 plus Autosampler (Waters Corporation, Milford, MA, USA). Samples were detected at wavelength of 254 nm. All samples were analysed using acetonitrile (80%):water (20%) as the mobile phase over 10 min and a flow rate of 1 mL/min. Stock solutions are prepared by dissolving 5mg of compound in 10 mL of mobile phase. Phosphate buffers at the desired pH values (4, 7.4, and 9) were prepared in accordance with the British Pharmacopoeia monograph 2015. 30 µL of stock solution was diluted with 1 mL of appropriate buffer, shaken and injected immediately. Samples were withdrawn and analysed at time intervals of t = 0 min, 5 min, 30 min, 60 min, 90 min, 120 min, 24 h and 48 h.
2.2. Biochemical Evaluation
2.2.1. MTT Assay for Measurement of Antiproliferative Effects in MCF-7 and MDA-MB-231 Cell Lines
The human breast carcinoma cell line, MCF-7, was purchased from the European Collection of Animal Cell Cultures (ECACC). The cells were maintained in MCF-7 complete medium; consisting of Eagle’s Minimum Essential Medium (MEM) supplemented with 10% (v/v) Foetal Bovine Serum (FBS), 2 mM l-glutamine, 100 μg/mL penicillin/streptomycin and 1% (v/v) non-essential amino acids. Cell cultures were maintained at 37 °C under a humidified atmosphere of 5% CO2/95% O2. The human breast carcinoma cell line, MDA, was purchased from the European Collection of Animal Cell Cultures (ECACC). The cells were maintained in MDA complete medium; consisting of Dulbecco’s Modified Eagle’s Medium (D-MEM) supplemented with 10% (v/v) Foetal Bovine Serum (FBS), 2 mM l-glutamine, 100 μg/mL penicillin/streptomycin. Cell cultures were maintained at 37 °C under a humidified atmosphere of 5% CO2/95% O2. The MTT assay was performed according to the reported protocol. The tetrazolium salt, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) is taken up only by metabolically active cells and cleaved to form a formazan dye by mitochondrial dehydrogenases [30]. Assays where repeated in three experiments performed in triplicate (unless otherwise stated) and reported results represent the mean value ± standard error mean. Graphs of percentage cell viability versus concentration of the subject compound were processed using PRISM [31].
2.2.2. Lactate Dehydrogenase Assay for Measurement of Cytotoxicity
In this assay, the release of cytoplasmic lactate dehydrogenase (LDH) is used as a measure of cell lysis. MCF-7 and MDA-MB-231 cells were seeded at a density of 1 × 104 cells/well in a 96-well plate and incubated for 24 h. The cells were then dosed with 2 μL volumes of the test compounds, over the concentration range 1 nM–50 μM. Cytotoxicity was determined using the CytoTox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI, USA) following the manufacturer’s protocol [32].
2.2.3. Estrogen Receptor Fluorescent Polarisation Assay
Competitive binding affinity experiments were carried out using purified baculovirus-expressed human ERα and ERβ and fluoromone, a fluorescein labelled estrogen ligand. Estrogen receptor binding ability of the selected compounds was investigated using ERα and ERβ fluorescence polarisation-based estrogen receptor competitive assay kits supplied by Invitrogen [33,34]. The assay was performed usibg a protocol described by the manufacturer. The assay allows for high throughput screening of potential ER-subtype ligands. The selected compounds were screened using both the ERα and ERβ competitive assay kits. The protocol for carrying out the assay is similar for both ER subtypes. Principally, the main difference between the kits relates to the functional receptor concentration and the specific activity of the different ERs [33,34].
2.2.4. Ishikawa Cell Line Study
The Ishikawa assay is used to measure estrogen stimulation of alkaline phosphatase enzyme activity (AlkP) by the Ishikawa cell line of human endometrial adenocarcinoma cells. The Ishikawa assay provides a measure of the agonist activity of a compound. The assay was carried out on the lead conjugate, 28. The assay was carried out following the method of Littlefield et al. [35]. The batch of Ishikawa cells were obtained as a gift from Professor R. Hochberg—who developed the alkaline phosphatase assay in Yale University, CT, USA. Tamoxifen was used as a reference compound in the assay.
2.2.5. NCI One-Dose and Five-Dose Screen Output
The one-dose screen output is reported as a mean graph of the percent growth of treated cells and is similar in appearance to mean graphs generated in the 5-dose assay. The value reported for the one-dose assay is growth relative to the no-drug control and relative to the time zero number of cells. The one-dose assay allows detection of growth inhibition (values between 0 and 100) and lethality (values less than 0). For example, a value of 100 means no growth inhibition. A value of 40 would mean 60% growth inhibition. A value of zero means no net growth over the course of the experiment. A value of −40 would mean 40% lethality. A value of −100 means all cells are dead. The results from the one-dose screen for 28 were manually entered into the COMPARE analysis software via an on-line submission form [36]. The results from the COMPARE analysis are retrievable on-line by searching using the relevant JobID reference number. The COMPARE analysis was run on a database of common anti-cancer agents (JobID: 37472) and the larger more comprehensive database including natural products and other submitted agents (JobID: 37473). Similarly, the results from the five-dose screen for 28 were also manually entered into the COMPARE analysis software via an on-line submission form. The COMPARE analysis was run on a database of common anti-cancer agents (JobID: 37885) and the larger more comprehensive database including natural products and other submitted agents (JobID: 37886).
2.3. Molecular Modelling
The lowest energy conformer produced [37] using MACROMODEL [38] was used to generate an ensemble of low energy conformations of 28 in OMEGA [39]. Fifty conformers were generated for the lead conjugate 28 using default parameters and saved as a .pdb file. The resulting .pdb file generated by OMEGA was then utilised by FRED [40] to dock and score the different compound conformers. The protein used to dock the conformers was 3ERT [41] (containing 4-hydroxytamoxifen bound in the human ERα LBD) and 1QKN [42] (containing raloxifene bound in rat ERβ LBD). The active site was defined by a three-dimensional box incorporating the 4-hydroxytamoxifen or raloxifene bound ligand. This box was also extended by five angstroms in each dimension to create additional space to allow for the docking procedure. Each conformer was docked and scored using three functions: Piecewise Linear Potential (PLP), Chemgauss and an updated version, Chemgauss2 [40].
PLP is a heavy atom scoring function; all potentials are based on distances from heavy atom centers (i.e., hydrogen position is irrelevant, although the presence or absence of hydrogen is not, as it can affect the atom typing). PLP recognises atom types such as hydrogen bond donors (i.e., primary and secondary amines), hydrogen bond acceptors (i.e., oxygen and nitrogen atoms with no bound hydrogens), hydroxyl groups (treated as both acceptors and donors) and non-polar entities (i.e., carbon, halogens and nitrogen or sulfur with more than two attached hydrogens). The Chemgauss scoring function combines the Shapegauss scoring function with additional potentials near specific functional groups. The Shapegauss scoring function represents all atoms as smooth Gaussian functions. A pairwise potential between ligand and protein atoms is applied that attempts to maximize their surface contact and minimize their volume overlap. Therefore, the potential is most favourable when the atoms are touching but not overlapping. A correction term is then applied to further penalize atoms that significantly overlap the protein. A consensus of the separate scoring functions is determined and the conformers are ranked accordingly.
The crystal structure of raloxifene, an antagonistic ligand, in rat ERβ isoform (pdb: 1QKN) was used due in this study due to the lack of reported determined co-crystallised antagonist ligands in the human ERβ isoform.
3. Results
3.1. Synthesis of Endoxifen-Combretastatin Conjugates
Many ER-ligand conjugates reported in the literature contain an agonistic ER-ligand analogue such as estrogen, in their conjugate structure [14,15]. As the goal in our investigation is to develop ER-antagonistic conjugates, possessing minimal agonist activity, only antagonistic ER-ligands were incorporated in the conjugate structural backbone. Endoxifen 11 which together with 4-hydroxytamoxifen is a significant metabolite of tamoxifen, was chosen as a suitable ER-ligand candidate for this study based on its potent ER-binding affinity and antiestrogenic properties [43]; it was also effective in degrading the estrogen receptor [44] (Figure 1). It has also been shown to inhibit aromatase [45] in the MCF-7 human cancer cell line [46]. The structurally related hydroxyendoxifen analogue 12, previously detected as a metabolite of tamoxifen [47], was also investigated as a potential ER ligand for conjugate design(Figure 1).
A modified multi-step route to the protected endoxifen scaffolds 9 and 10 was developed and is shown in Scheme 1. Initially, the phenolic 4,4′-dihydroxybenzophenone (1) was monoprotected as the tert-butyldimethylsilyl-ether (2); the 4-hydroxypropiophenone (3) was similarly protected to afford (4b). The benzophenone (2) and propiophenone starting materials (4a) and (4b) were coupled via a zinc/titanium tetrachloride/tetrahydrofuran McMurray reaction system to afford alkene products 5 and 6 in high yields (93%–98%) containing the triarylethylene ring motif predominant OH directed E-isomer [48]. A bromoethylation reaction was carried out using an excess of dibromoethane in the presence of a phase-transfer catalyst((nBu)4NHSO4) in basic conditions in order to introduce a bromoethylether functionality at the hydroxyl group position on the triarylethylene backbone in moderate yields (52%–54%). Methylamine undergoes reaction with the relevant bromide analogues 7 and 8 in a sealed tube to form the silyl-protected endoxifen 9 [16] and the disilylated endoxifen analogue 10 in moderate to high yields (55%–93%). The silyl ether protecting groups were removed using TBAF to afford the endoxifen 11 and hydroxyendoxifen 12 in high yields (80%–93%). Most of the E/Z isomeric mixture ratios of the analogues synthesised were calculated based on the basic side chain OCH2 or NCH2 signals. Unambiguous assignment of E/Z stereochemistry is confirmed using through Nuclear Overhauser Effect (NOE) NMR. In the 1H-NMR spectrum of the silyl ether 9 (Z:E ratio 1:1.3) the E-isomer CH2N signals are observed as a broad triplet at 2.99 ppm while the Z-isomer CH2N signals are observed as a triplet further upfield at 2.89 ppm. The E-isomer CH2O signals are observed as a triplet at 4.11 ppm(J = 5 Hz) while the Z-isomer CH2O signals are observed as a triplet further upfield at 3.95 ppm(J = 5 Hz). The relative chemical shifts assigned for the OCH2 and NCH2 signals for the protons in the basic side chain in the isomeric mixtures are in agreement with reported values for similar compounds [29]. The spectral data confirm that the trans-isomer OCH2 (and NCH2) signals are found further downfield when compared with the cis-isomer. Previous studies have demonstrated that 4-hydroxy substituted triarylethylenes such as endoxifen may isomerise under physiological conditions and have little impact on ER binding activity [49,50].
Scheme 1.

Synthesis of endoxifen 11 and hydroxyendoxifen derivative 12. Reagents and conditions: (i) TBDMS-Cl, Imidazole, DMF; (ii) Zn, TiCl4, THF; (iii) BrCH2CH2Br, NaOH (aq.), (nBu)4NHSO3; (iv) CH3NH2, sealed tube, 60 °C; (v) TBAF, THF. (TBDMS = tert-Butyldimethylsilyl).
Combretastatin A-4 is an important lead compound in drug development due to its potent antimitotic activity and ability to inhibit the depolymerisation of tubulin(Figure 1) [51]. To date, much work has been carried out in developing combretastatin analogues with potential anticancer applications [52]. The conjugation of combretastatin A4 analogues on steroidal scaffolds and their proapoptotic effects in MCF-7 cells has been recently reported [53]. In this study, a selection of combretastatin acrylic acid derivatives were synthesised using a reported Perkin condensation reaction route [21]. The common structural motif amongst the acrylic acid derivatives synthesised was the presence of a carboxylic acid group at either carbon position of the double bond between the two ring systems of the combretastatin core structure. Importantly, the carboxylic acid group allows for further chemical manipulation, such as the formation of amide and ester linkages, which is of interest in our conjugation strategy.
The combretastatin acrylic acids chosen for synthesis were selected based on the biochemical activity available for the “parent” combretastatin analogues [52]—and which differed in structure only by the absence of the acrylic acid’s carboxylic acid functional group [22,54,55,56,57,58,59,60,61,62,63,64,65]. By varying the benzaldehyde and phenylacetic acid in the Perkin condensation reaction, a series of combretastatin acrylic acid analogues 13–24 were prepared in yields of 36%–71% (see Scheme 2). In our investigation, combretastatin A-4 was used as a comparison standard for our biochemical evaluation and was synthesised according to a Wittig reaction route reported by Pettit et al. [20].
Scheme 2.

Synthesis of combretastatin acrylic acid analogues. Reagents and conditions: (i) (CH3CO)2O, Et3N, reflux, 3 h followed by conc HCl.
From the panel of combretastatin type acrylic acids 13–24 initially prepared, a subset of the more potent compounds was selected for the synthesis of the direct amide conjugates with endoxifen 11 and hydroxyendoxifen 12. The amine functional group of the silyl-protected ER ligands 9 and 10 and the carboxylic acid functional group of the combretastatin acrylic acid analogues 13–24 can undergo coupling reactions using DCC and HOBt, forming an amide linkage to afford silyl-ether protected conjugated compounds in high yields (See Scheme 3). The silyl-ether protecting groups were then removed using TBAF to afford the phenolic conjugates 27–46 as ~1:1 (E/Z) isomeric mixtures in high yield (88%–94%) following chromatographic purification. In the 1H NMR spectrum the characteristic protons of the ethyl group are observed in the region 0.90–0.96 ppm (CH3) and 2.40–2.50 ppm (CH2), the amine methyl group is found between 3.04 and 3.25 ppm, while the methylene protons of the basic side chain are identified at 3.53–4.20 ppm. The amide compounds 47–53 were also prepared by reaction of the acrylic acids 13, 14, 21 and 23 with pyrrolidine and piperidine using the Mukaiyama reagent (2-chloro-1-methylpyridinium iodide) as the coupling reagent. The amines 54 and 55 were obtained by reduction of the corresponding nitro compounds 50 and 51 using iron in hydrochloric acid.
Scheme 3.

Synthesis of endoxifen-combretastatin hybrid conjugates 27–46 and amides 47–55. Reagents and conditions: (i) DCC, HOBt, CH2Cl2, 20 °C, 24–48 h; (ii) TBAF, THF, 20 °C, 24 h (R1–R8: see Table 2); (iii) Pyrrolidine or piperidine, 2-chloro-1-methylpyridinium iodide, CH2Cl2, Et3N, 20 °C, 1 h; (iv) Fe, HCl, CH3CO2H, EtOH, reflux, 12 h.
3.2. Biochemical Studies
3.2.1. Antiproliferation and Cytotoxicity Studies
The ability of the compounds 27–46 synthesised to inhibit the proliferation of the human breast tumour MCF-7 cell line was investigated using a standard MTT cell viability assay while the compounds were concurrently tested to assess the extent of their cytotoxicity using a LDH assay [66]. The MCF-7 is an ER-positive human cancer cell line; where ER is overexpressed in the cell line [67]. Selected conjugates were also evaluated using ER-negative MDA human cancer cell-line in order to assess any possible ER-selectivity of the conjugates. The IC50 value obtained for the control 26 (CA4) in this assay is 0.008 μM for MCF-7 and is in good agreement with the reported values for CA4 using the MTT assay on human MCF-7 breast cancer cell lines [59,68,69]; the IC50 value obtained for the control endoxifen 11 in this assay is 0.029 nM while the reported IC50 value for endoxifen in the MCF-7 cell line is 50 nM [70].
In general, combretastatin acrylic acid derivatives have a lower antiproliferative potency compared to the lead compound, combretastatin A-4 (CA4), 26 (IC50 = 8 nM), (see Table 1). The acrylic acid 13 displayed the highest antiproliferative action in the series, with IC50 value of 0.120 μM while all the analogues tested showed negligible cytotoxic affects in the LDH assay, (0% cell death at 10 μM). The substitution pattern in the A and B rings of 13 are similar to that of the potent combretastatin CA4. It is interesting that the compound 23, in which the A ring having the 3,4,5-trimethoxyphenyl substituent is positioned on the carbon β to the acrylic acid is inactive with IC50 > 50 μM. The acrylic acids 14, 16, 21 and the ester 24 all demonstrated IC50 values less than 10 μM against MCF-7 cell line. The antiproliferative activity of acrylic acids has been previously reported [23]. The acrylic acid secondary amides 47–55 prepared from pyrrolidine and piperidine were also evaluated for antiproliferative activity in the MCF-7 cell line (Table 1). The compounds were found to be of low potency, e.g., the amide 49 (IC50 = >50 μM in comparison with the acrylic acid 13 (IC50 = 0.120 μM) which suggested that a more bulky amide such as endoxifen may be required for activity in MCF-7 cells.
Table 1.
Antiproliferative effects of acrylic acids 13–25 in MCF-7 human breast cancer cells a.
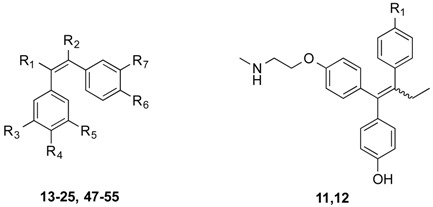 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | R7 | IC50 (μM) b |
| 13 | COOH | H | OMe | OMe | OMe | OMe | OH | 0.120 |
| 14 | COOH | H | OMe | OMe | OMe | OMe | H | 7.80 |
| 15 | COOH | H | OMe | OMe | OMe | OMe | F | 55.1 |
| 16 | COOH | H | OMe | OMe | OMe | OMe | NH2 | 6.70 |
| 17 | COOH | H | OMe | OMe | OMe | Br | H | >50 |
| 18 | COOH | H | OMe | OMe | OMe | F | H | >50 |
| 19 | COOH | H | OMe | OMe | OMe | Me | H | 26.1 |
| 20 | COOH | H | OMe | OMe | OMe | SMe | H | 10.6 |
| 21 | COOH | H | OMe | OMe | OMe | OMe | NO2 | 2.50 |
| 22 | COOH | H | OMe | OMe | OMe | -C4H4- | 15.3 | |
| 23 | H | COOH | OMe | OMe | OMe | OMe | H | >50 |
| 24 | CO2Me | H | OMe | OMe | OMe | OMe | OH | 2.80 |
| 25 | H | COOH | -OCH2O- | H | OMe | H | >50 | |
| 47 | H | CO-Pi | OMe | OMe | OMe | OMe | H | > 50 |
| 48 | H | CO-Py | OMe | OMe | OMe | OMe | H | >50 |
| 49 | CO-Pi e | H | OMe | OMe | OMe | OMe | OH | >50 |
| 52 | CO-Py f | H | OMe | OMe | OMe | OMe | H | >50 |
| 53 | CO-Pi e | H | OMe | OMe | OMe | OMe | H | g |
| 54 | CO-Py f | H | OMe | OMe | OMe | OMe | NH2 | >50 |
| 55 | CO-Pi e | H | OMe | OMe | OMe | OMe | NH2 | h |
| 26 (CA4) c | H | H | OMe | OMe | OMe | OMe | OH | 0.008 |
| 11 d | H | - | - | - | - | - | - | 0.029 |
| 12 | OH | - | - | - | - | - | - | 0.028 |
a Mean IC50 values of compounds for their antiproliferative effects and percent cytotoxicity on a human MCF-7 breast cancer cell line. b IC50 values are half maximal inhibitory concentrations required to block the growth stimulation of MCF-7 cells. Values are an average of at least three experiments performed in triplicate with typical standard errors below 15%. c The IC50 value obtained for 26 (CA4) in this assay is 8 nM for MCF-7 and is in good agreement with the reported values for CA4 using the MTT assay on human MCF-7 breast cancer cell lines(see refs. [59,68,69]). d The reported IC50 value for endoxifen in the MCF-7 cell line is 50 nM [70]. e Pi piperidine, f Py = pyrrole. g 35% growth inhibition for compound 53 at 10 μM. h 34% growth inhibition for compound 55 at 10 μM.
The conjugates 27–46 displayed a wide range of antiproliferative activity with IC50 values in the range 0.005–6.15 µM (see Table 2). In all examples, the coupling of the acrylic acid to the endoxifen 11 or the novel hydroxyendoxifen 12 resulted in an increase in the antiproliferative activity when compared to the corresponding acrylic acid and amides 47–55. The most potent conjugate 28 displayed antiproliferative action in the MCF-7 cell-line with an IC50 value of 5 nM—a value greater than either of the values for combretastatin A-4 (IC50 = 8 nM), endoxifen 11 (29 nM) and the novel hydroxyendoxifen analogue 12 (IC50 = 28 nM). In many examples, coupling of the acrylic acid to the novel hydroxyendoxifen 12 resulted in a more potent conjugate product than the coupling of the same acid with endoxifen 11, (e.g., IC50 of compound 27 = 0.033 nM, IC50 of compound 28 = 0.005 nM). Since there is much interest in combretastatin analogues having the 3-amino-4-methoxy substituion patern in Ring B [52] it was of interest in this work to evaluate the conjugates 45 and 46. While moderate activity was observed, (IC50 = 0.959 and 6.15 for compounds 45 and 46 respectively, these compounds were not as potent as the corresponding Ring B 3-hydroxy-4-methoxy substitute compounds 27 and 28. The ring B 3-fluoro-4-methoxy substituted compounds 43 and 44 displayed impressive antiproliferative activity with IC50 values of 180 and 194 nM respectively.
Table 2.
Antiproliferative effects of conjugate compounds 27–46 in MCF-7 human breast cancer cells a.
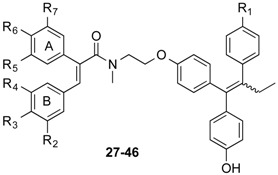 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | R7 | IC50 (μM) b | % Cell Death at 10 μM c |
| 27 | H | OH | OMe | H | OMe | OMe | OMe | 0.033 | 10 |
| 28 | OH | OH | OMe | H | OMe | OMe | OMe | 0.005 | 17 |
| 29 | H | H | OMe | H | OMe | OMe | OMe | 0.087 | 12 |
| 30 | OH | H | OMe | H | OMe | OMe | OMe | 0.043 | 19 |
| 31 | H | OMe | OMe | OMe | H | OMe | H | 2.08 | 1 |
| 32 | OH | OMe | OMe | OMe | H | OMe | H | 0.041 | 16 |
| 33 | H | -OCH2O- | H | H | OMe | H | 1.47 | 8 | |
| 34 | OH | -OCH2O- | H | H | OMe | H | 0.049 | 6 | |
| 35 | H | -C4H4- | H | OMe | OMe | OMe | 1.51 | 0 | |
| 36 | OH | -C4H4- | H | OMe | OMe | OMe | 3.62 | 3 | |
| 37 | H | H | Me | H | OMe | OMe | OMe | 1.75 | 2 |
| 38 | OH | H | Me | H | OMe | OMe | OMe | 0.372 | 12 |
| 39 | H | H | SMe | H | OMe | OMe | OMe | 0.790 | 3 |
| 40 | OH | H | SMe | H | OMe | OMe | OMe | 0.064 | 2 |
| 41 | H | H | Br | H | OMe | OMe | OMe | 1.03 | 5 |
| 42 | OH | H | Br | H | OMe | OMe | OMe | 2.35 | 7 |
| 43 | H | F | OMe | H | OMe | OMe | OMe | 0.180 | 2 |
| 44 | OH | F | OMe | H | OMe | OMe | OMe | 0.194 | 10 |
| 45 | H | NH2 | OMe | H | OMe | OMe | OMe | 0.959 | 8 |
| 46 | OH | NH2 | OMe | H | OMe | OMe | OMe | 6.15 | 6 |
| 26 (CA4) d | - | - | - | - | - | - | - | 0.008 | 13 |
| 11 e | - | - | - | - | - | - | - | 0.029 | 23 |
| 12 | - | - | - | - | - | - | - | 0.028 | 11 |
a Mean IC50 values of compounds for their antiproliferative effects and percent cytotoxicity on a human MCF-7 breast cancer cell line using the MTT assay. b IC50 values are half maximal inhibitory concentrations required to block the growth stimulation of MCF-7 cells. Values are an average of at least three experiments performed in triplicate with typical standard errors below 15%. c Lactate Dehydrogenase assay: Following treatment of the cells, the amount of LDH was determined using LDH assay kit from Promega. Data is presented as % cell lysis at compound concentration of 10 µM [66]. d The IC50 value obtained for 26 (CA4) in this assay is 8 nM for MCF-7 and is in good agreement with the reported values for CA4 using the MTT assay on human MCF-7 breast cancer cell lines ([59,68,69]). e The reported IC50 value for endoxifen 11 in the MCF-7 cell line is 50 nM [70].
Conjugate compounds 29 and 30 substituted with 4-methoxy group in Ring B of the acrylic acid component, also display potent activity with IC50 values of 87 nM and 43 nM respectively. Compound 31 which contains 3,4,5-trimethoxy substitution pattern in the Ring B acrylic acid component and the 4-methoxy substituent on Ring A demonstrate a reduced activity (IC50 = 2.08 µM), however in compound 32 which is the corresponding conjugate with hydroxyendoxifen, the antiprolifertive activity is determined at 41 nM, indicating the potent effect on activity of conjugation with hydroxyendoxifen. Inclusion of the 4-thiomethyl substituent or the 3,4-methylenedioxy substitution on Ring B of the acrylic acid component resulted in potent activity for the hydroxyendoxifen conjugated compounds 34 and 40 (IC50 = 49 nM and 64 nM respectively). Introduction of the 2-naphthyl Ring B as the acrylic acid component resulted in a reduced antiproliferative effect for compounds 36 and 37 (IC50 values = 1.51 and 3.62 nM respectively). It would have been interesting to determine whether the addition of estradiol would alter the IC50 values in Table 2 and thus definitively determine whether the antiproliferative effects observed had a ER-mediated component or whether these effects were solely mediated through the combretastatin component of the conjugate.
The most active compounds from the MCF-7 assay were selected for evaluation in the ER-negative MDA-MB-231 human breast cancer cell line. The selected conjugates (27–36, 41–46) as expcted displayed reduced antiproliferative activity (micromolar IC50 values) compared with results obtained for the ER-positive MCF-7 cell line, indicating that the conjugates show a selectivity for the ER expressing cell line (see Table 3). The most active compound was identified as conjugate 34 with IC50 value of 0.68 µM, while compounds compounds 27, 28, 35 and 44 displayed moderate activity with IC50 values of 2.67, 2.48, 2.16 and 1.85 µM, respectively, all of which are more potent than tamoxifen, (IC50 = 20 µM) and 4-hydroxytamoxifen (IC50 = 18 µM) in this ER negative cell line.
Table 3.
Antiproliferative effects of selected conjugate compounds in MDA-MB-231 human breast cancer cells a.
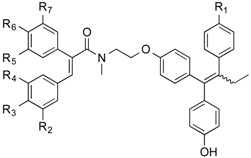 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | R7 | IC50 (μM) b | % Cell Death at 10 μM c |
| 27 | H | OH | OMe | H | OMe | OMe | OMe | 2.67 | 17 |
| 28 | OH | OH | OMe | H | OMe | OMe | OMe | 2.48 | 15 |
| 29 | H | H | OMe | H | OMe | OMe | OMe | 30.6 | 2 |
| 30 | OH | H | OMe | H | OMe | OMe | OMe | 6.29 | 7 |
| 31 | H | OMe | OMe | OMe | H | OMe | H | 51.0 | 0 |
| 32 | OH | OMe | OMe | OMe | H | OMe | H | 4.09 | 0 |
| 33 | H | OCH2O | H | H | OMe | H | 5.23 | 0 | |
| 34 | OH | OCH2O | H | H | OMe | H | 0.68 | 16 | |
| 35 | H | -C4H4- | H | OMe | OMe | OMe | 2.16 | 0 | |
| 36 | OH | -C4H4- | H | OMe | OMe | OMe | 4.32 | 0 | |
| 41 | H | H | Br | H | OMe | OMe | OMe | >50 | 0 |
| 42 | OH | H | Br | H | OMe | OMe | OMe | >50 | 9 |
| 43 | H | F | OMe | H | OMe | OMe | OMe | 30.3 | 0 |
| 44 | OH | F | OMe | H | OMe | OMe | OMe | 1.85 | 14 |
| 45 | H | NH2 | OMe | H | OMe | OMe | OMe | 7.11 | 0 |
| 46 | OH | NH2 | OMe | H | OMe | OMe | OMe | 49.7 | 0 |
| Tamoxifen | 20 | 0 | |||||||
| Hydroxytamoxifen | 18 | 0 | |||||||
| 26 (CA4) | 0.043 | 4 | |||||||
a Mean IC50 values of compounds for their antiproliferative effects and percent cytotoxicity on a human MDA breast cancer cell line. b IC50 values are half maximal inhibitory concentrations required to block the growth stimulation of MDA-MB-231 cells. Values are an average of at least three experiments performed in triplicate with typical standard errors below 15%. c Lactate Dehydrogenase assay: Following treatment of the cells, the amount of LDH was determined using LDH assay kit from Promega. Data is presented as % cell lysis at compound concentration of 10 μM [66].
3.2.2. Estrogen Receptor Binding
The receptor binding affinity for the lead conjugate 28 was investigated in both ERα and ERβ using a fluorescence polarisation based competitive binding assay. Competitive binding affinity experiments were carried out using purified baculovirus-expressed human ERα and ERβ and fluoromone, a fluorescein labelled estrogen ligand [33,34]. The endogenous ligand β-estradiol was used as the positive control in the experiments. The lead conjugate displayed potent binding in both ER isoforms (IC50 value for ERα: 0.9 nM; IC50 value for ERβ: 4.7 nM). These binding values are greater than the endogenous ligand estradiol, 4-hydroxytamoxifen, endoxifen 11 and significantly greater than that of the parent ER-ligand, hydroxyendoxifen 12 (ERα: IC50 44.1 nM; ERβ: IC50 39.7 nM) (see Table 4).
Table 4.
Estrogen receptor binding affinities for compounds 11, 12 and 28 a.
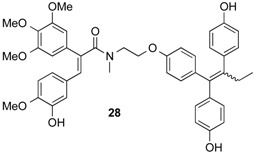 | |||
|---|---|---|---|
| Compound | IC50 (nM) a,b | Selectivity (α/β) | |
| ERα | ERβ | ||
| 28 | 1 | 5 | 5.22 |
| 11 (Endoxifen) | 47 | 118 | 2.52 |
| 12 (Hydroxyendoxifen) | 44 | 40 | 0.90 |
| 4-Hydroxytamoxifen | 30 | 26 | 0.88 |
| Estradiol | 6 | 6 | 0.98 |
a Mean IC50 values of compounds for their competitive binding in ERα and ERβ isoforms. b Values are an average of at least nine replicate experiments, for ERα with typical standard errors below 15%, and six replicate experiments for ERβ, with typical standard errors below 15%.
3.2.3. NCI 60 Cell Line Screen
On the basis of potency, compound 28 was evaluated in the National Cancer Institute (NCI, Bethesda, MD, USA) Division of Cancer Treatment and Diagnosis(DCTD)/Developmental Therapeutics Programme(DTP) in which the activity of the compound is determined using a 60-cell line screen facility of different cancer cell lines of diverse tumour origin [71]. Compound was tested for inhibition of growth(GI50) and cytotoxicity(LC50). These studies were performed at the NCI as part of their drug screening programme. Initially, the compound was evaluated against the 60 cell lines at a single dose of 10 μM; if significant growth inhibition was exhibited the compound was evaluated against the 60 cell panel at a further five concentration levels, 0.01–100 μM, (see Table 5). In the one-dose screen, 28 displayed low lethality and a mean growth inhibition value of 98.73% over the 60 cell lines. 28 displayed very high growth inhibition in the cell lines of non-small cell lung cancer NCI-H23 (97%) and NCI-H460 (98%); colon cancer HCT-116 (98%); breast cancer MCF-7 (94%) and MDA-MB-435 (91%); ovarian cancer OVCAR-8 (97%) and SK-OV-3 (95%); leukaemia RPMI-8226 (91%); renal cancers ACHN (96%), CAKI-1 (91%), RXF 393 (95%) and SN12C (90%); melanoma SK-MEL-2 (97%) and CNS cancer U251 (93%). The compound caused between 80%–89% growth inhibition in a further 14 cell lines and was very toxic to the non-small cell lung cancer NCI-H226 (73% lethality) and melanoma SK-MEL-5 (60% lethality) cell lines.
Table 5.
Results of the in vitro human cancer cell growth inhibition for compound 28 a. Antiproliferative evaluation of compound 28 in the NCI60 cell line in vitro screen.
| Cell Line | Compound 28 | Compound 28 | CELL LINE | Compound 28 | Compound 28 |
|---|---|---|---|---|---|
| GI% (10−5 M) a | GI50 (μM) b | GI% (10−5 M) a | GI50 (μM) b | ||
| Leukemia | Melanoma | ||||
| CCRF-CEM | 72.48 | 1.77 | LOX IMV1 | 1.55 | 87.57 |
| HL-60 (TB) | 100 d,f | 1.4 | MALME-3M | 1.76 | 76.76 |
| K-562 | 86.1 | 2.57 | M14 | 1.68 | 100 d,p |
| MOLT-4 | 64.31 | 1.52 | MDA-MB-235 | 0.747 | 90.98 |
| RPMI-8226 | 90.65 | 0.896 | SK-MEL-2 | 8.67 | 97.42 |
| SR | 69.03 | 2.35 | SK-MEL-28 | 1.65 | 76.19 |
| Non-Small Cell Lung Cancer | SK-MEL-5 | 0.837 | 100 d,q | ||
| A549/ATCC | 86.97 | 2.88 | UACC-257 | 2.83 | 100 d,r |
| EKVX | 85.11 | 2.01 | UACC-62 | 0.922 | 100 d,s |
| HOP-62 | 81.99 | 2.86 | Ovarian Cancer | ||
| HOP-92 | 86.19 | 0.764 | IGROV1 | 2.3 | 100 d,t |
| OVCAR-3 | 1 | 100 d,u | |||
| NCI-H226 | 100 d,g | 1.18 | OVCAR-4 | 2.59 | 81.47 |
| NCI-H23 | 96.76 | 2.19 | OVCAR-5 | 3.21 | 82.75 |
| NCI-H332M | 81.7 | 2.02 | OVCAR-8 | 2.02 | 97.21 |
| NCI-H460 | 98.38 | 1.42 | NCI/ADR-RES | 6.02 | 21.31 |
| NCI-H552 | 100 d,h | 1.39 | SK-OV-3 | 1.66 | 95.08 |
| Colon Cancer | Renal Cancer | ||||
| COLO 205 | 100 d,i | 2.15 | 786-0 | 1.91 | 100 d,v |
| HCT-2998 | 100 d,j | 1.25 | A498 | 2.94 | 80.37 |
| HCT-116 | 97.57 | 1.8 | ACHN | 1.92 | 96.43 |
| HCT-15 | 100 d,k | 1.53 | CAKI-1 | 1 | 91.46 |
| HT29 | 100 d,l | 1.16 | RXF 393 | 1.89 | 95.02 |
| KM12 | 100 d,m | 1.26 | SN12C | 1.8 | 90.3 |
| SW-620 | 81.32 | 1.21 | TK-10 | 1.65 | 100 d,w |
| CNS Cancer | UO-31 | 2.66 | 89.77 | ||
| SF-268 | 84.22 | 1.69 | Breast Cancer | ||
| SF295 | 73.4 | 1.74 | MCF-7 | 0.00998 | 94.3 |
| SF539 | 100 d,n | 1.18 | MDA-MB-231/ATCC | 1.61 | 73.55 |
| SNB-19 | 81.49 | 1.72 | HS 578T | 2.04 | 64.8 |
| SNB-75 | 60.65 | 5.96 | BT-549 | 1.12 | 100 d,x |
| U251 | 93.37 | 1.74 | T-47D | Nd c | 100 d,y |
| Prostate cancer | MDA-MB-468 | 1.12 | 100 d,z | ||
| PC-3 | 86.32 | 1.73 | |||
| DU-145 | 100 d,o | 1.66 | |||
| MG-MID (µM) e | 1.45 |
a Data obtained from NCI in vitro human tumour cell screen at 10 µM concentration. b GI50 is the molar concentration of the compound causing 50% inhibition of growth of the tumour cells; c Nd: Not determined; d Cytotoxic effect; e MG-MID: the mean of GI50 values over all cell lines for the tested compound. f Cell Growth Percent −13.32%. g Cell Growth Percent −72.63%. h Cell Growth Percent −22.04%. i Cell Growth Percent −41.64%. j Cell Growth Percent −16.22%. k Cell Growth Percent −15.41%. l Cell Growth Percent −32.91%. m Cell Growth Percent −51.93%. nCell Growth Percent −6.56%. o Cell Growth Percent −19.73%. p Cell Growth Percent −20.28%. q Cell Growth Percent −59.57%. r Cell Growth Percent −0.60%. s Cell Growth Percent −19.77%. t Cell Growth Percent 35.74%. u Cell Growth Percent 30.95%. v Cell Growth Percent −26.91%. w Cell Growth Percent −29.83%. x Cell Growth Percent −23.96%. y Cell Growth Percent 9.09%. z Cell Growth Percent −34.28%.
In the five-dose screen, 28 displayed low micromolar GI50 (IC50) values for the majority of the 60 cancer cell lines. However, 28 demonstrated a high selectivity towards MCF-7 breast cancer with a GI50 (IC50) value of 9.5 nM and a LC50 value greater than 50 μM for this cell line. The mean GI50 value compound 28 across all 60 panel cell lines is 1.45 µM, (log GI50 = −5.79). As 28 has been shown to be a high-affinity ER-binding ligand, the high specificity towards MCF-7 cells is most probably due to its selective antagonistic effects on the over-expressed ER within this cell-line. This is an impressive and promising result as it confirms the suitability of the selected prototypes and project strategy for the possible therapeutic application of the ER-conjugates synthesised in the study. The LC50 value is a measure of the cytotoxicity of the compound in each cell line; the mean LC50 for compound 28 is 46.77 µM and was greater than 50 µM in all but 4 cell lines. (NCI-H23: 48.9 µM; COLO 205: 26.2 µM; SK-MEL-5: 34.5 µM; A498: 32.6 µM). The mean LC50 for 28 across all 60 panel cell lines is 46.77 µM; (see Table 5).
The NCI provide the pattern recognition algorithm COMPARE [72,73]. The unique complexity of a 60-cell line dose response produced by a given compound results in a biological response pattern which can be utilized in pattern recognition algorithms. Using the COMPARE algorithms, it is possible to assign a putative mechanism of action for the screened compound, or to determine that the response pattern is unique and not similar to that of any of the standard prototype compounds included in the NCI database. In addition, following characterization of various cellular molecular targets in the 60-cell lines, it may be possible to select compounds most likely to interact with a specific molecular target. Generally, a correlation coefficient greater than 0.6 is considered a positive correlation [74]. For the five-dose screen COMPARE analyses, the highest correlation coefficients achieved for compound 28, from both the common anticancer agent database and the comprehensive database, were 0.934 in relation to the potent antiviral and anticancer agent, Didemnin B [75] which selectively induces apoptosis through dual inhibition of PPT1 and EEF1A1, 0.879 for the antileukemic agent and protein synthesis inhibitor, Bruceantin [76] and 0.430 in relation to the antimitotic/anti-tumour agent Rhizoxin 47 [77,78]. Correlation values are Pearson correlation coefficients based on GI50 mean graphs. These correlation values demonstrate a high similarity of activity and suggest a common mechanism of action between the agents. The antiproliferative activity observed for the conjugate compound 28 indicated that there is a significant therapeutic window between the concentration required for cancer cell growth inhibition and the concentration that is toxic to MCF-7 cells.
3.2.4. Ishikawa Cell Line Study
The Ishikawa assay is used to measure estrogen stimulation of alkaline phosphatase enzyme activity (AlkP) by the Ishikawa cell line of human endometrial adenocarcinoma cells [35]. The Ishikawa assay provides a measure of the agonist activity of a compound and was carried out on the lead conjugate, 28. Tamoxifen was used as a reference compound in the assay. The effect of the compounds on estrogen stimulation within the Ishikawa cells can be seen in Figure 2, which is representative of an experiment that was carried out three times. Tamoxifen displays some estrogenic activity at higher concentrations. When Tamoxifen is dosed with a 1 nM estradiol spike, there is estrogen stimulation at low concentration while a reduction occurs as the concentration of Tamoxifen is increased. The conjugate was dosed in duplicate. The conjugate 28 is similar to Tamoxifen when dosed individually. However, the conjugate appears not to display any estrogenic activity at higher concentrations, which is desirable. This may suggest that the conjugate is acting as a full antagonist. When 28 was dosed as a mixture with estradiol, there is a reduction in estrogenic stimulation at low concentrations and a more pronounced reduction in estrogen stimulation over the range of concentrations when compared with the Tamoxifen mixture. This preliminary result is promising as it demonstrates that the conjugate 28 does not display estrogenic stimulation in the cell line and shows reduced estrogenic stimulation compared to Tamoxifen over the concentration range investigated.
Figure 2.

Effect on alkaline phosphatase activity of conjugate 28 and tamoxifen in Ishikawa cells.
3.2.5. Stability Studies
The stability of the selected target compounds 27, 31 and 32 was evaluated in phosphate buffer at pH values in the range 4–9 and the half-life was determined to be greater than 24 h for each compound at these pH values. The compounds were found to stable without any significant degradation of the conjugates suggesting that the combretastatin fragment remains intact at physiological pH; and indicating that the combretastatin component of the conjugate has a role in displacing helix-12 of the ER resulting in the potent (and possibly pure) antagonistic activity.
3.3. Molecular Modelling
The intricate molecular basis of estrogen receptor agonism and antagonism has been well studied [1,79,80,81]. Phenol groups, a common structural motif to many ER ligands, take part in direct hydrogen bonding with the same key residues in the ER (i.e., Glu353, Arg 394 and His524 in human ERα). Within the ER ligand-binding domain (LBD), ligand recognition is achieved through a combination of specific hydrogen bonds and the complementarity values of the binding cavity with the relevant ligands non-polar character. Antagonistic ligands such as Tamoxifen and 4-hydroxytamoxifen contain a basic side chain group too large to be accommodated in the binding cavity, resulting in the displacement of the Helix-12 in the ER protein structure. In particular, the positioning of Helix-12 in the ER is key to the recruitment of coactivators/corepressors and determining the overall nature of the ligand effect [81].
The interactions of compound 28 was modelled in both ERα and ERβ isoforms in order to rationalise the biochemical data obtained for this compound. The modelling strategy protocol involved calculating the appropriate protonation state at physiological pH using MARVIN [82]; the determination of the lowest energy conformer using MACROMODEL [38]; the generation of an ensemble of low-energy conformers using OMEGA [39], followed by the docking and scoring of these conformers using FRED [40]. Graphical manipulations were carried out using DS VISUALIZER [83]. High-ranking docking solutions were investigated—noting favourable interactions between the conjugate and the residues in the ER isoform ligand binding domains.
The top-ranking docking solution in human ERα for compound 28 is shown in Figure 3. For comparison, the 4-hydroxytamoxifen bound ligand (reference pdb: 3ERT) [41] is displayed in yellow while the docked confirmations are coloured by element type (carbon = grey, hydrogen = white, oxygen = red, nitrogen = blue). In Figure 3, the Glu353 and Arg394 residues form hydrogen bond interactions with the phenol group, similar to that of 4-hydroxytamoxifen. In this docked solution the additional phenol group of 28 is found 3.9 Å away from the His524 residue and does not form a strong H-bond. The Asp351 residue is located 3.7 Å away from the amide nitrogen of 28, which would translate to a weak interaction. The Cys530 residue forms a H-bond with the C-4 methoxy group on the A-ring of the combretastatin fragment as illustrated in Figure 3. The increased bulk of the ligand side chain of the conjugate 28 may displace Helix-12 to a significant extent and could explain the antagonistic activity and impressive binding affinity of this lead compound 28 (0.9 nM in ERα; 4.7 nM in ERβ).
Figure 3.

Predicted binding mode of compound 28 in the ligand binding domain of ERα. Highest-ranking docking orientation and predicted receptor interactions for 28 (ball-and-stick representation) in ERα compared to the crystal solution for 4-hydroxytamoxifen (yellow); pdb 3ERT [41]. (H-bond interactions are illustrated as broken lines).
The top-ranking docking solutions in rat ERβ for compound 28 is shown in Figure 4. For comparison, the raloxifene bound ligand (reference pdb: 1QKN) [42] is displayed in yellow while the docked confirmations are coloured by element type (carbon = grey, hydrogen = white, oxygen = red, nitrogen = blue). In Figure 4, the residues Glu260 and Arg301 form favourable hydrogen bond interactions with the phenol group, similar to that of 4-hydroxytamoxifen. The additional group present on 28 forms an additional hydrogen bond interaction with the His430 residue. There is good agreement/overlap of the raloxifene core and the endoxifen moiety of 28. A methoxy group present on the combretastatin moiety of 28 forms another favourable hydrogen bond through a water molecule with the Asp258 residue within the LBD. This interaction could help “lock in” the ligand within the cavity site, resulting in the good binding affinity. Additional waters unaccounted for in this determined crystal structure may introduce further favourable hydrogen bond interactions with the other methoxy groups of the combretastatin moiety through a network of water molecules. The alkyl groups of Ile328 are in close proximity to a phenyl group present on the endoxifen moiety, which could produce a lipophilic-lipophilic interaction. The bulky side chain of 28 would have a greater degree of rigidity due to the presence of the amide linkage and may displace Helix-12 significantly in ERβ to produce antiproliferative effects observed.
Figure 4.

Predicted binding mode of compound 28 in the ligand binding domain of ERβ. Highest-ranking docking orientation and predicted receptor interactions for 28 (ball-and-stick representation) in ERβ compared to the crystal solution for 4-hydroxytamoxifen (yellow) (pdb 1QKN) [42]. (H-bond interactions are illustrated as broken lines).
4. Conclusions
A series of prototype ER-ligand conjugates 27–46 were successfully synthesised incorporating an endoxifen-combretastatin hybrid scaffold with potential SERM properties. A number of these novel compounds displayed potent antiproliferation activity in the MCF-t human breast cancer cell line with low cytotoxicity values. The conjugate 28 in the series was the most promising compound in the study, with potent antiproliferative activity in MCF-7 human breast cancer cell line (IC50: 5 nM), low cytotoxicity and impressive ER competitive binding IC50 values (ERα: IC50 0.9 nM; ERβ: IC50 4.7 nM). A number of these conjugate compounds also display activity in the ER negative cell line MDA-MB-321 at low micromolar and sub-micromolar concentrations, e.g., compound 34 (IC50 = 0.68 µM). Preliminary stability studies show that these conjugates do not degrade significantly at physiological pH values. Interestingly, from a structural biology standpoint, this result indicates that the ER can tolerate a large substituent such as the combretastatin analogue, at the basic side chain of the triarylethylene ER ligand scaffold without detrimental effect on the overall antiproliferative and ER binding characteristics and biochemical activity. The lead conjugate compounds 27 and 28 are currently under further investigation by our research group to further explore its mechanism of action and potential applications in the development of useful ER ligands. These compounds demonstrate a novel and interesting class of ER ligand and may have potential future application as medicinal agents for anticancer therapy.
Acknowledgments
The Ishikawa cells were a kind gift from Richard Hochberg, Yale University. This study was supported by the Irish Research Council for Science, Engineering and Technology (IRCSET). Nuclear magnetic resonance spectroscopy was carried out by Manuel Ruethers and John O’Brien at the Department of Chemistry, Trinity College Dublin. High-resolution mass spectroscopy was carried out by Martin Feeney at the Department of Chemistry, Trinity College Dublin and Dilip Rai at the Centre for Synthesis and Chemical Biology, University College Dublin. The Trinity Biomedical Sciences Institute (TBSI) is supported by a capital infrastructure investment from Cycle 5 of the Irish Education Authority’s Programme for Research in Third Level Institutions(PRTLI).
Author Contributions
Niall O. Keely synthesised and characterised all the molecules in the studies according to Scheme 1, Scheme 2 and Scheme 3, performed the cell studies and generated the data in Table 1, Table 2, Table 3 and Table 4, performed the molecular modelling study and generated Figure 3 and Figure 4, performed data analysis and interpreted data and wrote draft of the manuscript. Gloria Ana synthesised additional compounds, characterised these compounds, performed the cell studies and generated data in Scheme 2 and Table 1. Miriam Carr performed the Ishikawa cell study and generated Figure 2. Bassem Yassin performed the HPLC analytical study and the stability study. David G. Lloyd assisted with the design of the molecular modelling studies. Daniela Zisterer assisted with the design of the biochemical studies. Mary J. Meegan designed the studies, wrote drafts of the manuscript and submitted the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Ascenzi P., Bocedi A., Marino M. Structure-function relationship of estrogen receptor alpha and beta: Impact on human health. Mol. Asp. Med. 2006;27:299–402. doi: 10.1016/j.mam.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Powles T.J. Anti-oestrogenic prevention of breast cancer—The make or break point. Nat. Rev. Cancer. 2002;2:787–794. doi: 10.1038/nrc908. [DOI] [PubMed] [Google Scholar]
- 3.Jordan V.C. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 1. Receptor interactions. J. Med. Chem. 2003;46:883–908. doi: 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- 4.Jordan V.C. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 2. Clinical considerations and new agents. J. Med. Chem. 2003;46:1081–1111. doi: 10.1021/jm020450x. [DOI] [PubMed] [Google Scholar]
- 5.Meegan M.J., Lloyd D.G. Advances in the science of estrogen receptor modulation. Curr. Med. Chem. 2003;10:181–210. doi: 10.2174/0929867033368501. [DOI] [PubMed] [Google Scholar]
- 6.Dodge J.A., Richardson T.I. Novel selective estrogen receptor modulators (serms) Annu. Rep. Med. Chem. 2007;42:147–160. [Google Scholar]
- 7.Keely N.O., Meegan M.J. Selective estrogen receptor modulators: Recent developments in design, structural studies and clinical applications. Curr. Trends Med. Chem. 2009;6:1–17. [Google Scholar]
- 8.Jordan V.C. New insights into the metabolism of tamoxifen and its role in the treatment and prevention of breast cancer. Steroids. 2007;72:829–842. doi: 10.1016/j.steroids.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pasqualini J.R., Chetrite G.S. Recent insight on the control of enzymes involved in estrogen formation and transformation in human breast cancer. J. Steroid Biochem. Mol. Biol. 2005;93:221–236. doi: 10.1016/j.jsbmb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Scott S.M., Brown M., Come S.E. Emerging data on the efficacy and safety of fulvestrant, a unique antiestrogen therapy for advanced breast cancer. Expert Opin. Drug Saf. 2011;10:819–826. doi: 10.1517/14740338.2011.595560. [DOI] [PubMed] [Google Scholar]
- 11.O’Boyle N.M., Pollock J.K., Carr M., Knox A.J., Nathwani S.M., Wang S., Caboni L., Zisterer D.M., Meegan M.J. Beta-lactam estrogen receptor antagonists and a dual-targeting estrogen receptor/tubulin ligand. J. Med. Chem. 2014;57:9370–9382. doi: 10.1021/jm500670d. [DOI] [PubMed] [Google Scholar]
- 12.Jaracz S., Chen J., Kuznetsova L.V., Ojima I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorgan. Med. Chem. 2005;13:5043–5054. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 13.Morphy R., Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 14.Keely N.O., Meegan M.J. Targeting tumors using estrogen receptor ligand conjugates. Curr. Cancer Drug Targets. 2009;9:370–380. doi: 10.2174/156800909788166628. [DOI] [PubMed] [Google Scholar]
- 15.Dao K.L., Hanson R.N. Targeting the estrogen receptor using steroid-therapeutic drug conjugates (hybrids) Bioconjug. Chem. 2012;23:2139–2158. doi: 10.1021/bc300378e. [DOI] [PubMed] [Google Scholar]
- 16.Keely N.O., Zisterer D.M., Meegan M.J. Design, synthesis and biochemical evaluation of estrogen receptor ligand conjugates as tumor targeting agents. Lett. Drug Des. Discov. 2012;9:295–304. doi: 10.2174/157018012799129864. [DOI] [Google Scholar]
- 17.Burke P.J., Koch T.H. Design, synthesis, and biological evaluation of doxorubicin-formaldehyde conjugates targeted to breast cancer cells. J. Med. Chem. 2004;47:1193–1206. doi: 10.1021/jm030352r. [DOI] [PubMed] [Google Scholar]
- 18.Knebel N., von Angerer E. Platinum complexes with binding affinity for the estrogen receptor. J. Med. Chem. 1988;31:1675–1679. doi: 10.1021/jm00117a002. [DOI] [PubMed] [Google Scholar]
- 19.Vogel A.I., Tatchell A.R., Furnis B.S., Hannaford A.J., Smith P.W.G. Vogel’s Textbook of Practical Organic Chemistry. 5th ed. Prentice Hall; Englewood Cliffs, NJ, USA: 1996. [Google Scholar]
- 20.Pettit G.R., Singh S.B., Boyd M.R., Hamel E., Pettit R.K., Schmidt J.M., Hogan F. Antineoplastic agents. 291. Isolation and synthesis of combretastatins A-4, A-5, and A-6(1A) J. Med. Chem. 1995;38:1666–1672. doi: 10.1021/jm00010a011. [DOI] [PubMed] [Google Scholar]
- 21.Gaukroger K., Hadfield J.A., Hepworth L.A., Lawrence N.J., McGown A.T. Novel syntheses of cis and trans isomers of combretastatin A-4. J. Org. Chem. 2001;66:8135–8138. doi: 10.1021/jo015959z. [DOI] [PubMed] [Google Scholar]
- 22.Borrel C., Thoret S., Cachet X., Guenard D., Tillequin F., Koch M., Michel S. New antitubulin derivatives in the combretastatin a4 series: Synthesis and biological evaluation. Bioorgan. Med. Chem. 2005;13:3853–3864. doi: 10.1016/j.bmc.2005.02.039. [DOI] [PubMed] [Google Scholar]
- 23.Yong Z., Xu X. Preparation of 2-(3,4,5-trimethoxyphenyl)-3-phenylpropenoic acid derivatives for blood-vessel targeted-agent drugs. WO 2009059448. 2009 May 4;
- 24.Nam N.H., Byung-Zun A. Combretastatin-chalcone hybrids: Synthesis and cytotoxicity. Med. Chem. 2007;3:373–377. doi: 10.2174/157340607781024366. [DOI] [PubMed] [Google Scholar]
- 25.Eaddy J.F., III, Heyer D., Katamreddy S.R., Martin M.T., McClure M.S., Randhawa A.S., Samano V., Ray J.A. Preparation of acyloxydiphenylbutenylcinnamates as estrogen receptor modulator prodrugs. WO 2005033056. 2005 Apr 14;
- 26.Hadfield J.A., Gaukroger K., Hirst N., Weston A.P., Lawrence N.J., McGown A.T. Synthesis and evaluation of double bond substituted combretastatins. Eur. J. Med. Chem. 2005;40:529–541. doi: 10.1016/j.ejmech.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 27.Lloyd D.G., Smith H.M., O’Sullivan T., Zisterer D.M., Meegan M.J. Synthesis, structure-activity relationships and antagonistic effects in human MCF-7 breast cancer cells of flexible estrogen receptor modulators. Med. Chem. 2005;1:335–353. doi: 10.2174/1573406054368666. [DOI] [PubMed] [Google Scholar]
- 28.Vessieres A., Top S., Pigeon P., Hillard E., Boubeker L., Spera D., Jaouen G. Modification of the estrogenic properties of diphenols by the incorporation of ferrocene. Generation of antiproliferative effects in vitro. J. Med. Chem. 2005;48:3937–3940. doi: 10.1021/jm050251o. [DOI] [PubMed] [Google Scholar]
- 29.Fauq A.H., Maharvi G.M., Sinha D. A convenient synthesis of (Z)-4-hydroxy-N-desmethyltamoxifen (endoxifen) Bioorgan. Med. Chem. Lett. 2010;20:3036–3038. doi: 10.1016/j.bmcl.2010.03.117. [DOI] [PubMed] [Google Scholar]
- 30.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 31.Prism version 4. Graphpad Software, Inc.; La Jolla, CA, USA: 2010. [Google Scholar]
- 32.Promega Corporation, Cytotox 96® Non-Radioactive Cytotoxicity Assay; Promega Cytotox 96 Nonradioactive Cytotoxicity Assay protocol.pdf; 2016. [(accessed on 26 April 2016)]. Available online: www.promega.com.
- 33.Polarscreen™ er Alpha Competitor Assay, Green. [(accessed on 21 March 2016)]. Available online: https://tools.Thermofisher.Com/content/sfs/manuals/polarscreen_er_alpha_green_man.Pdf.
- 34.Polarscreen™ er Beta Competitor Assay Kit, Green. [(accessed on 21 March 2016)]. Available online: https://tools.Thermofisher.Com/content/sfs/manuals/polarscreen_er_beta_green_man.pdf.
- 35.Littlefield B.A., Gurpide E., Markiewicz L., McKinley B., Hochberg R.B. A simple and sensitive microtiter plate estrogen bioassay based on stimulation of alkaline phosphatase in ishikawa cells: Estrogenic action of delta 5 adrenal steroids. Endocrinology. 1990;127:2757–2762. doi: 10.1210/endo-127-6-2757. [DOI] [PubMed] [Google Scholar]
- 36.Paull K.D., Shoemaker R.H., Hodes L., Monks A., Scudiero D.A., Rubinstein L., Plowman J., Boyd M.R. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: Development of mean graph and compare algorithm. J. Natl. Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 37.Meegan M.J., Hughes R.B., Lloyd D.G., Williams D.C., Zisterer D.M. Flexible estrogen receptor modulators: Design, synthesis, and antagonistic effects in human MCF-7 breast cancer cells. J. Med. Chem. 2001;44:1072–1084. doi: 10.1021/jm001119l. [DOI] [PubMed] [Google Scholar]
- 38.Schrödinger Inc.; New York, NY, USA: [(accessed on 26 April 2016)]. Macromodel version 6.5.24. Available online: http://www.schrodinger.com/ [Google Scholar]
- 39.Openeye Scientific Software, Inc.; Santa Fe, NM, USA: [(accessed on 22 March 2010)]. Omega version 2.3.2. Available online: http://www.eyesopen.com. [Google Scholar]
- 40.Openeye Scientific Software, Inc.; Santa Fe, NM, USA: [(accessed on 26 April 2016)]. FRED version 2.2.5. Available online: http://docs.eyesopen.com/oedocking/fred.html. [Google Scholar]
- 41.Shiau A.K., Barstad D., Loria P.M., Cheng L., Kushner P.J., Agard D.A., Greene G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/S0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 42.Pike A.C., Brzozowski A.M., Hubbard R.E., Bonn T., Thorsell A.G., Engstrom O., Ljunggren J., Gustafsson J.A., Carlquist M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu X., Hawse J.R., Subramaniam M., Goetz M.P., Ingle J.N., Spelsberg T.C. The tamoxifen metabolite, endoxifen, is a potent antiestrogen that targets estrogen receptor alpha for degradation in breast cancer cells. Cancer Res. 2009;69:1722–1727. doi: 10.1158/0008-5472.CAN-08-3933. [DOI] [PubMed] [Google Scholar]
- 44.Lim Y.C., Li L., Desta Z., Zhao Q., Rae J.M., Flockhart D.A., Skaar T.C. Endoxifen, a secondary metabolite of tamoxifen, and 4-oh-tamoxifen induce similar changes in global gene expression patterns in MCF-7 breast cancer cells. J. Pharmacol. Exp. Ther. 2006;318:503–512. doi: 10.1124/jpet.105.100511. [DOI] [PubMed] [Google Scholar]
- 45.Lu W.J., Desta Z., Flockhart D.A. Tamoxifen metabolites as active inhibitors of aromatase in the treatment of breast cancer. Breast Cancer Res. Treat. 2012;131:473–481. doi: 10.1007/s10549-011-1428-z. [DOI] [PubMed] [Google Scholar]
- 46.Lv W., Liu J., Lu D., Flockhart D.A., Cushman M. Synthesis of mixed (E,Z)-, (E)-, and (Z)-norendoxifen with dual aromatase inhibitory and estrogen receptor modulatory activities. J. Med. Chem. 2013;56:4611–4618. doi: 10.1021/jm400364h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu J., Wang X., Xu Y., Dong Y., Yang S., Wu Y., Qin Y., Wu M. Mass spectrometric characterization of toremifene metabolites in human urine by liquid chromatography-tandem mass spectrometry with different scan modes. Analyst. 2011;136:467–472. doi: 10.1039/C0AN00487A. [DOI] [PubMed] [Google Scholar]
- 48.Coe P.L., Scriven C.E. Crossed coupling of functionalized ketones by low valent titanium (the mcmurry reaction)—A new stereoselective synthesis of tamoxifen. J. Chem. Soc. Perkin Trans. 1. 1986:475–477. doi: 10.1039/p19860000475. [DOI] [Google Scholar]
- 49.Katzenellenbogen J.A., Carlson K.E., Katzenellenbogen B.S. Facile geometric isomerization of phenolic non-steroidal estrogens and antiestrogens: Limitations to the interpretation of experiments characterizing the activity of individual isomers. J. Steroid Biochem. 1985;22:589–596. doi: 10.1016/0022-4731(85)90210-9. [DOI] [PubMed] [Google Scholar]
- 50.Maximov P.Y., Myers C.B., Curpan R.F., Lewis-Wambi J.S., Jordan V.C. Structure-function relationships of estrogenic triphenylethylenes related to endoxifen and 4-hydroxytamoxifen. J. Med. Chem. 2010;53:3273–3283. doi: 10.1021/jm901907u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nam N.H. Combretastatin A-4 analogues as antimitotic antitumor agents. Curr. Med. Chem. 2003;10:1697–1722. doi: 10.2174/0929867033457151. [DOI] [PubMed] [Google Scholar]
- 52.Tron G.C., Pirali T., Sorba G., Pagliai F., Busacca S., Genazzani A.A. Medicinal chemistry of combretastatin a4: Present and future directions. J. Med. Chem. 2006;49:3033–3044. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- 53.Parihar S., Kumar A., Chaturvedi A.K., Sachan N.K., Luqman S., Changkija B., Manohar M., Prakash O., Chanda D., Khan F., et al. Synthesis of combretastatin a4 analogues on steroidal framework and their anti-breast cancer activity. J. Steroid Biochem. Mol. Biol. 2013;137:332–344. doi: 10.1016/j.jsbmb.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 54.Cushman M., Nagarathnam D., Gopal D., Chakraborti A.K., Lin C.M., Hamel E. Synthesis and evaluation of stilbene and dihydrostilbene derivatives as potential anticancer agents that inhibit tubulin polymerization. J. Med. Chem. 1991;34:2579–2588. doi: 10.1021/jm00112a036. [DOI] [PubMed] [Google Scholar]
- 55.Cushman M., Nagarathnam D., Gopal D., He H.M., Lin C.M., Hamel E. Synthesis and evaluation of analogues of (Z)-1-(4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)ethene as potential cytotoxic and antimitotic agents. J. Med. Chem. 1992;35:2293–2306. doi: 10.1021/jm00090a021. [DOI] [PubMed] [Google Scholar]
- 56.Pinney K.G., Mejia M.P., Villalobos V.M., Rosenquist B.E., Pettit G.R., Verdier-Pinard P., Hamel E. Synthesis and biological evaluation of aryl azide derivatives of combretastatin a-4 as molecular probes for tubulin. Bioorgan. Med. Chem. 2000;8:2417–2425. doi: 10.1016/S0968-0896(00)00176-0. [DOI] [PubMed] [Google Scholar]
- 57.Lawrence N.J., Hepworth L.A., Rennison D., McGown A.T., Hadfield J.A. Synthesis and anticancer activity of fluorinated analogues of combretastatin A-4. J. Fluor. Chem. 2003;123:101–108. doi: 10.1016/S0022-1139(03)00117-9. [DOI] [Google Scholar]
- 58.Pettit G.R., Rhodes M.R., Herald D.L., Hamel E., Schmidt J.M., Pettit R.K. Antineoplastic agents. 445. Synthesis and evaluation of structural modifications of (Z)- and (E)-combretastatin A-41. J. Med. Chem. 2005;48:4087–4099. doi: 10.1021/jm0205797. [DOI] [PubMed] [Google Scholar]
- 59.Flynn B.L., Flynn G.P., Hamel E., Jung M.K. The synthesis and tubulin binding activity of thiophene-based analogues of combretastatin A-4. Bioorgan. Med. Chem. Lett. 2001;11:2341–2343. doi: 10.1016/S0960-894X(01)00436-X. [DOI] [PubMed] [Google Scholar]
- 60.Maya A.B., del Rey B., de Clairac R.P.L., Caballero E., Barasoain I., Andreu J.M., Medarde M. Design, synthesis and cytotoxic activities of naphthyl analogues of combretastatin A-4. Bioorgan. Med. Chem. Lett. 2000;10:2549–2551. doi: 10.1016/S0960-894X(00)00506-0. [DOI] [PubMed] [Google Scholar]
- 61.Perez-Melero C., Maya A.B., del Rey B., Pelaez R., Caballero E., Medarde M. A new family of quinoline and quinoxaline analogues of combretastatins. Bioorgan. Med. Chem. Lett. 2004;14:3771–3774. doi: 10.1016/j.bmcl.2004.04.098. [DOI] [PubMed] [Google Scholar]
- 62.Maya A.B., Perez-Melero C., Mateo C., Alonso D., Fernandez J.L., Gajate C., Mollinedo F., Pelaez R., Caballero E., Medarde M. Further naphthylcombretastatins. An investigation on the role of the naphthalene moiety. J. Med. Chem. 2005;48:556–568. doi: 10.1021/jm0310737. [DOI] [PubMed] [Google Scholar]
- 63.Sanchez Maya A.B., Perez-Melero C., Salvador N., Pelaez R., Caballero E., Medarde M. New naphthylcombretastatins. Modifications on the ethylene bridge. Bioorgan. Med. Chem. 2005;13:2097–2107. doi: 10.1016/j.bmc.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 64.Olszewski J.D., Marshalla M., Sabat M., Sundberg R.J. Potential photoaffinity labels for tubulin—Synthesis and evaluation of diazocyclohexadienone and azide analogs of colchicine, combretastatin, and 3,4,5-trimethoxybiphenyl. J. Org. Chem. 1994;59:4285–4296. doi: 10.1021/jo00094a048. [DOI] [Google Scholar]
- 65.Ohsumi K., Nakagawa R., Fukuda Y., Hatanaka T., Morinaga Y., Nihei Y., Ohishi K., Suga Y., Akiyama Y., Tsuji T. Novel combretastatin analogues effective against murine solid tumors: Design and structure-activity relationships. J. Med. Chem. 1998;41:3022–3032. doi: 10.1021/jm980101w. [DOI] [PubMed] [Google Scholar]
- 66.Bidlack J.M., Lockshin R.A. Evolution of LDH isozymes during programmed cell death. Comp. Biochem. Physiol. B. 1976;55:161–166. doi: 10.1016/0305-0491(76)90223-6. [DOI] [PubMed] [Google Scholar]
- 67.Mor G., Sapi E., Abrahams V.M., Rutherford T., Song J., Hao X.Y., Muzaffar S., Kohen F. Interaction of the estrogen receptors with the Fas ligand promoter in human monocytes. J. Immunol. 2003;170:114–122. doi: 10.4049/jimmunol.170.1.114. [DOI] [PubMed] [Google Scholar]
- 68.Dark G.G., Hill S.A., Prise V.E., Tozer G.M., Pettit G.R., Chaplin D.J. Combretastatin A-4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer Res. 1997;57:1829–1834. [PubMed] [Google Scholar]
- 69.De Martino G., La Regina G., Coluccia A., Edler M.C., Barbera M.C., Brancale A., Wilcox E., Hamel E., Artico M., Silvestri R. Arylthioindoles, potent inhibitors of tubulin polymerization. J. Med. Chem. 2004;47:6120–6123. doi: 10.1021/jm049360d. [DOI] [PubMed] [Google Scholar]
- 70.Johnson M.D., Zuo H., Lee K.H., Trebley J.P., Rae J.M., Weatherman R.V., Desta Z., Flockhart D.A., Skaar T.C. Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Res. Treat. 2004;85:151–159. doi: 10.1023/B:BREA.0000025406.31193.e8. [DOI] [PubMed] [Google Scholar]
- 71.Shoemaker R.H. The NCI 60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 72.National Cancer Institute; Bethesda, MD, USA: [(accessed on 21 March 2016)]. Welcome to the Developmental Therapeutics Program. Available online: http://dtp.Cancer.Gov. [Google Scholar]
- 73.National Cancer Institute; Bethesda, MD, USA: [(accessed on 21 March 2016)]. National Cancer Institute Biological Testing Branch. Available online: https://dtp.Nci.Nih.Gov/branches/btb/hfa.Html. [Google Scholar]
- 74.Leese M.P., Leblond B., Smith A., Newman S.P., Di Fiore A., De Simone G., Supuran C.T., Purohit A., Reed M.J., Potter B.V. 2-substituted estradiol bis-sulfamates, multitargeted antitumor agents: Synthesis, in vitro sar, protein crystallography, and in vivo activity. J. Med. Chem. 2006;49:7683–7696. doi: 10.1021/jm060705x. [DOI] [PubMed] [Google Scholar]
- 75.Calvert A.H. Fishing for new drugs. J. Clin. Oncol. 2005;23:7780–7782. doi: 10.1200/JCO.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 76.Cuendet M., Pezzuto J.M. Antitumor activity of bruceantin: An old drug with new promise. J. Nat. Prod. 2004;67:269–272. doi: 10.1021/np030304+. [DOI] [PubMed] [Google Scholar]
- 77.McLeod H.L., Murray L.S., Wanders J., Setanoians A., Graham M.A., Pavlidis N., Heinrich B., ten Bokkel Huinink W.W., Wagener D.J., Aamdal S., et al. Multicentre phase II pharmacological evaluation of rhizoxin. Eortc early clinical studies (ECSG)/pharmacology and molecular mechanisms (PAMM) groups. Br. J. Cancer. 1996;74:1944–1948. doi: 10.1038/bjc.1996.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsuruo T., Oh-hara T., Iida H., Tsukagoshi S., Sato Z., Matsuda I., Iwasaki S., Okuda S., Shimizu F., Sasagawa K., et al. Rhizoxin, a macrocyclic lactone antibiotic, as a new antitumor agent against human and murine tumor cells and their vincristine-resistant sublines. Cancer Res. 1986;46:381–385. [PubMed] [Google Scholar]
- 79.Wurtz J.M., Bourguet W., Renaud J.P., Vivat V., Chambon P., Moras D., Gronemeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nat. Struct. Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- 80.Brzozowski A.M., Pike A.C., Dauter Z., Hubbard R.E., Bonn T., Engstrom O., Ohman L., Greene G.L., Gustafsson J.A., Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 81.Pike A.C.W. Lessons learnt from structural studies of the estrogen receptor. Best Pract. Res. Clin. Endocrinol. Metab. 2006;20:1–14. doi: 10.1016/j.beem.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 82.Marvin Version 4.1, Chemaxon. [(accessed on 28 April 2016)]. Available online: http://www.Chemaxon.Com/
- 83.Accelyrs Software, Inc.; San Diego, CA, USA: [(accessed on 26 April 2016)]. Ds Visualizer Version 1.7. Available online: http://accelrys.Com/products/discovery-studio/visualization/ [Google Scholar]